

Abstract
Pulmonary arterial hypertension (PAH) is a debilitating disease with a high mortality rate. A hallmark of PAH is plexiform lesions (PLs), complex vascular formations originating from remodeled pulmonary arteries. The development and significance of these lesions have been debated and are not yet fully understood. Some features of PLs resemble neoplastic disorders, and there is a striking resemblance to glomeruloid-like lesions (GLLs) in glioblastomas. To further elucidate PLs, we used in situ methods, such as (fluorescent) IHC staining, three-dimensional reconstruction, and laser microdissection, followed by mRNA expression analysis. We generated compartment-specific expression patterns in the lungs of 25 patients (11 with PAH associated with systemic shunts, 6 with idiopathic PAH, and 8 controls) and GLLs from 5 glioblastomas. PLs consisted of vascular channels lined by a continuously proliferating endothelium and backed by a uniform myogenic interstitium. They also showed up-regulation of remodeling-associated genes, such as HIF1a, TGF-β1, VEGF-α, VEGFR-1/-2, Ang-1, Tie-2, and THBS1, but also of cKIT and sprouting-associated markers, such as NOTCH and matrix metalloproteinases. The cellular composition and signaling seen in GLLs in neural neoplasms differed significantly from those in PLs. In conclusion, PLs show a distinct cellular composition and microenvironment, which contribute to the plexiform phenotype and set them apart from other processes of vascular remodeling in patients with PAH. Neoplastic models of angiogenesis seem to be of limited use in further study of plexiform vasculopathy.
Pulmonary arterial hypertension (PAH) may occur either as a primary disease of unknown cause [idiopathic PAH (IPAH)] or as an associated manifestation of other diseases or malformations [eg, congenital shunts between the systemic and pulmonary circulation; associated PAH (APAH)].1,2 Characteristic histologic findings of PAH include remodeling of small pulmonary arteries and arterioles with varying degrees of endothelial cell proliferation, muscular hypertrophy, and intimal fibrosis, ultimately leading to an obliteration of precapillary vessels. Morphologic hallmarks of severe PAH are the so-called plexiform lesions (PLs): complex, glomeruloid-like vascular structures originating from the pulmonary arteries.3,4 Whether PLs represent just a morphologic “indicator lesion” or play a role in the pathogenesis and/or progression of PAH has not yet been clarified. On a more basal level, even the actual cellular composition of PLs has not been conclusively determined: the high plasticity of the (mesenchymal) cells involved, ie, their ability to change their phenotype depending on the current local microenvironment, have hampered analysis.5,6 The favored consensus describes the PL as a proliferating network of endothelial-lined vascular channels supported by a core of specialized and apoptosis-resistant myofibroblasts, smooth muscle cells, or even undifferentiated mesenchymal cells.7
Animal models using chronic hypoxia combined with the application of SU5416 (semaxinib), a vascular endothelial growth factor receptor (VEGFR) blocker, have produced glomeruloid lesions that mimic the formation of PLs to a certain extent.8 These lesions develop early after treatment with SU5416 combined with hypoxia, a situation that does not fully mirror the situation in humans, where there is evidence that severe pulmonary arterial pressure over a longer period is needed to induce PLs.9 Moreover, neoangiogenesis in peritumoral tissue, such as the so-called glomeruloid-like lesions (GLLs) in high-grade neural tumors, have also been discussed as a putative model for PLs.10–13 This experimental approach is compatible with the concept of the PL, or rather the adjacent artery from which it arises, as a circumscript angiogenic reservoir or niche that accommodates endothelial cells with a “quasi neoplastic” behavior, which contributes to remodeling of the pulmonary vasculature.14
Thus, although extensive analyses have been performed in animal experiments or alternative, tumor-associated vascular models that show a degree of morphologic similarity, actual PLs in humans have not yet been studied in more detail.
In this work, we studied the cellular composition, architecture, and local signaling of PLs in human PAH lungs. We compared the microenvironment in PLs with that in tumor-associated GLLs to examine the relevance of neoplastic models as a research platform for PLs. Furthermore, we aimed to explore whether PLs represent an epiphenomenon in severe PAH and whether the “angiogenic niche” plays a major role in vascular remodeling.
Materials and Methods
Specimens
We chose to focus on vascular remodeling in APAH associated with congenital heart disease. To this end, we selected 11 bilateral lung explants from patients with Eisenmenger's syndrome (New York Heart Association class IV). Of these patients, 7 showed prominent PLs (age at transplantation: arithmetic mean ± SD, 37.4 ± 9.9 years; median, 38 years) and 4 did not (age at transplantation: arithmetic mean ± SD, 34.5 ± 19.3 years; median, 28.8 years). Unagitated, nonremodeled pulmonary arteries taken from downsized lung tissue of donor lungs (n = 8) resected immediately before transplantation served as a reference for mRNA and protein expression analyses (Table 1).
Table 1.
Characteristics of the 11 Study Patients and 8 Controls
| No. | Sex | APAH with PLs | APAH without PLs | Heath-Edwards grade | Controls | Age at Tx (years) | Underlying condition | NYHA class at Tx | Mean PAP at Tx (mmHg) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | X | 4 | 51 | Atrial septal defect | IV | 65 | ||
| 2 | M | X | 4 | 42 | Atrial septal defect | IV | 70 | ||
| 3 | F | X | 4 | 45 | Ventricular septal defect | IV | 79 | ||
| 4 | F | X | 4 | 31 | Ventricular septal defect | IV | 72 | ||
| 5 | M | X | 4 | 38 | Complex cardiac malformation | IV | 65 | ||
| 6 | F | X | 4 | 34 | Patent ductus arteriosus | IV | 62 | ||
| 7 | F | X | 4 | 21 | Univentricular heart | IV | 68 | ||
| 8 | M | X | 3 | 23 | Complex cardiac malformation | IV | 65 | ||
| 9 | M | X | 3 | 58 | Complex cardiac malformation | IV | 68 | ||
| 10 | F | X | 3 | 42 | Complex cardiac malformation | IV | 70 | ||
| 11 | M | X | 3 | 15 | Alström's syndrome | IV | 80 | ||
| 12 | F | X | 66 | NA | NA | NA | |||
| 13 | F | X | 56 | NA | NA | NA | |||
| 14 | F | X | 46 | NA | NA | NA | |||
| 15 | F | X | 42 | NA | NA | NA | |||
| 16 | M | X | 49 | NA | NA | NA | |||
| 17 | M | X | 43 | NA | NA | NA | |||
| 18 | M | X | 43 | NA | NA | NA | |||
| 19 | M | X | 20 | NA | NA | NA |
F, female; M, male; NA, not applicable; NYHA, New York Heart Association; PAP, pulmonary arterial pressure; Tx, treatment.
In addition, we selected six bilateral lung explants from patients with IPAH (New York Heart Association class IV), all of them exhibiting prominent PLs (age at transplantation: arithmetic mean ± SD, 32.7 ± 17.1 years; median, 34 years). One of these patients carried a mutation of the BMP receptor 2 (BMPR2) (see Supplemental Table S1 at http://ajp.amjpathol.org).
All the specimens were inflated with formalin by the main bronchi and were formalin-fixed overnight before being extensively sampled and paraffin-embedded (FFPE). Subsequently, they were histologically evaluated, graded according to the Heath-Edwards classification, and correlated with clinical data to confirm the (histopathologic) diagnosis.15 In addition, five high-grade glial neoplasms (glioblastomas multiforme) with prominent peritumoral neoangiogenesis forming so-called GLLs were examined. The FFPE samples were retrieved from the archives of the Institute of Pathology of Hannover Medical School and were handled anonymously, following the requirements of the local ethics committee.
Light Microscopy and IHC Staining
Serially cut slides were IHC stained for different markers of endothelial and smooth muscle differentiation, vascular remodeling, fibrosis, and inflammation-associated markers using monoclonal antibodies and following a standard ABC protocol [angiopoietin (Ang)-1, Ang-2, CD3, CD20, CD31, CD34, CD68 (KP1), CD117 (c-KIT), CD141 (thrombomodulin), desmin Ki-67, mast cell tryptase, myocardin, β-type platelet-derived growth factor receptor, podoplanin, SMA, smooth muscle myosin heavy chain (smmhc), transforming growth factor (TGF)-β1, thrombospondin (THBS)-1, and VEGF-α; Table 2]. The staining intensity in different compartments was assessed in at least two locations in both lobes of the lung specimens as no reactivity, barely visible reactivity at high magnification (“weak”), well-recognizable reactivity at medium magnification (“intermediate”), and high protein expression levels visible even at low magnification (“strong”).16 The range of positive cells in the different compartments was scored semiquantitatively as no apparent reaction (score 0), positivity in <30% (score 1), positivity in ≥30% and <60% (score 2), and positivity in ≥60% (score 3) of cells.17,18
Table 2.
Antibodies Used in the Study
| Protein | Company | Dilution |
|---|---|---|
| Ang-1 | R&D Systems, Minneapolis, MN | 1:15 |
| Ang-2 | R&D Systems | 1:50 |
| CD3 | Dako, Carpinteria, CA | 1:200 |
| CD20 | Dako | 1:50 |
| CD31 | Dako | 1:75 |
| CD34 | Leica Microsystems GmbH, Wetzlar, Germany | Ready to use |
| CD68 (KP1) | Dako | 1:100 |
| CD117 (c-KIT) | Zytomed Systems GmbH, Berlin, Germany | 1:50 |
| CD141 (thrombomodulin) | AbD Serotec, Raleigh, NC | 1:20 |
| Desmin | Dako | 1:75 |
| Ki-67 | Thermo Fisher Scientific, Waltham, MA | 1:100 |
| Mast cell tryptase | Leica Microsystems GmbH | 1:50 |
| Myocardin | Covalab S.A.S., Villeurbanne France | 1:200 |
| β-Type platelet-derived growth factor receptor | Acris Antibodies Inc., San Diego, CA | 1:50 |
| Podoplanin | Acris Antibodies Inc. | 1:25 |
| SMA | Dako | 1:25 |
| Smmhc | US Biological, Swampscott, MA | 1:100 |
| TGF-β1 | Acris Antibodies Inc. | 1:500 |
| THBS1 | Lifespan, Providence, RI | 1:20 |
| VEGF-α | Santa Cruz Biotechnology, Santa Cruz, CA | 1:50 |
The detection system was the DAB Zytomed HRP kit (Zytomed Systems GmbH) for all.
We examined PLs and the adjacent arteries from which they sprout: remodeled arteries ≤750 μm from the corresponding PL, with varying degrees of media hypertrophy and (concentric) intimal proliferation. In PAH lungs without PLs, we analyzed remodeled arteries, which showed concentric laminar intimal proliferation and fibrosis (“concentric lesions” with a diameter of ≤500 μm). Unagitated arterial vessels from downsized lung tissue of donor lungs served as controls.
Inflammatory cells in PLs and their adjacent vessels and in controls were quantitated by counting the number of positively marked cells per square millimeter (positivity for CD3, CD20, CD45, CD68, and mast cell tryptase; Table 2). Staining intensity, where applicable, was graded as weak, intermediate, or strong.17,18 For negative controls, the primary antibody was replaced by bovine serum albumin.
Laser-Assisted Microdissection and RNA Extraction
Serial sections (5-μm–thick FFPE tissue) were mounted on a poly-l-lysine–coated membrane fixed onto a metal frame. After deparaffinization and routine (hemalum) staining, the CellCut Plus system (MMI Molecular Machines & Industries AG, Glattbrugg, Switzerland) was used for laser-assisted microdissection. Pathologic and anatomic structures (PLs, adjacent arteries, concentric lesions in patients with APAH without PLs, and controls) were sampled from at least two locations in both lobes of the explants using a no-touch technique, as described elsewhere19 (Figure 1). These microdissected areas included the adventitia directly adjacent to the vascular structures (Figure 1). Approximately 1500 cells were harvested from each compartment. The microdissected material was subsequently suspended in a proteinase K digestion buffer by placing it directly in the adhesive cap. After overnight digestion, RNA was isolated using phenol-chloroform extraction and precipitation following the established modus operandi.20
Figure 1.
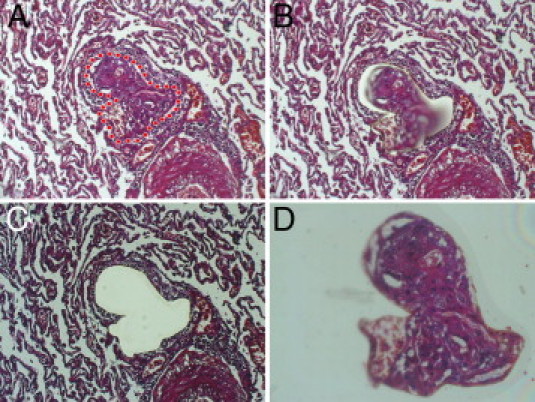
Laser-assisted microdissection. A–D: Isolation of a PL from the surrounding unagitated lung tissue by laser-assisted microdissection (the laser cut is visible in B; D shows the isolated PL in the cap). Note the adjacent artery from which the PL originates in the lower right corner in A–C. Original magnification: ×100 (A–C); ×200 (D). The IX71 microscope (Olympus Europa GmbH) with the CellCut Plus system was used for laser-assisted microdissection.
cDNA Synthesis and Preamplification
cDNA was synthesized using the High Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) and following the manufacturer's protocol. cDNA was preamplified in 14 PCR cycles with target gene–specific PCR primers, thus increasing the sensitivity of the subsequent real-time PCR analysis several thousand-fold (decrease of CT: 14 cycles; TaqMan PreAmp master mix kit, Applied Biosystems), as previously reported.18,20
Real-Time PCR and Expression Analysis
The preamplified cDNA from laser-microdissected samples was evaluated by real-time PCR (TaqMan 7500 real-time PCR system, Applied Biosystems, Carlsbad, CA). Quantification was performed in reactions containing preamplified cDNA, TaqMan Gene Expression Master Mix, and the individual TaqMan Gene Expression Assay (both from Applied Biosystems). A compilation of proliferation-, apoptosis-, fibrosis-, and inflammation-associated genes was selected and analyzed following the manufacturer's protocol (Table 3). For negative controls, the cDNA was replaced by water. CT values were calculated by normalization to the mean expression of three endogenous controls (POLR2A, β-GUS, and GAPDH) and were converted into 2−ΔCT values using Excel 8.0 (Microsoft Corp., Redmond, WA), which were then statistically analyzed using the Mann-Whitney U-test and GraphPad Prism, version 5.0 (GraphPad Software Inc., San Diego, CA).21 We considered P ≤ 0.05 to be statistically significant. Expression graphics were created using GraphPad Prism, version 5.0.
Table 3.
Target and Reference Genes
| No. | Target genes | Synonym/full name |
|---|---|---|
| 1 | Ang-1 | Angiopoietin-1 |
| 2 | Ang-2 | Angiopoietin-2 |
| 3 | BMP4 | Bone morphogenetic protein-4 |
| 4 | BMPR2 | Bone morphogenetic protein receptor type 2 |
| 5 | CASP9 | Caspase-9 |
| 6 | c-KIT | CD117 |
| 7 | des | Desmin |
| 8 | eNOS | Nitric oxide synthase 3, endothelial cell |
| 9 | FGF-2 | Fibroblast growth factor-2 |
| 10 | HIF1a | Hypoxia-inducible factor 1 |
| 11 | IL 1b | Interleukin-1b |
| 12 | IL 6 | Interleukin-6 |
| 13 | MMP9 | Matrix metalloproteinase 9 |
| 14 | MMP14 | Matrix metalloproteinase 14 |
| 15 | MYH11 | Smooth muscle myosin heavy chain 11 |
| 16 | MYOCD | Myocardin |
| 17 | NOTCH4 | Neurogenic locus notch homolog protein 4 |
| 18 | PECAM-1 | CD31 |
| 19 | PDGFRb | β-Type platelet-derived growth factor receptor |
| 20 | SMA | Smooth muscle actin |
| 21 | TGF-β1 | Transforming growth factor-beta 1 |
| 22 | THBS1 | Thrombospondin-1 |
| 23 | VEGF-α | Vascular endothelial growth factor alpha |
| 24 | VEGFR1 | Vascular endothelial growth factor receptor 1 |
| 25 | VEGFR2 | Vascular endothelial growth factor receptor 2 |
| Reference genes | ||
| 26 | POLR2A | RNA-polymerase 2 subunit A |
| 27 | GUSB | β-glucuronidase |
| 28 | GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
Immunofluorescence Double Staining
To confirm the findings of conventional IHC and compartment-specific mRNA expression analyses regarding endothelial and/or smooth muscle differentiation of PLs, we performed immunofluorescence double staining for CD31 and SMA, CD31 and Ki-67, and SMA and Ki-67. Serially cut 5-μm–thick FFPE slides were deparaffinized, and antigens were retrieved following the established procedure. Target structures were marked using monoclonal primary- and fluorescence-labeled secondary antibodies [Cy-conjugated AffiniPure F(ab')2 fragment antibodies (H+L); Jackson ImmunoResearch Laboratories Inc., West Grove, PA]. The nuclear structures were counterstained using DAPI (40 μg/mL; Carl Roth GmbH, Karlsruhe, Germany) before the slides were mounted.19
GLLs in Glioblastomas
Circumscribed peritumoral vascular formations, so-called GLLs in glioblastomas multiforme (n = 5), were identified by light microscopy. Vital nonnecrotic GLLs were stained by fluorescence IHC, followed by laser-microdissection and processing for RNA expression analysis as described previously herein for PLs.
Three-Dimensional Reconstruction of PLs
To further assess the structural composition of PLs, we used 30-μm-thick slides and (fluorescence) double stained them for CD31 and SMA as described previously herein. To generate three-dimensional (3-D) images of PLs, we used a microscope with an automated drive (Micro Focus, Rockville, MD) and special acquisition software (BX51 microscope and Cell P Software 3.3, Olympus Europa GmbH, Hamburg, Germany) and systematically scanned the whole width of the slides. The resulting 2-D images were merged into 3-D shapes. Unspecific, out-of-focus fluorescence was removed from the resulting 3-D images using deconvolution software (advanced maximum likelihood estimation). Images were displayed using the cell* Voxel Viewer (Olympus Europa GmbH).
Terminal Deoxynucleotidyl Transferase–Mediated dUTP Nick-End Labeling Assay
To analyze the role of apoptosis in the remodeling process in PLs beyond caspase mRNA expression (see previously herein), we assessed the content of fragmented DNA in PLs, adjacent vessels, and controls using an apoptosis detection kit (ApopTag plus peroxidase in situ; Millipore, Temecula, CA) on 5-μm FFPE samples following the manufacturer's protocol.
Results
Characterization of PLs and Neighboring Remodeled Arteries
PLs showed significant mRNA up-regulation of the endothelial markers CD31 and endothelial nitric oxide synthase and an increase in SMA expression compared with the adjacent arteries (Figure 2). Because PLs are mainly composed of “vascular channels,” (over)expression of markers of endothelial differentiation is not surprising, especially considering the comparable numbers of microdissected cells from the different compartments.
Figure 2.
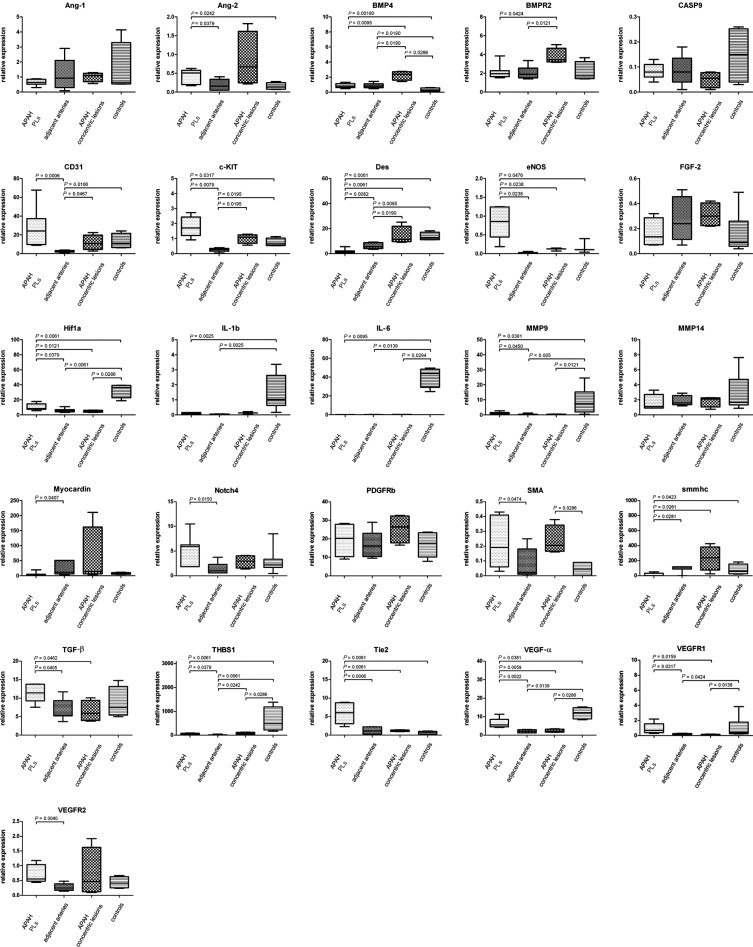
mRNA expression of target genes in PLs in APAH in alphabetical order. Comparison of mRNA expression characteristics of PLs in patients with congenital shunts between the systemic and pulmonary circulation (APAH), the adjacent arteries from which they sprout, concentric lesions in patients with APAH without PLs, and controls. Expression of target genes was calculated using the ΔCT method (see Materials and Methods for details). Middle horizontal lines indicate medians; error bars, percentiles. PDGFRb, β-type platelet-derived growth factor receptor.
The complementary IHC staining concurred with the results of mRNA expression analysis: CD31 showed strong laminar positivity in luminal cells of vascular channels in PLs and their adjacent arteries. The staining patterns of the endothelial markers CD34 and CD141 were comparable. SMA stained strongly in the slender, nonluminal interstitium, composed of a homogenous population of tightly layered mesenchymal/myogenic cells (Figure 3 and Table 3). These staining patterns in regular IHC were confirmed using immunofluorescence double staining: the CD31-positive endothelium of the vascular channels was continuously backed by SMA-positive cells, arranged in a uniform stratum less than or equal to four cell layers thick. 3-D reconstruction of PLs validated this cellular composition (Figure 4).
Figure 3.
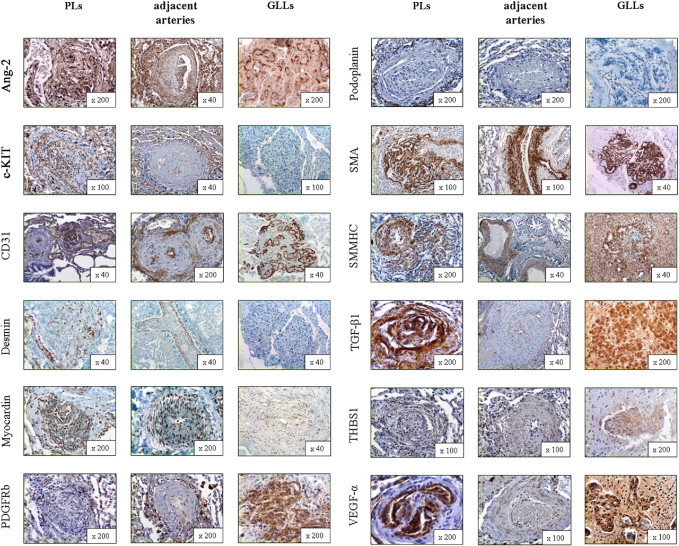
Protein expression of structural composition and tissue remodeling–associated markers in PLs, the adjacent arteries from which PLs originate, and GLLs in high-grade primary neural malignomas (in alphabetical order). Ang-2 showed strong cytoplasmic positivity in the endothelial/luminal compartment of PLs and also in the endothelial layer of the adjacent arteries and GLLs. c-KIT (CD117) stained positive in the endothelial/luminal compartment of PLs and in the endothelium of the adjacent arteries, whereas there was no delimitable staining in GLLs. CD31 staining was pronounced in the endothelial/luminal layers in PLs, adjacent arteries, and GLLs alike. Desmin showed only speckled cytoplasmic positivity in the interstitial/myogenic layer of PLs, whereas the media of the adjacent arteries was continuously positive and GLLs showed no staining reaction. Myocardin showed moderate cytoplasmic positivity in vascular structures in PLs and adjacent arteries, whereas GLLs stained only faintly. β-Type platelet-derived growth factor receptor (PDGFRb) stained only faintly in PLs, whereas the directly adjacent arteries showed moderate positivity, mainly at the border between the actual vascular wall and the neighboring connective tissue. GLLs showed only faint perivascular staining. Podoplanin showed no delimitable signal in PLs but a slender network of lymphatic vessels around them. There was also no positivity in the adjacent arteries or in GLLs. Staining for SMA showed slim but continuous positivity between the luminal/endothelial layers in PLs, whereas there was a prominent and homogenous staining pattern in the media of the adjacent arteries. In GLLs, the walls of the neoplastic blood vessels also showed strong cytoplasmic positivity, although it was less well organized compared with the nonneoplastic specimens/compartments. Smmhc stained weakly in the interstitium of PLs, whereas there was strong staining in the media of the adjacent arteries. GLLs displayed inhomogenous positivity of the neoplastic blood vessels. TGF-β1 stained strongly in the luminal and the interstitial compartment of PLs, whereas the adjacent arteries showed only faint positivity of the endothelium. Staining in GLLs was strong in the vascular structures. THBS1 showed inhomogenous cytoplasmic positivity in PLs, whereas the adjacent arteries showed only a faint reaction. GLLs showed inhomogenous moderate positivity. VEGF-α showed marked luminal/endothelial positivity, with only a faint endothelial signal in the adjacent arteries. The vessels in GLLs showed strong positivity. Original magnification is included in the lower right corner of each histologic image.
Figure 4.
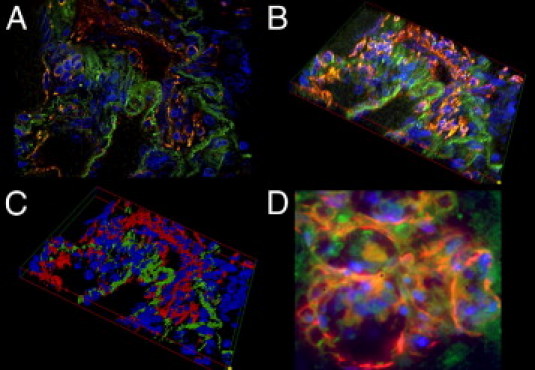
The-3-D reconstruction of a PL. A: A fluorescent double-stained PL: the luminal vascular channels stain positive for CD31 (red) and the adjacent interstitium shows homogenous positivity for SMA (green). B: Systematically scanned images of the whole width of the PL were merged into 3-D shapes. C: Images were skeletonized into a voxel model for increased clarity. Note the distinct separation of the endothelial layer and the interstitium in the PL. D: GLLs in high-grade neural tumors show a rather disorganized composition without an equally clear separation of endothelial layer and interstitium. Original magnification: ×630 (A–C); ×1000 (D).
The adjacent remodeled arteries with prominent media hypertrophy and intimal thickening showed significantly increased mRNA levels of desmin, myocardin, and smmhc compared with PLs (Figure 2), which was largely anticipated given the ratio of smooth muscle to endothelial cells exceeding 9:10 in these vessels. The corresponding IHC staining confirmed these expression patterns, with strong positivity in the prominent media layer for all three targets in pulmonary arteries but not in PLs. Whereas PLs, the adjacent arteries, concentric lesions in APAH without PLs, and controls did not stain for podoplanin, there was a marked accumulation of podoplanin-positive lymphatic vessels around PLs. Aside from the presence of PLs themselves, there were no reproducible morphologic differences between APAH specimens with and without PLs (Figure 3 and Tables 3 and 4).
Table 4.
IHC Staining Results in Vascular Compartments
| Protein | PLs | Adjacent arteries | Concentric lesions in APAH without PLs | Controls | GLLs |
|---|---|---|---|---|---|
| Ang-1 | |||||
| Semiquantitative score | 1 | 1 | 1 | 1 | 1 |
| Staining intensity | Weak | Weak | Weak | Weak | Intermediate |
| Ang-2 | |||||
| Semiquantitative score | 2 | 2 | 3 | 2 | 2 |
| Staining intensity | Strong | Intermediate | Intermediate | Weak | Strong |
| c-KIT (CD117) | |||||
| Semiquantitative score | 1 | 1 | 1 | 1 | 0 |
| Staining intensity | Intermediate | Weak | Weak | Weak | Absent |
| CD141 | |||||
| Semiquantitative score | 2 | 1 | 1 | 1 | 2 |
| Staining intensity | Strong | Strong | Strong | Strong | Strong |
| CD31 | |||||
| Semiquantitative score | 2 | 1 | 1 | 1 | 2 |
| Staining intensity | Strong | Strong | Strong | Strong | Strong |
| CD34 | |||||
| Semiquantitative score | 2 | 1 | 1 | 1 | 2 |
| Staining intensity | Strong | Intermediate | Strong | Strong | Strong |
| Desmin | |||||
| Semiquantitative score | 2 | 3 | 3 | 3 | 0 |
| Staining intensity | Intermediate | Strong | Strong | Strong | Absent |
| Myocardin | |||||
| Semiquantitative score | 2 | 2 | 3 | 2 | 1 |
| Staining intensity | Weak | Intermediate | Intermediate | Intermediate | Weak |
| PDGFRb | |||||
| Semiquantitative score | 2 | 2 | 2 | 2 | 1 |
| Staining intensity | Weak | Intermediate | Intermediate | Intermediate | Weak |
| Podoplanin | |||||
| Semiquantitative score | 0 | 0 | 0 | 0 | 0 |
| Staining intensity | Absent | Absent | Absent | Absent | Absent |
| SMA | |||||
| Semiquantitative score | 3 | 3 | 3 | 3 | 3 |
| Staining intensity | Strong | Strong | Strong | Intermediate | Strong |
| Smmhc | |||||
| Semiquantitative score | 3 | 3 | 3 | 3 | 2 |
| Staining intensity | Intermediate | Strong | Strong | Intermediate | Intermediate |
| TGF-β1 | |||||
| Semiquantitative score | 2 | 1 | 1 | 1 | 2 |
| Staining intensity | Strong | Weak | Weak | Weak | Strong |
| THBS1 | |||||
| Semiquantitative score | 2 | 1 | 2 | 2 | 2 |
| Staining intensity | Weak | Weak | Intermediate | Intermediate | Intermediate |
| VEGF-A | |||||
| Semiquantitative score | 2 | 2 | 2 | 2 | 2 |
| Staining intensity | Strong | Weak | Weak | Intermediate | Strong |
Semiquantitative scores indicate the range of positive cells (0, no apparent reaction; 1, positivity in <30%; 2, positivity in ≥30% and <60%; and 3, positivity in ≥60%). Staining intensity is indicated as absent, weak, intermediate, or strong.
PDGFRb, β-type platelet-derived growth factor receptor.
Together, PLs in IPAH and APAH showed only minor differences in composition-related mRNA and protein expression: whereas PLs in APAH showed significant up-regulation of CD31, those in IPAH showed a comparable trend toward up-regulation (Figures 2 and 3; also see Supplemental Figure S1 and Supplemental Table S2 at http://ajp.amjpathol.org) The height of the mean pulmonary arterial pressure or the pulmonary vascular resistance measured before transplantation had no effect on the composition of PLs.
Tissue Remodeling–Associated Markers in Pulmonary Vascular Compartments
PLs showed significantly increased mRNA levels of Ang-2 (but not Ang-1), c-KIT, HIF1a, matrix metalloproteinase (MMP) 9 (but not MMP14), NOTCH4, VEGF-α, VEGFR1, VEGFR2, and THBS1 compared with the adjacent remodeled pulmonary arteries. We also found up-regulation of Tie-2 and pivotal TGF-β1, which do not represent endothelial growth promoters per se compared with the adjacent remodeled pulmonary arteries.
IHC staining correlated with these findings, with strong luminal/endothelial positivity for Ang-2, c-KIT, VEGF-α, and TGF-β1 in PLs but not in the adjacent vessels. TGF-β1 also showed mild staining of the interstitium in PLs.
BMP-4 and BMPR2 were significantly up-regulated in concentric lesions in patients with APAH who did not exhibit PLs compared with other groups. All other markers did not show significant differences between compartments/groups (Figures 2 and 3 and Tables 3 and 4).
In summary, PLs in IPAH and APAH showed only minor differences in remodeling-associated gene regulation: cKIT, HIF1a, MMP9, TGF-β1, and THBS1 were up-regulated in PLs in APAH compared with the adjacent arteries, with a comparable but nonsignificant trend in PLs in IPAH (see Supplemental Figure S1 at http://ajp.amjpathol.org). Hemodynamic variables, such as the height of the mean pulmonary arterial pressure or pulmonary vascular resistance measured before transplantation, had no effect on remodeling-associated gene expression in PLs.
Proliferation and Apoptosis in PLs
Conventional IHC Ki-67 staining and immunofluorescence double staining for CD31/Ki-67 and SMA/Ki-67 showed a significant proliferation of CD31-positive endothelial and, to a lesser extent, SMA-positive interstitial cells in PLs compared with their adjacent pulmonary arteries (arithmetic mean ± SD: CD31+ cells in PLs, 8.2% ± 2.7%; SMA+ cells in PLs, 4.6% ± 2.2%; CD31+ cells in adjacent arteries, 1.1% ± 1.1%; and SMA+ cells in adjacent arteries, 1.2% ± 0.9%). We found no significant differences regarding apoptosis in mRNA expression analysis of caspases or terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling assays between PLs and the adjacent arteries or external controls (PLs, <0.5% positive; adjacent arteries, <0.5% positive) (Figure 5).
Figure 5.
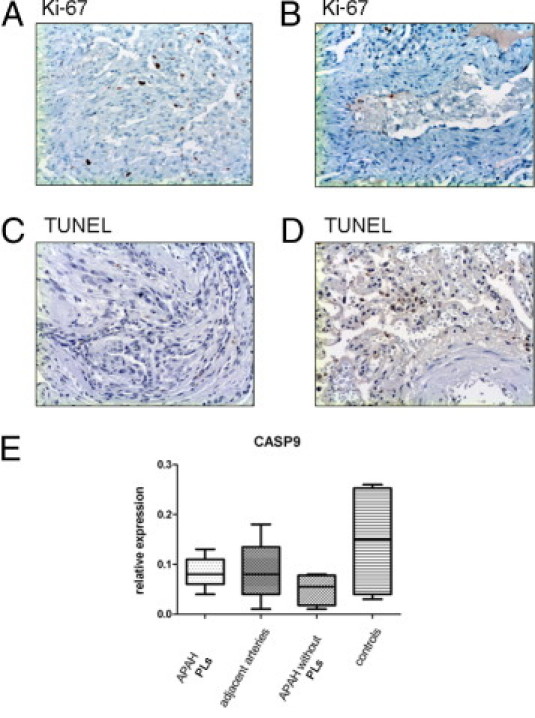
Proliferation and apoptosis in PLs in APAH. IHC staining for the proliferation marker Ki-67 in PLs (A) and the adjacent arteries (B). Whereas there is marked proliferation in PLs, only single cells of the endothelium stain positive in the adjacent arteries. Terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) assays do not show significant degrees of apoptosis in PLs (C) or the adjacent arteries (D), and neither is there significant mRNA up-regulation of apoptosis-associated caspase 9 (CASP9) (E). Middle horizontal lines indicate medians; error bars, percentiles. Original magnification, ×100 (A–D).
There were only minor differences regarding the ratio of proliferation and apoptosis between PLs in IPAH and APAH: caspase 9 showed barely significant down-regulation in PLs in IPAH cases compared with the adjacent arteries (see Supplemental Figure S1 at http://ajp.amjpathol.org), whereas mRNA expression levels in PLs and adjacent arteries were similar in APAH specimens (Figure 2).
Inflammatory Cells in PLs
CD3-positive T lymphocytes represented most inflammatory cells in PLs (arithmetic mean ± SD, 7.7% ± 6.7% of all cells in PLs). They also accumulated directly adjacent to PLs, where we found networks of podoplanin-positive lymphatic vessels (see previously herein). CD20-positive B lymphocytes accounted for only a few leukocytes in PLs (arithmetic mean ± SD: 1% ± 0.9% of all cells in PLs). CD68-positive macrophages (arithmetic mean ± SD: 6.2% ± 1.9% of all cells in PLs) and mast cells (arithmetic mean ± SD: 3.6% ± 3.4% of all cells in PL) showed an approximately equal distribution in PLs. The adjacent pulmonary arteries showed <1% CD3-, CD20-, CD68-, and mast cell tryptase–positive cells (Figure 6). PLs in IPAH and APAH showed no significant differences in concentrations of inflammatory cells.
Figure 6.
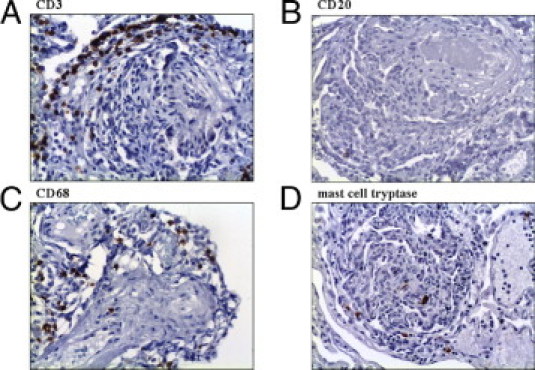
Inflammatory cells in PLs. Most inflammatory cells in PLs are CD3-positive T lymphocytes, which also accumulate around PLs (A). CD20-positive B lymphocytes account for only a few leukocytes in PLs (B). CD68-positive macrophages (C) and mast cells (D) show an almost equal distribution in PLs. Original magnification, ×100 (A–D).
PAH with BMPR2 Mutation and PLs
The PL in the one patient with a known BMPR2 mutation did not differ from the PL in the other patients with IPAH and APAH: no differences regarding expression patterns (mRNA and IHC analysis), structural composition, or cellularity could be found (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Characterization of GLLs
Whereas PLs in patients with PAH were well structured, with a layered composition of CD34-positive endothelium and SMA-positive interstitium ≤4 cell layers (see previously herein), the endothelium and media of the vessels that compose GLLs were irregularly shaped and contorted. The thickness of the media/smooth muscle interstitium in GLLs varied considerably and frequently exceeded six layers. PLs showed significant up-regulation of desmin, myocardin, and smmhc compared with GLLs. The corresponding IHC for all three proteins confirmed faint staining of desmin, myocardin, and smmhc in GLLs, whereas staining was moderate in PLs (Figures 3 and 4 and Table 4).
Comparison of Angiogenic Markers in PLs and GLLs in Glioblastomas
Compared with PLs, GLLs showed significant mRNA up-regulation of Ang-2, MMP9, and VEGFR2, whereas PLs showed up-regulation of Ang-2, BMPR2, endothelial nitric oxide synthase, and Tie-2. IHC analysis of Ang-1 and Ang-2 matched these findings, with strong expression of Ang-2 in PLs and intermediate staining intensity for Ang-1 in GLLs in glioblastomas. All other markers showed no significant differences between PLs and GLLs (Figure 7 and Tables 5 and 6).
Figure 7.
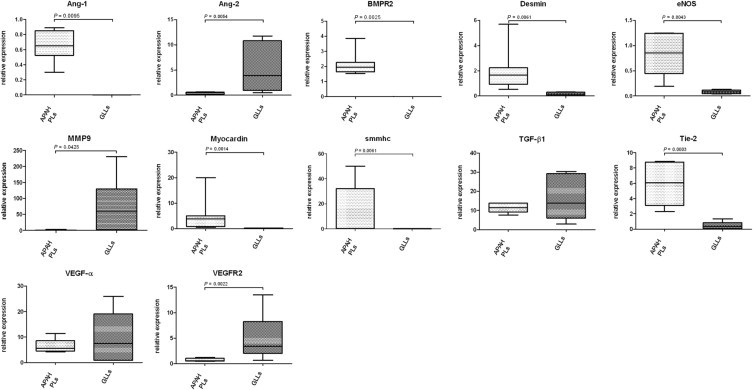
mRNA expression of target genes in PLs in APAH and GLLs in alphabetical order. Comparison of selected mRNA expression characteristics of PLs in patients with congenital shunts between the systemic and pulmonary circulation (APAH) and GLLs in high-grade primary neural tumors. Expression of target genes was calculated using the ΔCT method (see Materials and Methods for details). Lines indicate medians; error bars, percentiles.
Table 5.
mRNA Expression in PLs in APAH and the Adjacent Arteries from Which They Sprout
| Gene |
P value |
|
|---|---|---|
| PLs (APAH) | Adjacent arteries | |
| Ang-1 | NS | NS |
| Ang-2 | <0.05 | |
| BMP4 | NS | NS |
| BMPR2 | NS | NS |
| CASP9 | NS | NS |
| CD31 | <0.001 | |
| cKIT | <0.01 | |
| Des | <0.01 | |
| eNOS | <0.05 | |
| FGF-2 | NS | NS |
| HIF1a | <0.05 | |
| IL-1b | NS | NS |
| IL-6 | NS | NS |
| MMP9 | <0.05 | |
| MMP14 | NS | NS |
| Myocardin | <0.05 | |
| NOTCH4 | <0.05 | |
| PDGFRb | NS | NS |
| SMA | <0.05 | |
| Smmhc | <0.05 | |
| TGF-β1 | <0.05 | |
| THBS1 | <0.05 | |
| Tie2 | <0.001 | |
| VEGF-α | <0.01 | |
| VEGFR1 | <0.05 | |
| VEGFR2 | <0.01 | |
Table 6.
mRNA Expression in PLs in APAH and GLLs in High-Grade Glial Neoplasms
| Gene |
P value |
|
|---|---|---|
| PLs (APAH) | GLLs | |
| Ang-1 | <0.01 | |
| Ang-2 | <0.01 | |
| BMPR2 | <0.01 | |
| CD31 | NS | NS |
| cKIT | NS | NS |
| Des | <0.01 | |
| eNOS | <0.01 | |
| FGF-2 | NS | NS |
| HIF1a | NS | NS |
| IL-1b | NS | NS |
| IL-6 | NS | NS |
| MMP9 | <0.05 | |
| MMP14 | NS | NS |
| Myocardin | <0.01 | |
| NOTCH4 | NS | NS |
| PDGFRb | NS | NS |
| SMA | NS | NS |
| Smmhc | <0.01 | |
| TGF-β1 | NS | NS |
| THBS1 | NS | NS |
| Tie2 | <0.001 | |
| VEGF-α | NS | NS |
| VEGFR1 | NS | NS |
| VEGFR2 | <0.01 | |
Discussion
Complex vascular formations, so-called PLs, are histologic hallmarks of lungs from patients with PAH. Whether they represent morphologic sequelae of the raised intravascular pressure or actively contribute to the course of the disease is still debated; per se, both seem possible.22–24 PLs are generally thought to originate from a misled neoangiogenesis with a dysbalance between apoptosis/necrosis and subsequent proliferation of the endothelium.3 Although PLs and their development over time have been studied in animal experiments and in part in in vitro settings, knowledge about PLs in the human lung is still limited.
Although they are end-stage organs, recipient lung explants sampled at the date of transplantation represent the only feasible source of lung tissue with characteristic histologic changes of severe PAH in humans. Transbronchial or wedge biopsies are not obtained in day-to-day routine from patients with PAH; in fact, for the most part, the latter are contraindicated owing to considerable bleeding risk, and material taken during postmortem examinations is usually unsuitable for an extensive molecular workup.
From a morphologic point of view, using fluorescent double staining and regular IHC analysis and 3-D reconstruction, these results reveal that PLs are highly ordered structures. The thin, luminal layer around vascular channels was composed of a homogeneous population of CD31-, CD34-, and CD141-positive endothelial cells. This concurs with the mRNA up-regulation of CD31 and endothelial nitric oxide synthase, which suggests prominent endothelial differentiation and further extends previous findings describing endothelial cells as the predominant, if not only, cell type composing PLs. We found no thrombi in any of the PLs we examined. This suggests an intact intima/endothelium with a predominantly nonturbulent flow.5
We also found a distinct interstitial compartment in PLs, separating the individual blood channels from each other. It was composed of a thin, uniform layer of mesenchymal cells with myogenic differentiation (“interstitial cells of myogenic phenotype”). These cells showed prominent up-regulation of SMA, even compared with the adjacent remodeled arteries, which had prominent media and intima and, thus, consisted mainly of smooth muscle cells. There was, however, only limited expression of smmhc and, notably, desmin. As the concentration of these intermediary filament increases with advancing differentiation of smooth muscle cells toward a contractile phenotype,25,26 we consider the heterogeneous expression pattern an indicator of an intermediate state between the contractile and synthetic phenotypes, a finding that is consistent with IHC staining patterns described by others in postmortem case studies in lungs of patients with PAH.27 Whereas synthetic differentiation tends toward increased proliferation and migration, the contractile phenotype is associated with structural integrity. Both are needed for progressive vascular remodeling.25
Because remodeling in PAH lungs affects all precapillary vascular compartments, from the truncus pulmonalis down to the arterioles, the question arises as to why the remodeling process is especially pronounced and intricate in PLs. The low expression levels of myocardin and smmhc (but not of β-type platelet-derived growth factor receptor) in PLs might provide an answer: these three proteins are characteristic of pericytes. In this line, knockout experiments showed that the absence of these regulatory perivascular cells indeed promotes abundant endothelial hyperplasia in smaller vessels.28–30 A possible “outgrowth” of the vascular channels in PLs, with which the regulatory pericytes did not keep pace, might lead to a relative lack of pericytes in PLs. Thus, aside from local hypoxia and shear forces, lack of superordinate regulation might contribute to the ongoing remodeling in PAH lungs. Because pericytes possess considerable plasticity, they cannot be definitely characterized by mRNA and protein expression, thus severely hampering the analysis of their involvement in the formation of PLs.31–33
We repeatedly found delicate networks of lymphatic vessels surrounding the PLs, mainly populated by T lymphocytes, presumably identical to “clusters of inflammatory cells” identified by others.34,35 In the PL, no lymphatics could be shown. Although elevated serum levels of ILs in patients with PAH have been described, there was no increased expression of ILs in the vascular compartments examined.36
Although these specimens represent end-stage organs in severely ill patients, PLs still displayed a marked degree of ongoing remodeling. There was pronounced proliferation of endothelial cells, but we observed only a limited extent of cell division in the muscular interstitium. We found almost no detectable apoptosis in either cell type. This concurs with findings of others, who postulate PLs as a result of focal arterial wall apoptosis/necrosis. This initial selection process is thought to leave a circumscript aggregate or remnant of apoptosis-resistant endothelial cells, which compose and promote the growth and reshaping of PLs.3 This continuous proliferation with invagination and sprouting of vascular channels was mirrored by an increased expression of MMP9 and NOTCH4. Of these, the former is involved in the remodeling of the basement membrane during the sprouting process and the latter is involved in the cross talk with pivotal VEGF-α and TGF-β1, the smooth muscle layers and the endothelium, which also drives the reshaping of the local vasculature.37–39
The tissue remodeling–associated microenvironment in PLs in PAH is complex. We found significant up-regulation of hypoxia and shear stress–induced angiogenic mediators, such as HIF1a, VEGF-α and its corresponding receptors (VEGFR1 and VEGFR2), Ang-1, Tie-2, and THBS1 but also of c-KIT in PLs. Considering the importance of these pathways for angiogenesis in general, and particularly for the remodeling of the pulmonary blood vessels, up-regulation in PAH lungs per se is principally not surprising. These markers have been demonstrated to be elevated in different organs and the peripheral bloodstream of patients with PAH and in animal models, where especially the induction of the VEGF pathway correlated with the severity of PAH and, accordingly, survival.40–44 Their concerted up-regulation in PLs, especially compared with the adjacent remodeled arteries/concentric lesions, is, however, interesting. Although it is unlikely that lesions that arise as late in the course of a disease as PLs do in PAH,9 and by no means occur in every patient with PAH,4 are involved in the onset of the condition, they might well contribute to its sustainment as a source of mediators guiding vascular remodeling in other anatomic sites of the lung.
In contrast, the universally low mitotic rate and low expression of angiogenic/remodeling-associated markers in the arteries from which PLs arise do not support the concept of a circumscript “angiogenic niche” that harbors cells with a quasi neoplastic behavior in these adjacent vessels. Rather, we consider the sprouting of PLs to be an overshooting regenerative process and not a neoplastic event sensu stricto.
Significant up-regulation of hypoxemia-induced genes in the control group is explained by the cold ischemia to which the grafts, from which the downsizing-specimens were sampled before transplantation, were exposed while being transported to and prepared for the organ recipient.45,46 Recipient lungs with hypertension-induced changes, on the other hand, did not undergo cold ischemia but were fixated directly after explantation.
We found significant up-regulation of BMP-4 and BMPR2 in concentric lesions in remodeled pulmonary arteries of patients with APAH but not in cases with PLs. These proteins were originally named for their ability to facilitate bone tissue formation, but more recently their involvement in the signaling balance in lung development and fibrosis has been demonstrated: whereas the cross talk between BMPs and other members of the TGF superfamily is yet to be fully elucidated, various interactions, such as shared receptors and competition for subordinate SMADs, have been described, which for the most part serve the maintenance of tissue homeostasis.47 Elevated but also decreased levels of BMPs have been described in patients with tissue remodeling in different organs, and in vitro studies have attributed BMPs with a degree of protective properties.48,49 In the lung, aberrant BMP expression due to inactivation of the BMPR2 gene has been shown to lead to a disorganized proliferation of smooth muscle cells and to contribute to the development of PAH.50 Although the pathophysiologic significance of the BMPR2 mutation in IPAH has been proved, we could not find an effect on the morphology and composition of PLs.
Because neoangiogenesis in high-grade glial neoplasms (so-called GLLs) and PLs share a related morphology, they have been proposed as a putative model for PLs.10,51 In (fluorescent) IHC staining, we found blood vessels in GLLs to have a prominent variation in thickness and layering compared with PLs. A continuous myogenic layer separating the endothelium of individual channels was not always discernible in GLLs. Furthermore, compared with GLLs, PLs showed significant up-regulation of desmin, myocardin, and smmhc (but not of SMA). Thus, according to the yardstick discussed previously herein, smooth muscle cells in GLLs show a different phenotypic differentiation than do PLs.25 With an up-regulation of Ang-2 and Tie-2 in PLs, but Ang-2 and VEGFR2 in GLLs, remodeling-associated markers also differed significantly. Because of these apparent differences, we feel that neoplastic models, such as GLLs, will be of limited use in the further study of plexiform vasculopathy.
In conclusion, we found PLs to be well-organized structures with a distinct microenvironment that sets them apart from the remodeled arteries in PAH lungs. The present results imply that PLs represent a complex, multifactorial epiphenomenon with shared characteristics in APAH and IPAH. Aside from studies in the human setting, preference should be given to animal models, which, in contrast to neoplastic models, can depict the hemodynamic aspects of PAH.52
Acknowledgments
We thank Dr. Carsten in der Wiesche and Matthias Ernst (Olympus Europa GmbH) for their excellent technical support and Gillian Teicke for editing the text.
Footnotes
Supported by the “Integriertes Forschungs-und Behandlungszentrum Transplantation” [German Federal Ministry of Education (reference no. 01EO0802)], by the European Commission under the 6th Framework Program (reference no. LSHM-CT-2005-018725, PULMOTENSION), and by the Deutsche Forschungsgemeinschaft, SFB-Transregio-37/project B4.
D.J. and H.G. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.03.040.
Supplementary data
Comparison of mRNA expression characteristics of PLs in patients with IPAH, the adjacent arteries from which they sprout, concentric lesions in patients with APAH without PLs, and controls. Expression of target genes was calculated using the Δ CT method (see Materials and Methods for details). Thick dotted line indicates the expression levels of the one patient with a known BMPR2 mutation; thin lines, medians.
References
- 1.Simonneau G., Galie N., Rubin L.J., Langleben D., Seeger W., Domenighetti G., Gibbs S., Lebrec D., Speich R., Beghetti M., Rich S., Fishman A. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S–12S. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 2.Firth A.L., Mandel J., Yuan J.X. Idiopathic pulmonary arterial hypertension. Dis Model Mech. 2010;3:268–273. doi: 10.1242/dmm.003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakao S., Tatsumi K., Voelkel N.F. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res. 2009;10:95. doi: 10.1186/1465-9921-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pietra G.G., Edwards W.D., Kay J.M., Rich S., Kernis J., Schloo B., Ayres S.M., Bergofsky E.H., Brundage B.H., Detre K.M. Histopathology of primary pulmonary hypertension: a qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, Primary Pulmonary Hypertension Registry. Circulation. 1989;80:1198–1206. doi: 10.1161/01.cir.80.5.1198. [DOI] [PubMed] [Google Scholar]
- 5.Stevens T. Molecular and cellular determinants of lung endothelial cell heterogeneity. Chest. 2005;128:558S–564S. doi: 10.1378/chest.128.6_suppl.558S. [DOI] [PubMed] [Google Scholar]
- 6.Owens G.K., Kumar M.S., Wamhoff B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 7.Cool C.D., Stewart J.S., Werahera P., Miller G.J., Williams R.L., Voelkel N.F., Tuder R.M. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers: evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol. 1999;155:411–419. doi: 10.1016/S0002-9440(10)65137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abe K., Toba M., Alzoubi A., Ito M., Fagan K.A., Cool C.D., Voelkel N.F., McMurtry I.F., Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–2754. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 9.Abe M., Kimura T., Morimoto T., Taniguchi T., Yamanaka F., Nakao K., Yagi N., Kokubu N., Kasahara Y., Kataoka Y., Otsuka Y., Kawamura A., Miyazaki S., Horiuchi K., Ito A., Hoshizaki H., Kawaguchi R., Setoguchi M., Inada T., Kishi K., Sakamoto H., Morioka N., Imai M., Shiomi H., Nonogi H., Mitsudo K. Sirolimus-eluting stent versus balloon angioplasty for sirolimus-eluting stent restenosis: insights from the j-Cypher Registry. Circulation. 2010;122:42–51. doi: 10.1161/CIRCULATIONAHA.109.905802. [DOI] [PubMed] [Google Scholar]
- 10.Tuder R.M., Voelkel N.F. Plexiform lesion in severe pulmonary hypertension: association with glomeruloid lesion. Am J Pathol. 2001;159:382–383. doi: 10.1016/S0002-9440(10)61705-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skuli N., Liu L., Runge A., Wang T., Yuan L., Patel S., Iruela-Arispe L., Simon M.C., Keith B. Endothelial deletion of hypoxia-inducible factor-2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood. 2009;114:469–477. doi: 10.1182/blood-2008-12-193581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakao S., Tatsumi K. The effects of antiangiogenic compound SU5416 in a rat model of pulmonary arterial hypertension. Respiration. 2011;81:253–261. doi: 10.1159/000322011. [DOI] [PubMed] [Google Scholar]
- 13.Tuder R.M., Groves B., Badesch D.B., Voelkel N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 14.Rai P.R., Cool C.D., King J.A., Stevens T., Burns N., Winn R.A., Kasper M., Voelkel N.F. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:558–564. doi: 10.1164/rccm.200709-1369PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heath D., Edwards J.E. The pathology of hypertensive pulmonary vascular disease: a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation. 1958;18:533–547. doi: 10.1161/01.cir.18.4.533. [DOI] [PubMed] [Google Scholar]
- 16.Ruschoff J., Dietel M., Baretton G., Arbogast S., Walch A., Monges G., Chenard M.P., Penault-Llorca F., Nagelmeier I., Schlake W., Hofler H., Kreipe H.H. HER2 diagnostics in gastric cancer-guideline validation and development of standardized immunohistochemical testing. Virchows Arch. 2010;457:299–307. doi: 10.1007/s00428-010-0952-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jonigk D., Merk M., Hussein K., Maegel L., Theophile K., Muth M., Lehmann U., Bockmeyer C.L., Mengel M., Gottlieb J., Welte T., Haverich A., Golpon H., Kreipe H., Laenger F. Obliterative airway remodeling molecular evidence for shared pathways in transplanted and native lungs. Am J Pathol. 2011;178:599–608. doi: 10.1016/j.ajpath.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jonigk D., Theophile K., Hussein K., Bock O., Lehmann U., Bockmeyer C.L., Gottlieb J., Fischer S., Simon A., Welte T., Maegel L., Kreipe H., Laenger F. Obliterative airway remodelling in transplanted and non-transplanted lungs. Virchows Arch. 2010;457:369–380. doi: 10.1007/s00428-010-0949-x. [DOI] [PubMed] [Google Scholar]
- 19.Jonigk D., Lehmann U., Stuht S., Wilhelmi M., Haverich A., Kreipe H., Mengel M. Recipient-derived neoangiogenesis of arterioles and lymphatics in quilty lesions of cardiac allografts. Transplantation. 2007;84:1335–1342. doi: 10.1097/01.tp.0000287458.72440.75. [DOI] [PubMed] [Google Scholar]
- 20.Theophile K., Jonigk D., Kreipe H., Bock O. Amplification of mRNA from laser-microdissected single or clustered cells in formalin-fixed and paraffin-embedded tissues for application in quantitative real-time PCR. Diagn Mol Pathol. 2008;17:101–106. doi: 10.1097/PDM.0b013e318163f26e. [DOI] [PubMed] [Google Scholar]
- 21.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔ CT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Fishman A.P. Changing concepts of the pulmonary plexiform lesion. Physiol Res. 2000;49:485–492. [PubMed] [Google Scholar]
- 23.Crosby A., Jones F.M., Southwood M., Stewart S., Schermuly R., Butrous G., Dunne D.W., Morrell N.W. Pulmonary vascular remodeling correlates with lung eggs and cytokines in murine schistosomiasis. Am J Respir Crit Care Med. 2010;181:279–288. doi: 10.1164/rccm.200903-0355OC. [DOI] [PubMed] [Google Scholar]
- 24.Toshner M., Voswinckel R., Southwood M., Al-Lamki R., Howard L.S., Marchesan D., Yang J., Suntharalingam J., Soon E., Exley A., Stewart S., Hecker M., Zhu Z., Gehling U., Seeger W., Pepke-Zaba J., Morrell N.W. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2009;180:780–787. doi: 10.1164/rccm.200810-1662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rensen S.S., Doevendans P.A., van Eys G.J. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007;15:100–108. doi: 10.1007/BF03085963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerthoffer W.T. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100:607–621. doi: 10.1161/01.RES.0000258492.96097.47. [DOI] [PubMed] [Google Scholar]
- 27.Mitani Y., Ueda M., Komatsu R., Maruyama K., Nagai R., Matsumura M., Sakurai M. Vascular smooth muscle cell phenotypes in primary pulmonary hypertension. Eur Respir J. 2001;17:316–320. doi: 10.1183/09031936.01.17203160. [DOI] [PubMed] [Google Scholar]
- 28.Jones R., Capen D., Jacobson M. PDGF and microvessel wall remodeling in adult lung: imaging PDGF-Rβ and PDGF-BB molecules in progenitor smooth muscle cells developing in pulmonary hypertension. Ultrastruct Pathol. 2006;30:267–281. doi: 10.1080/01913120600820336. [DOI] [PubMed] [Google Scholar]
- 29.Assaad A.M., Kawut S.M., Arcasoy S.M., Rosenzweig E.B., Wilt J.S., Sonett J.R., Borczuk A.C. Platelet-derived growth factor is increased in pulmonary capillary hemangiomatosis. Chest. 2007;131:850–855. doi: 10.1378/chest.06-1680. [DOI] [PubMed] [Google Scholar]
- 30.Zhu P., Huang L., Ge X., Yan F., Wu R., Ao Q. Transdifferentiation of pulmonary arteriolar endothelial cells into smooth muscle-like cells regulated by myocardin involved in hypoxia-induced pulmonary vascular remodelling. Int J Exp Pathol. 2006;87:463–474. doi: 10.1111/j.1365-2613.2006.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kutcher M.E., Herman I.M. The pericyte: cellular regulator of microvascular blood flow. Microvasc Res. 2009;77:235–246. doi: 10.1016/j.mvr.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennedy A., Ng C.T., Biniecka M., Saber T., Taylor C., O'Sullivan J., Veale D.J., Fearon U. Angiogenesis and blood vessel stability in inflammatory arthritis. Arthritis Rheum. 2010;62:711–721. doi: 10.1002/art.27287. [DOI] [PubMed] [Google Scholar]
- 33.Raza A., Franklin M.J., Dudek A.Z. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am J Hematol. 2010;85:593–598. doi: 10.1002/ajh.21745. [DOI] [PubMed] [Google Scholar]
- 34.Angelini D.J., Su Q., Yamaji-Kegan K., Fan C., Teng X., Hassoun P.M., Yang S.C., Champion H.C., Tuder R.M., Johns R.A. Resistin-like molecule-β in scleroderma-associated pulmonary hypertension. Am J Respir Cell Mol Biol. 2009;41:553–561. doi: 10.1165/rcmb.2008-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carreira P.E. Pulmonary hypertension in autoimmune rheumatic diseases. Autoimmun Rev. 2004;3:313–320. doi: 10.1016/j.autrev.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Hecker M., Zaslona Z., Kwapiszewska G., Niess G., Zakrzewicz A., Hergenreider E., Wilhelm J., Marsh L.M., Sedding D., Klepetko W., Lohmeyer J., Dimmeler S., Seeger W., Weissmann N., Schermuly R.T., Kneidinger N., Eickelberg O., Morty R.E. Dysregulation of the IL-13 receptor system: a novel pathomechanism in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182:805–818. doi: 10.1164/rccm.200909-1367OC. [DOI] [PubMed] [Google Scholar]
- 37.van Hinsbergh V.W., Koolwijk P. Endothelial sprouting and angiogenesis: matrix metalloproteinases in the lead. Cardiovasc Res. 2008;78:203–212. doi: 10.1093/cvr/cvm102. [DOI] [PubMed] [Google Scholar]
- 38.Holderfield M.T., Hughes C.C. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-β in vascular morphogenesis. Circ Res. 2008;102:637–652. doi: 10.1161/CIRCRESAHA.107.167171. [DOI] [PubMed] [Google Scholar]
- 39.Merks R.M., Perryn E.D., Shirinifard A., Glazier J.A. Contact-inhibited chemotaxis in de novo and sprouting blood-vessel growth. PLoS Comput Biol. 2008;4:e1000163. doi: 10.1371/journal.pcbi.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ochoa C.D., Yu L., Al-Ansari E., Hales C.A., Quinn D.A. Thrombospondin-1 null mice are resistant to hypoxia-induced pulmonary hypertension. J Cardiothorac Surg. 2010;5:32. doi: 10.1186/1749-8090-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu A., Mosher D.F., Murphy-Ullrich J.E., Goldblum S.E. The counteradhesive proteins, thrombospondin 1 and SPARC/osteonectin, open the tyrosine phosphorylation-responsive paracellular pathway in pulmonary vascular endothelia. Microvasc Res. 2009;77:13–20. doi: 10.1016/j.mvr.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papaioannou A.I., Zakynthinos E., Kostikas K., Kiropoulos T., Koutsokera A., Ziogas A., Koutroumpas A., Sakkas L., Gourgoulianis K.I., Daniil Z.D. Serum VEGF levels are related to the presence of pulmonary arterial hypertension in systemic sclerosis. BMC Pulm Med. 2009;9:18. doi: 10.1186/1471-2466-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumpers P., Nickel N., Lukasz A., Golpon H., Westerkamp V., Olsson K.M., Jonigk D., Maegel L., Bockmeyer C.L., David S., Hoeper M.M. Circulating angiopoietins in idiopathic pulmonary arterial hypertension. Eur Heart J. 2010;31:2291–2300. doi: 10.1093/eurheartj/ehq226. [DOI] [PubMed] [Google Scholar]
- 44.Ito C., Akimoto T., Ioka T., Kobayashi T., Kusano E. TGF-β inhibits vascular sprouting through TGF-β type I receptor in the mouse embryonic aorta. Tohoku J Exp Med. 2009;218:63–71. doi: 10.1620/tjem.218.63. [DOI] [PubMed] [Google Scholar]
- 45.Jayle C., Favreau F., Zhang K., Doucet C., Goujon J.M., Hebrard W., Carretier M., Eugene M., Mauco G., Tillement J.P., Hauet T. Comparison of protective effects of trimetazidine against experimental warm ischemia of different durations: early and long-term effects in a pig kidney model. Am J Physiol Renal Physiol. 2007;292:F1082–F1093. doi: 10.1152/ajprenal.00338.2006. [DOI] [PubMed] [Google Scholar]
- 46.Lario S., Bescos M., Campos B., Mur C., Luque P., Alvarez R., Campistol J.M. Thrombospondin-1 mRNA expression in experimental kidney transplantation with heart-beating and non-heart-beating donors. J Nephrol. 2007;20:588–595. [PubMed] [Google Scholar]
- 47.Pegorier S., Campbell G.A., Kay A.B., Lloyd C.M. Bone morphogenetic protein (BMP)-4 and BMP-7 regulate differentially transforming growth factor (TGF)-β1 in normal human lung fibroblasts (NHLF) Respir Res. 2010;11:85. doi: 10.1186/1465-9921-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S., de Caestecker M., Kopp J., Mitu G., Lapage J., Hirschberg R. Renal bone morphogenetic protein-7 protects against diabetic nephropathy. J Am Soc Nephrol. 2006;17:2504–2512. doi: 10.1681/ASN.2006030278. [DOI] [PubMed] [Google Scholar]
- 49.Reynolds A.M., Xia W., Holmes M.D., Hodge S.J., Danilov S., Curiel D.T., Morrell N.W., Reynolds P.N. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1182–L1192. doi: 10.1152/ajplung.00020.2006. [DOI] [PubMed] [Google Scholar]
- 50.Machado R.D., Eickelberg O., Elliott C.G., Geraci M.W., Hanaoka M., Loyd J.E., Newman J.H., Phillips J.A., III, Soubrier F., Trembath R.C., Chung W.K. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S32–S42. doi: 10.1016/j.jacc.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ricci-Vitiani L., Pallini R., Biffoni M., Todaro M., Invernici G., Cenci T., Maira G., Parati E.A., Stassi G., Larocca L.M., De Maria R. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 52.Stenmark K.R., Meyrick B., Galie N., Mooi W.J., McMurtry I.F. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–L1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of mRNA expression characteristics of PLs in patients with IPAH, the adjacent arteries from which they sprout, concentric lesions in patients with APAH without PLs, and controls. Expression of target genes was calculated using the Δ CT method (see Materials and Methods for details). Thick dotted line indicates the expression levels of the one patient with a known BMPR2 mutation; thin lines, medians.