

Abstract
Metagenomics is the study of microbial communities sampled directly from their natural environment, without prior culturing. Among the computational tools recently developed for metagenomic sequence analysis, binning tools attempt to classify the sequences in a metagenomic dataset into different bins (i.e., species), based on various DNA composition patterns (e.g., the tetramer frequencies) of various genomes. Composition-based binning methods, however, cannot be used to classify very short fragments, because of the substantial variation of DNA composition patterns within a single genome. We developed a novel approach (AbundanceBin) for metagenomics binning by utilizing the different abundances of species living in the same environment. AbundanceBin is an application of the Lander-Waterman model to metagenomics, which is based on the l-tuple content of the reads. AbundanceBin achieved accurate, unsupervised, clustering of metagenomic sequences into different bins, such that the reads classified in a bin belong to species of identical or very similar abundances in the sample. In addition, AbundanceBin gave accurate estimations of species abundances, as well as their genome sizes—two important parameters for characterizing a microbial community. We also show that AbundanceBin performed well when the sequence lengths are very short (e.g., 75 bp) or have sequencing errors. By combining AbundanceBin and a composition-based method (MetaCluster), we can achieve even higher binning accuracy. Supplementary Material is available at www.liebertonline.com/cmb.
Key words: binning, EM algorithm, metagenomics, Poisson distribution
1. Introduction
Metagenomic studies have resulted in vast amounts of sequence, sampled from a variety of environments, leading to new discoveries and insights into the uncultured microbial world (Galperin, 2004), such as the diversity of microbes in different environments (Tringe et al., 2005; Dinsdale et al., 2008b), microbial (and microbe-host) interactions (Turnbaugh et al., 2006, 2009), and the environmental and evolutionary processes that shape these communities (Dinsdale et al., 2008a). Current metagenomic projects are facilitated by the rapid advancement of DNA sequencing techniques. Recently developed next-generation sequencing (NGS) technologies (Hutchison, 2007)—such as Roche/454 (Margulies et al., 2005) and Illumina/Solexa (Bentley, 2006)—provide lower cost sequence, without the cloning step inherent in conventional capillary-based methods. These NGS technologies have increased the amount of sequence data obtained in a single run by several orders of magnitude.
One of the primary goals of metagenomic projects is to characterize the organisms present in an environmental sample and identify the metabolic roles that each organism plays. Many computational tools have been developed to infer species information from raw short reads directly, without the need for assembly; assembly of metagenomic sequences into genomes is extremely difficult, since the reads are often very short and are sampled from multiple genomes. We categorize the various computational tools for the estimation of taxonomic content into two basic classes and review them briefly.
The first class of computational tools maps metagenomic sequences to taxa with or without using phylogeny (often referred to as the phylotyping of metagenomic sequences), utilizing similarity searches of the metagenomic sequences against a database of known genes/proteins. MEGAN (Huson et al., 2007) is a representative similarity-based phylotyping tool, which applies a simple lowest common ancestor algorithm to assign reads to taxa, based on BLAST results. Phylogenetic analysis of marker genes—including 16S rRNA genes (Chakravorty et al., 2007), DNA polymerase genes (Monier et al., 2008), and the 31 marker genes defined by Ciccarelli et al. (2006)—is also applied to determining taxonomic distribution. MLTreeMap (von Mering et al., 2007) and AMPHORA (Wu and Eisen, 2008) are two phylogeny-based phylotyping tools that have been developed, using phylogenetic analysis of marker genes for taxonomic distribution estimation: MLTreeMap uses TreePuzzle (Schmidt et al., 2002), and AMPHORA uses PHYML (Guindon and Gascuel, 2003). CARMA (Krause et al., 2008) searches for conserved Pfam domains and protein families (Finn et al., 2006) in raw metagenomic sequences and classifies them into a higher-order taxonomy, based on the reconstruction of a phylogenetic tree for each matching Pfam family. These similarity-based and phylogeny-based phylotyping tools have a common limitation: they do not say much about the taxonomic distribution of the reads that do not match known genes/proteins, which may constitute the majority of the metagenomic sequences for some samples. A more recent approach, PhymmBL (Brady & Salzberg, 2009) combines similarity search and DNA composition patterns to map metagenomic sequences to taxa, achieving an improved phylogenetic classification for short reads.
A second class of computational tools attempts to solve a related but distinct problem, the binning problem, which is to cluster metagenomic sequences into different bins (species). Most existing computational tools for binning utilize DNA composition. The basis of these approaches is that genome G + C content, dinucleotide frequencies, and synonymous codon usage vary among organisms, and are generally characteristic of evolutionary lineages (Bentley and Parkhill, 2004). Tools in this category include TETRA (Teeling et al., 2004), MetaClust (Woyke et al., 2006), CompostBin (Chatterji et al., 2008), TACOA (Diaz et al., 2009), MetaCluster (Yang et al., 2010), and a genomic barcode based method (Zhou et al., 2008). Most composition-based methods achieve a reasonable performance only for long reads (at least 800 bp). TACOA is able to classify genomic fragments of length 800 and 1000 bp into the phylogenetic rank of class with high accuracy (accurate classification at the order and genus level requires fragments of ≥3 kb) (Diaz et al., 2009); CompostBin was tested on synthetic datasets of 1-kb reads (Chatterji et al., 2008). This length limitation (∼1 kb) will be difficult (if not impossible) to break, because of the local variation of DNA composition (Bentley and Parkhill, 2004). MetaCluster, one of the most recently developed composition-based methods, employs a different distance metric (Spearman footrule distance) to reduce the local variations for 4-mers so that it can bin reads of 300 bps. But reads with 50–150 bp lengths are still out of reach (Yang et al., 2010). Foerstner et al. (2005) reported that the GC content of complex microbial communities seems to be globally and actively influenced by the environment, suggesting that it may be even harder to distinguish fragments from different species living in the same environment, based on DNA composition.
In addition, metagenomic sequences may be sampled from species of very different abundances (e.g., the Acid Mine Drainage project [Tyson et al., 2004] found two dominant species, accompanied by several other rarer species in that environment), and the difference in abundances may affect the classification results for DNA-composition based approaches. For example, a weighted PCA was adopted instead of a standard PCA in CompostBin, to reduce the dimension of composition space, considering that the within-species variance in the more abundant species might be overwhelming, compared to between-species variance (Chatterji et al., 2008). MetaCluster also reported that the binning accuracy decreased when the abundance ratio increased, especially for closer species (Yang et al., 2010).
Here we report a novel binning tool, AbundanceBin, which can be used to classify very short sequences sampled from species with different abundance levels (Fig. 1a). The fundamental assumption of our method is that reads are sampled from genomes following a Poisson distribution (Lander and Waterman, 1988). In the context of metagenomics, we model the sequencing reads as a mixture of Poisson distributions. We propose an Expectation-Maximization (EM) algorithm to find parameters for the Poisson distributions, which reflect the relative abundance levels of the source species. We note that a similar method was first described by Li and Waterman (2003), for the purpose of modeling the repeat content in a conventional genome sequencing project, and Sharon et al. (2009) proposed a statistical framework for protein family frequency estimation from metagenomic sequences based on the Lander-Waterman model, given that different protein families are of different lengths. AbundanceBin assigns reads to bins using the fitted Poisson distribution. In addition, AbundanceBin gives an estimation of the genome size (or the concatenated genome size of species of the same or very similar abundances), and the coverage (which reflects the abundances of species) of each bin, all in an unsupervised manner. Since AbundanceBin is based on l-tuple content (not the composition), in principle it can be applied to classify reads that are as short as l bp. We report below first the algorithm and then tests of AbundanceBin on several synthetic metagenomic datasets and a real metagenomic dataset. We further combine AbundanceBin and another composition-based method (MetaCluster), and use this methodology to achieve even higher binning accuracy.
FIG. 1.

(a) A schematic illustration of AbundanceBin pipeline. (b) The recursive binning approach used to automatically determine the number of bins.
2. Methods
Randomized shotgun sequencing procedures result in unequal sampling of different genomes, especially when the species abundance levels differ. We seek to discover the abundance values as well as the genome sizes automatically and then bin reads accordingly. We assume that the distribution of sequenced reads follows the Lander-Waterman model (Lander and Waterman, 1988), which calculates the coverage of each nucleotide position using a Poisson distribution. We thus view the sequencing procedure in metagenomic projects as a mixture of m Poisson distributions, m being the number of species. The goal is to find the mean values λ1 to λm, which are the abundance levels of the species, of these Poisson distributions.
2.1. Mixed Poisson distributions
In random shotgun sequencing of a genome, the probability that a read starting from a certain position is N/(G − L + 1), where N is the number of reads, G is the genome size, and L is the length of reads. N/(G − L + 1) ≈ N/G, given G >> L. Assume x is a read and a l-tuple w belongs to x. The number of occurrences of w in the set of reads follows a Poisson distribution with parameter λ = N(L − l + 1)/(G − L + 1) ≈ NL/G in a random sampling process with read length L.
Similarly, for a metagnomic dataset, the number of occurrences of w in the set of reads also follows a Poisson distribution with parameter λ = N(L − l + 1)/(G − L + 1) ≈ NL/G, but G in this case is the total length of the genomic sequences contained in the metagenomic dataset. In metagenomic datasets, the reads are from species with different abundances. If the abundance of a species i is n, the total number of occurrences of w in the whole set of reads coming from this species should follow a Poisson distribution with parameter λi = nλ, due to the additivity of Poisson distribution. Now the problem of finding the relative abundance levels of different species is transformed to the modeling of mixed Poisson distributions.
2.2. Binning algorithm
Given a set of metagenomic sequences, the algorithm starts by counting l-tuples in all reads (Fig. 1a). Denote 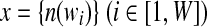 , where n(wi) is the observed count of tuple i and W is the total number of possible l-tuples. Denote S as the total number of bins (for inference of S, see Session 2.4). Denote g = {gi} and λ = {λi} (for
, where n(wi) is the observed count of tuple i and W is the total number of possible l-tuples. Denote S as the total number of bins (for inference of S, see Session 2.4). Denote g = {gi} and λ = {λi} (for 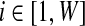 ), where gi and λi are the (collective) genome size and abundance level of bin i, respectively. Denote θ = {S, g, λ}. Then the goal of the binning algorithm is to optimize the logarithm of the joint probability (likelihood) of obtaining a particular vector of observed l-tuple counts x and the parameters θ, logP(x, θ). The hidden variables in the optimization problem are the bin identities of the l-tuples. We use an Expectation-Maximization (EM) algorithm to solve the optimization problem by marginalizing over the hidden variables. The EM steps are as follows:
), where gi and λi are the (collective) genome size and abundance level of bin i, respectively. Denote θ = {S, g, λ}. Then the goal of the binning algorithm is to optimize the logarithm of the joint probability (likelihood) of obtaining a particular vector of observed l-tuple counts x and the parameters θ, logP(x, θ). The hidden variables in the optimization problem are the bin identities of the l-tuples. We use an Expectation-Maximization (EM) algorithm to solve the optimization problem by marginalizing over the hidden variables. The EM steps are as follows:
1. Initialize the total number of bins S, their (collective) genome size gi, and abundance level λi for
 . We tested various initialization conditions and decide to set the abundance levels to the multiples of 10 (e.g., 1, 10, 20, 30, 40 for five bins) and set the genome sizes to 1,000,000 for all bins.
. We tested various initialization conditions and decide to set the abundance levels to the multiples of 10 (e.g., 1, 10, 20, 30, 40 for five bins) and set the genome sizes to 1,000,000 for all bins.2. Calculate the probability that the l-tuple wj (
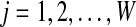 ; W is the total number of possible l-tuples) coming from ith species given its count n(wj) (for details of equation 1, see Supplementary Material, available at www.liebertonline.com/cmb).
; W is the total number of possible l-tuples) coming from ith species given its count n(wj) (for details of equation 1, see Supplementary Material, available at www.liebertonline.com/cmb).
 |
(1) |
3. Calculate the new values for each gi and λi.
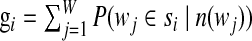 |
(2) |
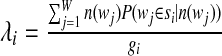 |
(3) |
4. Iterate steps 2 and 3 until the parameters converge or the number of runs exceeds a maximum number of runs. The convergence of parameters is defined as
 |
(4) |
where
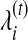 and
and 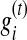 represent the abundance level and genome length of bin i at iteration t, respectively. The maximum number of runs is set to 100 (which is sufficient for the convergence of the EM algorithm for all the cases we have tested).
represent the abundance level and genome length of bin i at iteration t, respectively. The maximum number of runs is set to 100 (which is sufficient for the convergence of the EM algorithm for all the cases we have tested).
Once the EM algorithm converges, we can estimate the probability of a read assigned to a bin, based on its l-tuples binning results as,
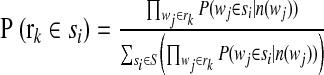 |
(5) |
where rk is a given read, wj is the l-tuples that belong to rk, and si is any bin. A read will be assigned to the bin with the highest probability among all bins. A read remains unassigned if 90% of its l-tuples are excluded (counts too low or too high; see Section 2.3), or if the highest probability is <50%.
2.3. Lower- and upper-limit for l-tuple counts
We set a lower- and upper-limit for l-tuple counts, as additional parameters, when we approximate λi and gi. The lower-limit is introduced to deal with sequencing errors, and the upper-limit is introduced to handle l-tuples with extremely high counts, such as those from vector sequences or repeats of high copy numbers. Let the lower-limit be Blower and the upper-limit be Bupper. Then the formula for calculating λi and gi is modified to
 |
(6) |
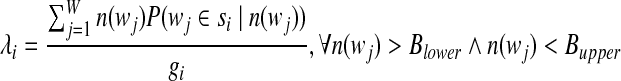 |
(7) |
2.4. Automatic determination of the total number of bins by a recursive binning approach
In the EM algorithm, we need to provide the number of bins as an input, in order to determine the parameters of the mixed Poisson distributions. However, this number is often unknown, as for most metagenomic projects. We implemented a recursive binning approach to determine the total number of bins automatically. The recursive binning approach works by separating the dataset into two bins and proceeds by further splitting bins (Fig. 1b)—it is a top-down approach. The recursive binning approach was motivated by the observation that reads from genomes with higher abundances are better classified than those with lower abundance. The recursive procedure continues if (1) the predicted abundance values of two bins differ significantly, i.e., |λi − λj|/min(λi, λj) ≥ 1/2; (2) the predicted genome sizes are larger than a certain threshold (currently set to 400,000, considering that the smallest genomes of living organisms yet found are about 500,000 bp—Nanoarchaeum equitans has a genome of 490,885 bp, and Mycoplasma genitalium has a genome of 580,073 bp); and (3) the number of reads associated with each bin is larger than a certain threshold proportion (3%) of the total number of reads classified in the parent bin.
2.5. Combination of AbundanceBin and MetaCluster, a composition-based method
Short reads sampled from species of similar abundances will be classified into the same bin by AbundanceBin. Therefore, these reads can only be further classified into different bins by other binning approaches that utilize species-specific patterns, such as DNA compositions. We combine AbundanceBin and MetaCluster, one of the most recently developed DNA composition-based binning approachs, as follows. Given a metagenomic dataset, AbundanceBin is first used to classify reads into different bins (abundance bins), and then MetaCluster is used to further classify reads in each abundance bin into species bins, each containing reads sampled from a species. We expect that such a two-step approach may achieve higher binning accuracy than using composition-based methods alone, because composition-based methods are less likely to be affected by the different abundance levels of the reads when the reads are classified into different abundance bins in advance. Note that in MetaCluster the desired number of bins needs to be defined by prior knowledge, which limits the practical application of our integrated approach. But our proof-of-concept experiments show that AbundanceBin can be used to improve the composition-based binning of reads, especially when the reads are short.
3. Results
We tested AbundanceBin on various synthetic metagenomic datasets with short (∼400 bp) and very short (∼75 bp) sequence lengths, and the results show that AbundanceBin gives both accurate classification of reads to different bins and precise estimation of the abundances—as well as the genome sizes—in each bin. Since these parameters are usually unknown in real metagenomic datasets, we focused on synthetic datasets for benchmarking. We also applied AbundanceBin to the actual AMD dataset and revealed a relatively clear picture of the complexity of the microbial community in that environment, consistent with the analysis reported in Tyson et al. (2004).
3.1. Tests of abundance differences and the length of l-tuples
We did a series of experiments to test the abundance ranges of species required for accurate binning of reads. Figure 2a shows the binning results for simulated short reads sampled from two genomes (Mycoplasma genitalium G37 and Buchnera aphidicola str. BP) at abundance ratios, 4:1, 3:1, 2.5:1, 2:1, 1.5:1, and 1:1 (with 50,000 simulated reads of ∼400 bases for each setting). The classification error rate is low if the abundance ratio is 2.0 (0.1% and 4.7% for ratio 4:1 and 2:1, respectively), but rises dramatically when the abundance ratio drops to 1.5:1 (the error rate is 20.6% for abundance ratio 1.5:1). We conclude that the abundance ratio needs to reach at least 2:1 for a good classification by AbundanceBin.
FIG. 2.

(a) The classification error rates for classifying reads sampled from two genomes versus their abundance differences. (b) The error rates for different l-tuple lengths. These error rates are averaged over four test cases, including three two-genome cases (one test case with species differ in phylum level and the other two test cases with species differ in species level) and one three-genome case.
We also test different lengths of l-tuples on several test cases, including three two-genome cases (one case with species differ in phylum level and the other two cases with species differ in species level) and one three-genome case with species differ in phylum level. The averaged error rates are shown in Figure 2b. The results show that when l drops to 16, the binning performance dropped significantly. The performance improve gradually when l increases to 20. Considering the performance on the tested cases, we choose to use l = 20 for the following experiments.
3.2. AbundanceBin achieves accurate binning, estimation of species abundance, and genome size
The binning results on several synthetic datasets of short reads are summarized in Table 1. AbundanceBin achieved both accurate estimation of species abundances, and accurate assignment of reads to bins of different abundances. The classification error rates are 0.10% and 0.64% for the classification of reads of length 400 bp and 75 bp, respectively, sampled from two genomes (cases A and C in Table 1). The error rates for the classification of reads sampled from more genomes are slightly higher than for two-genome scenarios (e.g., the classification error rates for two synthetic metagenomic datasets with reads of length 400 bp and 75 bp, sampled from three genomes, are 3.10% and 6.18%, respectively, as shown in Table 1). For the classification of reads sampled from more than two genomes, most of the errors occur in the least abundant bin. But AbundanceBin was still able to classify the reads from species of higher abundances correctly for all the tested synthetic metagenomic datasets, including one with reads sampled from six different genomes (Table 2).
Table 1.
Tests of AbundanceBin on Synthetic Metagenomic Datasets (A–D without Sequencing Errors, and E–F with Sequencing Errorsa)
| |
|
|
|
|
Abundance |
Genome size |
|
||
|---|---|---|---|---|---|---|---|---|---|
| ID | Speb | Lenc | Total reads | Bin | Real | Predicted | Real | Predicted | Error rate (%) |
| A | 2 | 400 bp | 50,000 | 1 | 27.23 | 26.27 | 580,076 | 570,859 | 0.10 (0.20d) |
| 2 | 6.83 | 6.49 | 615,980 | 614,605 | |||||
| B | 3 | 400 bp | 50,000 | 1 | 24.64 | 23.78 | 580,076 | 568,549 | 3.10 (6.64) |
| 2 | 6.13 | 6.02 | 615,980 | 517,110 | |||||
| 3 | 1.8 | 2.39 | 1,072,950 | 941,425 | |||||
| C | 2 | 75 bp | 200,000 | 1 | 20.47 | 15.66 | 580,076 | 562,584 | 0.64 (1.07) |
| 2 | 5.08 | 3.92 | 615,980 | 608,401 | |||||
| D | 3 | 75 bp | 200,000 | 1 | 27.6 | 20.93 | 580,076 | 565,859 | 6.18 (11.74) |
| 2 | 6.93 | 5.99 | 615,980 | 368,836 | |||||
| 3 | 2.07 | 2.43 | 1,072,950 | 1,100,309 | |||||
| E | 2 | 297 bp | 50,000 | 1 | 20.21 | 11.63 | 580,076 | 521,168 | 1.12 (0.99) |
| 2 | 5.07 | 3.01 | 615,980 | 945,435 | |||||
| F | 3 | 297 bp | 150,000 | 1 | 55.48 | 30.58 | 580,076 | 559,395 | 8.20 (11.41) |
| 2 | 13.98 | 9.6 | 615,980 | 341,290 | |||||
| 3 | 3.50 | 2.72 | 1,072,950 | 3,064,199 | |||||
The average sequencing error rate introduced is 3%, higher than the error rate of recent 454 machines (e.g., the accuracy rate reported in Huse et al. [2007] is 99.5%). A 3% sequencing error can reduce the l-tuple counts by about half (i.e., about 1 − 0.9720 = 0.46 of expected 20-mers without sequencing errors), which makes accurate estimation of abundance and genome size difficult.
The number of species used in simulating each metagenomic dataset. The genomes used in these tests are Mycoplasma genitalium G37, Buchnera aphidicola str. BP, and Chlamydia muridarum Nigg. The first two genomes are used for the two species cases.
The average length of the simulated reads.
Normalized error rates (see Methods for details).
Table 2.
Comparison of Binning Performance Using the Recursive Binning Approach (“Recursive”) versus the Binning When the Total Number of Bins Is Given (“Predefined”)
| |
Error rate (normalized error rate) |
|
|---|---|---|
| Test cases | Predefined | Recursive |
| Three genomes (no error model; 400 bp) | 3.10% (6.64%) | 3.24% (7.47%) |
| Three genomes (no error model; 75 bp) | 6.18% (11.74%) | 4.84% (9.31%) |
| Three genomes (454 error model; 297 bp) | 8.21% (11.41%) | 2.29% (4.21%) |
| Four genomes (no error model; 400 bp) | 1.12% (5.16%) | 2.96% (6.96%) |
| Six genomes (no error model; 400 bp) | 2.50% (9.23%) | 3.73% (13.07%) |
We emphasize here that AbundanceBin can bin reads as short as 75 bases with reasonable classification error rates (Table 1). As discussed above, binning of very short reads, such as 75 bases, is extremely difficult and cannot be achieved by any of the existing composition based binning approaches, due to the substantial variation in DNA composition within a single genome. AbundanceBin will also give an estimation of the genome size for each bin. As shown in Table 1, for most of the tested cases, the estimated genome sizes are very close to the real ones. We note that AbundanceBin will classify reads from different species of similar abundances into a single bin. In this case, the predicted genome size for that bin is actually the sum of the genome sizes of the species classified into that bin.
AbundanceBin also works well on binning closely related species (closely related species often have similar genomes, and therefore it is often very difficult to separate reads sampled from closely related species). For the synthetic metagenomic datasets we tested, most reads from species that differ at only the species level can still be classified into correct bins with very low error rates. For examples, for two datasets, the error rates for binning with AbundanceBin are 0.96% and 0.68% for the dataset simulated from the genomes of Corynebacterium efficiens YS-314 and Corynebacterium glutamicum ATCC 13032, and the dataset simulated from the genomes of Helicobacter hepaticus ATCC 51449 and Helicobacter pylori 26695 (both sets of genomes only differ at the species level), respectively. These results demonstrate the ability of our algorithm to separate short reads from closely related species, even if the species are of the same genus. (Note that AbundanceBin cannot separate reads from different strains of the same species into different bins.)
3.3. AbundanceBin can handle sequencing errors
As mentioned above, AbundanceBin can be configured to ignore l-tuples that only appear once to deal with sequencing errors, considering that those l-tuples are likely to be contributed by reads with sequencing errors and that the chance of having reads with sequencing errors at the same position will be extremely low. This may exclude some genuine l-tuples, but our tests reveal that AbundanceBin achieves even better performance if all l-tuples of count 1 are discarded for classifying reads with sequencing errors (data not shown). AbundanceBin achieves slightly worse classification of reads when reads contain sequencing errors, as compared to the classification of simulated reads without sequencing errors (see cases E and F in Table 1). This is expected, given that many spurious l-tuples are generated with a 454 sequencing error model. For example, 12,901,691 20-tuples can be found in a dataset of simulated reads from two genomes with sequencing errors (case E in Table 1), five times more than the case without error models (2,370,720).
3.4. AbundanceBin doesn't require prior knowledge of the total number of bins
Table 2 compares the performance of AbundanceBin using the recursive binning approach on several synthetic metagenomic datasets to that of AbundanceBin given the total number of bins. Overall the performances of the recursive binning approach are comparable to the cases with predefined bin numbers. Figure 3 depicts the recursive binning results of the classification of one of the synthetic metagenomic datasets (which has reads sampled from six genomes) into six bins of different abundances (with classification error rate = 3.73%), starting with a bin that includes all the reads and ending with six bins each having reads correctly assigned to them. It is interesting that the recursive binning approach achieves even better performance for some cases. A simple explanation to this observation is that the recursive binning strategy may create bigger abundance differences, especially at the beginning of the binning process, and AbundanceBin works better at separating reads from species with greater abundance differences (Fig. 2a). We note again that the high abundant bins are classified relatively well. The majority of errors occur in low abundant bins.
FIG. 3.

The recursive binning of a read dataset into six bins of different abundances (each box represents a bin with the numbers indicating the abundance of the reads classified to that bin; e.g., the bin on the top has all the reads, which will be divided into two bins, one with reads of abundances 1.5, 4, 8, and 64, and the other bin with reads of abundances 32 and 64).
3.5. Binning of acid mine drainage (AMD) datasets
The AMD microbial community was reported to consist of two species of high abundance and three other less abundant species (Tyson et al., 2004). With the difference of two abundance levels in this environment, we expect that the algorithm could classify the AMD dataset into two bins. We applied AbundanceBin to a synthetic AMD dataset (so that we have correct answers for comparison) and the real AMD dataset from Tyson et al. (2004).
The synthetic AMD dataset contains 150,000 reads from five genomes, with abundances 4:4:1:1:1. The lengths of reads is 400 bp in average. Our recursive binning approach automatically classified the reads into two bins with an error rate of 1.03% (Fig. 4a). Note here that each bin has reads sampled from multiple species. We consider that a read is classified correctly if it is classified into the bin of the correct abundance. (See session 3.6 for binning results at individual species level.) The binning accuracy dropped only slightly (with an error rate of 2.25%) for the synthetic AMD dataset when sequencing errors are introduced into the dataset.
FIG. 4.

The binning results for a simulated (a) and the actual (b) AMD datasets. The histogram shows the total number of reads from different genomes classified to each bin.
We also applied AbundanceBin to reads from the actual AMD dataset (downloaded from NCBI trace archive; 13696_environmental_sequence.007). AbundanceBin successfully classified these reads into exactly two bins (one of high abundance and one of low abundance) using the recursive binning approach (Fig. 4b). Note the reads in this dataset have vector sequences, which result in a very small number of l-tuples of extremely high abundance (the highest count is 50,720)—this phenomenon has been utilized for vector sequence removal, as described in White et al. (2008). Two approaches were employed to avoid the influences of the vector sequences: (1) we used the Figaro software package (White et al., 2008) to trim the vector sequences, and (2) we set an upper-limit for the count of all l-tuples, ignoring l-tuples with counts larger than the upper-limit (200 by default). We also downloaded the sequences of five scaffolds of the five partial genomes assembled from the AMD dataset, so that we can estimate the classification accuracy of AbundanceBin. The classification error rate of the AMD sequences is ∼14.38%. Note this error rate only gives us a rough estimation of the classification accuracy, since only 58% of the AMD reads can be mapped back to the assembled scaffolds based on similarity searches by BLAST—we mapped a read to a scaffold if the read matches the scaffold with BLAST E-value of ≤1e-50, sequence similarity of ≥95%, and a matched length of ≥70% of the read length. We emphasize that AbundanceBin achieved a much better classification (with an error rate of 1.03%) for the synthetic AMD reads, for which we have correct answers to compare with.
3.6. AbundanceBin complements composition-based binning approaches
AbundanceBin can achieve accurate binning of very short metagenomic reads by utilizing abundance differences of the source species of the reads as shown above. However, it cannot be used to separate reads sampled from species of similar abundances. On the other hand, the performance of composition-based binning approaches drops for binning reads with abundance differences. We combine AbundanceBin (an abundance-based binning approach) and MetaCluster (a composition-based binning approach), so that reads of different abundances can first be separated and then reads of similar abundances can be further classified. We apply this methodology to classify both the synthetic and real AMD datasets. As shown in Table 3, the classification results of this combined approach are better than those of MetaCluster. For the synthetic AMD dataset with 400 bp reads, the error rate of the combined approach is 4.72%, much lower than 26.82% by using MetaCluster alone. Similar trend also exists for the real AMD dataset: the error rate of the combined approach (21.76%) is lower than that of MetaCluster (51.15%).
Table 3.
Comparison of Binning Performance Using the Combined Approach of AbundanceBin and MetaCluster and Using MetaCluster Alone
| |
Error rate (normalized error rate) |
|
|---|---|---|
| Test cases | Combined approach | MetaCluster |
| Synthetic AMD dataset (400 bp) | 4.72% (5.71%) | 26.82% (24.23%) |
| Synthetic AMD dataset (250 bp) | 9.74% (11.48%) | 29.83% (36.75%) |
| Synthetic AMD dataset (125 bp) | 22.22% (25.29%) | 40.63% (50.53%) |
| Synthetic AMD dataset (75 bp) | 37.71% (36.25%) | 59.16% (63.09%) |
| Real AMD dataset | 21.76% (36.81%) | 51.15% (66.24%) |
Current composition-based approaches cannot classify very short reads. To test to what extent AbundantBin can help composition-based methods for binning, we simulated AMD datasets with different read lengths ranging from 75 bp to 250 bp—current Illumina sequencers are able to generate longer reads (>75 bp; see http://illumina.com/systems/hiseq_2000.ilmn), which are still shorter than pyrosequences and Sanger sequences. The binning results are summarized in Table 3. The classification error rates for the combined approach in all test cases are significantly lower than MetaCluster. The majority of errors are caused by the inability of MetaCluster to separate very short sequences (due to the local variation of DNA composition patterns). Overall, these results demonstrate that a better binning can be achieved to separate metagenomic reads by combining orthogonal information, the abundance differences of the source species and their different composition patterns.
4. Conclusion
We have shown that our abundance-based algorithm for binning has the ability to classify short reads from species with different abundances. Our approach has two unique features. First, our method is “unsupervised” (i.e., it doesn't require any prior knowledge for the binning). Second, our method is especially suitable for short reads, as long as the length of reads exceeds the length of the l-tuple (e.g., 20). AbundanceBin can in principle be applied to any metatagenomic sequences acquired by current NGS, without human interpretation.
Since the initialization conditions of the EM algorithm could have an impact on the convergence, we test various initialization conditions and finally decide to set the abundance levels to the multiples of 10 and the genome sizes to 1,000,000 for all species. The advantage of this setting is that the abundance differences are big enough so that each bin will converge to the correct direction. This setting works well for all datasets that we tested, including synthetic datasets and real datasets.
We implemented a simple strategy—excluding l-tuples that are counted only once from the abundance estimation—to handle sequencing errors, and tests have showed that AbundanceBin achieved better classification if all l-tuples of single count are discarded for the test cases that contain sequencing errors. One potential problem of discarding l-tuples of low counts is that some genuine l-tuples will be discarded as well, which results in a lower abundance estimation and a worse prediction of genome sizes, especially for the species with low abundance, as shown in Table 1. But we argue that AbundanceBin can still capture the relative abundances of different bins correctly, which is more important than the absolute values. Another potential problem is that reads from low abundant genomes may not be classified when sequencing errors are introduced in the reads. For example, the number of unclassified reads in a two-genome case (metagenomic dataset E in Table 1) is 12, and 389 in a three-genome case (metagenomic dataset F in Table 1). All unclassified reads in both cases belong to the least abundant species, indicating that the abundance values greatly affect the predicted results, especially when sequencing errors are present. We expect that both problems will become less problematic as sequencing coverage is increased, which is possible with massive throughput NGS techniques. As for the abundance ratio required for successful classification, we find that the ratio should be at least 2:1 to obtain an acceptable result. The required ratio, of course, is also affected by several other factors, such as the actual abundance level, the average length of reads, and the sequencing error rate. Our tests intentionally use well-classified datasets to allow us to follow changes in classification error resulting from abundance differences, but other factors besides the abundance ratio must also be considered.
AbundanceBin runs fast, and all the tests shown in the paper were completed within 1 hour (using a single CPU on Intel® Xeon® 2.00 GHz) with very moderate memory usage. For example, binning of the synthetic metagenomic dataset A (Table 1) requires 100 MB memory and takes less than 2 minutes; binning of dataset B requires 150 MB memory and also takes less than 2 minutes. Even for larger dataset such as the simulated AMD dataset, which contains 150,000 reads, the binning process needs only 300 MB memory and takes about 7 minutes. Therefore, AbundanceBin requires only modest amount of memory, unless it is dealing with very large datasets.
Since AbundanceBin employs a unique feature—species abundance levels—to achieve binning of reads, it can be used to assist other tools to analyze metagenomic datasets. To demonstrate this usage, we combine the power of AbundanceBin and MetaCluster to separate datasets with species abundance differences. We apply this methodology on the synthetic and the real AMD dataset, and the results are satisfactory: the error rates of this combined approach are much lower than those of MetaCluster for both tests. These results confirm our hypothesis that, by separating the whole dataset into several sub-datasets, each containing reads with similar abundance level, the composition-based approach can be applied to each sub-dataset, without being influenced by the differences in abundance levels. We will test different strategies for determining the number of bins for MetaCluster, by either using phylogenetic marker genes for assessing the total number of species as in Chatterji et al. (2008), or testing different clustering algorithms that can automatically determine the total number of clusters (Teeling et al., 2004; Woyke et al., 2006; Chatterji et al., 2008; Diaz et al., 2009; Yang et al., 2010). Our tests show that by integrating different information, we may improve binning accuracy. But binning problem for very short reads (e.g., of 75 bp) is still an extremely difficult problem to solve. We also expect that applying AbundanceBin to separate reads into bins of different abundances (coverages), prior to the assembly of metagenomic sequences, will improve the quality of genome assembly.
Supplementary Material
Acknowledgments
We thank Dr. Haixu Tang for helpful discussions and Dr. Thomas G. Doak for reading the manuscript. This research was supported by NIH grant 1R01HG004908-02 and NSF CAREER award DBI-084568.
Disclosure Statement
No competing financial interests exist.
References
- Bentley D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 2006;16:545–552. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Bentley S.D. Parkhill J. Comparative genomic structure of prokaryotes. Annu. Rev. Genet. 2004;38:771–792. doi: 10.1146/annurev.genet.38.072902.094318. [DOI] [PubMed] [Google Scholar]
- Brady A. Salzberg S.L. Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat. Methods. 2009;6:673–676. doi: 10.1038/nmeth.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty S. Helb D. Burday M., et al. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods. 2007;69:330–339. doi: 10.1016/j.mimet.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterji S. Yamazaki I. Bai Z., et al. CompostBin: a DNA composition-based algorithm for binning environmental shotgun reads. Proc. RECOMB 2008. 2008:17–28. [Google Scholar]
- Ciccarelli F.D. Doerks T. von Mering C., et al. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- Diaz N.N. Krause L. Goesmann A., et al. TACOA: taxonomic classification of environmental genomic fragments using a kernelized nearest neighbor approach. BMC Bioinformatics. 2009;10:56. doi: 10.1186/1471-2105-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinsdale E.A. Edwards R.A. Hall D., et al. Functional metagenomic profiling of nine biomes. Nature. 2008a;452:629–632. doi: 10.1038/nature06810. [DOI] [PubMed] [Google Scholar]
- Dinsdale E.A. Pantos O. Smriga S., et al. Microbial ecology of four coral atolls in the Northern Line Islands. PLoS ONE. 2008b;3:e158. doi: 10.1371/journal.pone.0001584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D. Mistry J. Schuster-Bockler B., et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foerstner K.U. von Mering C. Hooper S.D., et al. Environments shape the nucleotide composition of genomes. EMBO Rep. 2005;6:1208–1213. doi: 10.1038/sj.embor.7400538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin M. Metagenomics: from acid mine to shining sea. Environ. Microbiol. 2004;6:543–545. doi: 10.1111/j.1462-2920.2004.00652.x. [DOI] [PubMed] [Google Scholar]
- Guindon S. Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Huse S.M. Huber J.A. Morrison H.G., et al. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D.H. Auch A.F. Qi J., et al. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison C.A., 3rd DNA sequencing: bench to bedside and beyond. Nucleic Acids Res. 2007;35:6227–6237. doi: 10.1093/nar/gkm688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause L. Diaz N.N. Goesmann A., et al. Phylogenetic classification of short environmental DNA fragments. Nucleic Acids Res. 2008;36:2230–2239. doi: 10.1093/nar/gkn038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E.S. Waterman M.S. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics. 1988;2:231–239. doi: 10.1016/0888-7543(88)90007-9. [DOI] [PubMed] [Google Scholar]
- Li X. Waterman M.S. Estimating the repeat structure and length of DNA sequences using l-tuples. Genome Res. 2003;13:1916–1922. doi: 10.1101/gr.1251803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M. Egholm M. Altman W.E., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monier A. Claverie J.M. Ogata H. Taxonomic distribution of large DNA viruses in the sea. Genome Biol. 2008;9:R106. doi: 10.1186/gb-2008-9-7-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H.A. Strimmer K. Vingron M., et al. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Sharon I. Pati A. Markowitz V.M., et al. A statistical framework for the functional analysis of metagenomes. Proc. RECOMB 2009. 2009:496–511. [Google Scholar]
- Teeling H. Waldmann J. Lombardot T., et al. TETRA: a web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinformatics. 2004;5:163. doi: 10.1186/1471-2105-5-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe S.G. von Mering C. Kobayashi A., et al. Comparative metagenomics of microbial communities. Science. 2005;308:554–557. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- Turnbaugh P.J. Hamady M. Yatsunenko T., et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P.J. Ley R.E. Mahowald M.A., et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1131. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Tyson G.W. Chapman J. Hugenholtz P., et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- von Mering C. Hugenholtz P. Raes J., et al. Quantitative phylogenetic assessment of microbial communities in diverse environments. Science. 2007;315:1126–1130. doi: 10.1126/science.1133420. [DOI] [PubMed] [Google Scholar]
- White J.R. Roberts M. Yorke J.A., et al. Figaro: a novel statistical method for vector sequence removal. Bioinformatics. 2008;24:462–467. doi: 10.1093/bioinformatics/btm632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyke T. Teeling H. Ivanova N.N., et al. Symbiosis insights through metagenomic analysis of a microbial consortium. Nature. 2006;443:950–955. doi: 10.1038/nature05192. [DOI] [PubMed] [Google Scholar]
- Wu M. Eisen J.A. A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 2008;9:R151. doi: 10.1186/gb-2008-9-10-r151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B. Peng Y. Leung H.C.M., et al. MetaCluster: unsupervised binning of environmental genomic fragments and taxonomic annotation. ACM-BCB 2010. 2010:17–28. [Google Scholar]
- Zhou F. Olman V. Xu Y. Barcodes for genomes and applications. BMC Bioinformatics. 2008;9:546. doi: 10.1186/1471-2105-9-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.