

Abstract
The release of neurotransmitters, neurotrophins, and neuropeptides is modulated by Ca2+ mobilization from the endoplasmic reticulum (ER) and activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII). Furthermore, when neuronal cultures are subjected to prolonged depolarization, presynaptic CaMKII redistributes from the cytoplasm to accumulate near active zones (AZs), a process that is reminiscent of CaMKII translocation to the postsynaptic side of the synapse. However, it is not known how presynaptic CaMKII activation and translocation depend on neuronal activity and ER Ca2+ release. Here these issues are addressed in Drosophila motoneuron terminals by imaging a fluorescent reporter of CaMKII activity and subcellular distribution. We report that neuronal excitation acts with ER Ca2+ stores to induce CaMKII activation and translocation to a subset of AZs. Surprisingly, activation is slow, reflecting T286 autophosphorylation and the function of presynaptic ER ryanodine receptors (RyRs) and inositol trisphosphate receptors (IP3Rs). Furthermore, translocation is not simply proportional to CaMKII activity, as T286 autophosphorylation promotes activation, but does not affect translocation. In contrast, RNA interference-induced knockdown of the AZ scaffold protein Bruchpilot disrupts CaMKII translocation without affecting activation. Finally, RyRs comparably stimulate both activation and translocation, but IP3Rs preferentially promote translocation. Thus, Ca2+ provided by different presynaptic ER Ca2+ release channels is not equivalent. These results suggest that presynaptic CaMKII activation depends on autophosphorylation and global Ca2+ in the terminal, while translocation to AZs requires Ca2+ microdomains generated by IP3Rs.
Introduction
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is implicated in learning, memory, and synaptic plasticity. These effects may involve T286 autophosphorylation, which generates autonomous Ca2+-independent activity, and postsynaptic translocation to interact with target proteins (Merrill et al., 2005). In addition, CaMKII regulates presynaptic excitability, development, vesicle dynamics, and neurosecretion (Griffith et al., 1994; Wang et al., 1994; Shakiryanova et al., 2007; Wang, 2008; de Jong and Verhage, 2009; Wong et al., 2009; Carrillo et al., 2010). CaMKII-regulated release of classical neurotransmitters, neuropeptides, and neurotrophins depends on Ca2+ release from endoplasmic reticulum (ER) (He et al., 2000; Jin and Hawkins, 2003; Kubota et al., 2005; Kolarow et al., 2007; Shakiryanova et al., 2007). However, the basis of this finding remains unclear because the effect of ER Ca2+ release on CaMKII activity in the nerve terminal has not been measured directly. Presynaptic regulation may also involve CaMKII redistribution, as electron microscopy reveals CaMKII accumulation near active zones (AZs) following prolonged K+-induced depolarization of neuronal cultures (Tao-Cheng et al., 2006). However, it is not known whether this presynaptic translocation occurs in native nerve terminals during physiological activity. Determining how presynaptic CaMKII activation and translocation depend on nerve activity and ER Ca2+ release requires a method for measuring CaMKII catalytic activity and localization in the living nerve terminal.
CaMKII activity has been detected optically in the neuronal soma and dendrites with a CaMKII reporter called Camui (Takao et al., 2005). Camui is a fluorescence resonance energy transfer (FRET)-based indicator that reports the transition from the basal state to the activated conformation as an increase in CFP/YFP (yellow fluorescent protein) ratio (Takao et al., 2005). In principle, because GFP-tagged CaMKII reports postsynaptic CaMKII translocation (Merrill et al., 2005), Camui might report presynaptic translocation as well as activation. Therefore, we set out to image Camui in a living nerve terminal amenable to optical studies. The Drosophila larval neuromuscular junction (NMJ) features large (i.e., >2 μm in diameter) type Ib synaptic boutons in which neuropeptide-containing dense-core vesicles (DCVs) and FRET signaling indicators have been resolved optically (Levitan et al., 2007). Furthermore, presynaptic DCV mobility, accumulation, and exocytosis are regulated by ER Ca2+ release by ryanodine receptors (RyRs) and CaMKII (Shakiryanova et al., 2007; Wong et al., 2009). Although most CaMKII at the larval NMJ is postsynaptic (Koh et al., 1999), it is easy in Drosophila to target expression of transgenic FRET indicators specifically to neurons so that signaling in the nerve terminal can be followed in real time. Therefore, we hypothesized that the Drosophila NMJ would be well suited for studying the effect of activity on presynaptic CaMKII using the Camui FRET indicator.
Here Camui is expressed and imaged in Drosophila motoneuron type Ib synaptic boutons. Consistent with predictions from previous findings, electrical activity and ER Ca2+ release stimulate CaMKII activation. Furthermore, CaMKII translocation from the cytoplasm to accumulate near AZs is detected in response to nerve stimulation and endogenous nerve activity within the intact animal. Finally, it is shown that CaMKII activation and translocation in the nerve terminal are subject to differential control by autophosphorylation, Bruchpilot (BRP) levels, and ER Ca2+ channels.
Materials and Methods
Flies.
Camui (Takao et al., 2005) was excised by NheI and XbaI from the plasmid kindly provided by Dr. Y. Hayashi (Massachusetts Institute of Technology, Cambridge, MA) and inserted at the XbaI site of pUAST. Mutations (T286A, T305/306D) were introduced by PCR. Expression of the UAS constructs in transgenic animals was induced with the pan-neuronal driver elav-gal4 or the motoneuron driver ok6-gal4. RNA interference (RNAi) lines, which were crossed with the sensor-expressing flies, were obtained from the TRiP (Transgenic RNAi Project) stock center [inositol trisphosphate receptor (IP3R), 25937; BRP, 25891], the Vienna Drosophila RNAi Center (BRP, 107748; RyR, 109631), and the National Institute of Genetics of Japan (RyR, 10844R-3). Flies expressing strawberry fluorescent protein-tagged BRP-short have been described previously (Schmid et al., 2008).
Imaging.
Single-plane-of-focus wide-field imaging of type Ib boutons located on muscles 6 and 7 of filleted wandering third instar larvae of either sex was performed in glutamate-supplemented HL3 (containing, in mm: 70 NaCl, 5 KCl, 1.5 CaCl2, 20 MgCl2, 10 NaHCO3, 5 trehalose, 115 sucrose, 10 sodium glutamate, and 5 sodium HEPES, pH 7.2) with an Olympus 1.1 numerical aperture 60× water-immersion objective. The depth of field of this objective ensures that most of the bouton volume is in focus and that the rest of the bouton will be only slightly out of focus. This is evident from our measurement of Nc82-labeled AZs, which is comparable to that obtained with stacks of confocal images (Kim et al., 2009). FRET measurements of CaMKII activity (i.e., the CFP/YFP ratio), which were performed every 3 s, used a 440DF20 excitation filter, a 455DRLP dichroic mirror, and alternating 480DF30 (for CFP) and 535DF25 (for YFP) emission filters on a filter wheel (Sutter Instruments) as described previously (Shakiryanova and Levitan, 2008). To follow translocation without ratiometric FRET assays, Camui was imaged with standard fluorescein optics every 3 s. Fura-2 ratiometric Ca2+ measurements were performed as described previously (Chouhan et al., 2010). For imaging in the living intact animal, a larva was placed in a drop of saline between a glass slide and a coverslip with cut coverslip spacers and viewed with a 1.45 numerical aperture oil-immersion 60× objective using fluorescein optics. For immunostaining BRP, the semi-intact preparation was fixed with 4% paraformaldehyde for 15 min and incubated with Nc82 monoclonal antibody (1:100) in PBS with 0.3% Triton X-100 and 5% goat serum overnight at 4°C. The secondary Texas Red-labeled anti-mouse antibody was applied for 2 h at room temperature. Protocols for the application of thapsigargin (Tg), ryanodine, KN92, and KN93 have been described previously (Shakiryanova et al., 2007). Xestospongin C (Xesto) was applied in a 20 min preincubation. For experiments with two experimental conditions, the t test was used for determining statistical significance. Otherwise, statistical analysis was performed with ANOVA followed by Dunnett's post hoc test. With drugs that were dissolved in DMSO, an equivalent amount of the vehicle was used as the matched control.
Reagents.
KN-92, KN-93, xestospongin C, and ryanodine (high purity) were obtained from EMD Chemicals. Thapsigargin was obtained from Alomone Labs. Plectreurys toxin II (PLTX) was obtained from the Peptide Institute (Osaka, Japan). Dextran-conjugated Fura-2 was obtained from Invitrogen Corp.. The Nc82 monoclonal antibody was obtained from the Developmental Studies Hybridoma Bank (Iowa City, Iowa).
Results
CaMKII activation and translocation in Drosophila motoneuron terminals
Presynaptic CaMKII activation in single type Ib boutons was measured in a high-Mg2+ saline supplemented with glutamate to suppress NMJ contraction. Initially, motor nerves were stimulated at 70 Hz for 15 s, which induces CaMKII-dependent presynaptic DCV mobilization, DCV capture, and potentiation of neuropeptide release in type Ib boutons (Shakiryanova et al., 2005, 2006, 2007; Wong et al., 2009). This stimulus increased the CFP/YFP ratio, which reports CaMKII activity (Takao et al., 2005), throughout the bouton (Fig. 1A). CaMKII activation was inhibited by the CaMKII-specific calmodulin-binding site inhibitor KN93, but not the inactive analog KN92 (Fig. 1B). The calmodulin site was further implicated by showing that the Camui T305/306D mutant, which mimics inhibitory phosphorylation of the calmodulin-binding site (Colbran and Soderling, 1990), abolished the response (Fig. 1B). Finally, PLTX, a blocker of presynaptic voltage-gated Ca2+ (Cav) channels (Kuromi et al., 2010), also abolished CaMKII activation (Fig. 1B). Thus, activity-evoked Ca2+ influx through Cav channels induces calmodulin activation of presynaptic CaMKII.
Figure 1.

Activation of CaMKII in synaptic boutons. A, Pseudo-color image showing the change in the CFP/YFP FRET ratio in a type Ib bouton induced by nerve stimulation for 15 s at 70 Hz. An increase in CaMKII activity is shown as a shift to warmer colors. Scale bar, 2 μm. B, Quantification of FRET responses to stimulation for 15 s at 70 Hz in control boutons (Con) (n = 8), control boutons treated with 10 μm KN92 (n = 5), 10 μm KN93 (n = 9), or 20 nm PLTX (n = 6), and in T305/306D mutants (n = 6). *p < 0.05, ***p < 0.001. C, FRET responses to 70 Hz nerve stimulation with 100 μm ryanodine (Ryan) (n = 6) or expression of RyR-targeted RNAi constructs RyR109631 (n = 7) and RyR10844R-3 (n = 11). Note that an increase in the plotted ratio corresponds to activation.
CaMKII-dependent regulation of peptidergic DCVs in Drosophila motoneurons requires Ca2+-induced Ca2+ release by presynaptic ER RyRs (Shakiryanova et al., 2007; Wong et al., 2009). Therefore, to determine whether Camui is an effective reporter of presynaptic CaMKII activity in Drosophila motoneuron terminals, RyRs were inhibited. Null mutants could not be used because of lethality. Therefore, RyRs were targeted by neuronal expression of two RNAi constructs. Also, to further exclude off-target and developmental effects, RyRs were inhibited acutely by applying ryanodine, which binds to both mammalian and Drosophila RyRs (Vázquez-Martínez et al., 2003). With each of these independent means of inhibiting RyRs, CaMKII did not fully activate (Fig. 1C). Thus, in accordance with indirect studies, Camui verified that presynaptic CaMKII is activated by surface and ER Ca2+ channels.
In vitro ultrastructure experiments have shown that native CaMKII translocates toward AZs in response to prolonged depolarization (Tao-Cheng et al., 2006). Because this stimulus can mimic tetanic stimulation, Camui was imaged as motor nerves were stimulated at 70 Hz for 1 min. This produced Camui redistribution into puncta, which were visualized with fluorescein optics after fixing the NMJ and immunostaining for the AZ marker BRP with the Nc82 monoclonal antibody (Fig. 2A, Camui in green, Nc82 in magenta). The 1.46 ± 0.35 Camui puncta per bouton (n = 21 boutons from 3 preparations) were always located near a subset of the 11.45 ± 0.32 AZs per bouton (n = 191 boutons from 22 preparations) (Fig. 2A). However, because Camui puncta often did not persist during fixation, puncta formation was also monitored with fluorescein optics in living terminals from animals also expressing the strawberry fluorescent protein-tagged BRP-short construct (Schmid et al., 2008), which labeled 10.35 ± 0.73 AZs per bouton, n = 82 boutons from 13 preparations (i.e., ∼10% fewer than detected with Nc82). In the absence of fixation, more CaMKII clusters were evident: the maximum number of Camui puncta per bouton (Cmax) was 3.31 ± 0.35 (n = 59 boutons from 11 preparations). Furthermore, 85% of puncta (i.e., 55 of 65) were located near strawberry-labeled AZs (Fig. 2B). Given the ∼90% efficiency of AZ labeling by the strawberry construct, these live imaging data are consistent with the immunofluorescence data: presynaptic CaMKII usually clusters near a subset of AZs. The agreement of Camui experiments with previous electron microscopy, along with the expected activation by surface and ER Ca2+ channels (see above), establish that the FRET indicator can be used in the Drosophila NMJ to probe the dynamics of presynaptic CaMKII activation and translocation.
Figure 2.

Localization of CaMKII clusters and active zones. A, Wide-field fluorescence images of fixed preparations showing Camui localization before (Con) and after stimulation for 60 s at 70 Hz (Stim), BRP immunofluorescence detected with Nc82 antibody, and a superposition of CaMKII (green) and BRP (magenta) signals (Merged). B, Wide-field images of a live bouton showing CaMKII indicator (green) before (Con) and after stimulation (Stim), strawberry-labeled Brp-short (magenta, BRP), and merged superposition of CaMKII and BRP signals acquired after stimulation (Merged). Scale bars, 2 μm.
Autophosphorylation promotes presynaptic CaMKII activation
We then focused on the kinetics of presynaptic CaMKII activation. Time-lapse studies revealed that, in contrast to rapid Ca2+/calmodulin activation of CaMKII in vitro (De Koninck and Schulman, 1998; Bradshaw et al., 2002), full activity-dependent stimulation of presynaptic CaMKII occurred slowly and required >30 Hz stimulation, with the rate increasing with frequency (Fig. 3A). Furthermore, CaMKII deactivation depended on the duration of stimulation: a longer bout of stimulation slowed deactivation and induced residual CaMKII activation that persisted after electrical stimulation ceased (Fig. 3B). To determine whether slow CaMKII activation and deactivation reflect presynaptic cytoplasmic Ca2+ kinetics, ratiometric Ca2+ measurements were conducted with dextran-conjugated fura-2. In contrast to CaMKII (Fig. 3C, closed circles), Ca2+ responses increased and decreased quickly (Fig. 3C, gray trace). Rapid Ca2+ kinetics was also evident in experiments with transgenic expression of TN-XXL and GCaMP3 Ca2+ indicators (data not shown), further verifying that Ca2+ alone does not explain the slow time course of presynaptic CaMKII activation. Therefore, the role of T286 autophosphorylation, which generates autonomously active CaMKII (Merrill et al., 2005; Wang, 2008), was examined. Replacing Camui threonine 286 with alanine (i.e., T286A) to preclude its autophosphorylation reduced CaMKII activation and made the response more transient (Fig. 3C, compare open and closed circles). Hence, presynaptic CaMKII activation is enhanced and sustained by autophosphorylation.
Figure 3.

Presynaptic CaMKII activity kinetics. A, CaMKII activation in response to stimulation at 30 Hz (n = 5), 50 Hz (n = 5), 70 Hz (n = 8), and 100 Hz (n = 7). B, Deactivation time course of CaMKII stimulated at 70 Hz for 15 s (n = 4) and 60 s (n = 8). For 15 s, τ = 5.38 s. For 60 s, τ = 8.93 s. Note persistent activity after 60 s stimulation. C, Time course of intracellular Ca2+ (gray trace shows mean from 10 experiments) and CaMKII activity in controls (closed circles, n = 8) and T286A mutants (open circles, n = 6) during 1 min of 70 Hz activity. Error bars indicate SEM.
Presynaptic CaMKII translocation and dispersal occurs in vivo
Time-lapse experiments were also used to study the kinetics of CaMKII redistribution induced with 70 Hz nerve stimulation (Fig. 4A; see Notes). While the time course of formation of individual puncta was variable, on average they became evident at 16.8 ± 2.7 s (n = 15). Puncta formation was accompanied by a decrease in the diffuse signal evident before stimulation (Fig. 4B,C; see Notes). Puncta could appear and then aggregate (Fig. 4B) or grow (Fig. 4C) near AZs. Once fully formed, puncta either were immobile or moved modestly (Fig. 4D). Thus, CaMKII translocated from the cytoplasm to ultimately reside in clusters near AZs.
Figure 4.
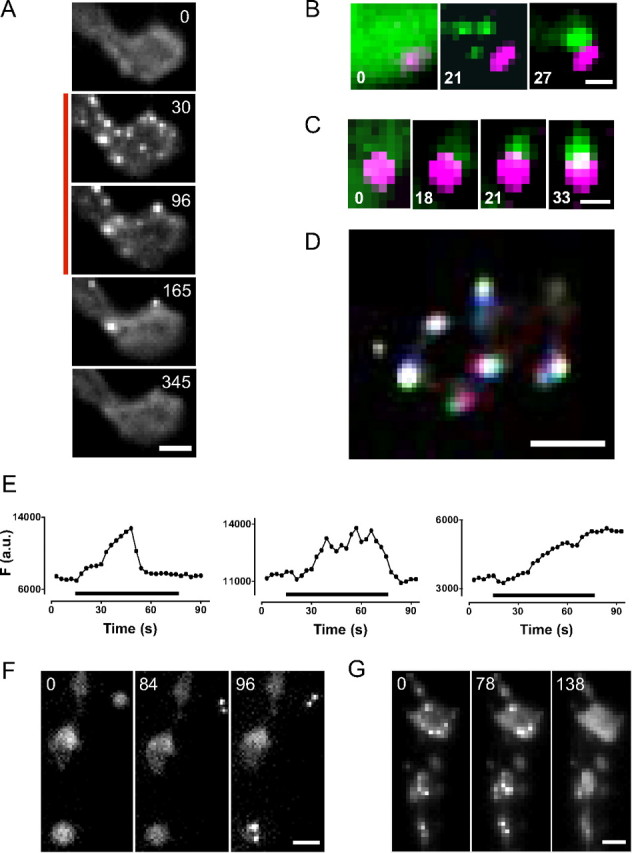
Activity-induced translocation of presynaptic CaMKII. A, Sequence of images of a type Ib bouton showing the localization of Camui before, during, and after stimulation at 70 Hz for 120 s (indicated by bar on left). Fluorescein optics were used. Scale bar, 2 μm. B, Appearance and aggregation of Camui puncta (green) adjacent to an AZ marked with the strawberry-tagged BRP-short (magenta). Scale bar, 0.5 μm. Only part of a bouton is shown. C, Appearance and growth of a Camui punctum near an AZ. Scale bar, 0.5 μm. Only part of a bouton is shown. D, Movement of Camui puncta. Three sequential time-lapse images taken 3 s apart were presented in different colors (red, green, and blue) and then superimposed so that immobile signal is white and motion is indicated by other colors. Scale bar, 2 μm. E, Time courses for three CaMKII clusters from a single bouton induced by 60 s stimulation at 70 Hz. F (a.u.), Punctum fluorescence in arbitrary units. F, G, Sequences of fluorescence images acquired through the cuticle of undissected larvae show CaMKII clustering (F) and dispersal (G) in different boutons. Scale bars, 2 μm. For all images, numbers show time in seconds.
Imaging also revealed decreases in the number of clusters (Fig. 4A). The disappearance of puncta could not be attributed to movement out of the plane of focus because the depth of field of our optics encompasses most of the bouton volume and lost puncta never reappeared. Hence, clusters must have dispersed. Interestingly, even in one bouton, dispersal kinetics could vary (Fig. 4A,E): individual clusters could disperse either during stimulation (Fig. 4E, left) when CaMKII is active, shortly after stimulation ceased (Fig. 4E, middle) when the kinase is deactivating, or long after the cessation of stimulation (i.e., clusters persisted beyond the time frame of an experiment) (Fig. 4A,E right) when kinase activity is low. Hence, presynaptic CaMKII translocation is not a simple consequence of activation.
To determine whether CaMKII translocation could be induced by physiological electrical activity, imaging was performed in live intact larvae. Viewing Camui in motoneuron boutons through the cuticle showed that presynaptic CaMKII is typically diffuse after halothane anesthesia (data not shown). In the absence of anesthesia, muscle contractions associated with attempted movement by larvae sandwiched between a slide and a coverslip made continuous imaging through the cuticle difficult. Nevertheless, time-lapse imaging between contractions generated by the animals themselves revealed formation and dispersal of CaMKII clusters (Fig. 4F,G) (n = 5). Hence, the translocation of diffuse presynaptic CaMKII into puncta originally detected with nerve stimulation in high Mg2+ in the semi-intact preparation occurs in vivo in intact animals with endogenously generated electrical activity.
Divergent roles of RyRs and autophosphorylation in CaMKII translocation
Because FRET measurements showed that RyRs and autophosphorylation promote CaMKII activation (Figs. 1C, 3C), the effects of RyRs and autophosphorylation on translocation during a 1 min bout of 70 Hz stimulation were examined by quantifying maximum number of fully formed clusters (Cmax), a parameter that can be measured reliably despite the variation in the dynamics of puncta formation and dispersal. Inhibiting RyRs with ryanodine or presynaptic expression of RyR RNAi reduced CaMKII translocation (Fig. 5A). Interestingly, this reduction was comparable to the effect on activation, which was measured at the peak for controls (i.e., at 33 s) (Fig. 5A, compare open and closed bars). Indeed, plotting Cmax versus FRET showed that the data obtained with RyR inhibition (blue circles) fell within the 95% confidence limits (dashed lines) for a linear regression line derived from controls (Con), DMSO, and the inactive analog KN92, and conditions that yielded inactive CaMKII [unstimulated (Unstim) and PLTX] (Fig. 5B). Therefore, while RyRs are not absolutely required for the effects of electrical activity on presynaptic CaMKII, presynaptic CaMKII activation and translocation are proportionally facilitated by RyRs.
Figure 5.
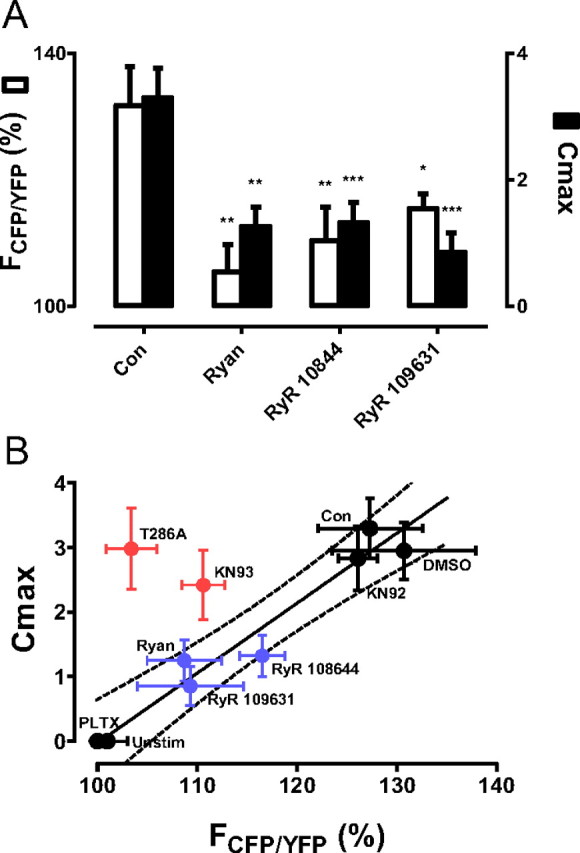
Presynaptic CaMKII regulation by the RyR and T286 autophosphorylation. A, FRET responses (open bars) after 33 s of 70 Hz stimulation, which produces peak activation in control boutons (Con) (n = 8), for boutons treated with 100 μm ryanodine (Ryan) (n = 6) and boutons expressing RyR-targeted RNAi constructs RyR 109631 (n = 7), and RyR 10844 (n = 11) are compared with Cmax (closed bars) for Con (n = 11), Ryan (n = 8), RyR 109631 (n = 7), and RyR 10844 (n = 12). *p < 0.05, **p < 0.01, ***p < 0.001. B, Cmax is plotted versus FRET at 30 s. The line and 95% confidence limits (dashed lines) were generated by linear regression between Con, DMSO, and KN92 data points (upper right black circles) and unstimulated (Unstim) and PLTX-treated data points (lower left black circles). Red circles show data from T286A and KN93, which inhibit autophosphorylation. Note that inhibiting RyRs (blue circles) produces results that fall near the line, but that inhibiting autophosphorylation produces results in the upper left quadrant beyond the confidence limits.
In contrast, while autophosphorylation promoted CaMKII activation (Fig. 3C), translocation did not depend on autophosphorylation. This was evident in experiments in which translocation was quantified in the presence of KN93, which reduces kinase activation and hence autophosphorylation, its inactive analog KN92, and the T286A mutation, which precludes autophosphorylation. Even though KN93 and T286A reduce activation (Fig. 1B, 3C), neither significantly affected Cmax compared with control or KN92 (p = 0.72, ANOVA). The preferential effect of autophosphorylation on activation is evident on the Cmax versus FRET plot, where KN93 and T286A results (red circles) appear beyond the 95% confidence limits of the control line in the upper left quadrant (Fig. 5B).
Inhibition of CaMKII translocation by BRP knockdown
Preferential regulation of translocation without altering activation is also possible. This type of result was found in experiments probing whether BRP is more than a simple marker of CaMKII translocation. Because the attempt to recombine the Camui insert on the second chromosome with a BRP mutant was unsuccessful, RNAi expression in neurons was used to target BRP. Immunostaining confirmed that two different RNAi constructs decreased BRP protein in type Ib boutons without noticeably affecting bouton size (Fig. 6A). Despite this knockdown, CaMKII activation was preserved: neither construct produced a statistically significant effect on FRET (Fig. 6B, white bars). Yet, CaMKII translocation was nearly abolished (Fig. 6B, black bars). The similar results obtained with two different BRP RNAi constructs minimizes the probability of an off-target effect, suggesting that BRP specifically influences CaMKII translocation. More generally, these results establish that CaMKII activation can occur normally even when translocation to AZs is disrupted.
Figure 6.
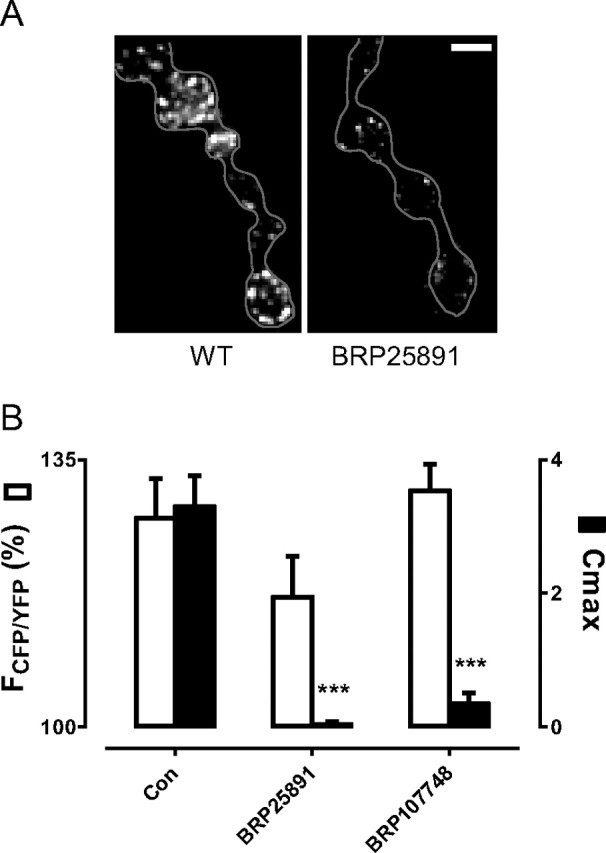
Inhibition of activity-induced CaMKII translocation by RNAi-induced knockdown of BRP. A, BRP immunofluorescence in control boutons and boutons expressing RNAi BRP 25891. Similar results were obtained with BRP 107748. Scale bar, 2 μm. Gray line shows terminal profile. B, FRET responses at 30 s (white bars) and Cmax (black bars) in control boutons (Con, n = 8) and boutons expressing BRP RNAi constructs BRP25891 (n = 9) and BRP107748 (n = 8). ***p < 0.001. FRET responses did not change significantly with BRP knockdown, but Cmax was reduced significantly.
IP3Rs preferentially induce presynaptic CaMKII translocation
To date, RyRs have fully accounted for the participation of the ER Ca2+ in presynaptic cGMP signaling and regulation of DCV mobility, exocytosis, and capture at the Drosophila NMJ (Shakiryanova et al., 2007; Shakiryanova and Levitan, 2008; Wong et al., 2009). These precedents predict that depleting ER Ca2+ stores with the sarco/endoplasmic reticulum Ca2+-ATPase inhibitor Tg would be equivalent to inhibiting RyRs. However, with Tg, activation was inhibited to a greater extent and CaMKII translocation was essentially abolished (Fig. 7A, closed circles; B, bars labeled Tg). The dramatic response to ER Ca2+ depletion when compared with RyR inhibition suggests that an ER Ca2+ release channel in addition to RyRs influences presynaptic CaMKII. Therefore, the role of IP3R, which functions in type Ib boutons (Klose et al., 2010), was investigated.
Figure 7.

Regulation of CaMKII activation and translocation by IP3Rs. A, FRET responses in the presence of 0.1% DMSO (n = 8), 20 μm thapsigargin in DMSO (Tg, n = 5), 0.1 μm xestospongin C in DMSO (Xesto, n = 9), or with neuronal expression of IP3R RNAi (n = 10). B, FRET induced by 30 s of stimulation at 70 Hz (white bars) is compared with Cmax. *p < 0.05, ***p < 0.001. C, Cmax versus FRET plot showing that IP3R inhibition (shaded triangles) or BRP knockdown (open circles) produces results that fall in the lower right quadrant. Thus, IP3Rs and BRP preferentially affect CaMKII translocation.
Because of the lethality of IP3R-null mutants, neuronally expressed RNAi was used to genetically test for a role of presynaptic IP3Rs and acute application of a specific inhibitor of the Drosophila IP3R, Xesto (Vázquez-Martínez et al., 2003) was used to preclude off-target and developmental effects. Each of these independent approaches for reducing IP3R activity lowered FRET responses, particularly at later times (Fig. 7A). Thus, the impact of IP3Rs on activation, both in terms of magnitude and time course, is reminiscent of RyRs. Hence, Ca2+ provided from each of these ER Ca2+ channels contributes to presynaptic CaMKII activation. However, inhibiting IP3Rs nearly eliminated CaMKII translocation (Fig. 7B). This preferential effect is obvious on a Cmax versus FRET plot (Fig. 7C), where Xesto and the IP3R RNAi data (shaded triangles) appear in the lower right quadrant beyond the 95% confidence limits of the line along with the effects of BRP knockdown (open circles). Thus, in contrast to Ca2+ supplied by RyRs, Ca2+ supplied by IP3Rs preferentially induces CaMKII translocation toward AZs.
Discussion
Imaging presynaptic CaMKII activation and translocation in living terminals
Although CaMKII alters nerve terminal excitability, neurosecretion, and development, direct studies of CaMKII activation and translocation in living nerve terminals are lacking because of technical limitations. For example, the low level of CaMKII in the nerve terminal compared with the postsynaptic density, as well as the small size of most boutons, hinders light level in immunohistochemical assays of presynaptic CaMKII translocation. Electron microscopy coupled with immunodetection overcomes limitations in spatial resolution, but cannot follow CaMKII dynamics in a living preparation. Thus, the dependence of presynaptic CaMKII enzymatic activity and translocation on stimulation frequency, ER Ca2+ channels, and autophosphorylation was unknown. Here a FRET indicator that detects both activation and translocation was shown to reproduce known features of presynaptic CaMKII behavior and then used to determine how CaMKII dynamics is controlled in an intact living nerve terminal.
Presynaptic CaMKII activation by autophosphorylation and global Ca2+
First, stimulation of presynaptic CaMKII by electrical stimulation, autophosphorylation, and ER Ca2+ channels was quantified in real time. These experiments established that CaMKII activation is surprisingly slow and frequency dependent (Fig. 3). This property likely reflects the fact that Ca2+ entry via presynaptic Cav channels alone is not optimal for activating CaMKII. Rather, stimulation of CaMKII also is promoted by presynaptic ER Ca2+ release and autophosphorylation (Figs. 1, 3, 7). The similarity of CaMKII kinetics induced by disrupting either of the latter processes suggests autophosphorylation is promoted by the increased activation of CaMKII by ER Ca2+. Furthermore, the participation of RyRs and IP3Rs (Figs. 1C, 7), the requirement for Cav channels (Fig. 1B), and the insensitivity to knockdown of BRP (Fig. 6), which enhances Cav channel clustering at AZs (Kittel et al., 2006), imply that presynaptic CaMKII activation is driven by global presynaptic Ca2+ elevation induced by ER and surface Ca2+ channels.
Compartmentalized control of CaMKII translocation toward AZs
Imaging experiments also revealed that CaMKII translocation toward AZs detected by immunoelectron microscopy in depolarized cultured neurons also occurs with native electrical activity in vivo (Fig. 4F). Experiments that used BRP immunolocalization or somewhat less efficient transgenic fluorescent protein labeling showed that presynaptic CaMKII clusters usually accumulate at AZs (Figs. 2, 4B,C). This does not reflect a simple aggregation of CaMKII already localized near AZs because CaMKII initially localized away from AZs was depleted. Furthermore, once fully formed CaMKII clusters localized near AZs, their motion was limited (Fig. 4D). Therefore, it seems that initially diffuse presynaptic CaMKII forms clusters that are tethered near AZs.
Strikingly, disrupting autophosphorylation, which reduces activation, does not affect presynaptic CaMKII translocation (Fig. 5B). This might reflect that activating a subset of the subunits in the CaMKII dodecamer is sufficient for translocation. However, translocation is not a simple consequence of limited activation because, even though CaMKII activation tends to be greater after inhibiting IP3Rs than RyRs, translocation is more markedly disrupted by IP3R inhibition (Fig. 7C). Furthermore, CaMKII clustering at AZs can long outlast stimulated elevation of presynaptic Ca2+ (compare Figs. 4A,E to 3C, red trace), implying that Ca2+ alone cannot account for persistent translocation. Finally, CaMKII translocates only to a subset of AZs, suggesting heterogeneity between AZs. Differential release between AZs in a single type Ib bouton has recently been demonstrated (Peled and Isacoff, 2011). Hence, it is possible that a single mechanism induces heterogeneity in AZ-mediated transmitter release and recruitment of CaMKII.
Our results suggest that presynaptic CaMKII translocation is preferentially induced by IP3R-generated Ca2+ microdomains located at a subset of AZs (Fig. 8). Consistent with this proposal, CaMKII translocation requires the AZ-associated scaffold protein BRP, which is long enough to separately integrate signals from Ca2+ microdomains generated by AZ Cav channel clusters at its C terminus and IP3Rs potentially located at its N terminus. BRP might affect CaMKII translocation also because it enhances Cav channel clustering (Kittel et al., 2006) and it is a potential CaMKII substrate (Obenauer et al., 2003). In contrast, RyRs may be positioned further away from BRP so that their local Ca2+ release cannot mimic the action of IP3Rs (Fig. 8). Such a differential distribution of ER Ca2+ channels has not been studied in Drosophila nerve terminals, but is consistent with independent localization and function of RyRs and IP3Rs detected in other cell types and neuronal compartments (Blaustein and Golovina, 2001). According to this view, diffuse CaMKII samples global Ca2+ changes induced by Cav and all ER Ca2+ channels to activate, but local Ca2+ changes generated by IP3Rs are needed for recruitment of CaMKII. With such a model, depletion of the IP3-sensitive Ca2+ pool would account for the observed dispersal of clusters that sometimes occurs during stimulation (Fig. 4A,E).
Figure 8.

Model of CaMKII activation and translocation in the nerve terminal. Global Ca2+ from Cav channels, IP3Rs, and RyRs combine to activate presynaptic CaMKII (red arrows). Then, CaMKII dodecamers cluster near BRP-containing AZs (black arrows), where Ca2+ microdomains from Cav channels and ER IP3Rs are integrated.
Our experiments suggest that translocation is limited to a subset of AZs and is delayed compared with CaMKII activation and synaptic transmission. Thus, it is unlikely that translocation occurs for rapid acute regulation of transmitter release. Indeed, the protocols typically used for quantal analysis (e.g., low-frequency action potentials in the presence of low Ca2+) are unlikely to induce much CaMKII signaling. However, motor neurons fire in bursts that in vivo imaging experiments show are sufficient to translocate presynaptic CaMKII (Fig. 4F). Thus, future experiments will have to use more physiological stimuli to probe whether CaMKII is recruited to AZs to regulate synaptic transmission. Such effects could be selective, possibly because CaMKII is recruited to AZs that are most active and/or positioned near ER IP3Rs (Fig. 8). Interestingly, another consequence of translocation to AZs is that CaMKII is depleted from the presynaptic cytoplasm. Hence, CaMKII activity can be redirected away from substrates that are excluded from AZs (e.g., neuropeptide vesicles and K+ channels) to substrates preferentially localized at AZs. In this way, the role of CaMKII in the nerve terminal changes dynamically depending both on its enzymatic activity and location.
Notes
Supplemental material for this article is available at http://www.pharmacology.us/LevitanVideo. Activity-dependent clustering of CaMKII. Time-lapse movie acquired with fluorescein optics with an interval of 3 s between frames. A 70 Hz nerve stimulation induces diffuse Camui to form puncta in type Ib boutons. This material has not been peer reviewed.
Footnotes
This work was funded by NIH Grants R01 NS32385 to E.S.L. and R01 NS061914 to G.T.M. We thank Dr. Yasunori Hayashi (Massachusetts Institute of Technology, Cambridge, MA) for the FRET sensor plasmid.
References
- Blaustein MP, Golovina VA. Structural complexity and functional diversity of endoplasmic reticulum Ca2+ stores. Trends Neurosci. 2001;24:602–608. doi: 10.1016/s0166-2236(00)01891-9. [DOI] [PubMed] [Google Scholar]
- Bradshaw JM, Hudmon A, Schulman H. Chemical quenched flow kinetic studies indicate an intraholoenzyme autophosphorylation mechanism for Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2002;277:20991–20998. doi: 10.1074/jbc.M202154200. [DOI] [PubMed] [Google Scholar]
- Carrillo RA, Olsen DP, Yoon KS, Keshishian H. Presynaptic activity and CaMKII modulate retrograde semaphorin signaling and synaptic refinement. Neuron. 2010;68:32–44. doi: 10.1016/j.neuron.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouhan AK, Zhang J, Zinsmaier KE, Macleod GT. Presynaptic mitochondria in functionally different motor neurons exhibit similar affinities for Ca2+ but exert little influence as Ca2+ buffers at nerve firing rates in situ. J Neurosci. 2010;30:1869–1881. doi: 10.1523/JNEUROSCI.4701-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran RJ, Soderling TR. Calcium/calmodulin-independent autophosphorylation sites of calcium/calmodulin-dependent protein kinase II. Studies on the effect of phosphorylation of threonine 305/306 and serine 314 on calmodulin binding using synthetic peptides. J Biol Chem. 1990;265:11213–11219. [PubMed] [Google Scholar]
- de Jong AP, Verhage M. Presynaptic signal transduction pathways that modulate synaptic transmission. Curr Opin Neurobiol. 2009;19:245–253. doi: 10.1016/j.conb.2009.06.005. [DOI] [PubMed] [Google Scholar]
- De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- Griffith LC, Wang J, Zhong Y, Wu CF, Greenspan RJ. Calcium/calmodulin-dependent protein kinase II and potassium channel subunit eag similarly affect plasticity in Drosophila. Proc Natl Acad Sci U S A. 1994;91:10044–10048. doi: 10.1073/pnas.91.21.10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Yang F, Xie Z, Lu B. Intracellular Ca2+ and Ca2+/calmodulin-dependent kinase II mediate acute potentiation of neurotransmitter release by neurotrophin-3. J Cell Biol. 2000;149:783–792. doi: 10.1083/jcb.149.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin I, Hawkins RD. Presynaptic and postsynaptic mechanisms of a novel form of homosynaptic potentiation at Aplysia sensory-motor neuron synapses. J Neurosci. 2003;23:7288–7297. doi: 10.1523/JNEUROSCI.23-19-07288.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Kumar V, Lin YQ, Karunanithi S, Ramaswami M. Fos and Jun potentiate individual release sites and mobilize the reserve synaptic vesicle pool at the Drosophila larval motor synapse. Proc Natl Acad Sci U S A. 2009;106:4000–4005. doi: 10.1073/pnas.0806064106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A, Wagh DA, Pawlu C, Kellner RR, Willig KI, Hell SW, Buchner E, Heckmann M, Sigrist SJ. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science. 2006;312:1051–1054. doi: 10.1126/science.1126308. [DOI] [PubMed] [Google Scholar]
- Klose MK, Dason JS, Atwood HL, Boulianne GL, Mercier AJ. Peptide-induced modulation of synaptic transmission and escape response in Drosophila requires two G-protein-coupled receptors. J Neurosci. 2010;30:14724–14734. doi: 10.1523/JNEUROSCI.3612-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YH, Popova E, Thomas U, Griffith LC, Budnik V. Regulation of DLG localization at synapses by CaMKII-dependent phosphorylation. Cell. 1999;98:353–363. doi: 10.1016/s0092-8674(00)81964-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarow R, Brigadski T, Lessmann V. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J Neurosci. 2007;27:10350–10364. doi: 10.1523/JNEUROSCI.0692-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota M, Narita K, Murayama T, Suzuki S, Soga S, Usukura J, Ogawa Y, Kuba K. Type-3 ryanodine receptor involved in Ca2+-induced Ca2+ release and transmitter exocytosis at frog motor nerve terminals. Cell Calcium. 2005;38:557–567. doi: 10.1016/j.ceca.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Ueno K, Kidokoro Y. Two types of Ca2+ channel linked to two endocytic pathways coordinately maintain synaptic transmission at the Drosophila synapse. Eur J Neurosci. 2010;32:335–346. doi: 10.1111/j.1460-9568.2010.07300.x. [DOI] [PubMed] [Google Scholar]
- Levitan ES, Lanni F, Shakiryanova D. In vivo imaging of vesicle motion and release at the Drosophila neuromuscular junction. Nat Protoc. 2007;2:1117–1125. doi: 10.1038/nprot.2007.142. [DOI] [PubMed] [Google Scholar]
- Merrill MA, Chen Y, Strack S, Hell JW. Activity-driven postsynaptic translocation of CaMKII. Trends Pharmacol Sci. 2005;26:645–653. doi: 10.1016/j.tips.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled ES, Isacoff EY. Optical quantal analysis of synaptic transmission in wild-type and rab3-mutant Drosophila motor axons. Nat Neurosci. 2011;14:519–526. doi: 10.1038/nn.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A, Hallermann S, Kittel RJ, Khorramshahi O, Frölich AM, Quentin C, Rasse TM, Mertel S, Heckmann M, Sigrist SJ. Activity-dependent site-specific changes of glutamate receptor composition in vivo. Nat Neurosci. 2008;11:659–666. doi: 10.1038/nn.2122. [DOI] [PubMed] [Google Scholar]
- Shakiryanova D, Klose MK, Zhou Y, Gu T, Deitcher DL, Atwood HL, Hewes RS, Levitan ES. Presynaptic ryanodine receptor-activated calmodulin kinase II increases vesicle mobility and potentiates neuropeptide release. J Neurosci. 2007;27:7799–7806. doi: 10.1523/JNEUROSCI.1879-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiryanova D, Levitan ES. Prolonged presynaptic posttetanic cyclic GMP signaling in Drosophila motoneurons. Proc Natl Acad Sci U S A. 2008;105:13610–13613. doi: 10.1073/pnas.0802131105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiryanova D, Tully A, Hewes RS, Deitcher DL, Levitan ES. Activity-dependent liberation of synaptic neuropeptide vesicles. Nat Neurosci. 2005;8:173–178. doi: 10.1038/nn1377. [DOI] [PubMed] [Google Scholar]
- Shakiryanova D, Tully A, Levitan ES. Activity-dependent synaptic capture of transiting peptidergic vesicles. Nat Neurosci. 2006;9:896–900. doi: 10.1038/nn1719. [DOI] [PubMed] [Google Scholar]
- Takao K, Okamoto K, Nakagawa T, Neve RL, Nagai T, Miyawaki A, Hashikawa T, Kobayashi S, Hayashi Y. Visualization of synaptic Ca2+/calmodulin-dependent protein kinase II activity in living neurons. J Neurosci. 2005;25:3107–3112. doi: 10.1523/JNEUROSCI.0085-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao-Cheng JH, Dosemeci A, Winters CA, Reese TS. Changes in the distribution of calcium calmodulin-dependent protein kinase II at the presynaptic bouton after depolarization. Brain Cell Biol. 2006;35:117–124. doi: 10.1007/s11068-007-9012-5. [DOI] [PubMed] [Google Scholar]
- Vázquez-Martínez O, Cañedo-Merino R, Díaz-Muñoz M, Riesgo-Escovar JR. Biochemical characterization, distribution and phylogenetic analysis of Drosophila melanogaster ryanodine and IP3 receptors, and thapsigargin-sensitive Ca2+ ATPase. J Cell Sci. 2003;116:2483–2494. doi: 10.1242/jcs.00455. [DOI] [PubMed] [Google Scholar]
- Wang J, Renger JJ, Griffith LC, Greenspan RJ, Wu CF. Concomitant alterations of physiological and developmental plasticity in Drosophila CaM kinase II-inhibited synapses. Neuron. 1994;13:1373–1384. doi: 10.1016/0896-6273(94)90422-7. [DOI] [PubMed] [Google Scholar]
- Wang ZW. Regulation of synaptic transmission by presynaptic CaMKII and BK channels. Mol Neurobiol. 2008;38:153–166. doi: 10.1007/s12035-008-8039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MY, Shakiryanova D, Levitan ES. Presynaptic ryanodine receptor-CaMKII signaling is required for activity-dependent capture of transiting vesicles. J Mol Neurosci. 2009;37:146–150. doi: 10.1007/s12031-008-9080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]