

Abstract
Alzheimer's disease (AD) is a neurodegenerative pathology characterized by the presence of senile plaques and neurofibrillary tangles, accompanied by synaptic and neuronal loss. The major component of senile plaques is an amyloid β protein (Aβ) formed by pathological processing of the Aβ precursor protein. We assessed the time-course and regional effects of a single intracerebroventricular injection of aggregated Aβ fragment 25–35 (Aβ25-35) in rats. Using a combined biochemical, behavioral, and morphological approach, we analyzed the peptide effects after 1, 2, and 3 weeks in the hippocampus, cortex, amygdala, and hypothalamus. The scrambled Aβ25-35 peptide was used as negative control. The aggregated forms of Aβ peptides were first characterized using electron microscopy, infrared spectroscopy, and Congo Red staining. Intracerebroventricular injection of Aβ25-35 decreased body weight, induced short- and long-term memory impairments, increased endocrine stress, cerebral oxidative and cellular stress, neuroinflammation, and neuroprotective reactions, and modified endogenous amyloid processing, with specific time-course and regional responses. Moreover, Aβ25-35, the presence of which was shown in the different brain structures and over 3 weeks, provoked a rapid glial activation, acetylcholine homeostasis perturbation, and hippocampal morphological alterations. In conclusion, the acute intracerebroventricular Aβ25-35 injection induced substantial central modifications in rats, highly reminiscent of the human physiopathology, that could contribute to physiological and cognitive deficits observed in AD.
Alzheimer's disease (AD) is a chronic neurodegenerative pathology characterized by the presence of senile plaques and neurofibrillary tangles, accompanied by synaptic and neuronal loss in brain areas responsible for learning and memory impairments.1 The major component of senile plaques is an amyloid β protein (Aβ) derived from amyloid precursor protein (APP). Genetic, cell biological, and postmortem studies on AD brain, together with Aβ neurotoxicity findings, gave rise to the amyloid cascade hypothesis to explain Aβ-associated neurodegenerative processes.2 In normal healthy individuals, Aβ peptides are present only in small quantities, as soluble monomers that circulate in cerebrospinal fluid and blood. In AD patients, Aβ peptides, which vary in length from 40 to 43 amino acids, accumulate as insoluble fibrillar deposits.3
When cultured rat hippocampal neurons are exposed to aggregated Aβ peptides, their neurites adopt a dystrophic appearance and become comparable to those observed surrounding and infiltrating senile plaques. This observation suggested that Aβ is responsible for the neuritic abnormalities in AD pathology.4 Structure-activity studies revealed that peptides containing the highly hydrophobic 25–35 region formed stable aggregates and mediated neuronal death by necrosis or apoptosis.5,6,7 The truncated Aβ25-35 fragment includes extracellular and intramembrane residues that have been reported to represent an active region of Aβ.8
In vivo, two nontransgenic rodent models of AD have been studied to analyze the molecular, morphological, and behavioral consequences of amyloid toxicity: the infusion of Aβ1-40/42 protein and the injection of preaggregated Aβ25-35 peptide. These models have led to highly pertinent pathomimetic observations and provide complementary approaches to the numerous transgenic mouse lines developed as AD models. In particular, at 1 week after Aβ25-35 injection, reactive gliosis was observed in the rat hippocampus,9 caspase-3 activity was induced in the hippocampus and cortex,9 a reduction in the number of neurons was measured in the CA1 or CA3 hippocampal area,9–11 and significant oxidative stress was observed.11–13
Nonetheless, this amyloid toxicity model remains controversial with respect to AD. The grounds for objection center on three points. First, although the presence of Aβ25-35 has been repeatedly demonstrated in brains of AD patients,14,15 this fragment appears to be a minor peptide in plaques and its contribution to the toxicity induced by all amyloid-related species remains unclear. Second, the intracerebroventricular injection of Aβ25-35 induces marked toxicity within days or weeks, which is not in accord with the long-term progression of AD in humans (an argument that is unrelated to the nature of the toxicity). Third, it is possible that peptide injection mimics only the amyloid toxicity observed in AD physiopathology, but not the processes involved in Aβ formation or Tau protein hyperphosphorylation. Klementiev et al16 recently documented some evidence that contradicts this latter reservation. Moreover, very recently, Chavant et al17 reported that, at 2 weeks after injection, Aβ25-35 significantly increased APP processing in the hippocampus of mice.
To realize the potential of such a nontransgenic model for the physiopathological processes occurring in AD, the extent and time course of the toxicity induced after amyloid peptide injection and the differential vulnerability of brain structures need to be established. We therefore analyzed the consequences of intracerebroventricular injection of aggregated Aβ25-35 in terms of time-course changes and in different brain structures through several morphological, hormonal, biochemical, and behavioral parameters, as well as addressing the modifications in central APP processing, brain-derived neurotrophic factor (BDNF) levels, cerebral inflammation, and hippocampus vulnerability. We also evaluated over time the distribution within brain structures of the injected Aβ25-35 fragment.
Materials and Methods
Animals
Adult male Sprague-Dawley rats (Depré, Saint-Doulchard, France) aged 6 to 7 weeks and weighing 260 to 280 g at the beginning of the experiments were housed under standard laboratory conditions (12 hours/12 hours light/dark cycle with lights on at 7:00 AM; 21 ± 1°C; food and water ad libitum). The animals were treated in accordance with the European Community Council Directive 86/609 on the protection of animals used for experimental and other scientific purposes. All experiments were performed on conscious rats between 9:00 AM and 2:00 PM (ie, during the diurnal trough of the circadian rhythm). Animals were weighed daily.
Amyloid β Peptide
Amyloid β fragment 25–35 (Aβ25-35) (Figure 1A) and scrambled Aβ25-35 peptides (NeoMPS, Strasbourg, France) were dissolved in sterile bidistilled water at 1 μg/μL concentration (soluble form) and were stored at −20°C. The scrambled Aβ25-35 and Aβ25-35 peptides were aggregated by in vitro incubation at 37°C for 4 days.18 The human Aβ1-42 protein, which was synthesized at the Keck Foundation at Yale University, was generously provided by Dr. Einar Sigurdsson.19
Figure 1.
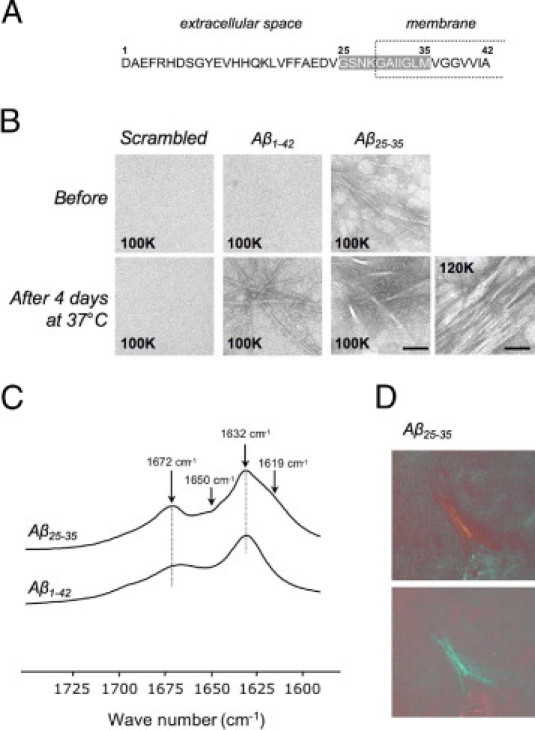
A: Amino acid sequence of Aβ1-42. Shading indicates the Aβ25-35 sequence; the intramembrane domain is indicated by a dashed box. B: Electron microscopy of scrambled Aβ25-35, Aβ1-42, and Aβ25-35 peptides before and after in vitro incubation at 37°C for 4 days. Aβ1-42 and Aβ25-35 formed an extensive network of fibrils after 4 days at 37°C, but scrambled peptide did not; the image at the far right presents a higher-magnification view for the Aβ25-35 after incubation. Original magnification, ×100,000 (100K; scale bar = 50 nm) or ×120,000 (120K; scale bar = 60 nm). C: Fourier-transformed IR spectra of Aβ1-42 and Aβ25-35 peptides preaggregated in vitro at 37°C for 4 days. The spectrum of Aβ25-35 aggregates revealed two additional bands at 1619 and 1650 cm−1, compared with the spectrum of Aβ1-42 aggregates. D: Congo Red staining of Aβ25-35 peptide preaggregated in vitro at 37°C for 4 days. The Congo Red-stained sample was viewed under polarized light. The green birefringence was observed in preaggregated Aβ25-35 samples.
Electron Microscopy
The ability of the scrambled Aβ25-35, Aβ1-42, and Aβ25-35 peptides to form fibrils after 4 days incubation at 37°C was assessed by electron microscopy. The peptide solutions were placed on a copper Formvar carbon-coated grid (Oxford Instruments, Abingdon, UK). After 2 minutes adsorption at room temperature, samples were counterstained with 2% uranyl acetate (Sigma-Aldrich, Saint-Quentin Fallavier, France) and then examined under a JEOL-1200EX2 transmission electron microscope (JEOL, Tokyo, Japan).
Fourier-Transformed IR Spectroscopy
Infrared spectra were obtained with a Vertex-70 Fourier-transformed IR spectrometer (Bruker, Ettlingen, Germany). The spectra (200 scan accumulation) were coadded after registration at a spectral resolution of 2 cm−1 and analyzed using Bruker OPUS 6.5 software. For comparison of β-amyloid aggregated peptides (Aβ1-42 and Aβ25-35), samples that had been incubated for 4 days at 37°C were deposited onto a CaF2 plate and the solvent was allowed to evaporate overnight at room temperature.20
Congo Red Staining
Congo Red staining was used to determine whether Aβ had a cross β-pleated sheet structure in Aβ (a typical feature of amyloid fibrils). Congo Red does not bind to dimers or trimers, but only to higher-order aggregates (the so-called protofibrils and amyloid-like fibrils). The complex of Congo Red and Aβ fibrils is seen as a green birefringence under polarized light. After 4 days of incubation at 37°C, the sample was put onto the bioadhesive hydrophobic printed slide and allowed to dry at room temperature (2 hours). Congo Red staining was obtained using a commercial kit in accordance with the manufacturer's protocol (Sigma-Aldrich). The slides were viewed under polarized light using a microscope equipped with polarizing filter (Leica DMR, Nanterre, France).
Experimental Procedures
Animals were divided into three groups. One group was left undisturbed (control rats), a second group received an injection of an incubated scrambled peptide (10 μg/rat i.c.v.), and a third group received an injection of aggregated Aβ25-35 peptide (10 μg/rat i.c.v.).21 For the intracerebroventricular injection through a Hamilton syringe (VWR International, Fontenay-sous-Bois, France), the animals were anesthetized with an intramuscular injection of 0.2 mL of a mixture of ketamine hydrochloride (80 mg/kg body weight) and xylazine (10 mg/kg body weight). They were then injected stereotactically directly into the lateral ventricles at the following coordinates: AP, −1 mm; L, ±1.5 mm; and DV, −3.5 mm.22
General Locomotor Activity and Body Temperature Variations
On the day of the intracerebroventricular injection and during the same anesthetic session, a single telemetric transmitter (PhysioTel; Data Sciences International–DSI, St. Paul, MN) was implanted intraperitoneally. The corresponding receiver (RA1010; DSI) was fixed under the animal's cage and was connected via a BMC100 consolidation matrix (DSI) to a Dataquest III computerized data analyzer (DSI). The animals were then recorded during the 3 weeks after peptide injection and the telemetric data were analyzed. This system allows measurement of continuous general locomotor activity and body temperature variations, as described previously.23
Spatial Short-Term Memory
The delayed alternation of rats was assessed in the T-maze test.24 The T-maze consisted of two short arms (A and B), extending from a longer alley and enclosed with high walls. The test involved two trials separated by 1 hour. During the training session, one short arm (B) was closed. Rats were placed at the end of the long alley, allowed to visit the maze for 10 minutes and then returned into their home cage. During the test session, animals were placed in the maze for 2 minutes, with free access to both arms. The number of visits and time spent in each arm were measured. The results were expressed as the ratio of the time spent in the initially closed novel arm to the time spent in the familiar arm, and as the ratio of the number of entries into the novel arm to the number of entries into the familiar arm.
Spatial Long-Term Memory
Place learning was tested in a water maze.21 The maze was a circular pool (diameter 150 cm, height 40 cm) divided into four quadrants. The water temperature (25 ± 2°C), light intensity, external cues in the room, and water opacity were rigorously reproduced. A transparent acrylic glass platform (diameter 10 cm) was immersed 2 cm under the water surface at the center of one quadrant (the training quadrant). Training consisted of three swims per day for 5 days. Each rat was allowed a 90-second swim to find the platform and was left on the platform for 30 seconds. The median latency was determined during each training sessions. A probe test was performed 4 hours after the last training session. The platform was removed and each rat was allowed a free 60-second swim. The percentage of time spent in the training quadrant was determined.
Endocrine Stress
Blood samples were collected after the rats were sacrificed by decapitation, as described previously.25 A radioimmunoassay kit (Biotrak; GE Healthcare, Little Chalfont, UK) was used to assay plasma corticosterone in a 50-μL plasma sample diluted (1:5) with the assay buffer. The intra- and interassay coefficients of variation were 5% and 7%, respectively. The assay sensitivity was 0.6 ng/mL.
Oxidative Stress
Quantification of lipid peroxidation in tissue extracts was based on Fe(III) Xylenol Orange complex formation according to the Hermes-Lima method,26 as described previously.12
Cellular Stress
Glial fibrillary acidic protein, caspase-9, caspase-12, and caspase-3 were used as measures of cellular stress.
Rats were sacrificed by decapitation and structures of interest were weighed, immediately frozen in liquid nitrogen, and stored at −20°C. Tissues were sonicated with a VibraCell ultrasonic processor (Sonics & Materials, Newtown, CT) in 2% SDS. To detect glial fibrillary acidic protein (GFAP), homogenates of pro- and cleaved caspase-9, pro- and cleaved caspase-12, and pro-caspase-3 were then boiled for 5 minutes and centrifuged for 30 minutes at 14,000 × g. To detect cleaved caspase-3, tissues were homogenized using a specific lysis buffer (Triton X100 1%; Tris-HCl pH 7.5, 20 mmol/L; NaCl 150 mmol/L; EDTA 10 mmol/L; Na3VO4 100 μmol/L), as described previously.27 Supernatants were collected and the protein concentration was measured using a Pierce BCA protein assay kit (Thermo Fisher Scientific, Brebieres, France) and 20 to 40 μg from each sample was taken for Western blot analysis, depending on the structure and the antigen involved. Samples were boiled for 5 minutes, separated in SDS polyacrylamide gel (12%), and transferred to a Whatman nitrocellulose membrane (GE Healthcare). The membrane was incubated overnight (4°C) with either a mouse anti-GFAP (1:1000; Sigma-Aldrich), a rabbit anti-procaspase-3 or a rabbit monoclonal anti-caspase-3 cleaved form (1:1000 and 1:200, respectively; Cell Signaling Technology; Ozyme, Saint Quentin en Yvelines, France), a rabbit anti-caspase-9 (pro- and cleaved forms; 1:1000; Cell Signaling Technology), a rat anti-caspase-12 (pro- and cleaved forms; 1:5000; Sigma-Aldrich), or a mouse anti-β-tubulin (1:5000; Sigma-Aldrich). The membrane was then rinsed and incubated for 2 hours with the appropriate horseradish peroxidase-conjugated secondary antibodies (Sigma-Aldrich). Peroxidase activity was revealed by using enhanced-chemiluminescence (ECL; Millipore, Saint Quentin en Yvelines, France) reagents. The intensity of peroxidase activity was quantified using ImageJ software version 1.44 (NIH, Bethesda, MD). The β-tubulin was taken as loading control for all immunoblotting experiments.
APP Processing
To determine the effect of Aβ25-35 intracerebroventricular injection on APP processing, 60 μg from each sample was taken for Western blot analysis according to the procedures detailed above. The primary antibody used to detect APP (125 kDa) and C99 fragment (13 kDa) was a rabbit anti-amyloid precursor protein (PA1-84165; 1:750; Thermo Fisher Scientific).
BDNF Content
Rats were sacrificed by decapitation and structures of interest were weighed, immediately frozen in liquid nitrogen, and stored at −20°C until assayed. BDNF content was measured with a conventional enzyme-linked immunosorbent assay (BDNF-Emax immunoassay system; Promega, Madison, WI; Charbonnières-les-Bains, France), as described previously.25 The assay sensitivity was 15 pg/tube. The BDNF concentration was expressed as pg/g wet weight. The intra- and interassay coefficients of variation were 3% and 6%, respectively.
Histology
For histological analyses, we used cresyl violet staining and immunolabeling with GFAP, ionized calcium binding adaptor molecule 1 (Iba-1), poly-sialic acid neural cell adhesion molecule (PSA-NCAM), and vesicular acetylcholine transporter (VAChT).
Animals were anesthetized using an intramuscular injection of ketamine/xylazine solution and were perfused intracardially with 50 mL of NaCl 0.9% followed by 100 mL phosphate buffer 0.2 mol/L containing 4% paraformaldehyde. Brains were removed and postfixed in the same fixative for 48 hours (4°C) and then in a sucrose solution (30%) for 3 days. Thereafter, the tissues were included in a block of optimal cutting temperature compound (Tissue-Tek; Sakura Finetek, Torrance, CA) and quickly frozen in acetone chilled on dry ice. Frozen brains were mounted on a cryostat (Leica) and serially cut into 10-μm coronal sections. For histology, sections were stained with 0.2% cresyl violet reagent, dehydrated, and mounted. Four sections were studied from each brain, taken from the anterior hippocampus level (−3.0 to −4.0 from the bregma),22 with intervals of 250 μm. Sections were selected on a subjective random basis. Numbers of neurons in the different hippocampal fields were counted as described previously.9,10,13 Counting was performed using a light microscope (Leica DMR). Digitized images were acquired using a 40× objective and were analyzed with ImageJ software. Neuron densities on slices (number of neurons in optical field expressed as the number of cells per square millimeter) were calculated as the arithmetic mean number of neurons in the two hemispheres and, for each animal, as the arithmetic mean of results obtained for each of the four slices.
Analyses of glial (GFAP) and acetylcholine (VAChT) markers were conducted according to a diaminobenzidine immunohistochemistry (IHC) approach; analyses of microglial (Iba-1) and neurogenesis (PSA-NCAM) markers were conducted according to a fluorescent IHC approach.28 Sections were incubated overnight at room temperature with a mouse anti-GFAP (1:1000, Sigma-Aldrich), a rabbit anti-Iba-1 (1:500; Wako Pure Chemical Industries, Osaka, Japan), a rabbit anti-VAChT (1:500; Sigma-Aldrich), or a rabbit anti-PSA-NCAM antibody (1:200; AbCys, Paris, France). Sections were then incubated for 2 hours with the appropriate biotinylated (anti-GFAP and anti-VAChT) or fluorescent (Alexa Fluor 488; anti-Iba-1 and anti-PSA-NCAM) secondary antibodies (Sigma-Aldrich or Molecular Probes, Leiden, The Netherlands). Biotinylated sections were incubated for 1 hour in avidin-biotin complex (ABC kit; Vector Laboratories, Burlingame, CA). The signal was detected with a diaminobenzidine kit (Vector Laboratories) according to the manufacturer's instructions, and the nuclei of fluorescent sections were counterstained with DAPI (Molecular Probes). Immunostaining specificity was determined by incubating control sections with the secondary antiserum alone.
Aβ25-35 Distribution
To analyze the distribution within brain structures of the injected Aβ25-35 fragment, we used a peptide tagged with a fluorescent dye: Aβ25-35 HiLyte Fluor 488 (Aβ25-35-HLF; AnaSpec/Eurogentec, Angers, France). The ability of Aβ25-35-HLF to form amyloid fibrils after 4 days of incubation at 37°C was assessed by electron microscopy and Congo Red staining, as described previously. At different times (1, 3, and 12 hours; 1 and 3 days; and 1, 2, and 3 weeks) after the intracerebroventricular Aβ25-35-HLF injection, the animals were anesthetized and perfused and their brains were removed and postfixed, as described previously. Coronal sections (30 μm) were prepared with a vibrating-blade microtome (Leica). Before being mounted, sections were counterstained with DAPI to visualize nuclei.
Statistical Analysis
Data are expressed as means ± SEM. Comparisons were performed using one-way or two-way analysis of variance, followed by a Fisher's multiple comparison test or using a Mann-Whitney nonparametric test. P < 0.05 was considered significant.
Results
Molecular Properties of Aβ25-35
No aggregate of negatively stained scrambled or Aβ1-42 peptides was observed in electron micrographs immediately after suspension (Figure 1B). In contrast, dense and heterogeneous aggregates were detected in Aβ25-35 solution. The morphology of Aβ aggregates changed after incubation. Over the entire microscope grid, Aβ1-42 aggregation exhibited long, straight fibrils. Some fibrils were associated together. For Aβ25-35 aggregates, higher-order structures composed of large bundles and sharp fibrils of various length and order dominated.
Secondary aggregated peptide structures were analyzed using Fourier-transformed IR spectroscopy (Figure 1C). In both spectra, strong amide I bands were characterized by two peaks centered at 1632 and 1672 cm−1, typical of the antiparallel β-sheet structure of amyloid fibrils.29 The Aβ25-35 spectrum revealed two additional features. The contribution of the band at 1619 cm−1, which is characteristic of intermolecular β-sheet structures and/or aggregated strands,30 reflected the propensity of Aβ25-35 to form bundles (Figure 1B). The weak band at approximately 1650 cm−1 could be assigned to non-β-sheet structures.31
Using Congo Red staining, green birefringent material was observed in preaggregated Aβ25-35 peptide samples under polarized light, indicative of the presence of amyloid-like structures (Figure 1D). This material was absent in preaggregated scrambled Aβ25-35 peptide (data not shown).
Regional Distribution of Injected Aβ25-35
Because previous studies did not examine the regional distribution of Aβ25-35 peptide after intracerebroventricular injection, we first addressed this point using a HiLyte Fluor 488-tagged peptide (Aβ25-35-HLF). After having verified the ability of Aβ25-35-HLF to form amyloid fibrils after 4 days of incubation at 37°C by electron microscopy (Figure 2A) and Congophilic staining (Figure 2B), the localization of the peptide was evaluated in the brain at 1, 3, and 12 hours, 1 and 3 days, and 1, 2, and 3 weeks after intracerebroventricular injection. The observations are presented in Figure 2 and summarized in Table 1. Control rat sections were treated and examined under the same conditions as injected rat sections and displayed no specific labeling. Aβ25-35-HLF was present over time at the injection site (Figure 2, C–G). At this site, Aβ25-35-HLF accumulated and was progressively trapped by local cells (Figure 2, F and G).
Figure 2.
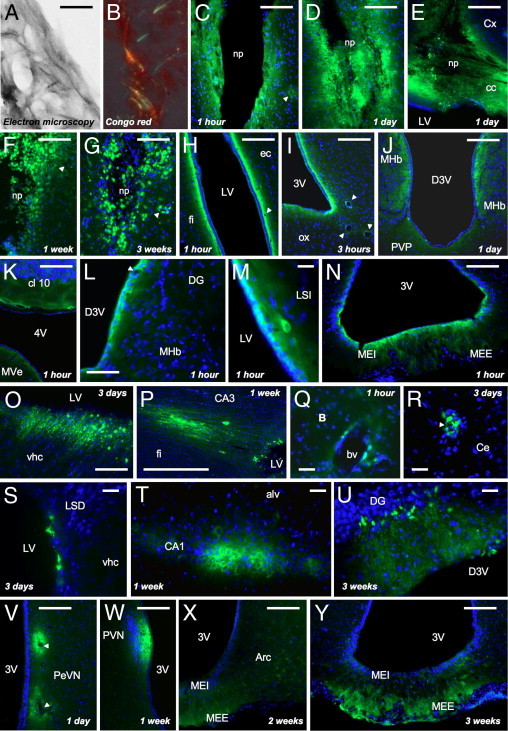
A: Electron microscopy of Aβ25-35-HLF peptide after in vitro incubation at 37°C for 4 days. Aβ25-35-HLF formed an extensive network of fibrils after 4 days at 37°C. Scale bar = 120 nm. B: Congo Red staining of Aβ25-35-HLF peptide preaggregated in vitro at 37°C for 4 days. Green birefringence was observed in preaggregated Aβ25-35-HLF. C–Y: Localization within brain structures of Aβ25-35-HLF, determined at 1, 3, and 12 hours, 1 and 3 days, and 1, 2, and 3 weeks after intracerebroventricular injection. Aβ25-35-HLF was visualized in green; the nucleus was counterstained with DAPI (blue). alv, alveus of the hippocampus; Arc, arcuate hypothalamic nucleus; B, basal nucleus of Meynert; bv, blood vessel; CA1, field CA1 of hippocampus; CA3, field CA3 of the hippocampus; cc, corpus callosum; Ce, central amygdala nucleus; cl 10, cerebellar lobule 10; Cx, cerebral cortex; D3V, dorsal third ventricle; DG, dentate gyrus; ec, external capsule; fi, fimbria of the hippocampus; LSD, lateral septal nucleus–dorsal part; LSI, lateral septal nucleus–intermediate part; LV, lateral ventricle; MEE, median eminence–external part; MEI, median eminence–internal part; MHb, medial habenular nucleus; MVe, medial vestibular nucleus; np, needle path; ox, optic chiasma; PeVN, periventricular hypothalamic nucleus; PVN, paraventricular hypothalamic nucleus; PVP, paraventricular thalamic nucleus–posterior part; vhc, ventral hippocampal commissure; 3V, third ventricle; 4V, fourth ventricle. Arrowheads indicate blood vessels. Scale bars = 100 μm.
Table 1.
Brain Localization of Preaggregated Aβ25-35-HLF
| Localization | Photo⁎ | Aβ25-35-HLF labeling |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 hour | 3 hours | 12 hours | 1 day | 3 days | 1 week | 2 weeks | 3 weeks | ||
| Needle path and injection site | C–G | + | + | + | + | + | + | + | + |
| Ventricles | |||||||||
| Lateral ventricle | H, M, O–P, S | + | + | + | + | + | + | + | + |
| Third ventricle (3V) | I, N, V–Y | + | + | + | + | + | + | + | + |
| Dorsal 3V (D3V) | J, L, U | + | + | + | + | + | + | + | + |
| Fourth ventricle (4V) | K | + | + | + | + | − | − | − | − |
| Ventricle bordering alv | − | − | − | + | + | + | + | + | |
| Brain structures | |||||||||
| Hippocampus | L, P, T, U | − | − | − | − | bv/+ | bv/+ | bv/+ | bv/+ |
| Fimbria | H, P | − | − | − | − | + | + | + | + |
| Septum | M, S | + | + | + | + | + | + | + | + |
| Hypothalamus | I, N, V–Y | bv/+ | bv/+ | bv/+ | bv/+ | bv/+ | bv/+ | bv/+ | bv/+ |
| Median eminence | N, V, Y | + | + | + | + | + | + | + | + |
| Basal nucleus of Meynert | Q | bv | bv | bv | bv | bv | bv | bv | bv |
| Amygdala | R | bv | bv | bv | bv | bv | bv | bv | bv |
| Frontal cortex | bv | bv | bv | bv | bv | bv | bv | bv | |
| Brainstem/cerebellum | K | + | + | + | + | − | − | − | − |
| Cells | |||||||||
| Glial cell | S | − | − | − | + | + | + | + | + |
| Neuron | M, T, U | + | + | + | + | + | + | + | + |
| Ependymocyte | H–N, S, V–Y | + | + | + | + | + | + | + | + |
Aβ25-35-HLF, Aβ25-35 HiLyte Fluor 488; alv, alveus of the hippocampus; bv, blood vessels and surrounding area; bv/+, blood vessel and brain structure; −, absence; +, presence.
Photos are presented in Figure 2, C–Y.
Aβ25-35-HLF was found at each of the time points analyzed in the different brain ventricles (Table 1). This was particularly evident in the lateral ventricle (Figure 2H) and the third ventricles, at the dorsal and ventral parts (Figure 2, I and J). Aβ25-35-HLF was also found in the fourth ventricle, at the level of the brainstem (Figure 2K), but only during the first 24 hours after injection (Table 1). More discretely and only from 1 day after injection, Aβ25-35-HLF was found in ventricle bordering the hippocampus alveus (Table 1). In ventricles, Aβ25-35-HLF was first trapped by ependymal cells bordering ventricles (Figure 2, O and P) before it gradually penetrated the surrounding structures (Figure 2, L–X).
At all time points analyzed, Aβ25-35-HLF was also found in the walls of blood vessels, in almost the entire brain (Table 1 and Figure 2, C, F–I, L, Q, R, and V). In addition, Aβ25-35-HLF was particularly localized in all structures of interest: the frontal cortex (Table 1), the amygdala (Figure 2R), the hippocampus (only from 3 days) (Figure 2, L, O, P, T, and U), and the hypothalamus (Figure 2, N and V–Y), but also the basal nuclei of Meynert (Figure 2Q) and the septum (Figure 2, M and S). Aβ25-35-HLF exhibited a diffusion gradient over time from the ventricles or blood vessels to the brain cells (neuronal and glial cells). In addition to ependymal cells, Aβ25-35-HLF was found in neurons (Figure 2, M and T–V) and in glial cells (Figure 2S), particularly in the hypothalamic median eminence (Figure 2, N, V, and Y), but also in nerve fibers at the level of the corpus callosum (Figure 2E) and the hippocampal commissure and fimbria (Figure 2, O and P).
In all structures of interest, Aβ25-35-HLF was found in blood vessels, ependymal cells, neurons, and/or glial cells from the first hours after the intracerebroventricular injection, except in the hippocampus, where Aβ25-35-HLF was found only from 3 days (Table 1).
Body Weight and Physiological Rhythms
Intracerebroventricular injection of Aβ25-35 significantly decreased the body weight of animals. Two-way analysis of variance revealed significant effects of treatment, time, and their interaction. At 5 days after peptide injection, however, the treated rats showed a weight gain similar to that of control and scrambled peptide-treated animals (Figure 3A).
Figure 3.
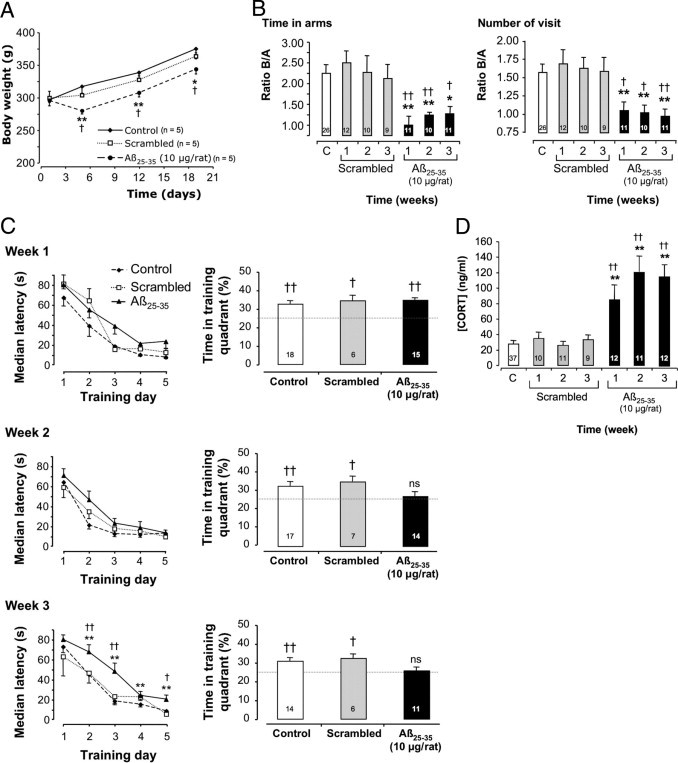
A: Time-course effects of Aβ25-35 injection (10 μg/rat i.c.v.) on body weight. The injection of the scrambled Aβ25-35 peptide (10 μg/rat i.c.v.) served as negative control. Results are expressed as means ± SEM (n = 5 per group). Two-way repeated-measures analysis of variance: F2,36 = 4.19 (P < 0.05) for treatment; F3,36 = 203.9 (P < 0.0001) for time; and F6,36 = 7.58 (P < 0.0001) for the interaction. *P < 0.05, **P < 0.01 versus control value at the same time; †P < 0.05 versus scrambled group value at the same time. B: Time-course effects of Aβ25-35 injection (10 μg/rat i.c.v.) on the ability of rats to perform a spatial short-term memory task (T-maze). During training, only arm A was available for exploration. During retention, performed with an intertrial time interval of 10 minutes, both arms were available. Results show the time spent and number of visits, in terms of arm B/arm A ratios. The injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v.) served as negative control. Results are expressed as means ± SEM. One-way analysis of variance: F6,82 = 3.00 (P < 0.01) for time; F6,82 = 3.04 (P < 0.01) for number of visits. *P < 0.05, **P < 0.01 versus noninjected rats [control group (C)]; †P < 0.05, ††P < 0.01 versus respective scrambled peptide-treated rats. The number of animals per group is indicated on data bars. C: Time-course effects of Aβ25-35 intracerebroventricular injection on rat behavior in a spatial long-term memory test (water maze). Animals were subjected to three swims per day for 5 days (90 seconds duration with an intertrial time interval of 20 minutes) to find the platform. The injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v.) served as negative control. Results are expressed as means ± SEM. Two-way repeated-measures analysis of variance: at week 1, F2,260 = 10.2 (P < 0.0001) for treatment, F4,260 = 133 (P < 0.0001) for trials, and F8,260 = 1.85 (P > 0.05) for the interaction; at week 2, F2,268 = 2.44 (P > 0.05) for treatment, F4,268 = 123 (P < 0.0001) for trials, and F8,268 = 0.73 (P > 0.05) for the interaction; and at week 3, F2,268 = 20.8 (P < 0.0001) for treatment, F4,268 = 125 (P < 0.0001) for trials, and F8,268 = 2.37 (P < 0.05) for the interaction. *P < 0.05, **P < 0.01 versus control noninjected rats; †P < 0.05, ††P < 0.01 versus respective scrambled peptide treated rats. The probe test was performed 4 hours after the last training trial in a single swim (60 seconds duration) without platform. The presence in the training quadrant was analyzed versus the chance level (25%). †P < 0.05, ††P < 0.01; ns, nonsignificant. The number of animals per group is indicated within the probe test data bars. D: Variations in plasma corticosterone (CORT) levels determined in rats at 1, 2, and 3 weeks after injection of Aβ25-35 scrambled peptide (10 μg/rat i.c.v., negative control) or Aβ25-35 (10 μg/rat i.c.v.). The values are expressed as means ± SEM. One-way analysis of variance: F6,95 = 22.36 (P < 0.0001). **P < 0.01 versus control noninjected rats [control group (C)]; ††P < 0.01 versus respective scrambled peptide-treated rats. The number of animals per group is indicated on data bars.
General locomotor activity and body temperature rhythms were continuously recorded using telemetric transmitters and revealed normal circadian differences but not among groups (see Supplemental Figure S1 at http://ajp.amjpathol.org). The three-way repeated-measures analysis of variance for general locomotor activity and body temperature data revealed significant differences. To simplify interpretation, we calculated the significance for each week independently, using a two-way repeated-measures analysis of variance. This analysis revealed a significant difference between night and day values, but no difference induced by Aβ25-35 injection; for example, for general locomotor activity at week 1, F1,276 = 0.76 (P > 0.05) for treatment, F23,276 = 12.89 (P < 0.0001) for time, and F23,276 = 0.87 (P > 0.05) for the interaction; and for body temperature at week 3, F1,276 = 0.07 (P > 0.05) for treatment, F23,276 = 60.66 (P < 0.0001) for time, and F23,276 = 1.05 (P > 0.05) for the interaction.
Learning and Memory Capacities
Spatial short-term memory was examined using delayed alternation in a T-maze. Analysis of variance for ratios of time spent and of number of visits in the novel arm versus the familiar one revealed significant effects, because Aβ25-35-injected rats exhibited deficits, compared with control and scrambled peptide-injected rats, at each time point measured (Figure 3B).
The spatial reference memory was analyzed using a water-maze procedure. When rats started training at 1, 2, or 3 weeks after peptide injection, acquisition profiles decreased with training (Figure 3C). Two-way repeated-measures analysis of variance showed an effect of training trials for each week and an effect of treatment for weeks 1 and 3. Animals tested at 3 weeks after Aβ25-35 injection showed significant increases in latencies during trials 2 to 5, compared with scrambled peptide-treated or control rats, indicating a significant alteration of acquisition performance (Figure 3C). Probe test analysis (Figure 3C) revealed that Aβ25-35-treated rats at weeks 2 and 3 showed no preferential presence in the training quadrant.
Endocrine Stress
Intracerebroventricular injection of Aβ25-35 progressively increased plasma corticosterone concentration after 1, 2, and 3 weeks, compared with control and scrambled peptide-injected rats (Figure 3D).
Oxidative Stress
In the frontal cortex, amygdala, and hippocampus, Aβ25-35 intracerebroventricular injection induced a significant increase in lipid peroxidation levels after 1 week, which was more pronounced in the hippocampus (+54% versus control group value) than in the frontal cortex or amygdala (+33% or +25%, respectively). At 2 and 3 weeks after Aβ25-35 injection, a progressive decrease in lipid peroxidation levels was observed in the frontal cortex and amygdala (−44% and −32%, respectively), whereas in the hippocampus, after a return to basal value at 2 weeks, lipid peroxidation levels again increased significantly at 3 weeks (+23% versus control group value). In the hypothalamus, oxidative stress seemed to be differentially regulated. After an initial and significant decrease in peroxidized lipid levels 1 week after Aβ25-35 injection (−55%), a progressive increase was observed, reaching +23% at 3 weeks after injection (Figure 4).
Figure 4.
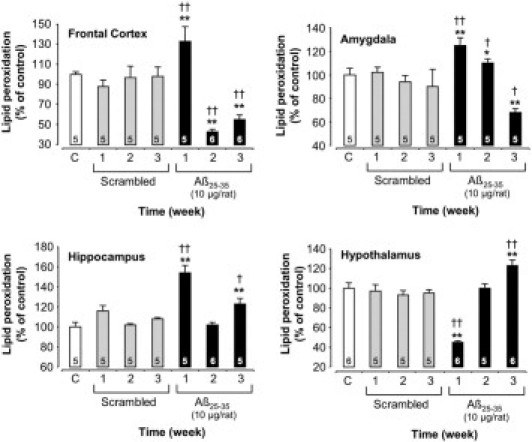
Variations in lipid peroxidation levels in the frontal cortex, amygdala, hippocampus, and hypothalamus determined in rats at 1, 2, and 3 weeks after injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v., negative control) or Aβ25-35 (10 μg/rat i.c.v.). Results are expressed as means ± SEM. One-way analysis of variance: frontal cortex, F6,30 = 8.37 (P < 0.0001); amygdala, F6,28 = 8.17 (P < 0.0001); hippocampus, F6,29 = 24.42 (P < 0.0001); and hypothalamus, F6,31 = 16.08 (P < 0.0001). *P < 0.05, **P < 0.01 versus control noninjected rats [control group (C)]; †P < 0.05, ††P < 0.01 versus respective scrambled peptide treated rats. The number of animals per group is indicated on data bars.
Neuroprotective Reaction
At 1 week after Aβ25-35 injection, BDNF content was significantly increased in the frontal cortex (+33%) and amygdala (+39%), before a progressive reduction to 44% in the frontal cortex and 32% in the amygdala at 3 weeks. In the hippocampus and hypothalamus, a significant increase in BDNF content was observed 1 week after Aβ25-35 injection (+20% and +28%, respectively), which was augmented after 3 weeks (+49% and +43%, respectively) (Figure 5).
Figure 5.
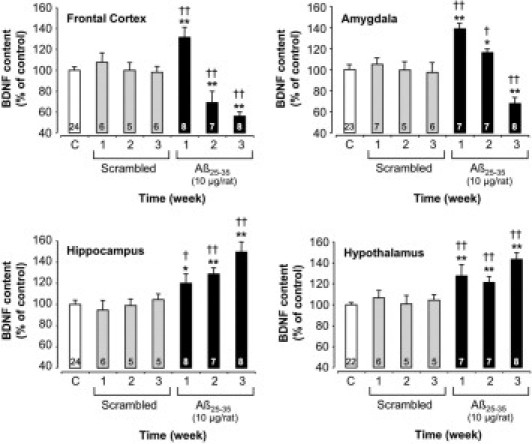
Variations in BDNF content in the frontal cortex, amygdala, hippocampus, and hypothalamus determined in rats at 1, 2, and 3 weeks after injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v., negative control) or Aβ25-35 (10 μg/rat i.c.v.). Results are expressed as means ± SEM. One-way analysis of variance: frontal cortex, F6,57 = 9.05 (P < 0.0001); amygdala, F6,56 = 8.58 (P < 0.0001); hippocampus, F6,56 = 8.03 (P < 0.0001); and hypothalamus, F6,54 = 8.65 (P < 0.0001). *P < 0.05, **P < 0.01 versus control noninjected rats [control group (C)]; †P < 0.05, ††P < 0.01 versus respective scrambled peptide treated rats. The number of animals per group is indicated on data bars.
Mitochondrial Stress
We assessed the level of mitochondrial stress using Western blotting to measure levels of pro- and activated caspase-9 in the cerebral structures of interest (Figure 6). Scrambled peptide injection did not affect pro- and cleaved caspase-9 levels, and no change was observed in the hypothalamus after Aβ25-35 injection. In the frontal cortex and hippocampus, the peptide induced an increase in activated caspase-9 after 1 week (+36% and +56%, respectively). After 3 weeks, increases in pro- and cleaved caspase-9 levels were measured (frontal cortex, +77% and +99%; hippocampus, +26% and +49%), respectively. In the amygdala, no significant increase in pro- and cleaved caspase-9 was observed before week 3 (+44% and +77%, respectively) (Figure 6).
Figure 6.
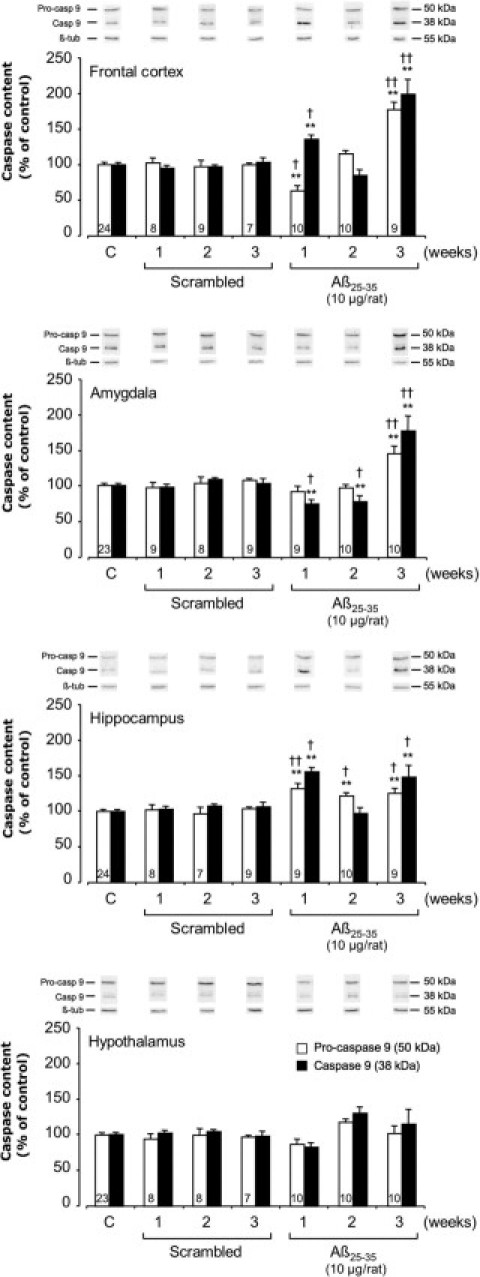
Variations in pro- and activated caspase-9 levels in the frontal cortex, amygdala, hippocampus, and hypothalamus determined in rats by Western blotting at 1, 2, and 3 weeks after injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v., negative control) or Aβ25-35 (10 μg/rat i.c.v.). Pro-caspase-9 (50 kDa) and activated caspase-9 (38 kDa) variations were normalized with those of β-tubulin (β-tub; 55 kDa) and were compared with noninjected rats [control group (C)]. Results are expressed as means ± SEM. One-way analysis of variance: frontal cortex, F6,71 = 9.51 (P < 0.0001) for pro-caspase-9 and F6,71 = 8.12 (P < 0.0001) for cleaved caspase-9; amygdala, F6,72 = 7.06 (P < 0.0001) and F6,72 = 17.08 (P < 0.0001) for the two caspase-9 forms, respectively; hippocampus, F6,70 = 4.36 (P < 0.001) and F6,70 = 3.56 (P < 0.01) for the two forms, respectively; and hypothalamus, F6,71 = 0.99 (P > 0.05) and F6,71 = 2.09 (P > 0.05) for the two forms. **P < 0.01 versus control noninjected rats [control group (C)]; †P < 0.05, ††P < 0.01 versus respective scrambled peptide treated rats. The number of animals per group for both caspase forms is indicated on white data bars.
Endoplasmic Reticulum Stress
To characterize endoplasmic reticulum stress, we measured pro- and cleaved caspase-12 levels by Western blotting (Figure 7). Scrambled peptide injection did not affect pro- and cleaved caspase-12 levels. By contrast, in the frontal cortex and amygdala, Aβ25-35 injection induced an increase in pro- and cleaved caspase-12 after 1 week (frontal cortex, +159% and +122%; amygdala, +83% and +36%, respectively), which was globally sustained after 3 weeks. By contrast, in the hippocampus, pro- and cleaved caspase-12 levels were significantly increased at 2 and 3 weeks after Aβ25-35 injection (+150% and +58% at week 2; +58% and +73% at week 3, respectively), and no variation was observed in the hypothalamus (Figure 7).
Figure 7.
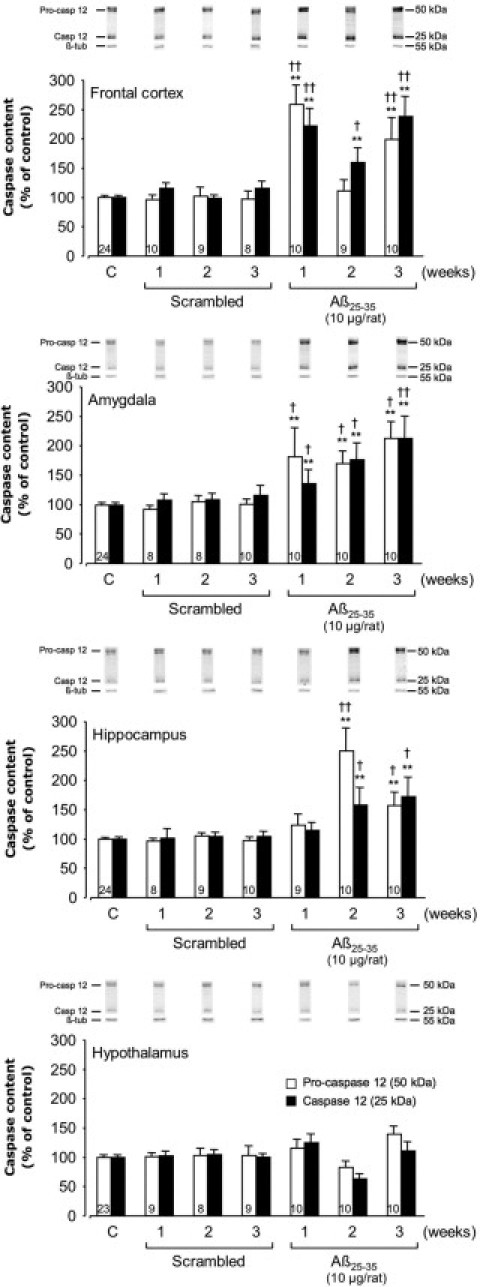
Variations in pro- and activated caspase-12 levels in the frontal cortex, amygdala, hippocampus, and hypothalamus determined in rats by Western blotting at 1, 2, and 3 weeks after injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v., negative control) or Aβ25-35 (10 μg/rat i.c.v.). Pro-caspase-12 (50 kDa) and activated caspase-12 (25 kDa) variations were normalized with those of β-tubulin (β-tub, 55 kDa) and were compared with untreated rats [control group (C)]. Results are expressed as means ± SEM. One-way analysis of variance: frontal cortex, F6,74 = 12.93 (P < 0.0001) for pro-caspase-12 and F6,74 = 14.85 (P < 0.0001) for cleaved caspase-12; amygdala, F6,74 = 4.84 (P < 0.001) and F6,74 = 6.58 (P < 0.0001) for the two caspase-12 forms, respectively; hippocampus, F6,74 = 10.42 (P < 0.0001) and F6,74 = 3.56 (P < 0.01) for the two forms, respectively; and hypothalamus, F6,73 = 1.92 (P > 0.05) and F6,73 = 1.80 (P > 0.05) for the two forms. **P < 0.01 versus control noninjected rats [control group (C)]; †P < 0.05, ††P < 0.01 versus respective scrambled peptide treated rats. The number of animals per group for both caspase forms is indicated on white data bars.
Apoptosis
To characterize apoptosis, we measured pro- and cleaved caspase-3 levels by Western blotting. The scrambled peptide injection did not affect levels of pro- and cleaved caspase-3. The effects of Aβ25-35 injection on cleaved caspase-3 were limited to the amygdala, hippocampus, and hypothalamus; no effect was observed in the frontal cortex (Figure 8). In the frontal cortex and hippocampus, Aβ25-35 injection provoked a sustained increase in pro-caspase-3 levels over the 3-week experimental period, with respective increases of 54% and 107% at week 1, 98% and 56% at week 2, and 85% and 42% at week 3. In the amygdala, a significant increase in pro-caspase-3 levels was observed, beginning after 2 weeks (+103% at week 2 and +66% at week 3), whereas in the hypothalamus, pro-caspase-3 levels were significantly increased only during the first 2 weeks after Aβ25-35 injection (+33% at week 1 and +67% at week 2) (Figure 8). Cleaved caspase-3 levels were significantly increased only at weeks 1 and 3 in the amygdala (+34% and +35%, respectively), but only at week 2 in the hippocampus (+17%). By contrast, in the hypothalamus, cleaved caspase-3 levels were significantly decreased at 2 and 3 weeks after Aβ25-35 injection (−36% and −29%, respectively) (Figure 8).
Figure 8.
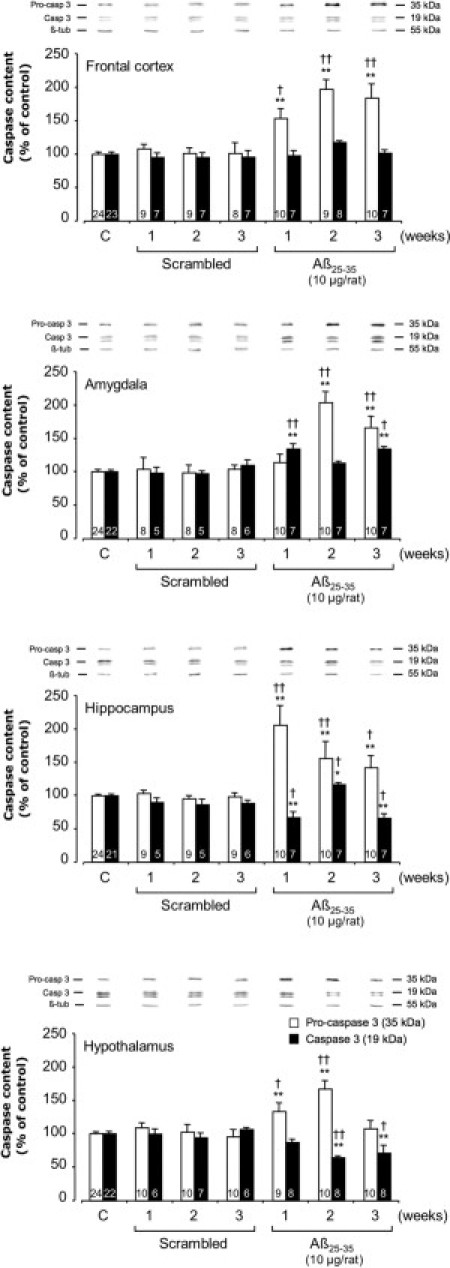
Variations in pro- and activated caspase-3 levels in the frontal cortex, amygdala, hippocampus, and hypothalamus determined in rats by Western blotting at 1, 2, and 3 weeks after injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v., negative control) or Aβ25-35 (10 μg/rat i.c.v.). Pro-caspase-3 (35 kDa) and activated caspase-3 (19 kDa) variations were normalized with those of β-tubulin (β-tub, 55 kDa) and were compared with untreated rats [control group (C)]. Results are expressed as means ± SEM. One-way analysis of variance: frontal cortex, F6,72 = 15.9 (P < 0.0001) for pro-caspase-3 and F6,60 = 1.06 (P > 0.05) for cleaved-caspase-3; amygdala, F6,72 = 14.0 (P < 0.0001) and F6,53 = 7.42 (P < 0.0001) for the two caspase-3 forms, respectively; hippocampus, F6,75 = 9.91 (P < 0.0001) and F6,52 = 8.01 (P < 0.0001) for the two forms, respectively; and hypothalamus, F6,77 = 8.74 (P < 0.0001) and F6,59 = 6.22 (P < 0.0001) for the two forms. *P < 0.05, **P < 0.01 versus control noninjected rats [control group (C)]; †P < 0.05, ††P < 0.01 versus respective scrambled peptide treated rats. The number of animals per group is indicated on data bars.
APP Processing
To determine whether the Aβ25-35 injection affected APP processing, we measured APP and C99 levels by Western blotting (Figure 9). The scrambled peptide injection did not affect levels of either APP or C99. By contrast, Aβ25-35 injection provoked a sustained increase in APP levels from week 2 in the frontal cortex (+62% at week 2 and +32% at week 3) (Figure 9) and over the 3-week experimental period in the hippocampus (+49% at week 1, +29% at week 2, and +28% at week 3). In the amygdala, a significant decrease in APP levels was observed after 1 and 2 weeks (−32% at week 1 and −55% at week 2), whereas after 3 weeks the APP levels were significantly increased (+30%). In the hypothalamus, APP levels were significantly increased after the first week after Aβ25-35 injection (+78%), but were significantly decreased thereafter (−42% at week 2 and −69% at week 3) (Figure 9). In the frontal cortex and hippocampus, Aβ25-35 injection provoked a sustained increase in C99 levels over the first 2 weeks (+22% and +41% at week 1; +30% and +21% at week 2). However, although in the frontal cortex C99 levels were again increased after 3 weeks (+23%), in the hippocampus a significant decrease of C99 levels was observed (−61%). In the amygdala, C99 levels were significantly decreased during the first 2 weeks after Aβ25-35 injection (−33% at week 1 and −35% at week 2), but after 3 weeks were increased (+23% versus control group value). In the hypothalamus, no variation of C99 levels was observed at 1 and 2 weeks after Aβ25-35 injection, but after 3 weeks the C99 levels were significantly decreased (−70%) (Figure 9).
Figure 9.
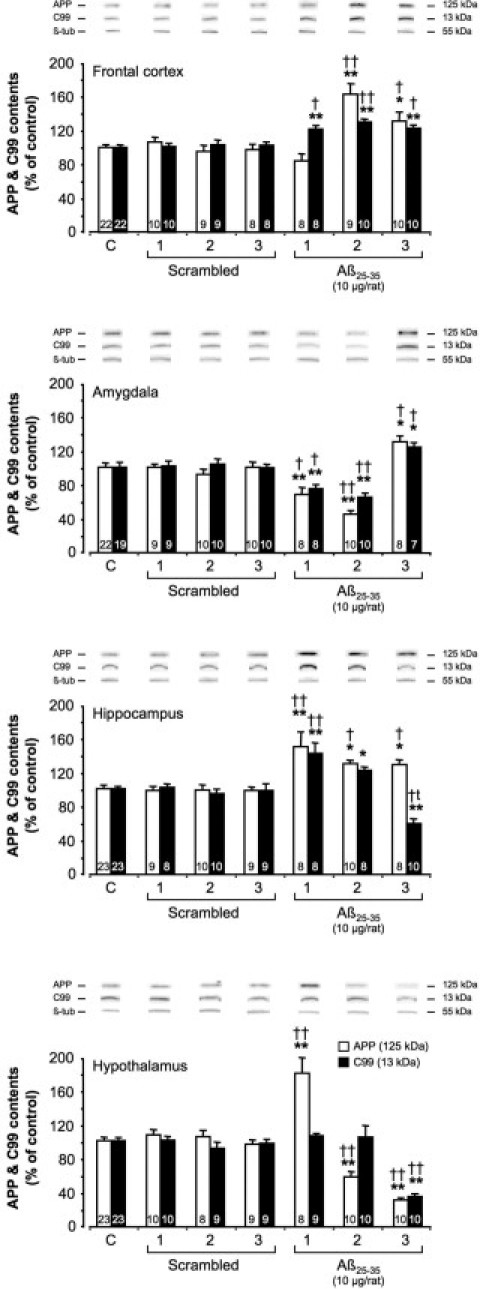
Variations in APP and C99 levels in the frontal cortex, amygdala, hippocampus, and hypothalamus determined in rats by Western blotting at 1, 2, and 3 weeks after injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v., negative control) or Aβ25-35 (10 μg/rat i.c.v.). APP (125 kDa) and C99 (13 kDa) variations were normalized with those of β-tubulin (β-tub, 55 kDa) and compared with noninjected rats [control group (C)]. Results are expressed as means ± SEM. One-way analysis of variance: frontal cortex, F6,69 = 12.3 (P < 0.0001) for APP and F6,70 = 9.72 (P < 0.0001) for C99; amygdala, F6,70 = 16.9 (P < 0.0001) for APP and F6,66 = 13.9 (P < 0.0001) for C99; hippocampus, F6,70 = 8.21 (P < 0.001) for APP and F6,69 = 16.1 (P < 0.0001) for C99; and hypothalamus, F6,71 = 37.3 (P < 0.0001) for APP and F6,73 = 15.0 (P < 0.0001) for C99. *P < 0.05, **P < 0.01 versus control noninjected rats [control group (C)]; †P < 0.05, ††P < 0.01 versus respective scrambled peptide-treated rats. The number of animals per group is indicated on data bars.
Neuroinflammation
The intracerebroventricular injection of Aβ25-35 significantly modified astroglial activity (as indicated by GFAP level) over the study period in all structures of interest, compared with control and scrambled peptide intracerebroventricularly injected rats (Figure 10A). At 3 weeks after Aβ25-35 injection, GFAP levels reached 58% in the cortex and 73% in the amygdala. In the hippocampus, a marked increase (+42%) in astroglial activity was observed after 1 week, followed by a progressive decrease in GFAP levels (−26% at week 3). In the hypothalamus, after an initial increase (+41% at week 1), a ricochet increase was observed 3 weeks after Aβ25-35 injection (+27%) (Figure 10A).
Figure 10.
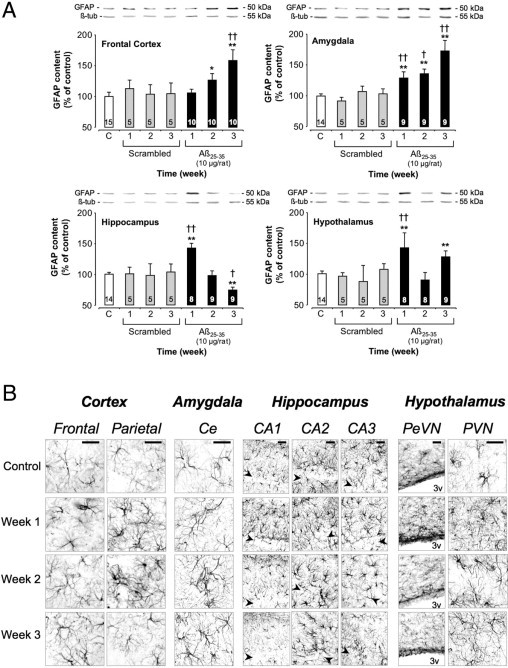
A: Variations in GFAP levels in the frontal cortex, amygdala, hippocampus, and hypothalamus determined in rats by Western blotting at 1, 2, and 3 weeks after injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v., negative control) or Aβ25-35 (10 μg/rat i.c.v.). GFAP (50 kDa) variations were normalized with those of β-tubulin (β-tub, 55 kDa) and were compared with untreated rats [control group (C)]. Results are expressed as means ± SEM. One-way analysis of variance: frontal cortex, F6,53 = 5.28 (P < 0.001); amygdala, F6,49 = 12.5 (P < 0.0001); hippocampus, F6,48 = 5.21 (P < 0.001); and hypothalamus, F6,47 = 4.98 (P < 0.001). *P < 0.05, **P < 0.01 versus control group; †P < 0.05, ††P < 0.01 versus respective scrambled peptide treated rats. The number of animals per group is indicated on data bars. B: Time-course effects of Aβ25-35 (10 μg/rat) intracerebroventricular injection on astrocyte reaction using GFAP immunolabeling in the frontal and parietal cortex, amygdala, hippocampus, and hypothalamus determined in control untreated rats and at 1, 2, and 3 weeks after Aβ25-35 injection. The injection of the Aβ25-35 scrambled peptide (10 μg/rat i.c.v.) served as negative control and induced no modifications in the GFAP signal. CAn, hippocampal subfields; Ce, central amygdaloid nucleus; PeVN, periventricular nucleus; PVN, paraventricular nucleus; 3v: third ventricle. Arrowheads indicate the hippocampus layer of granular cells. Scale bars = 100 μm.
By using GFAP immunoreactivity, which is indicative of astrogliosis, a more qualitative examination could be performed. Indeed, GFAP immunolabeling was observed throughout the frontal and parietal cortex, amygdala, hippocampus, and hypothalamic nuclei in Aβ25-35 intracerebroventricularly injected rats (Figure 10B), whereas no modifications were observed after the scrambled peptide intracerebroventricular injection (data not shown). Strongly labeled astrocytes with the typical morphology of their activated state were differentially observed, depending on the time point and brain region of interest. Astrogliosis was more pronounced in the frontal cortex during week 3, whereas an increase in activated astrocytes was noted in the parietal cortex at 1 and 2 weeks after intracerebroventricular injection of Aβ25-35. In the amygdala and hypothalamic nuclei, astrogliosis increased from week 1 to week 3 after Aβ25-35 injection. In the hippocampus, GFAP immunoreactivity was increased in areas adjacent to the pyramidal neuron layers, with the most pronounced astrogliosis occurring from week 1 to week 3 in the CA2 subfield, where relative astrocytes had infiltrated the pyramidal layers. In CA1 and CA3, astrogliosis was maximal at 1 week after Aβ25-35 injection (Figure 10B).
Iba-1 immunoreactivity indicative of microgliosis was observed throughout the frontal cortex, hippocampus, parietal cortex, amygdala, and hypothalamus of Aβ25-35-injected rats (Figure 11, A–E). No modification was observed after injection of scrambled peptide (data not shown). Strongly labeled microglial cells with the typical morphology of their activated form were differentially observed, depending on the time point and brain region. Progressive hypertrophy and hyper-ramification of microglia was observed 1 week after Aβ25-35 injection, followed by progressive recruitment of new activated microglial cells from 2 weeks. This phenomenon was more pronounced in the frontal and parietal cortex and amygdala (Figure 11, A, C, and D) than in the hippocampus and hypothalamus (Figure 11, B and E).
Figure 11.
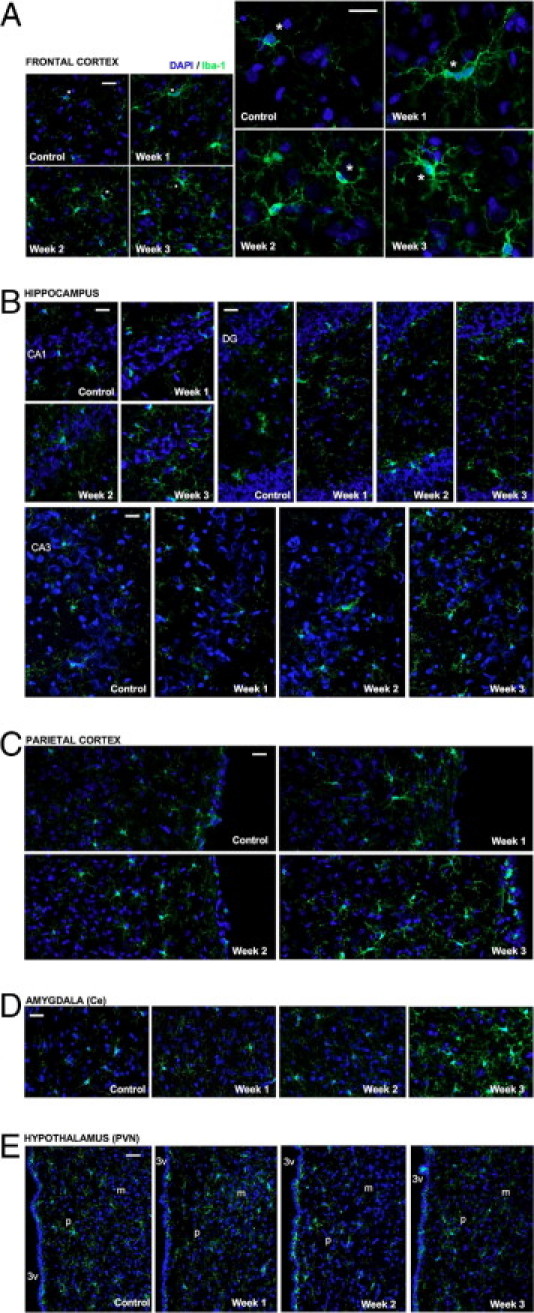
Time-course effects of Aβ25-35 (10 μg/rat) intracerebroventricular injection on the microglial reaction using Iba-1 immunolabeling in the frontal cortex (A), hippocampus CA1, CA3, and DG subfields (B), parietal cortex (C), amygdala (D), and hypothalamus paraventricular nucleus (E) determined in untreated rats [control group (C)] and at 1, 2, and 3 weeks after Aβ25-35 injection. The injection of the Aβ25-35 scrambled peptide (10 μg/rat i.c.v.) served as negative control and induced no modifications of the Iba-1 signal. Activated microglia were visualized with Alexa Fluor 488-labeled specific antibody against Iba-1 (green); the nucleus was counterstained with DAPI (blue). In A, asterisks indicate matching locations in corresponding images presented at both lower and higher magnification. CAn, hippocampal subfields; Ce, central amygdaloid nucleus; DG, dentate gyrus; m, magnocellular; p, parvocellular; PVN, paraventricular hypothalamic nucleus; 3v, third ventricle. Scale bars = 100 μm.
Cholinergic System
Within the basal forebrain of control rats, large numbers of VAChT-positive cell bodies were seen in the nucleus basalis of Meynert (Figure 12A). In the hypothalamus, a very dense plexus of VAChT-immunoreactive fibers were present in the external layer of the median eminence and weakly VAChT-positive cell bodies were noted in the arcuate nucleus (Figure 12B). A dense network of VAChT-positive nerve fibers was seen in the parietal cortex, with the highest density found in layers I, IV, and V (Figure 12C). In hippocampal formation (Figure 12D), the highest density of VAChT-positive fibers was seen in the pyramidal cell layer of the CA3 subfield, whereas in the dentate gyrus (DG) the highest density was observed around granular cell layers. No modification was observed after scrambled peptide injection (data not shown). By contrast, Aβ25-35 injection induced a progressive decrease in VAChT immunolabeling, which was more pronounced at 3 weeks after injection in the nucleus basalis (Figure 12A), parietal cortex (Figure 12C), and hippocampus (Figure 12D). No effect was induced by Aβ25-35 in the hypothalamus (Figure 12B).
Figure 12.
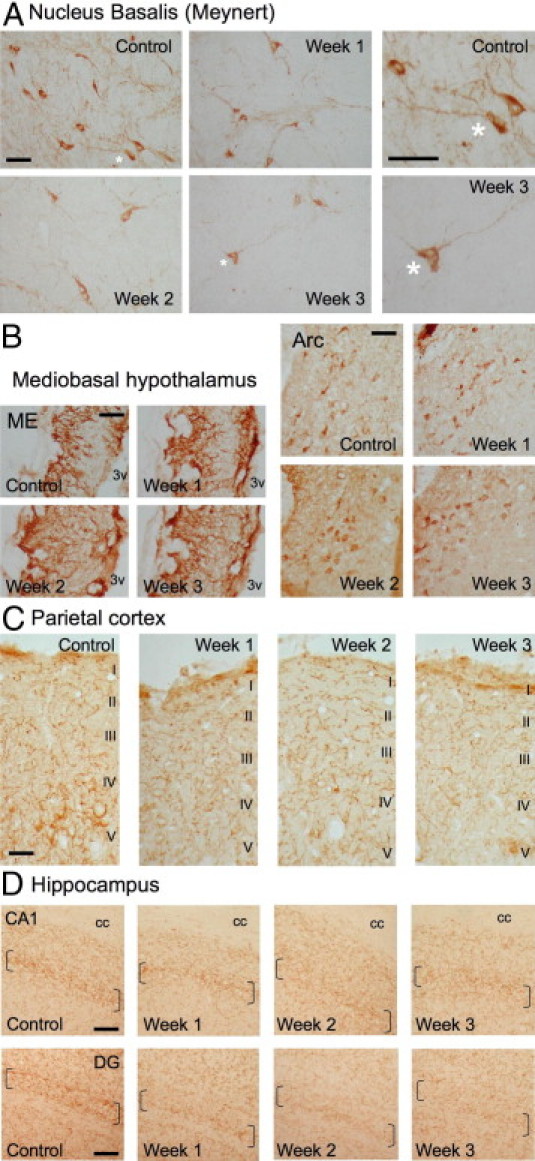
Time-course effects of Aβ25-35 (10 μg/rat) intracerebroventricular injection on VAChT immunolabeling within the nucleus basalis of Meynert (A), mediobasal hypothalamus (B), parietal cortex with levels I to V cortical layers indicated (C), and hippocampus (D) were determined in control untreated rats and at 1, 2, and 3 weeks after Aβ25-35 injection. In A, asterisks indicate matching locations in corresponding images presented at both lower and higher magnification. In D, brackets locate the hippocampus granular cell layers. Arc, arcuate nucleus; cc, corpus callosum; ME, median eminence; 3v, third ventricle. Scale bars = 100 μm.
Hippocampus Integrity
Loss of pyramidal cells in the hippocampus layers was measured using cresyl violet staining (Figure 13, A and B). No significant modification was observed after scrambled peptide injection. By contrast, Aβ25-35 induced a significant decrease in stained cells in the CA1, CA2, CA3, and DG hippocampus subfields from week 1 to week 3. This decrease was more marked in the CA1 and CA2 subfields (−30% in CA1 at week 1 and −37% in CA2 at week 2) (Figure 13B).
Figure 13.
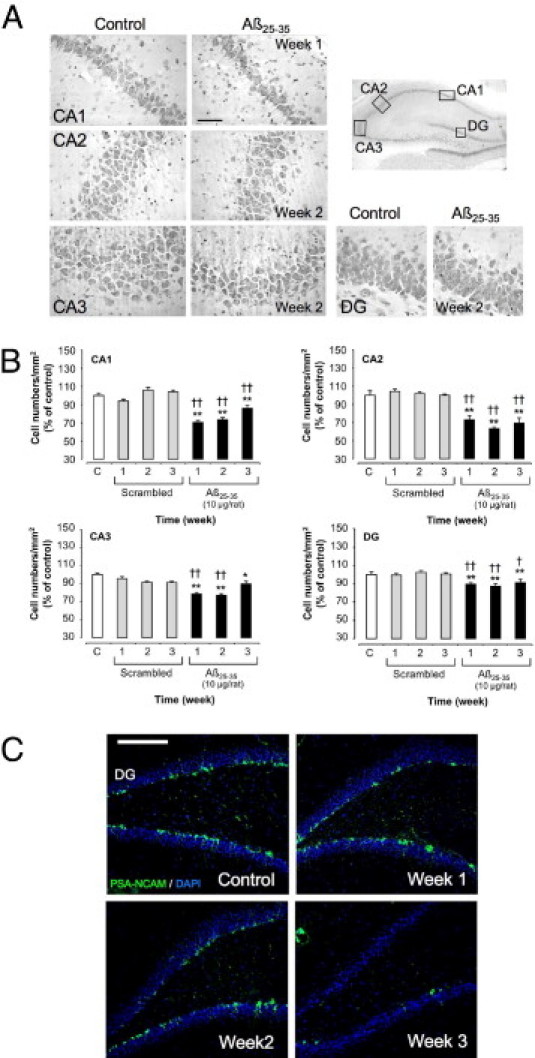
A: Time-course effects of Aβ25-35 injection (10 μg/rat i.c.v.) on hippocampus pyramidal cell numbers. Representative microphotographs of coronal sections of cresyl violet-stained hippocampus CA1, CA2, CA3, and DG subfields, obtained in control untreated rats and after Aβ25-35 intracerebroventricular injection. Scale bar = 100 μm. B: Average numbers of hippocampus pyramidal cells determined in untreated rats [control group (C)] and at 1, 2, and 3 weeks after the injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v., negative control) or Aβ25-35 (10 μg/rat i.c.v.). Results are expressed as means ± SEM (n = 4 per group). One-way analysis of variance: CA1, F6,21 = 39.6 (P < 0.0001); CA2, F6,21 = 29.1 (P < 0.0001); CA3, F6,21 = 11.8 (P < 0.0001); and DG, F6,21 = 8.40 (P < 0.001). *P < 0.05, **P < 0.01 versus control rats; †P < 0.05, ††P < 0.01 versus scrambled peptide treated rats. C: Effects of Aβ25-35 (10 μg/rat) intracerebroventricular injection on hippocampus DG neurogenesis using PSA-NCAM immunolabeling, determined in untreated control rats and at 1, 2, and 3 weeks after Aβ25-35 intracerebroventricular injection. The injection of scrambled amyloid peptide (10 μg/rat i.c.v.) served as negative control and induced no modifications in the PSA-NCAM signal. Neurogenesis was visualized in coronal sections of the DG with Alexa Fluor 488-labeled specific antibody against PSA-NCAM (green); the nucleus was counterstained with DAPI (blue). Scale bar = 200 μm.
PSA-NCAM-positive cells indicative of neurogenesis were revealed within the DG (Figure 13C). A progressive decline in PSA-NCAM labeling was observed after Aβ25-35 injection, whereas no modification was observed after scrambled peptide injection.
Discussion
In the present study, we thoroughly analyzed the time-dependent and regional pathophysiological effects of Aβ25-35 injection in rats. First, we showed that Aβ25-35 peptide formed a β-sheet structure and amyloid-like fibrils. This short fragment has been identified in AD patient brains and is likely produced endogenously by enzymatic cleavage of Aβ1-40.15,32 Indeed, findings from the IHC study of Kubo et al15 support the idea that Aβ1-40 in senile plaques is truncated by brain proteases during aging to generate toxic Aβ25-35/40. Their use of two kinds of anti-Aβ25/26–35/40 antibodies demonstrated clearly that the fragments were present both in the core of senile plaques and extracellular neurofibrillary tangles (Aβ25/26–35) and in the degenerating hippocampal CA1 neurons with intracellular neurofibrillary tangles (Aβ25–40), in AD brains, but not in age-matched control subjects.15
Deposits of Aβ25-35 peptide after its injection have not yet been characterized by IHC, for lack of specific and selective antibodies. However, Klementiev et al16 and Chavant et al17 reported an accumulation of Aβ1-40 immunolabeling in the hippocampus and cerebral cortex, induced 4 and 2 weeks, respectively, after the Aβ25-35 injection. In the present study, we followed over time the cerebral localization of Aβ25-35 after injection into the lateral ventricle by using an Aβ25-35-HLF-tagged peptide. In a very short time, Aβ25-35-HLF was found throughout all ventricles and was picked up by the ependymal cells lining these ventricles. Progressively, Aβ25-35-HLF penetrated through the ependymal barrier and the surrounding structures and reaches the brain vasculature. Gradually, a diffusion gradient developed from the ventricles and/or the walls of blood vessels to the brain regions, particularly evident in the septum, hypothalamus, hippocampus, amygdala, the different cortical regions, and basal nucleus. Of note, Aβ25-35-HLF was still present 3 weeks after injection, demonstrating a long-lasting presence and an important lifespan for this fragment in brain tissues. It seems to be particularly trapped by specialized glial cells at the scar induced by the injection needle, the ventricles, the walls of blood vessels, and certain brain structures. The peptide was present within tanycyte cells at the hypothalamic median eminence level.
The evolution over time of fluorescence in this area could even reflect the ability of tanycytes to trap the peptide from the third ventricle and to clear it into the hypothalamo-pituitary portal blood system. Moreover, in the long term, the peptide is likely absorbed by nerve fibers and some neurons, where it seems to accumulate. The present descriptive study, showing that Aβ25-35 reached all brain areas examined, clearly reinforces the idea that there is a differential response to the toxicity, depending on the brain area. We note that Congo Red staining failed to reveal the presence of polarized aggregates (data not shown). The present observation that Aβ25-35-HLF resulted in a rather diffuse labeling, after penetration through the ependymocyte barrier and diffusion into the structure, suggests that the peptide and/or aggregate concentration within tissues remains relatively low, preventing its detection under polarizing filters.
Analysis of APP processing after Aβ25-35 injection confirmed previous data obtained in mice 2 weeks after injection.17 We observed increases in APP processing and amyloidogenic pathway (C99 levels) in the frontal cortex at all time points, in the amygdala after 3 weeks, and in the hippocampus mainly after 1 and 2 weeks. We therefore confirmed the Aβ25-35 influence on seeding of endogenous Aβ1–40/42 proteins in the rodent brain, particularly highlighting differences over time and among structures. These Aβ1–40/42 proteins synthesized de novo could contribute to the global toxicity measured in this model. We plan future studies to address this point.
A rapid inflammatory response occurred after Aβ25-35 injection. After 1 week, the peptide induced reactive gliosis, by up-regulation of GFAP expression, and hypertrophic astrocytes were observed in the hippocampus (present study and Ref. 13); in the present study, hypertrophic astrocytes were observed also in the frontal and parietal cortex, amygdala, and hypothalamus. Moreover, we observed that Aβ25-35 injection also increased Iba-1 immunoreactivity, a marker of activated microglia, and the increase was associated with progressive hypertrophy and hyper-ramification of these cells.33 This phenomenon was more pronounced in the frontal and parietal cortex and amygdala (structures that are very rapidly reached by Aβ25-35) than in the hippocampus and hypothalamus.
Aβ-induced toxicity was also associated with histological changes in the hippocampus. Aβ25-35 injection resulted in a moderate but significant reduction in the granule cell number in all hippocampus areas from 1 week to 1 month after injection in rats (present study and Refs. 9 and 10) and after 1 week in mouse.11 The extent of cell loss was limited to a 30% to 40% decrease in the number of viable cells in the most vulnerable areas, whereas a significantly greater loss was noted in all pyramidal cell layers. The neurodegenerative process thus appeared to be generalized, contrary to what is usually observed in excitotoxic or chemical neurotoxicity models.34–36 This pattern is, however, consistent with hippocampus damage observed in AD brains.37,38
Amyloid toxicity directly affects neuronal physiology, particularly the cholinergic system. After 8 days, Aβ25-35 injection decreased choline acetyltransferase activity in the medial septum, cortex, and hippocampus and decreased the number of choline acetyltransferase-immunoreactive cells in the medial septum.39 In the present study, we identified the presence of the peptide within cholinergic structures and we observed that VAChT immunoreactivity was progressively decreased in the hippocampus, parietal cortex, and basal nuclei of Meynert, but not in the hypothalamus. The cholinergic deficits induced by Aβ25-35 injection therefore appear to be in accord with the well-characterized pathological hallmarks described in AD.37,38
Oxidative stress contributed to the in vivo Aβ-induced toxicity. At 1 week after injection, Aβ25-35 induced significant oxidative stress in the hippocampus, as reflected by measured increases in lipid peroxidation, protein nitration, and superoxide generation.11–13,40 In the present study, the early increases in lipid peroxidation levels observed after peptide injection were gradually reduced, particularly in the rat frontal cortex and amygdala, suggesting that oxidative stress could be alleviated by endogenous protective systems (putatively neurotrophins, such as BDNF).
Most studies describing the apoptotic effects of amyloid peptides have been based on in vitro cell culture models. They demonstrated that Aβ1-42 and Aβ25-35 induce apoptosis through caspase-dependent pathways.41–47 In vivo, 1 week after Aβ25-35, an increase in caspase-3 activity in the hippocampus of rats9 and increases in levels of pro-caspases 9, 12, and 3 in the hippocampus of mice11 were observed. In the present study, we observed increases in pro- and cleaved forms for caspase-9 (mitochondrial stress marker) and caspase-12 (endoplasmic reticulum stress marker) in all structures except the hypothalamus. These biochemical measures confirmed that the hypothalamus is highly resistant to Aβ toxicity, despite the penetration of the peptide into the paraventricular, periventricular, and arcuate hypothalamic nuclei and also the median eminence of the hypothalamus. Moreover, increases in pro-caspase-3 expression were measured in almost all structures at any time point, but increases in cleaved caspase-3 were measured mainly in the hippocampus after 2 weeks and in the amygdala after 1 and 3 weeks. Cleaved caspase-3 expression levels appeared to be relatively limited, compared with the increased levels of mitochondrial and endoplasmic reticulum stress markers and pro-caspase-3.
Apoptosis induction could be more thoroughly analyzed using alternative approaches, including cytochrome-c release, caspase-6 expression, or the terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) method. As an alternative to apoptosis, necrosis cell death could also be considered. In vivo, the complete elimination of apoptotic cells prevents an inflammatory response, whereas necrosis often results in inflammatory reactions.48 Moreover, in cell culture models, Aβ25-35 was reported to induce apoptosis at lower concentrations (5 and 10 μmol/L)5 and necrosis at higher concentrations (20 and 40 μmol/L).49
Oxidative stress, apoptosis, and morphological damage induced by amyloid peptides suggest deficits in endogenous neuroprotective mechanisms. Among these, BDNF is known to protect neurons against various types of brain insult. Moreover, in rat cortical neuron cultures, at sublethal concentrations Aβ1-42 interferes with BDNF signaling, thus increasing neuron vulnerability and abrogating BDNF protection against apoptosis induced by DNA damage or by trophic deprivation.50 In vivo, Aβ1-40 and Aβ25-35 infusion also triggered BDNF mRNA expression 7 days after the start of peptide infusion.51 In the present study, spatiotemporal examination of the influence of Aβ25-35 on BDNF protein expression revealed a sustained BDNF increase in the hippocampus and hypothalamus over time from weeks 1 to 3. By contrast, Aβ25-35 induced a transitory BDNF increase in the frontal cortex and amygdala 1 week after injection, followed by a decrease 3 weeks after injection in both structures. Notably, these profiles appeared to be very consistent with those of other toxicity markers (ie, neuroinflammation, oxidative stress, and caspase induction). The sustained increase in BDNF levels in the hypothalamus was correlated with low oxidative stress, no endoplasmic reticulum or mitochondrial stress, and down-regulation of activated caspase-3. These observations suggest the existence within the hypothalamus of protective mechanisms that could in part be related to BDNF. In contrast, however, the caspase activation observed after 2 weeks in the amygdala and frontal cortex could result in part from BDNF deficits. The expression of BDNF receptors, TrkB isoforms and p75, and other neurotrophins, particularly nerve growth factor (NGF), must therefore be further analyzed to clarify the involvement of trophic factors in the differential vulnerability of brain structures to amyloid toxicity.
Because glucocorticoids act synergistically with excitatory amino acids, particularly with glutamate,52,53 chronic overstimulation could be extremely toxic, particularly in the hippocampus,54 and thus could participate in the etiology of AD. Although no data are available on the effects of Aβ peptide injection on hypothalamic-pituitary-adrenal axis activity, several studies have demonstrated that glucocorticoids modulate APP processing,55 increase Aβ1-42-induced neurodegeneration in basal nuclei of Meynert,56 and increase Aβ25-35 toxicity in hippocampus neurons.57 Moreover, in vivo chronic corticosterone administration was shown to increase Aβ1-42 and N-methyl-D-aspartate-induced neurodegeneration in cholinergic neurons from the nucleus basalis in the rat.56 In the present study, we obtained clear evidence that Aβ25-35 increased the blood concentration of corticosterone from the first week and at least up to the third week. Note that plasma corticosterone levels observed in Aβ25-35-treated rats were in the same range as observed in chronically stressed rats,58 which is consistent with the long-term hyperactivity of the hypothalamic-pituitary-adrenal axis. These findings suggest, as postulated in AD patients, that glucocorticoids may be an important contributor to the onset and progression of the AD pathology.59,60
Neurogenesis in the adult DG subfield of the hippocampus occurs constitutively throughout postnatal life, and the rate of neurogenesis within the DG can be altered under various physiological and pathophysiological conditions.61,62 Few data have been published on the effects of Aβ peptide injection on hippocampus neurogenesis. Several controversial in vitro findings seem to be dependent on the doses and oligomer structure of the Aβ peptide used.63–65 We clearly observed that Aβ25-35 injection progressively impaired the immunoreactivity of the PSA-NCAM neurogenesis marker in hippocampus DG cells. Aβ25-35 injection decreased the number of newly generated and developing granule cells. The exact effect of Aβ25-35 on proliferation and differentiation stages could be further investigated with adequate markers.66 Moreover, this observation suggests in particular that the pyramidal cell loss observed after Aβ25-35 injection could have been in part due to a deficit in neurogenesis processes.
Remarkably, the brain structures of interest showed a differential vulnerability to Aβ25-35 toxicity. These differential effects observed after Aβ25-35 injection seem to be more likely due to the particular sensitivity of each region to the toxicity, induced directly or indirectly by Aβ25-35, rather than to difference in the penetration of the peptide within the structures. Indeed, the peptide was present at all time points examined, with a penetration gradient from the ventricles and blood vessels into the deep structures. For instance, the hypothalamus, rapidly and long-lastingly reached by Aβ25-35, is poorly responsive with respect to caspase expression and activation. This difference could have several explanations.
First, no excitotoxic process associated with neural death or apoptosis has been observed in the hypothalamus (in contrast to the hippocampus, for instance). Second, neuroprotective systems, and particularly the BDNF system, are not regulated in the same manner in the hypothalamus as in other brain areas (for a review, see Ref. 67). Indeed, we have reported previous studies addressing the BDNF involvement in the regulation of the hypothalamic-pituitary-adrenal axis23,25,28,68,69 and more recently reported that, after a chronic stress, BDNF is a key factor involved in the regulation and adaptive strategy of the hypothalamic-pituitary-adrenal axis, and particularly regulates activity of hypothalamic neurons.58 The BDNF system helps maintain adequate reactivity, mostly under chronic stimulation, but this could in turn weaken other brain regions and, in particular, could weaken the neuronal integrity of the hippocampus. This specific regulation of BDNF level in the hypothalamus could in part explain the relatively high resistance of this cerebral region to amyloid toxicity.
Third, the influence of Aβ25-35 on APP processing in the hypothalamus resulted in a transitory increase in APP expression, not associated with an increase in C99, suggesting an inhibition of amyloidogenic pathways. This regional difference in sensitivity has been observed also in AD patients. In postmortem studies, amyloid deposits were detected in the hypothalamus only in late phases of AD.70 All Aβ plaques identified in the hypothalamus were of the Congo Red-negative amorphic type71,72 and were comparable to the morphology of amyloid deposits observed in hippocampal and cortical structures more precociously in AD patients,73 suggesting that different protective mechanisms are involved in the hypothalamus.
Finally, other neuropeptides (eg, somatostatin or pituitary adenylate cyclase-activating polypeptide) and neurohormones (eg, dehydroepiandrosterone and pregnenolone sulfates) with demonstrated neuroprotective action are particularly concentrated within the hypothalamus.74–78 These hypothalamic neuropeptides and neurohormones likely contribute to the particular resistance of this structure to the amyloid toxicity.
In summary, Aβ25-35 injection results in a massive toxicity with biochemical and behavioral alterations, neuroinflammation, deregulation of endogenous neuroprotective systems, and neurodegeneration. This model also results in modified APP processing and in increased pathological Tau phosphorylation through activation of glycogen synthase kinase-3β (GSK-3β).16 These symptomatic and pathophysiological similarities between the Aβ25-35 model and AD bring clear face validity and construct validity to the model. The model could therefore be particularly suitable for developing and evaluating potential new drugs against AD and for identifying new pathological mechanisms. Moreover, although there is no doubt that progressive Aβ accumulation contributes to the AD pathology and that extracellular amyloid deposits are a hallmark of AD, it is conceivable that these deposits may be only one aspect of a larger pathological cascade and so may be indirect consequences of protective responses geared toward sequestering toxic soluble Aβ molecules within plaques, from which oligomeric toxic fragments could be released by proteolysis.79–82 Thus, the body of data accumulated on this animal model, together with the evidence of Aβ25–35/40 presence in AD patient brains, clearly suggests that the peptide role in the evolution and diagnosis of AD could be largely underestimated.
Acknowledgments
The authors thank Edmond Savary for assistance in data collection and Dr. Susanna Malmström for improving the English in the manuscript.
Footnotes
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.03.021.
Supplementary data
Variations of general locomotor activity and body temperature determined at 1, 2, and 3 weeks after injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v., scrambled group; n = 7) or Aβ25-35 (10 μg/rat i.c.v., Aβ25-35 group; n = 7). Locomotor activity and body temperature were monitored using telemetric sensors as described under Materials and Methods. The thick black line indicates the nocturnal phase (7:00 PM to 7:00 AM). Results are expressed as means/hour for 1, 2, and 3 weeks after the intracerebroventricular injection.
References
- 1.Selkoe D.J. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399(6738 Suppl):A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 2.Yankner B.A. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 4.Pike C.J., Cummings B.J., Cotman C.W. Beta-amyloid induces neuritic dystrophy in vitro: similarities with Alzheimer pathology. Neuroreport. 1992;3:769–772. doi: 10.1097/00001756-199209000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Pike C.J., Walencewicz-Wasserman A.J., Kosmoski J., Cribbs D.H., Glabe C.G., Cotman C.W. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- 6.Behl C., Davis J.B., Klier F.G., Schubert D. Amyloid beta peptide induces necrosis rather than apoptosis. Brain Res. 1994;645:253–264. doi: 10.1016/0006-8993(94)91659-4. [DOI] [PubMed] [Google Scholar]
- 7.Ivins K.J., Thornton P.L., Rohn T.T., Cotman C.W. Neuronal apoptosis induced by beta-amyloid is mediated by caspase-8. Neurobiol Dis. 1999;6:440–449. doi: 10.1006/nbdi.1999.0268. [DOI] [PubMed] [Google Scholar]
- 8.Yankner B.A., Duffy L.K., Kirschner D.A. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 9.Stepanichev M.Y., Zdobnova I.M., Yakovlev A.A., Onufriev M.V., Lazareva N.A., Zarubenko I.I., Gulyaeva N.V. Effects of tumor necrosis factor-alpha central administration on hippocampal damage in rat induced by amyloid beta-peptide (25–35) J Neurosci Res. 2003;71:110–120. doi: 10.1002/jnr.10469. [DOI] [PubMed] [Google Scholar]
- 10.Stepanichev M.Y., Zdobnova I.M., Zarubenko I.I., Lazareva N.A., Gulyaeva N.V. Studies of the effects of central administration of beta-amyloid peptide (25–35): pathomorphological changes in the hippocampus and impairment of spatial memory. Neurosci Behav Physiol. 2006;36:101–106. doi: 10.1007/s11055-005-0167-1. [DOI] [PubMed] [Google Scholar]
- 11.Villard V., Espallergues J., Keller E., Alkam T., Nitta A., Yamada K., Nabeshima T., Vamvakides A., Maurice T. Antiamnesic and neuroprotective effects of the aminotetrahydrofuran derivative ANAVEX1-41 against amyloid beta(25–35)-induced toxicity in mice. Neuropsychopharmacology. 2009;34:1552–1566. doi: 10.1038/npp.2008.212. [DOI] [PubMed] [Google Scholar]
- 12.Meunier J., Ieni J., Maurice T. The anti-amnesic and neuroprotective effects of donepezil against amyloid beta25-35 peptide-induced toxicity in mice involve an interaction with the sigma1 receptor. Br J Pharmacol. 2006;149:998–1012. doi: 10.1038/sj.bjp.0706927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stepanichev M.Y., Zdobnova I.M., Zarubenko I.I., Moiseeva Y.V., Lazareva N.A., Onufriev M.V., Gulyaeva N.V. Amyloid-beta(25–35)-induced memory impairments correlate with cell loss in rat hippocampus. Physiol Behav. 2004;80:647–655. doi: 10.1016/j.physbeh.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Gruden M.A., Davidova T.B., Malisauskas M., Sewell R.D.E., Voskresenskaya N.I., Wilhelm K., Elistratova E.I., Sherstnev V.V., Morozova-Roche L.A. Differential neuroimmune markers to the onset of Alzheimer's disease neurodegeneration and dementia: autoantibodies to Abeta((25–35)) oligomers, S100b and neurotransmitters. J Neuroimmunol. 2007;186:181–192. doi: 10.1016/j.jneuroim.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 15.Kubo T., Nishimura S., Kumagae Y., Kaneko I. In vivo conversion of racemized beta-amyloid ([D-Ser 26]A beta 1–40) to truncated and toxic fragments ([D-Ser 26]A beta 25–35/40) and fragment presence in the brains of Alzheimer's patients. J Neurosci Res. 2002;70:474–483. doi: 10.1002/jnr.10391. [DOI] [PubMed] [Google Scholar]
- 16.Klementiev B., Novikova T., Novitskaya V., Walmod P.S., Dmytriyeva O., Pakkenberg B., Berezin V., Bock E. A neural cell adhesion molecule-derived peptide reduces neuropathological signs and cognitive impairment induced by Abeta25-35. Neuroscience. 2007;145:209–224. doi: 10.1016/j.neuroscience.2006.11.060. [DOI] [PubMed] [Google Scholar]
- 17.Chavant F., Deguil J., Pain S., Ingrand I., Milin S., Fauconneau B., Perault-Pochat M.C., Lafay-Chebassier C. Imipramine, in part through tumor necrosis factor inhibition, prevents cognitive decline and -amyloid accumulation in a mouse model of Alzheimer's Disease. J Pharmacol Exp Ther. 2010;332:505–514. doi: 10.1124/jpet.109.162164. [DOI] [PubMed] [Google Scholar]
- 18.Maurice T., Lockhart B.P., Su T.P., Privat A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- 19.Sigurdsson E.M., Scholtzova H., Mehta P.D., Frangione B., Wisniewski T. Immunization with a nontoxic/nonfibrillar amyloid-beta homologous peptide reduces Alzheimer's disease-associated pathology in transgenic mice. Am J Pathol. 2001;159:439–447. doi: 10.1016/s0002-9440(10)61715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marchal S., Shehi E., Harricane M.C., Fusi P., Heitz F., Tortora P., Lange R. Structural instability and fibrillary aggregation of non-expanded human Ataxin-3 revealed under high pressure and temperature. J Biol Chem. 2003;278:31554–31563. doi: 10.1074/jbc.M304205200. [DOI] [PubMed] [Google Scholar]
- 21.Delobette S., Privat A., Maurice T. In vitro aggregation facilitates beta-amyloid peptide-(25–35)-induced amnesia in the rat. Eur J Pharmacol. 1997;319:1–4. doi: 10.1016/s0014-2999(96)00922-3. [DOI] [PubMed] [Google Scholar]
- 22.Paxinos G., Watson C. Academic Press; San Diego, CA: 1997. The rat brain in stereotaxic coordinates: The compact 3rd edition. [Google Scholar]
- 23.Naert G., Ixart G., Tapia-Arancibia L., Givalois L. Continuous i.c.v. infusion of brain-derived neurotrophic factor modifies hypothalamic-pituitary-adrenal axis activity, locomotor activity and body temperature rhythms in adult male rats. Neuroscience. 2006;139:779–789. doi: 10.1016/j.neuroscience.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 24.Meunier J., Gue M., Récasens M., Maurice T. Attenuation by a sigma1 (sigma1) receptor agonist of the learning and memory deficits induced by a prenatal restraint stress in juvenile rats. Br J Pharmacol. 2004;142:689–700. doi: 10.1038/sj.bjp.0705835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Givalois L., Naert G., Rage F., Ixart G., Arancibia S., Tapia-Arancibia L. A single brain-derived neurotrophic factor injection modifies hypothalamo-pituitary-adrenocortical axis activity in adult male rats. Mol Cell Neurosci. 2004;27:280–295. doi: 10.1016/j.mcn.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Hermes-Lima M., Willmore W.G., Storey K.B. Quantification of lipid peroxidation in tissue extracts based on Fe(III)xylenol orange complex formation. Free Radic Biol Med. 1995;19:271–280. doi: 10.1016/0891-5849(95)00020-x. [DOI] [PubMed] [Google Scholar]
- 27.Cotrufo T., Viegi A., Berardi N., Bozzi Y., Mascia L., Maffei L. Effects of neurotrophins on synaptic protein expression in the visual cortex of dark-reared rats. J Neurosci. 2003;23:3566–3571. doi: 10.1523/JNEUROSCI.23-09-03566.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Givalois L., Arancibia S., Alonso G., Tapia-Arancibia L. Expression of BDNF and its receptors in the median eminence cells with sensitivity to stress. Endocrinology. 2004;135:4737–4747. doi: 10.1210/en.2004-0616. [DOI] [PubMed] [Google Scholar]
- 29.Halverson K.J., Sucholeiki I., Ashburn T.T., Lansbury P.T. Location of beta-sheet-forming sequences in amyloid proteins by FTIR. J Am Chem Soc. 1991;113:6701–6703. [Google Scholar]
- 30.Janek K., Behlke J., Zipper J., Fabian H., Georgalis Y., Beyermann M., Bienert M., Krause E. Water-soluble beta-sheet models which self-assemble into fibrillar structures. Biochemistry. 1999;38:8246–8252. doi: 10.1021/bi990510+. [DOI] [PubMed] [Google Scholar]
- 31.Surewicz W.K., Mantsch H.H., Chapman D. Determination of protein secondary structure by Fourier transform infra-red spectroscopy: a critical assessment. Biochemistry. 1993;32:389–394. doi: 10.1021/bi00053a001. [DOI] [PubMed] [Google Scholar]
- 32.Kaneko I., Morimoto K., Kubo T. Drastic neuronal loss in vivo by beta-amyloid racemized at Ser(26) residue: conversion of non-toxic [D-Ser(26)]beta-amyloid 1–40 to toxic and proteinase-resistant fragments. Neuroscience. 2001;104:1003–1011. doi: 10.1016/s0306-4522(01)00155-5. [DOI] [PubMed] [Google Scholar]
- 33.Streit W.J., Walter S.A., Pennel N.A. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 34.Hagan J.J., Jansen J.H., Broekkamp C.L. Selective behavioural impairment after acute intoxication with trimethyltin (TMT) in rats. Neurotoxicology. 1988;9:53–74. [PubMed] [Google Scholar]
- 35.Nabeshima T., Katoh A., Ishimaru H., Yoneda Y., Ogita K., Murase K., Ohtsuka H., Inari K., Fukuta T., Kameyama T. Carbon monoxide-induced delayed amnesia, delayed neuronal death and change in acetylcholine concentration in mice. J Pharmacol Exp Ther. 1991;256:378–384. [PubMed] [Google Scholar]
- 36.Ordy J.M., Thomas G.J., Volpe B.T., Dunlap W.P., Colombo P.M. An animal model of human-type memory loss based on aging, lesion, forebrain ischemia, and drug studies with the rat. Neurobiol Aging. 1988;9:667–683. doi: 10.1016/s0197-4580(88)80131-3. [DOI] [PubMed] [Google Scholar]
- 37.Blennow K., de Leon M.J., Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 38.Jakob-Roetne R., Jacobsen H. Alzheimer's disease: from pathology to therapeutic approaches. Angew Chem Int Ed Engl. 2009;48:3030–3059. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi Y., Kawashima S. Effects of amyloid-beta-(25–35) on passive avoidance, radial-arm maze learning and choline acetyltransferase activity in the rat. Eur J Pharmacol. 2001;412:265–272. doi: 10.1016/s0014-2999(01)00730-0. [DOI] [PubMed] [Google Scholar]
- 40.Alkam T., Nitta A., Mizoguchi H., Itoh A., Murai R., Nagai T., Yamada K., Nabeshima T. The extensive nitration of neurofilament light chain in the hippocampus is associated with the cognitive impairment induced by amyloid beta in mice. J Pharmacol Exp Ther. 2008;327:137–147. doi: 10.1124/jpet.108.141309. [DOI] [PubMed] [Google Scholar]
- 41.Allen J.W., Eldadah B.A., Faden A.I. Beta-amyloid-induced apoptosis of cerebellar granule cells and cortical neurons: exacerbation by selective inhibition of group I metabotropic glutamate receptors. Neuropharmacology. 1999;38:1243–1252. doi: 10.1016/s0028-3908(99)00044-1. [DOI] [PubMed] [Google Scholar]
- 42.Casley C.S., Land J.M., Sharpe M.A., Clark J.B., Duchen M.R., Canevari L. Beta-amyloid fragment 25–35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol Dis. 2002;10:258–267. doi: 10.1006/nbdi.2002.0516. [DOI] [PubMed] [Google Scholar]
- 43.Lu D.C., Soriano S., Bredesen D.E., Koo E.H. Caspase cleavage of the amyloid precursor protein modulates amyloid beta-protein toxicity. J Neurochem. 2003;87:733–741. doi: 10.1046/j.1471-4159.2003.02059.x. [DOI] [PubMed] [Google Scholar]
- 44.Mattson M.P., Partin J., Begley J.G. Amyloid beta-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 1998;807:167–176. doi: 10.1016/s0006-8993(98)00763-x. [DOI] [PubMed] [Google Scholar]
- 45.Movsesyan V.A., Stoica B.A., Faden A.I. MGLuR5 activation reduces beta-amyloid-induced cell death in primary neuronal cultures and attenuates translocation of cytochrome c and apoptosis-inducing factor. J Neurochem. 2004;89:1528–1536. doi: 10.1111/j.1471-4159.2004.02451.x. [DOI] [PubMed] [Google Scholar]
- 46.Wei W., Norton D.D., Wang X., Kusiak J.W. Abeta17-42 in Alzheimer's disease activates JNK and caspase-8 leading to neuronal apoptosis. Brain. 2002;125:2036–2043. doi: 10.1093/brain/awf205. [DOI] [PubMed] [Google Scholar]
- 47.Yan X.Z., Qiao J.T., Dou Y., Qiao Z.D. Beta-amyloid peptide fragment 31–35 induces apoptosis in cultured cortical neurons. Neuroscience. 1999;92:177–184. doi: 10.1016/s0306-4522(98)00727-1. [DOI] [PubMed] [Google Scholar]
- 48.Sutton E.T., Hellermann G.R., Thomas T. Beta-amyloid-induced endothelial necrosis and inhibition of nitric oxide production. Exp Cell Res. 1997;230:368–376. doi: 10.1006/excr.1996.3440. [DOI] [PubMed] [Google Scholar]
- 49.Geci C., How J., Alturaihi H., Kumar U. Beta-amyloid increases somatostatin expression in cultured cortical neurons. J Neurochem. 2007;101:664–673. doi: 10.1111/j.1471-4159.2006.04415.x. [DOI] [PubMed] [Google Scholar]
- 50.Tong L., Balazs R., Thornton P.L., Cotman C.W. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang Y., Yamada K., Kanou Y., Miyazaki T., Xiong X., Kambe F., Murata Y., Seo H., Nabeshima T. Spatiotemporal expression of BDNF in the hippocampus induced by the continuous intracerebroventricular infusion of beta-amyloid in rats. Mol Brain Res. 2000;80:188–197. doi: 10.1016/s0169-328x(00)00158-3. [DOI] [PubMed] [Google Scholar]
- 52.Krugers H.J., Koolhaas J.M., Bohus B., Korf J. A single social stress-experience alters glutamate receptor-binding in rat hippocampal CA3 area. Neurosci Lett. 1993;154:73–77. doi: 10.1016/0304-3940(93)90174-j. [DOI] [PubMed] [Google Scholar]
- 53.Lowy M.T., Gault L., Yamamoto B.K. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem. 1993;61:1957–1960. doi: 10.1111/j.1471-4159.1993.tb09839.x. [DOI] [PubMed] [Google Scholar]
- 54.McEwen B.S. Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol. 2008;583:174–185. doi: 10.1016/j.ejphar.2007.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Catania C., Sotiropoulos I., Silva R., Onofri C., Breen K.C., Sousa N., Almeida O.F. The amyloidogenic potential and behavioral correlates of stress. Mol Psychiatry. 2009;14:95–105. doi: 10.1038/sj.mp.4002101. [DOI] [PubMed] [Google Scholar]
- 56.Abrahám I., Harkany T., Horvath K.M., Veenema A.H., Penke B., Nyakas C., Luiten P.G. Chronic corticosterone administration dose-dependently modulates Abeta(1–42)- and NMDA-induced neurodegeneration in rat magnocellular nucleus basalis. J Neuroendocrinol. 2000;12:486–494. doi: 10.1046/j.1365-2826.2000.00475.x. [DOI] [PubMed] [Google Scholar]
- 57.Goodman Y., Bruce A.J., Cheng B., Mattson M.P. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 58.Naert G., Ixart G., Maurice T., Tapia-Arancibia L., Givalois L. Brain-derived neurotrophic factor and hypothalamic-pituitary-adrenal axis adaptation processes in a depressive-like state induced by chronic restraint stress. Mol Cell Neurosci. 2011;46:55–66. doi: 10.1016/j.mcn.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 59.Davis K.L., Davis B.M., Greenwald B.S., Mohs R.C., Mathé A.A., Johns C.A., Horvath T.B. Cortisol and Alzheimer's disease: I: basal studies. Am J Psychiatry. 1986;143:300–305. doi: 10.1176/ajp.143.3.300. [DOI] [PubMed] [Google Scholar]
- 60.Masugi F., Ogihara T., Sakaguchi K., Otsuka A., Tsuchiya Y., Morimoto S., Kumahara Y., Saeki S., Nishide M. High plasma levels of cortisol in patients with senile dementia of the Alzheimer's type. Methods Find Exp Clin Pharmacol. 1989;11:707–710. [PubMed] [Google Scholar]
- 61.Ming G.L., Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–250. doi: 10.1146/annurev.neuro.28.051804.101459. [DOI] [PubMed] [Google Scholar]
- 62.Taupin P. Neurogenesis in the adult central nervous system. C R Biol. 2006;329:465–475. doi: 10.1016/j.crvi.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 63.Chen Y., Dong C. Abeta40 promotes neuronal cell fate in neural progenitor cells. Cell Death Differ. 2009;16:386–394. doi: 10.1038/cdd.2008.94. [DOI] [PubMed] [Google Scholar]
- 64.López-Toledano M.A., Shelanski M.L. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J Neurosci. 2004;24:5439–5444. doi: 10.1523/JNEUROSCI.0974-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mazur-Kolecka B., Golabek A., Nowicki K., Flory M., Frackowiak J. Amyloid-beta impairs development of neuronal progenitor cells by oxidative mechanisms. Neurobiol Aging. 2006;27:1181–1192. doi: 10.1016/j.neurobiolaging.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 66.von Bohlen Und Halbach O. Immunohistological markers for staging neurogenesis in adult hippocampus. Cell Tissue Res. 2007;329:409–420. doi: 10.1007/s00441-007-0432-4. [DOI] [PubMed] [Google Scholar]
- 67.Tapia-Arancibia L., Rage F., Givalois L., Arancibia S. Physiology of BDNF: focus on hypothalamic function. Front Neuroendocrinol. 2004;25:77–107. doi: 10.1016/j.yfrne.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 68.Givalois L., Marmigère F., Rage F., Ixart G., Arancibia S., Tapia-Arancibia L. Immobilization stress rapidly and differentially modulates BDNF and TrkB mRNA expression in the pituitary gland of adult male rats. Neuroendocrinology. 2001;74:148–159. doi: 10.1159/000054681. [DOI] [PubMed] [Google Scholar]
- 69.Marmigère F., Givalois L., Rage F., Arancibia S., Tapia-Arancibia L. Rapid induction of BDNF expression in the hippocampus during immobilization stress challenge in adult rats. Hippocampus. 2003;13:646–655. doi: 10.1002/hipo.10109. [DOI] [PubMed] [Google Scholar]
- 70.Thal D.R., Rüb U.M., Orantes M.M., Braak H.M. Phases of Abeta-deposition in the human brain and its relevance for the development of Alzheimer's disease. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 71.Van de Nes J.A., Kamphorst W., Ravid R., Swaab D.F. Comparison of beta-protein/a4 deposits and Alz-50-stained cytoskeletal changes in the hypothalamus and adjoining areas of Alzheimer's disease patients: amorphic plaques and cytoskeletal changes occur independently. Acta Neuropathol. 1998;96:129–138. doi: 10.1007/s004010050872. [DOI] [PubMed] [Google Scholar]
- 72.Van de Nes J.A., Konermann S., Nafe R., Swaab D. Beta-protein/a4 deposits are not associated with hyperphosphorylated Tau in somatostatin neurons in the hypothalamus of Alzheimer's disease patients. Acta Neuropathol. 2006;111:126–138. doi: 10.1007/s00401-005-0018-8. [DOI] [PubMed] [Google Scholar]
- 73.Standaert D.G., Lee V.M., Greenberg B.D., Lowery D.E., Trojanowski J.Q. Molecular features of hypothalamic plaques in Alzheimer's disease. Am J Pathol. 1991;139:681–691. [PMC free article] [PubMed] [Google Scholar]
- 74.Arancibia S., Silhol M., Moulière F., Meffre J., Höllinger I., Maurice T., Tapia-Arancibia L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis. 2008;31:316–326. doi: 10.1016/j.nbd.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 75.Duclot F., Meffre J., Jacquet C., Gong ora C., Maurice T. Mice knock out for the histone acetyltransferase p300/CREB binding protein-associated factor develop a resistance to amyloid toxicity. Neuroscience. 2010;167:850–863. doi: 10.1016/j.neuroscience.2010.02.055. [DOI] [PubMed] [Google Scholar]
- 76.Kojro E., Postina R., Buro C., Meiringer C., Gehrig-Burger K., Fahrenholz F. The neuropeptide PACAP promotes the alpha-secretase pathway for processing the Alzheimer amyloid precursor protein. FASEB J. 2006;20:512–514. doi: 10.1096/fj.05-4812fje. [DOI] [PubMed] [Google Scholar]
- 77.Bourgault S., Vaudry D., Dejda A., Doan N.D., Vaudry H., Fournier A. Pituitary adenylate cyclase-activating polypeptide: focus on structure-activity relationships of a neuroprotective peptide. Curr Med Chem. 2009;16:4462–4480. doi: 10.2174/092986709789712899. [DOI] [PubMed] [Google Scholar]
- 78.Weill-Engerer S., David J.P., Sazdovitch V., Liere P., Eychenne B., Pianos A., Schumacher M., Delacourte A., Baulieu E.E., Akwa Y. Neurosteroid quantification in human brain regions: comparison between Alzheimer's and nondemented patients. J Clin Endocrinol Metab. 2002;87:5138–5143. doi: 10.1210/jc.2002-020878. [DOI] [PubMed] [Google Scholar]
- 79.Watson D., Castano E., Kokjohn T.A., Kuo Y.M., Lyubchenko Y., Pinskyz D., Connolly E.S., Esh C., Luehrs D.C., Stine W.B., Rowse L.M., Emmerling M.R., Roher A.E. Physicochemical characteristics of soluble oligomeric Abeta and their pathologic role in Alzheimer's disease. Neurol Res. 2005;27:869–881. doi: 10.1179/016164105X49436. [DOI] [PubMed] [Google Scholar]
- 80.Ferreira S.T., Vieira M.N., De Felice F.G. Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. IUBMB Life. 2007;59:332–345. doi: 10.1080/15216540701283882. [DOI] [PubMed] [Google Scholar]
- 81.Walsh D.M., Selkoe D.J. A beta oligomers–a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 82.Millucci L., Ghezzi L., Bernardini G., Santucci A. Conformations and biological activities of amyloid beta peptide 25–35. Curr Protein Pept Sci. 2010;11:54–67. doi: 10.2174/138920310790274626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Variations of general locomotor activity and body temperature determined at 1, 2, and 3 weeks after injection of scrambled Aβ25-35 peptide (10 μg/rat i.c.v., scrambled group; n = 7) or Aβ25-35 (10 μg/rat i.c.v., Aβ25-35 group; n = 7). Locomotor activity and body temperature were monitored using telemetric sensors as described under Materials and Methods. The thick black line indicates the nocturnal phase (7:00 PM to 7:00 AM). Results are expressed as means/hour for 1, 2, and 3 weeks after the intracerebroventricular injection.