

Abstract
Vascular endothelial growth factor A (VEGF-A) can play both beneficial and deleterious roles in renal diseases, where its specific function might be determined by nitric oxide bioavailability. The complexity of VEGF-A in renal disease could in part be accounted for by the distinct roles of its two receptors; VEGFR1 is involved in the inflammatory responses, whereas VEGFR2 predominantly mediates angiogenesis. Because nondiabetic chronic renal disease is associated with capillary loss, we hypothesized that selective stimulation of VEGFR2 could be beneficial in this setting. However, VEGFR2 activation may be deleterious in the presence of nitric oxide deficiency. We systematically overexpressed a mutant form of VEGF-A binding only VEGFR2 (Flk-sel) using an adeno-associated virus-1 vector in wild-type and eNOS knockout mice and then induced renal injury by uninephrectomy. Flk-sel treatment increased angiogenesis and lowered blood pressure in both mouse types. Flk-sel overexpression caused mesangial injury with increased proliferation associated with elevated expression of PDGF, PDGF-β receptor, and VEGFR2; this effect was greater in eNOS knockout than in wild-type mice. Flk-sel also induced tubulointerstitial injury, with some tubular epithelial cells expressing α-smooth muscle actin, indicating a phenotypic evolution toward myofibroblasts. In conclusion, prestimulation of VEGFR2 can potentiate subsequent renal injury in mice, an effect enhanced in the setting of nitric oxide deficiency.
Vascular endothelial growth factor A (VEGF-A) plays a key role in maintaining peritubular and glomerular capillary integrity in the normal kidney. In renal disease, however, the actions of VEGF-A are more complicated. A reduction in renal VEGF-A is observed in acute and chronic nondiabetic renal disease, mostly associated with a loss of glomerular and peritubular capillaries.1–5 In these situations, administration of VEGF-A has been shown to improve renal histology and function.3,4,6,7 In contrast, levels of both circulating and local VEGF-A are high in diabetes, and excessive VEGF-A has been shown to have a role in mediating glomerular hypertrophy, proteinuria, and retinopathy,8,9 indicating that VEGF-A is deleterious in this unique situation. Given the fact that diabetic conditions are associated with a lower bioavailability of nitric oxide (NO), we have proposed a hypothesis that the deleterious effect of VEGF-A in diabetes could be attributed to a reduced bioavailability of endothelial NO in the kidney. Consistent with this hypothesis, diabetic nephropathy is worsened in the setting of endothelial NO deficiency and the effects of VEGF-A to induce angiogenesis and inflammation are enhanced in this setting.10,11
The actions of VEGF-A are mediated through its two receptors, Flt-1 (VEGFR1) and Flk-1 (VEGFR2). VEGFR1 is involved in the inflammatory response by stimulating macrophage chemotaxis,11,12 as well as vascular permeability and vessel stabilization.13,14 In contrast, VEGFR2 mediates angiogenesis15 and reduces blood pressure.16 The specific roles of VEGFR1 and VEGFR2 in nondiabetic and diabetic kidney disease are not well understood. One could postulate that VEGFR2 (but not VEGFR1) might have a key role in protecting the kidney from nondiabetic injury by preserving capillary number, because of its angiogenic effects. In diabetic nephropathy, however, VEGFR2 likely has a causal role, as is suggested by studies showing up-regulation of VEGFR2 mRNA expression within weeks of the onset of diabetes in the rat, whereas VEGFR1 was undetectable.17 In this situation, a reduction in endothelial NO, which is often observed in diabetic patients, might be a key factor to enhance VEGFR2 signaling for excessive angiogenesis.18,19
To begin to understand the roles of VEGFR1 and VEGFR2 in diabetic and nondiabetic kidney diseases, we generated an adeno-associated virus containing a mutant form of VEGF-A that binds only to VEGFR2 (rAAV1-Flk-sel) and injected these vectors in normal wild-type (WT) mice and in eNOS-knockout mice (eNOSKO) lacking endothelial NO synthase. Because the role of a specific factor can be more evident in the presence of renal injury than in normal kidney, uninephrectomy (UNx) was performed to accelerate renal injury. We hypothesized that administration of rAAV1-Flk-sel in UNx-WT mice would be protective by enhancing angiogenesis of glomerular and peritubular capillaries. In contrast, we predicted that overexpression of rAAV1-Flk-sel in UNx-eNOSKO mice would be deleterious. Overexpression of the VEGF-A mutant for VEGFR1 was unsuccessful in our hands; therefore, here we present the results only for overexpression of Flk-sel.
Materials and Methods
Experimental Protocol
Eight-week-old male C57BL/6J-Nos3tm1Unc mice (eNOSKO mice) and background strain C57BL6/J mice (WT mice) weighing 20 to 25 g were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were housed under a 12-hour light/dark cycle with free access to food and water. All experiments were performed in accordance with the Animal Care and Use Committee of the University of Florida. Mice (n = 10 in each group) were injected intramuscularly with 1.0 × 1010 viral particles of either rAAV1-empty vector (EV) or rAAV1-Flk-sel. This level was chosen because a lower dose of 1.0 × 109 viral particles failed to increase serum VEGF-A levels in pilot studies. Flk-sel was provided by Dr. Napoleon Ferrara (Genentech, South San Francisco, CA). The construction and characterization of Flk-sel has been described previously,20 with mutations in human VEGF165 at positions D63S/G65M/L66R.16,21,22 The rAAV vectors expressing Flk-sel were generated and purified in the Vector Core Laboratory at the University of Florida, using techniques described previously.23 Three months later, UNx was used to induce kidney injury. Renal histology was examined at 4 months with blood and urine collected to measure blood urea nitrogen and urinary albumin/creatinine ratio, as described previously.24 The scheme of the experimental design is presented in Figure 1.
Figure 1.

Study protocol. Three months after adeno-associated virus (AAV) injection, UNx was used to induce kidney injury. Mice were sacrificed at 4 months.
Blood Pressure Measurements
Systolic blood pressure (BP) was assessed every month using a tail-cuff sphygmomanometer (Visitech BP2000; Visitech Systems, Apex, NC). Animals were accustomed to the machine by training, and all measurements were performed at the same time of day.
Serum VEGF-A Measurement
Serum VEGF-A levels were measured at 3 and 4 months using a commercial human enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. We had previously confirmed that Flk-sel can be detected by this kit.10
IHC
Kidneys were fixed in neutral buffered formalin (10%), embedded in paraffin, and sectioned at 2-μm thickness. Tubulointerstitial injury was assessed by immunohistochemistry (IHC) with goat anti-human collagen III (SouthernBiotech, Birmingham, AL) and rat anti-mouse F4/80 (Serotec, Oxford, UK) which detects a subtype of macrophages present predominately in the interstitium.11,25 Phenotypic changes in tubular epithelial cells were evaluated using rabbit anti-human α-SMA (Abcam, Cambridge, MA) and rabbit anti-human transforming growth factor β1 (TGF-β1) (Santa Cruz Biotechnology, Santa Cruz, CA). Rabbit anti-mouse collagen IV (Chemicon International, Temecula, CA), rabbit anti-human fibronectin (Sigma-Aldrich, St. Louis, MO), and mouse anti-human CD68 (Abcam; performed on frozen kidney), a marker of macrophage subtypes predominantly expressed in glomeruli,11,25 were used to evaluate glomerular injury. For vascular assessment of angiogenic responses, capillary endothelial cells were detected by a rabbit anti-rat thrombomodulin26 and rabbit monoclonal human VEGFR2 (Cell Signaling Technology, Danvers, MA). Immunostaining on methyl Carnoy's fixed tissue sections was performed with either rabbit anti-human platelet-derived growth factor receptor-β (PDGFRβ) (Santa Cruz Biotechnology), rabbit anti-human PDGF-B (Ab-1; Oncogene Research Products, La Jolla, CA), or anti-human PDGF-D (kindly provided by ZymoGenetics, Seattle, WA) to examine mesangial damage, followed by ImmPress anti-rabbit Ig polymer detection system (Vector Laboratories, Burlingame CA). To examine cell proliferation, the thymidine analog 5-bromo-2′-deoxyuridine (BrdU) (50 mg/kg; Sigma-Aldrich) was intraperitoneally administered to mice 2 hours before sacrifice. BrdU-positive cells were detected with a rat monoclonal BrdU antibody (Serotec). For all antibodies, color was developed using 3,3′-diaminobenzidine (Dako, Carpinteria, CA). For negative controls, primary antibodies were replaced with species-matched antibodies.
Morphological Quantification
Periodic acid-Schiff was used to detect morphological characteristics. All glomeruli (>50 glomerular cross-sections per biopsy) and the whole cortical tubulointerstitial area in the axial plane were scanned with the AxioVision image analyzer (Carl Zeiss, Thornwood, NY) and examined. The total dimension of the glomerulus was determined by tracing the outline of the glomerular tuft. The percentage of mesangial matrix (defined as mesangial area) was determined by assessing the PAS-positive area and dividing by the total area of the glomerular tuft. Mesangiolysis was determined as presence of dissolution of the mesangial matrix.24 Mesangial cell number was quantified using the Oxford classification,27 in which each glomerulus is assessed by the most cellular mesangial area, omitting the mesangial area immediately adjacent to the vascular stalk. In addition, capillary lumen number per glomerulus was also counted, to evaluate angiogenic responses. To examine glomerular lesions, we measured the positive immunostained area in each glomerular cross section for collagen IV, fibronectin, thrombomodulin, and VEGFR2 as an overall percentage of positive area of the glomerular tuft, using computerized image analysis. To quantify endothelial proliferation, the numbers of BrdU-positive cells attached to positive signals for thrombomodulin were counted. Similarly, percentage of positive area for thrombomodulin, collagen III, F4/80, VEGFR2, TGF-β1, and PDGF-B was examined in whole renal cortex, to assess tubulointerstitial injury. Glomerular CD68+ cells per square millimeter were counted by examining 50 glomeruli, tubules, and interstitial tissues. in the renal cortex. All quantifications were performed in a blinded manner by two independent investigators (W.S. and T.K.).
Electron Microscopy
Kidneys were fixed in glutaraldehyde, postfixed with osmium tetroxide, and embedded in Epon 812 medium (Nisshin EM, Tokyo, Japan). Ultrathin sections were examined with a H7100 electron microscope (Hitachi, Ibaraki, Japan).
Western Blot Analysis
Mouse kidney tissues were snap-frozen in liquid nitrogen for protein isolation. Western blot analysis was performed with 10 to 30 μg protein/lane. Blots were incubated with either rabbit monoclonal VEGFR1 antibody (Epitomics, Burlingame, CA), rabbit monoclonal VEGFR2 antibody (Cell Signaling), mouse monoclonal β-actin antibody (Sigma-Aldrich), rabbit anti-mouse Akt (both total Akt and phosphorylated Ser473) (Cell Signaling), mouse monoclonal anti-human GAPDH (Santa Cruz Biotechnology), rabbit anti-TGF-β1 (Cell Signaling), rabbit anti-HIF1α (Novus Biologicals, Littleton, CO), a rabbit anti-human α-SMA antibody (Abcam), or a mouse anti-human E-cadherin antibody (BD Transduction Laboratories, San Diego, CA), followed by incubation with peroxidase-conjugated rabbit IgG or mouse IgG (DakoCytomation, Carpinteria, CA). Receptor phosphorylation of VEGFR1 was detected using a rabbit anti-phospho-Flt-1 (pY1213) antibody (Millipore–Chemicon International, Temecula, CA). Phosphorylated VEGFR2 was detected by a rabbit anti-mouse-pY949-VEGF R2 antibody (BioSource, Camarillo, CA). Proteins were visualized with an enhanced chemiluminescence detection system (Amersham–GE Healthcare, Piscataway NJ). The density of each band was measured using public domain ImageJ software version 1.41 (NIH, Bethesda, MD).
Cell Culture
Human renal proximal tubular (HK2) cells were cultured in keratinocyte-SFM medium containing epidermal growth factor, bovine pituitary extracts, and 10% serum (Invitrogen, Carlsbad, CA). Ten percent serum was added in order to keep cells healthy during stimulation for 72 hours. After subconfluence, cells were stimulated by recombinant human VEGF-A 165 (100 ng/mL; PeproTech, Rocky Hill, NJ) in the presence of 10% serum to examine if VEGF alters the expressions of E-cadherin and α-smooth muscle actin (α−SMA), analyzed by Western blotting at 24, 48 and 72 hours as described above.
Statistical Analysis
All values are expressed as means ± SD. Statistical analysis was performed with unpaired, two-tailed Student's t-tests for single comparisons or analysis of variance with post hoc testing using Tukey's method for multiple comparisons. A P value of <0.05 was taken to indicate a significant difference.
Results
Systemic Overexpression of rAAV1-Flk-sel in WT and eNOSKO Mice
Initial experiments were performed to determine whether rAAV1-Flk-sel had any effect on WT and eNOSKO mice without renal disease. We administered rAAV1-Flk-sel intramuscularly to both WT and eNOSKO mice and examined them 3 months later. Injection of rAAV1-Flk-sel led to significant elevation of systemic human VEGF-A at 3 months after injection in both mouse types (P < 0.01 versus rAAV1-EV in WT and eNOSKO mice) (Figure 2A), and levels were similar in both mouse types. eNOSKO mice exhibited mild mesangial expansion (Figure 2D), compared with WT mice (Figure 2B), probably because of eNOS deficiency at this age.28 rAAV1-Flk-sel did not alter renal morphology (Figure 2, C and E) or kidney weight in either mouse type (Table 1). Unfortunately, neither blood urea nitrogen nor urine albumin excretion was examined at this point. No remarkable morphological abnormalities were found in spleen, heart, or liver in the mice at this point (data not shown).
Figure 2.
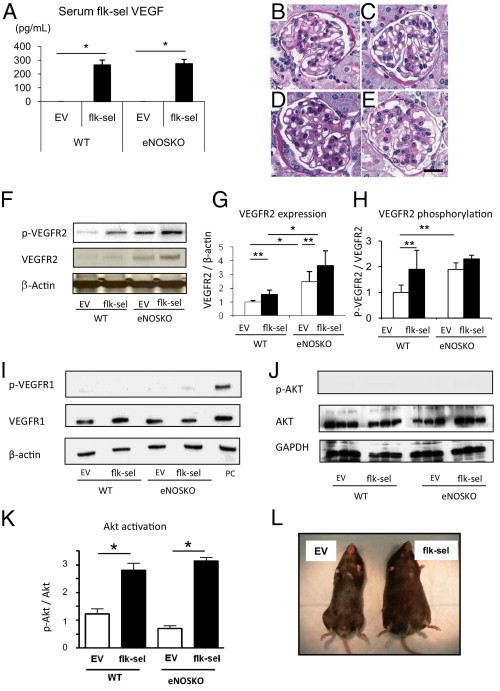
Systemic overexpression of rAAV1-Flk-sel. A: Serum human VEGF-A concentrations in WT and eNOSKO mice treated with rAAV1-EV or rAAV1-Flk-sel at 3 months (before UNx). B–E: Representative PAS staining of glomerulus at 3 months from WT mice treated with rAAV1-EV (B) or with rAAV1-Flk-sel (C) and from eNOSKO mice treated with rAAV1-EV (D) or with rAAV1-Flk-sel (E). Flk-sel treatment did not change glomerular injury in either type of mouse at 3 months (C and E), compared with control EV treatment (B and D), whereas eNOSKO mice exhibited mild mesangial expansion with either treatment (D and E). Scale bar = 20 μm. F: Western blot for renal VEGFR2 in whole kidneys of WT and eNOSKO mice treated with rAAV1-EV or rAAV1-Flk-sel 1 month after UNx. G and H: Quantitative analysis for VEGFR2 expression (VEGFR2/β-actin ratio) (G) and phosphorylation (phosphorylated VEGFR2/total VEGFR2) (H). I: Western blot for VEGFR1 expression and phosphorylation. J: Western blot for phosphorylated Akt. K: quantification of phosphorylated Akt. L: Representative photographs of eNOSKO mice treated with rAAV1-EV or rAAV1-Flk-sel after UNx. *P < 0.01; **P < 0.05. Data are expressed as means ± SD; n = 10 in each group.
Table 1.
General Characteristics of Mice at 3 Months after rAAV Injection
| Characteristic | Wild type |
eNOSKO |
||
|---|---|---|---|---|
| EV | Flk-sel | EV | Flk-sel | |
| Body weight B (g) | 28.0 ± 2.0 | 30.0 ± 2.3⁎ | 27.3 ± 1.7 | 31.1 ± 3.3† |
| Kidney weight K (g) | 0.16 ± 0.014 | 0.16 ± 0.021 | 0.14 ± 0.011 | 0.15 ± 0.011 |
| K/B (10−2) | 0.57 ± 0.05 | 0.55 ± 0.08 | 0.52 ± 0.05 | 0.49 ± 0.06 |
eNOSKO, eNOS knockout; EV, empty vector; Flk-sel, VEGFR2-selective; rAAV, recombinant adeno-associated virus.
P < 0.05 versus WT-EV.
P < 0.01 versus eNOSKO-EV.
Given these findings, we then induced renal injury by performing UNx in both WT and eNOSKO mice at 3 months after rAAV1-Flk-sel injection and examined the mice 1 month later. Both UNx-WT and UNx-eNOSKO mice treated with rAAV1-Flk-sel exhibited an increase in renal total and phosphorylated VEGFR2 (Figure 2, F–H). We did not observe any changes in VEGFR1 levels, and there was no evidence of VEGFR1 phosphorylation in any of the groups of mice (Figure 2I). In further confirmation of VEGFR2 activation, we observed elevated Akt phosphorylation, a downstream target of VEGFR2 signaling,29 after Flk-sel treatment in both WT and eNOSKO mice (Figure 2, J and K). These data suggest that renal VEGFR2 was successfully stimulated by rAAV1-Flk-sel administration in this model.
rAAV1-Flk-sel overexpression was associated with increased body weight in both UNx-WT and UNx-eNOSKO mice, compared with rAAV1-EV treatment (Figure 2L and Table 2). Although previous studies indicated that VEGF-A causes renal hypertrophy,30,31 rAAV1-Flk-sel treatment did not alter kidney size after UNx; instead, the ratio of kidney to body weight was reduced (Table 2). Although overexpression of rAAV1-Flk-sel was well tolerated by both WT and eNOSKO animals, it did lead to some changes in renal function after UNx (Table 2). UNx-WT mice treated with rAAV1-Flk-sel had elevated blood urea nitrogen and urinary albumin excretion, compared with those treated with rAAV1-EV. Blood urea nitrogen was higher in UNx-eNOSKO than in UNx-WT animals, and rAAV1-Flk-sel treatment further enhanced levels. rAAV1-Flk-sel administration to UNx-eNOSKO mice did not lead to alterations in albumin excretion.
Table 2.
General Characteristics of Mice at 1 Month after Uninephrectomy
| Characteristic | Wild type |
eNOSKO |
||
|---|---|---|---|---|
| EV | Flk-sel | EV | Flk-sel | |
| Body weight B (g) | 27.2 ± 1.8 | 31.6 ± 3.2⁎⁎⁎ | 26.9 ± 2.0 | 36.0 ± 5.8‡ |
| Kidney weight K (g) | 0.20 ± 0.014 | 0.20 ± 0.0088 | 0.17 ± 0.020 | 0.19 ± 0.012† |
| K/B (10−2) | 0.75 ± 0.05 | 0.65 ± 0.05⁎⁎ | 0.62 ± 0.06⁎⁎ | 0.53 ± 0.09 |
| BUN (mg/dL) | 28.7 ± 3.4 | 34.2 ± 3.9⁎ | 37.8 ± 2.1⁎⁎⁎⁎ | 41.9 ± 3.8† |
| u-Alb/u-Cre | 0.12 ± 0.07 | 0.48 ± 0.07⁎⁎⁎⁎ | 0.88 ± 0.39⁎⁎⁎ | 0.70 ± 0.50 |
BUN, blood urea nitrogen; eNOSKO, eNOS knockout; EV, empty vector; Flk-sel, VEGFR2-selective; u-Alb/u-Cre, urine-albumin/urine creatinine ratio; WT, wild type.
P < 0.05,
P < 0.01,
P < 0.005, and
P < 0.001 versus WT-EV.
P < 0.05 and
P < 0.001 versus eNOSKO-EV.
Role of VEGFR2 in Blood Pressure in WT and eNOSKO Mice
Consistent with reports that VEGFR2 is responsible for reducing BP,16 we found rAAV1-Flk-sel WT mice had lower systemic BP than rAAV1-EV WT mice (Figure 3), both before UNx and at 1 month after. Given that the effect of VEGF-A on lowering BP is thought to be secondary to stimulating endothelial NO,32 we had hypothesized that BP would not be lowered in the eNOSKO mouse. However, BP was unexpectedly lowered in eNOSKO mice, with rAAV1-Flk-sel overexpression before and at 1 month after UNx (Figure 3).
Figure 3.
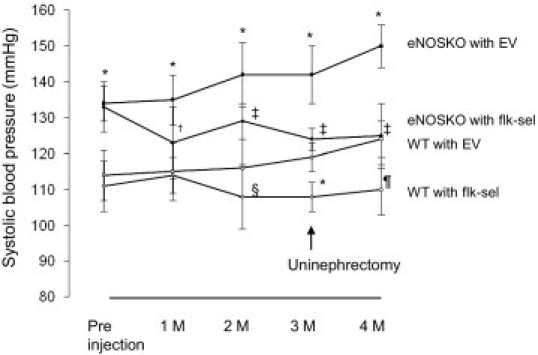
Time course of systolic blood pressure. The rAAV1-Flk-sel treatment lowered blood pressure (BP) beginning at 2 months after injection in WT mice and the change was sustained in UNx-WT mice. BP was significantly higher in eNOSKO mice, compared with WT mice before treatment, but administration of rAAV1-Flk-sel led to a significant decrease 1 month later. The lowering of BP by rAAV1-Flk-sel was also observed in UNx-eNOSKO mice. Solid symbols, eNOSKO mice; open symbols, WT mice. Squares, rAAV1-EV; circles, rAAV1-flk-sel. *P < 0.001 versus WT mice treated with rAAV1-EV; †P < 0.01 versus eNOSKO mice treated with rAAV1-EV; ‡P < 0.001 versus eNOSKO mice treated with rAAV1-EV; §P < 0.05 versus WT mice treated with rAAV1-EV; ¶P < 0.01 versus WT mice treated with rAAV1-EV. Data are expressed as means ± SD; n = 10 in each group.
Increased Angiogenesis in the Kidneys of Flk-sel Overexpressing Mice
One of the key actions of VEGFR2 is to enhance endothelial cell proliferation. We therefore examined BrdU incorporation, as a marker of cell proliferation,33 and thrombomodulin, as a specific endothelial cell marker24,34 (Figure 4). Administration of rAAV1-Flk-sel tended to increase thrombomodulin-positive endothelial cells in glomeruli and tubulointerstitium in UNx-WT mice. This effect was significantly pronounced in the UNx-eNOSKO mice (Figure 4, G and J). The increase in endothelial cells was associated with induction of proliferating cells labeled with BrdU (Figure 4, H and K). Double staining of thrombomodulin with BrdU confirmed an increase in proliferating endothelial number by rAAV1-Flk-sel, which was enhanced in UNx-eNOSKO animals (Figure 4I). Counts of glomerular capillaries were also elevated by rAAV1-Flk-sel in both UNx-WT and UNx-eNOSKO mice, but a lack of eNOS had no effect on this parameter (Figure 5H).
Figure 4.
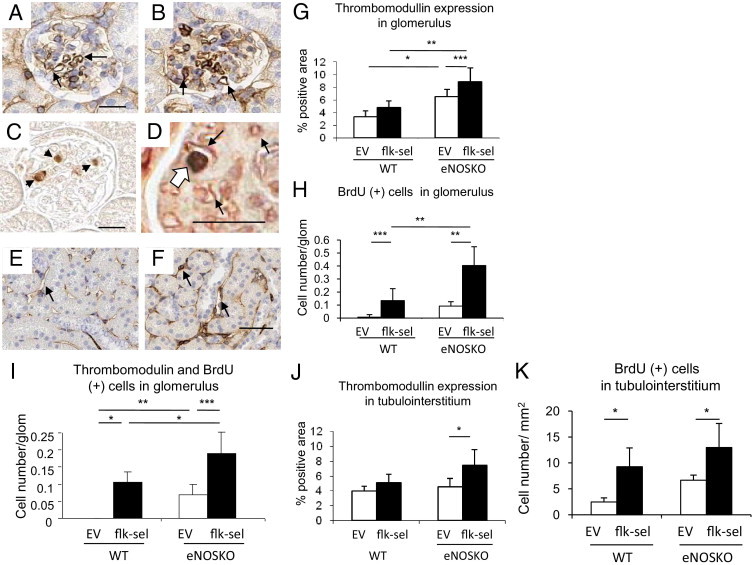
Angiogenic response induced by rAAV1-Flk-sel treatment. A–F: IHC in the glomerulus (A–D) and tubulointerstitium (E and F) for thrombomodulin (black arrows in A, B, E, and F), BrdU (arrow in C) and double staining of thrombomodulin (black arrows) with BrdU (white arrow) (D) at 1 month after UNx in eNOSKO mice treated with rAAV1-EV (A and E) or with rAAV1-Flk-sel (B, C, D, and F). Scale bars = 20 μm. G–K: Quantitative analysis for thrombomodulin expression (G and J) and number of BrdU-positive cells (H and K) in the glomerulus (G and H) and tubulointerstitium (J and K), and for double staining of thrombomodulin with BrdU in the glomerulus (I). *P < 0.01, **P < 0.001, and ***P < 0.05. Data are expressed as means ± SD; n = 10 in each group.
Figure 5.
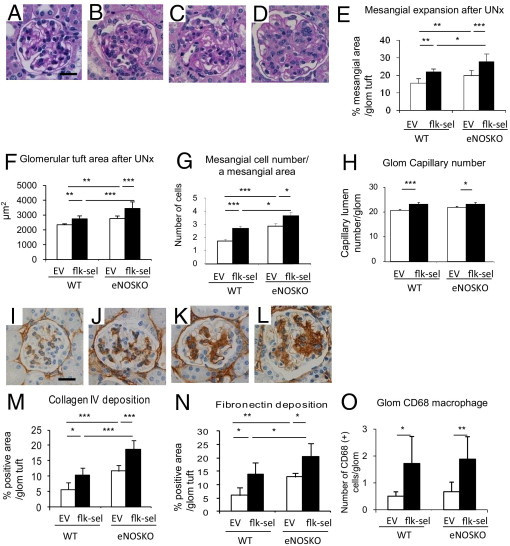
Mesangial expansion in rAAV1-Flk-sel treated mice. A–D: Representative PAS staining of the glomerulus after UNx: UNx-WT mice treated with rAAV1-EV (A) or with rAAV1-Flk-sel (B); UNx-eNOSKO mice treated with rAAV1-EV (C) or with rAAV1-Flk-sel (D). Scale bar = 20 μm. E–H: Quantitative analysis for mesangial expansion (E), glomerular area (F), mesangial cellularity (G), and glomerular (Glom) capillary number (H). I–K: IHC for collagen IV deposition in glomeruli after UNx: UNx-WT mice treated with rAAV1-EV (I) or with rAAV1-Flk-sel (J); UNx-eNOSKO mice treated with rAAV1-EV (K) or with rAAV1-Flk-sel (L). Scale bar = 20 μm. M–O: Quantitative analysis for % positive area of deposition of glomerular collagen IV (M) and fibronectin (N), with number of CD68+ cells in the glomerulus (Glom) (O). *P < 0.01, **P < 0.05, and ***P < 0.001. Data are expressed as means ± SD; n = 10 in each group.
De Novo Mesangial Proliferative Glomerular Injury
We had postulated that overexpression of rAAV1-Flk-sel would be beneficial in UNx-WT mice, because of its ability to lower BP and stimulate angiogenesis; however, unexpectedly, a marked mesangial expansion and proliferation were induced. rAAV1-Flk-sel induced mesangial expansion, glomerular hypertrophy, and mesangial proliferation in UNx-WT and UNx-eNOSKO mice (Figure 5, A–G), but there was no alteration in mesangiolysis (data not shown). Enhanced mesangial expansion was accompanied by elevation in collagen IV and fibronectin deposition (Figure 5, I–N). An increase in CD68+ glomerular macrophages after rAAV1-Flk-sel treatment in both UNx-WT and UNx-eNOSKO mice also likely contributed to the high cellularity in the mesangium. However, a lack of eNOS had no effect on this response (Figure 5O).
Electron microscopy confirmed these findings, with a marked increase of mesangial cells (Figure 6A) and subendothelial vacuolization (Figure 6B) in UNx-eNOSKO mice overexpressing rAAV1-Flk-sel. Narrowing of the capillary lumen was concomitant with endothelial swelling and mesangial cell proliferation and interposition in UNx-eNOSKO mice treated with rAAV1-Flk-sel (Figure 6C). Podocyte cell bodies had a normal appearance, whereas focal foot process fusion was occasionally observed in UNx-eNOSKO mice, with or without treatment (Figure 6, D and E).
Figure 6.
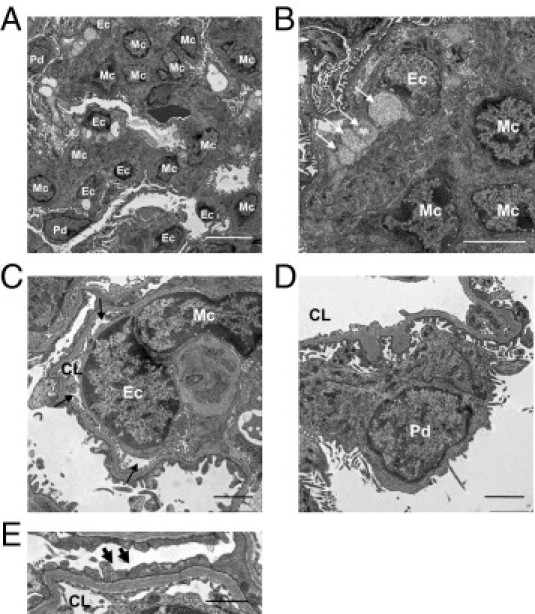
Glomerular ultrastructure in UNx-eNOSKO mice treated with rAAV1-Flk-sel. Representative electron micrographs demonstrate increased mesangial cell (Mc) number (A) and extracellular matrix deposition (A and B) in glomeruli of UNx-eNOSKO mice treated with rAAV1-Flk-sel. B: Vacuolization is observed in the subendothelial space (arrows). C: A narrowing of glomerular capillary lumen (CL) (arrows) due to swelling of the endothelial cell (Ec) with partial mesangial interposition is present in UNx-eNOSKO mice treated with rAAV1-Flk-sel treatment. In contrast, podocytes (Pd) appear normal (D), although some foot processes are fused (arrows) (E).
The PDGF-B and -D isoforms are key cytokines in regulating mesangial cell proliferation.35 We found that PDGF-D was barely expressed in glomeruli of UNx-WT mice but was significantly induced by rAAV1-Flk-sel treatment (Figure 7, A and B). A lack of eNOS itself also induced mesangial PDGF-D expression, compared with nontreated UNx-WT mice, and this was greater in the presence of rAAV1-Flk-sel overexpression (Figure 7, C and D). PDGF-B and PDGF-βR expression mirrored the PDGF-D staining patterns in these mice (Figure 7, E–L). The observation that overexpression of rAAV1-Flk-sel resulted in mesangial expansion and proliferation led us to closely evaluate the sites of expression of VEGFR2 in this model. Although it is predominantly expressed in endothelial cells,36 VEGFR2 has been reported in mesangial cells of diseased glomeruli,37 and VEGFR2 was found to be induced in the mesangial cells of UNx-WT mice after rAAV1-Flk-sel treatment (Figure 7, M, N, and Q). Furthermore, VEGFR2 was also expressed in UNx-eNOSKO mice and was greatly enhanced with rAAV1-Flk-sel treatment (Figure 7, O, P, and Q). Study with isolated glomeruli would be required to confirm these findings.
Figure 7.
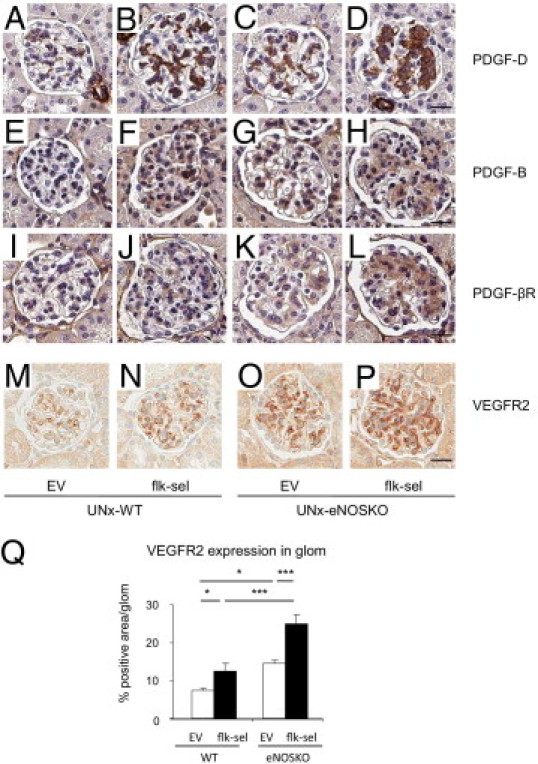
Glomerular expression of PDGF family and VEGFR2. A–P: Staining (brown) for PDGF-D, PDGF-B, PDGF-βR, and VEGFR2 in glomeruli of four groups of mice 1 month after UNx: UNx-WT mice treated with rAAV1-EV (A, E, I, and M) or with rAAV1-Flk-sel (B, F, J, and N); UNx-eNOSKO mice treated with rAAV1-EV (C, G, K, and O) or with rAAV1-Flk-sel (D, H, L, and P). Scale bars = 20 μm. Q: Quantitative analysis for % positive VEGFR2 expression in glomerulus (glom). *P < 0.01, ***P < 0.001. Data are expressed as means ± SD; n = 10 in each group.
Tubulointerstitial Injury
Mild tubulointerstitial injury was observed in UNx-eNOSKO mice treated with rAAV1-EV, as evidenced by tubular dilation (Figure 8, A and B) and atrophy accompanied by detachment of tubular epithelial cells (data not shown); the injury was more prominent in UNx-eNOSKO mice treated with rAAV1-Flk-sel. Collagen III deposition was more severe in UNx-eNOSKO mice treated with rAAV1-EV than in UNx-WT mice treated with rAAV1-EV, whereas rAAV1-Flk-sel treatment significantly induced interstitial collagen III deposition in UNx-eNOSKO mice (Figure 8, C–E). F4/80-positive macrophage infiltration was induced by rAAV1-Flk-sel treatment in both eNOSKO and WT mice (Figure 8F).
Figure 8.
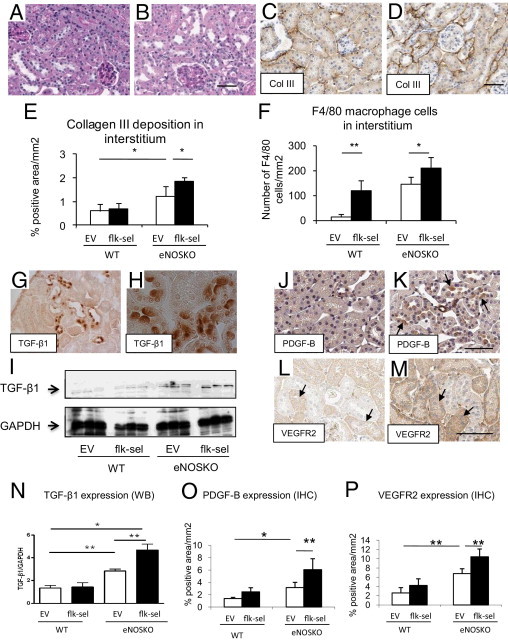
Tubulointerstitial injury. A: UNx-eNOSKO mice treated with rAAV1-EV shows no evidence of tubular injury. B: Tubular injury is apparent under PAS staining in UNx-eNOSKO mice treated with rAAV1-Flk-sel. C and D: IHC for collagen III in UNx-eNOSKO mice treated with rAAV1-EV (C) or with rAAV1-Flk-sel (D). E and F: Quantitative analysis of collagen III deposition (E) and F4/80-positive macrophage infiltration (F). G and H: TGF-β1 expression in UNx-eNOSKO mice treated with rAAV1-Flk-sel is detected in some tubular epithelial cells under low-power magnification (G, ×200), whereas a high power view revealed TGF-β1 located in the apical epithelial membrane (H, ×630). I and N: Western blotting (WB) of renal cortex for TGF-β1 (H) and its quantification (N). In terms of PDGF-B expression, compared with UNx-eNOSKO mice treated with rAAV1-EV (J), PDGF-B staining (arrow) is enhanced in UNx-eNOSKO mice treated with rAAV1-Flk-sel (K), as shown by its quantification (O). VEGFR2 is expressed (arrows) in UNx-eNOSKO mice treated with rAAV1-EV (L), and expression is pronounced in tubular cells of UNx-eNOSKO mice treated with rAAV1-Flk-sel (M). P: Quantification of IHC for VEGFR2. *P < 0.05, **P < 0.001. Data are expressed as means ± SD; n = 10 in each group. Scale bars = 50 μm.
Expression of TGF-β1 (Figure 8, G and H), PDGF-B (Figure 8, J, K, and O), and VEGFR2 (Figure 8, L, M, and P) was increased in damaged tubular epithelial cells in UNx-eNOSKO and was enhanced with rAAV1-Flk-sel treatment. In particular, TGF-β1 expression was located in apical membrane of damaged tubular cells (Figure 8H); total protein levels of TGF-β1 were increased in whole kidney lysates of UNx-eNOSKO animals and were further enhanced by Flk-sel stimulation (Figure 8, I and N).
Strikingly, the damaged tubular epithelial cells expressed α-SMA, a marker of myofibroblast in UNx-eNOSKO mice, which was further enhanced by rAAV1-Flk-sel treatment (Figure 9, A and B). Consistently, total α-SMA expression in the kidney as indicated by Western blotting was elevated in these groups (Figure 9, C and D). To determine whether VEGF directly induces α-SMA on tubular epithelial cells, we conducted an in vitro study with HK2 cells. Consistent with our in vivo data, we found that VEGF-A induced α-SMA at 48 and 72 hours. In addition, VEGF-A also reduced E-cadherin expression, an epithelial cell marker (Figure 9E).
Figure 9.
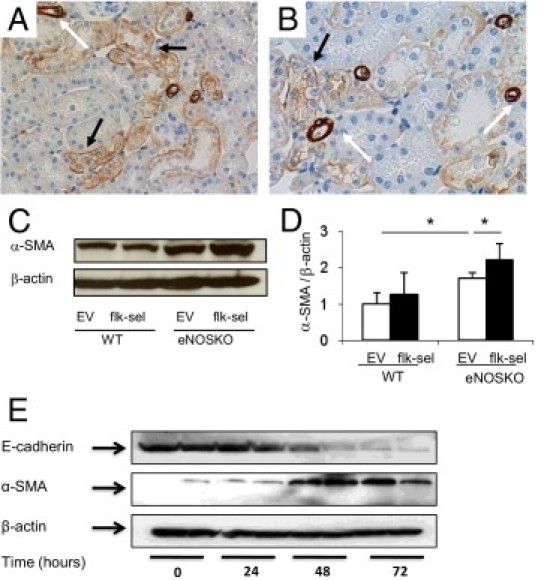
Expression of myofibroblasts markers in tubular epithelial cells. A and B: Expression of α-SMA in renal cortex (A, original magnification ×400) and medulla (B, original magnification ×400) of UNx-eNOSKO mice treated with rAAV1-Flk-sel is detected by IHC. α-SMA is strongly expressed in vascular smooth muscle cells in arterioles and arteries (white arrow), whereas weaker signal (black arrow) is seen in tubular epithelial cells. C and D: Western blotting for α-SMA and its quantification. E: VEGF (100 ng/mL) induces α-SMA expression at 48 and 72 hours, accompanied by a reduction in E-cadherin in HK2 cells, confirming the in vivo findings. *P < 0.05. Data are expressed as means ± SD; n = 10 in each group.
A proposed mechanism accounting for effects of specific VEGFR-2 stimulation is presented in Figure 10.
Figure 10.
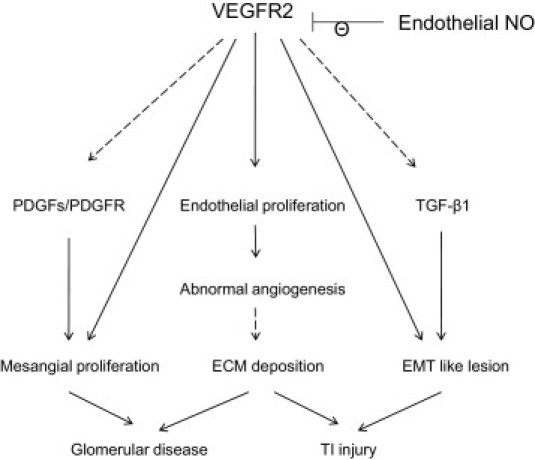
A proposed mechanism by which VEGFR2 stimulation predisposes to renal injury in the mouse kidney. EMC, extracellular matrix; EMT, epithelial-mesenchymal transition; NO, nitric oxide; PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; TGF-β1, transforming growth factor β1; TI, tubulointerstitial; VEGFR2, vascular endothelial growth factor receptor 2. Dashed lines indicate a proposed pathway that was not demonstrated in the present study.
Discussion
In the present study, the primary finding was that supraphysiological activation of VEGFR2 before UNx predisposed WT mouse kidneys to renal injury. This finding was surprising, because VEGF-A administration has been reported to be beneficial in several types of renal injury.3,4,6,7 A potential explanation for this discrepancy is the timing of VEGF-A treatment and whether this maneuver occurs before or after onset of renal injury. The progression of renal injury is usually associated with a loss of capillaries, and so preventing capillary loss by VEGF-A might be beneficial. In contrast, the present study examined the effect of VEGF-A therapy performed before the onset of renal injury. Here, VEGF-A overexpression has the potential to lead to excessive numbers of vessels, which could be structurally and functionally immature38 and the presence of which may augment renal disease progression.
We found that stimulation of VEGFR2 caused abnormal angiogenesis in the kidney, and VEGF-A also contributed to the development of other renal injury features, including mesangial expansion, macrophage influx, and tubulointerstitial injury, in particular in the presence of NO deficiency. Of note, VEGF-A acting through the activation of VEGFR2, but not VEGFR1, is known to play a key role in diabetic nephropathy,17 which is characterized by phenotypes similar to those observed in the present study. Others have shown that VEGF-A inhibition prevents an increase in glomerular volume in diabetic animals.8,9,30 It is therefore possible that high VEGF-A, in addition to high glucose, could potentially contribute not only to the development of angiogenesis but to other features of diabetic glomerular injury. In this regard, giving Flk-sel therapy to diabetic patients might lead to further deterioration in diabetic nephropathy.
There are several potential mechanisms for how VEGF-A can alter mesangial cell biology. VEGF-A may directly bind to VEGFR2 expressed on mesangial cells. Previous studies have shown that VEGFR2 is exclusively expressed in endothelial cells in the normal mouse glomerulus,36 but we found VEGFR2 expression in the mesangial area of UNx-WT mice after Flk-sel treatment. Furthermore, eNOS deficiency also induced VEGFR2 expression, which was enhanced with Flk-sel treatment in UNx-eNOSKO mice. Given that Thomas et al37 also demonstrated VEGFR2 expression in mesangium in human proliferative glomerulonephritis, these cells may express VEGFR2 under certain conditions. The precise mechanism for elevated VEGFR2 in this model is unclear, but one possibility is that UNx leads to a positive feedback loop and hence to autocrine stimulation of VEGFR-2.39
Mesangial proliferation induced by Flk-sel therapy was associated with alterations in expression of PDGF/PDGFR, which are potent mitotic factors for mesangial cells.40 This finding is consistent with previous report.41 Notably, VEGF-A is capable of signaling through PDGFRs,42 and so our VEGF-A mutant might directly stimulate PDGF pathways in our animals, leading to alterations in mesangial cell biology. Future studies are required to address this issue.
The role of VEGFR2 in podocytes is controversial. Veron et al43 documented that overexpression of VEGF-A in podocytes stimulates VEGFR2 expression in autocrine fashion, whereas our experiments showed that VEGFR2 stimulation did not affect podocyte structure. The most likely explanation for this discrepancy is that VEGF-A was locally overexpressed in podocytes themselves in the Veron et al43 study, but Flk-sel levels were increased in the circulation in the present study. The concentration of Flk-sel could presumably be reduced when it reaches podocyte cells. Alternatively, our findings are supported also by a recent study showing that VEGFR2-knockout podocytes do not lead to any glomerular abnormalities in mice up to 6 months of age, indicating that podocyte VEGFR2 is biologically inactive.44
A striking finding in the present study was that Flk-sel overexpression led to expression of α-SMA in some epithelial cells in damaged tubules. This was confirmed by our in vitro study, which showed that VEGF-A caused a reduction of the E-cadherin expression (a marker of epithelial cells) and an induction of α-SMA expression (a marker of mesenchymal cells), suggesting that VEGF might cause a phenotypic change in tubular cells. Caution is advised in interpreting these data, however, because recent studies have challenged the existence of this epithelial-mesenchymal transition in kidney disease.45
In opposition to our predictions, and despite the fact that inflammatory reactions after VEGF-stimulation are thought to be attributed to VEGFR1, not VEGFR2,11,12 selective VEGFR2 stimulation elevated macrophage infiltration as assessed by F4/80 and CD68 staining, regardless of eNOS expression, The renal macrophage infiltration observed in the present study was unlikely to be due to direct VEGFR2 stimulation, but probably was due to an indirect effect from other cytokines that may be expressed in injured kidneys.
We found that stimulation of VEGFR2 caused renal injury, but without aggravation of albuminuria. This discordance between urine albumin excretion and renal function has been found also in several clinical studies. The ONTARGET study showed that stronger inhibition of the renin-angiotensin system accelerated renal disease progression, whereas urinary protein excretion was reduced.46 Given these facts, perhaps urine albumin excretion and renal injury are not always associated. Future study would be warranted to address this issue.
VEGFR2 stimulation has been shown to lower BP by stimulating endothelial NO production.16,32 In contrast, the present study documented that overexpression of Flk-sel reduced systemic BP, despite eNOS deficiency. This suggests that alternative factors that contribute to BP regulation, including endothelial-derived hyperpolarizing factor,47–50 could be altered by Flk-sel.
Finally, we noted that VEGFR2 stimulation resulted in an increase in weight in our mice. Although the present study did not address the mechanism or assess lipid profiles, perhaps an increase in angiogenesis on fat tissue was involved, in addition to adipogenesis.51,52 Because adipose tissue is known to produce cytokines and adipokines, it remains possible that such factors may even contribute to renal disease progression.
There are some caveats for the present study. First, serum levels of Flk-sel could be too high to provide insights into the actual pathological role of VEGFR2 in kidney disease. Second, we need to consider whether our animal model replicates human renal disease. However, given that in diabetic nephropathy VEGF-A expression is up-regulated early,17 before renal disease progression, the insights provided by the present study might help us understand pathogenesis of the development of diabetic nephropathy and potentially other renal diseases if they are associated with a reduction of NO bioavailability. Third, discrepancies in endothelial cell analysis should be noted. Although thrombomodulin staining was significantly greater in Flk-sel eNOSKO mice than in Flk-sel WT mice, this difference was not observed in glomerular capillary number. It is possible that small changes in endothelial cell proliferation might not necessarily translate to an increase in capillary number. Alternatively, thrombomodulin staining may be a more sensitive detector of endothelial cells, compared with counting capillary lumens. Finally, thrombomodulin could be expressed in nonendothelial cells in this model.53 This last possibility seems unlikely, however, given that thrombomodulin is associated with the endothelial marker CD34 in eNOSKO mice.24 Last of all, IHC was used to assess the levels of soluble factors such as PDGF. This may not be the most reliable method, because of secretion of these molecules; other strategies, such as glomerular isolation, should be considered in the future.
In conclusion, the present study documented that VEGFR2 stimulation before renal insult can predispose to an acceleration of renal disease, especially in the presence of endothelial NO deficiency.
Acknowledgments
We thank Mr. Norihiko Suzuki (Nagoya University, Nagoya, Japan) for excellent technical assistance and Dr. Richard J. Johnson (University of Colorado Denver, Denver, CO) for precious advice and critical review of this manuscript.
Footnotes
Supported by a Juvenile Diabetes Research Foundation–JDRF grant (T.N), a Bogue Research Fellowship from University College London (D.A.L), and a Kidney Research UK Senior Non-Clinical Fellowship (D.A.L).
References
- 1.Kang D.H., Anderson S., Kim Y.G., Mazzalli M., Suga S., Jefferson J.A., Gordon K.L., Oyama T.T., Hughes J., Hugo C., Kerjaschki D., Schreiner G.F., Johnson R.J. Impaired angiogenesis in the aging kidney: vascular endothelial growth factor and thrombospondin-1 in renal disease. Am J Kidney Dis. 2001;37:601–611. doi: 10.1053/ajkd.2001.22087. [DOI] [PubMed] [Google Scholar]
- 2.Kang D.H., Joly A.H., Oh S.W., Hugo C., Kerjaschki D., Gordon K.L., Mazzali M., Jefferson J.A., Hughes J., Madsen K.M., Schreiner G.F., Johnson R.J. Impaired angiogenesis in the remnant kidney model: I: Potential role of vascular endothelial growth factor and thrombospondin-1. J Am Soc Nephrol. 2001;12:1434–1447. doi: 10.1681/ASN.V1271434. [DOI] [PubMed] [Google Scholar]
- 3.Suga S., Kim Y.G., Joly A., Puchacz E., Kang D.H., Jefferson J.A., Abraham J.A., Hughes J., Johnson R.J., Schreiner G.F. Vascular endothelial growth factor (VEGF121) protects rats from renal infarction in thrombotic microangiopathy. Kidney Int. 2001;60:1297–1308. doi: 10.1046/j.1523-1755.2001.00935.x. [DOI] [PubMed] [Google Scholar]
- 4.Kang D.H., Hughes J., Mazzali M., Schreiner G.F., Johnson R.J. Impaired angiogenesis in the remnant kidney model: II: Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J Am Soc Nephrol. 2001;12:1448–1457. doi: 10.1681/ASN.V1271448. [DOI] [PubMed] [Google Scholar]
- 5.Yuan H.T., Li X.Z., Pitera J.E., Long D.A., Woolf A.S. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am J Pathol. 2003;163:2289–2301. doi: 10.1016/s0002-9440(10)63586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim Y.G., Suga S.I., Kang D.H., Jefferson J.A., Mazzali M., Gordon K.L., Matsui K., Breiteneder-Geleff S., Shankland S.J., Hughes J., Kerjaschki D., Schreiner G.F., Johnson R.J. Vascular endothelial growth factor accelerates renal recovery in experimental thrombotic microangiopathy. Kidney Int. 2000;58:2390–2399. doi: 10.1046/j.1523-1755.2000.00422.x. [DOI] [PubMed] [Google Scholar]
- 7.Shimizu A., Masuda Y., Mori T., Kitamura H., Ishizaki M., Sugisaki Y., Fukuda Y. Vascular endothelial growth factor165 resolves glomerular inflammation and accelerates glomerular capillary repair in rat anti-glomerular basement membrane glomerulonephritis. J Am Soc Nephrol. 2004;15:2655–2665. doi: 10.1097/01.ASN.0000141038.28733.F2. [DOI] [PubMed] [Google Scholar]
- 8.de Vriese A.S., Tilton R.G., Elger M., Stephan C.C., Kriz W., Lameire N.H. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol. 2001;12:993–1000. doi: 10.1681/ASN.V125993. [DOI] [PubMed] [Google Scholar]
- 9.Flyvbjerg A., Dagnaes-Hansen F., De Vriese A.S., Schrijvers B.F., Tilton R.G., Rasch R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes. 2002;51:3090–3094. doi: 10.2337/diabetes.51.10.3090. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa T., Sato W., Sautin Y.Y., Glushakova O., Croker B., Atkinson M.A., Tisher C.C., Johnson R.J. Uncoupling of vascular endothelial growth factor with nitric oxide as a mechanism for diabetic vasculopathy. J Am Soc Nephrol. 2006;17:736–745. doi: 10.1681/ASN.2005070759. [DOI] [PubMed] [Google Scholar]
- 11.Sato W., Kosugi T., Zhang L., Roncal C.A., Heinig M., Campbell-Thompson M., Yuzawa Y., Atkinson M.A., Grant M.B., Croker B.P., Nakagawa T. The pivotal role of VEGF on glomerular macrophage infiltration in advanced diabetic nephropathy. Lab Invest. 2008;88:949–961. doi: 10.1038/labinvest.2008.60. [DOI] [PubMed] [Google Scholar]
- 12.Barleon B., Sozzani S., Zhou D., Weich H.A., Mantovani A., Marmé D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- 13.Shih S.C., Ju M., Liu N., Smith L.E. Selective stimulation of VEGFR-1 prevents oxygen-induced retinal vascular degeneration in retinopathy of prematurity. J Clin Invest. 2003;112:50–57. doi: 10.1172/JCI17808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fong G.H., Rossant J., Gertsenstein M., Breitman M.L. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 15.Kiba A., Sagara H., Hara T., Shibuya M. VEGFR-2-specific ligand VEGF-E induces non-edematous hyper-vascularization in mice. Biochem Biophys Res Commun. 2003;301:371–377. doi: 10.1016/s0006-291x(02)03033-4. [DOI] [PubMed] [Google Scholar]
- 16.Li B., Ogasawara A.K., Yang R., Wei W., He G.W., Zioncheck T.F., Bunting S., de Vos A.M., Jin H. KDR (VEGF receptor 2) is the major mediator for the hypotensive effect of VEGF. Hypertension. 2002;39:1095–1100. doi: 10.1161/01.hyp.0000018588.56950.7a. [DOI] [PubMed] [Google Scholar]
- 17.Cooper M.E., Vranes D., Youssef S., Stacker S.A., Cox A.J., Rizkalla B., Casley D.J., Bach L.A., Kelly D.J., Gilbert R.E. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 1999;48:2229–2239. doi: 10.2337/diabetes.48.11.2229. [DOI] [PubMed] [Google Scholar]
- 18.Nakagawa T. Uncoupling of the VEGF-endothelial nitric oxide axis in diabetic nephropathy: an explanation for the paradoxical effects of VEGF in renal disease. Am J Physiol Renal Physiol. 2007;292:F1665–F1672. doi: 10.1152/ajprenal.00495.2006. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa T., Kosugi T., Haneda M., Rivard C.J., Long D.A. Abnormal angiogenesis in diabetic nephropathy. Diabetes. 2009;58:1471–1478. doi: 10.2337/db09-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B., Fuh G., Meng G., Xin X., Gerritsen M.E., Cunningham B., de Vos A.M. Receptor-selective variants of human vascular endothelial growth factor: Generation and characterization. J Biol Chem. 2000;275:29823–29828. doi: 10.1074/jbc.M002015200. [DOI] [PubMed] [Google Scholar]
- 21.Gerber H.P., McMurtrey A., Kowalski J., Yan M., Keyt B.A., Dixit V., Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway: Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 22.Gille H., Kowalski J., Li B., LeCouter J., Moffat B., Zioncheck T.F., Pelletier N., Ferrara N. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2): A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276:3222–3230. doi: 10.1074/jbc.M002016200. [DOI] [PubMed] [Google Scholar]
- 23.Mu W., Long D.A., Ouyang X., Agarwal A., Cruz P.E., Roncal C.A., Nakagawa T., Yu X., Hauswirth W.W., Johnson R.J. Angiostatin overexpression is associated with an improvement in chronic kidney injury by an anti-inflammatory mechanism. Am J Physiol Renal Physiol. 2009;296:F145–F152. doi: 10.1152/ajprenal.90430.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawa T., Sato W., Glushakova O., Heinig M., Clarke T., Campbell-Thompson M., Yuzawa Y., Atkinson M., Johnson R.J., Croker B. Diabetic eNOS knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18:539–550. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- 25.Masaki T., Chow F., Nikolic-Paterson D.J., Atkins R.C., Tesch G.H. Heterogeneity of antigen expression explains controversy over glomerular macrophage accumulation in mouse glomerulonephritis. Nephrol Dial Transplant. 2003;18:178–181. doi: 10.1093/ndt/18.1.178. [DOI] [PubMed] [Google Scholar]
- 26.Yuzawa Y., Brentjens J.R., Brett J., Caldwell P.R., Esposito C., Fukatsu A., Godman G., Stern D., Andres G. Antibody-mediated redistribution and shedding of endothelial antigens in the rabbit. J Immunol. 1993;150:5633–5646. [PubMed] [Google Scholar]
- 27.Working Group of the International IgA Nephropathy Network and the Renal Pathology Society. Roberts I.S., Cook H.T., Troyanov S., Alpers C.E., Amore A., Barratt J. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. 2009;76:546–556. doi: 10.1038/ki.2009.168. [DOI] [PubMed] [Google Scholar]
- 28.Nakayama T., Sato W., Yoshimura A., Zhang L., Kosugi T., Campbell-Thompson M., Kojima H., Croker B.P., Nakagawa T. Endothelial von Willebrand factor release due to eNOS deficiency predisposes to thrombotic microangiopathy in mouse aging kidney. Am J Pathol. 2010;176:2198–2208. doi: 10.2353/ajpath.2010.090316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu L.W., Mayo L.D., Dunbar J.D., Kessler K.M., Baerwald M.R., Jaffe E.A., Wang D., Warren R.S., Donner D.B. Utilization of distinct signaling pathways by receptors for vascular endothelial cell growth factor and other mitogens in the induction of endothelial cell proliferation. J Biol Chem. 2000;275:5096–5103. doi: 10.1074/jbc.275.7.5096. [DOI] [PubMed] [Google Scholar]
- 30.Flyvbjerg A., Schrijvers B.F., De Vriese A.S., Tilton R.G., Rasch R. Compensatory glomerular growth after unilateral nephrectomy is VEGF dependent. Am J Physiol Endocrinol Metab. 2002;283:E362–E366. doi: 10.1152/ajpendo.00007.2002. [DOI] [PubMed] [Google Scholar]
- 31.Senthil D., Choudhury G.G., McLaurin C., Kasinath B.S. Vascular endothelial growth factor induces protein synthesis in renal epithelial cells: a potential role in diabetic nephropathy. Kidney Int. 2003;64:468–479. doi: 10.1046/j.1523-1755.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 32.Facemire C.S., Nixon A.B., Griffiths R., Hurwitz H., Coffman T.M. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. 2009;54:652–658. doi: 10.1161/HYPERTENSIONAHA.109.129973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iatropoulos M.J., Williams G.M. Proliferation markers. Exp Toxicol Pathol. 1996;48:175–181. doi: 10.1016/S0940-2993(96)80039-X. [DOI] [PubMed] [Google Scholar]
- 34.Ohashi R., Shimizu A., Masuda Y., Kitamura H., Ishizaki M., Sugisaki Y., Yamanaka N. Peritubular capillary regression during the progression of experimental obstructive nephropathy. J Am Soc Nephrol. 2002;13:1795–1805. doi: 10.1097/01.asn.0000018408.51388.57. [DOI] [PubMed] [Google Scholar]
- 35.Floege J., Eitner F., Alpers C.E. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol. 2008;19:12–23. doi: 10.1681/ASN.2007050532. [DOI] [PubMed] [Google Scholar]
- 36.Eremina V., Cui S., Gerber H., Ferrara N., Haigh J., Nagy A., Ema M., Rossant J., Jothy S., Miner J.H., Quaggin S.E. Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol. 2006;17:724–735. doi: 10.1681/ASN.2005080810. [DOI] [PubMed] [Google Scholar]
- 37.Thomas S., Vanuystel J., Gruden G., Rodriguez V., Burt D., Gnudi L., Hartley B., Viberti G. Vascular endothelial growth factor receptors in human mesangium in vitro and in glomerular disease. J Am Soc Nephrol. 2000;11:1236–1243. doi: 10.1681/ASN.V1171236. [DOI] [PubMed] [Google Scholar]
- 38.Pettersson A., Nagy J.A., Brown L.F., Sundberg C., Morgan E., Jungles S., Carter R., Krieger J.E., Manseau E.J., Harvey V.S., Eckelhoefer I.A., Feng D., Dvorak A.M., Mulligan R.C., Dvorak H.F. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest. 2000;80:99–115. doi: 10.1038/labinvest.3780013. [DOI] [PubMed] [Google Scholar]
- 39.Kremer C., Breier G., Risau W., Plate K.H. Up-regulation of flk-1/vascular endothelial growth factor receptor 2 by its ligand in a cerebral slice culture system. Cancer Res. 1997;57:3852–3859. [PubMed] [Google Scholar]
- 40.Hudkins K.L., Gilbertson D.G., Carling M., Taneda S., Hughes S.D., Holdren M.S., Palmer T.E., Topouzis S., Haran A.C., Feldhaus A.L., Alpers C.E. Exogenous PDGF-D is a potent mesangial cell mitogen and causes a severe mesangial proliferative glomerulopathy. J Am Soc Nephrol. 2004;15:286–298. doi: 10.1097/01.asn.0000108522.79652.63. [DOI] [PubMed] [Google Scholar]
- 41.Hakroush S., Moeller M.J., Theilig F., Kaissling B., Sijmonsma T.P., Jugold M., Akeson A.L., Traykova-Brauch M., Hosser H., Hähnel B., Gröne H.J., Koesters R., Kriz W. Effects of increased renal tubular vascular endothelial growth factor (VEGF) on fibrosis, cyst formation, and glomerular disease. Am J Pathol. 2009;175:1883–1895. doi: 10.2353/ajpath.2009.080792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ball S.G., Shuttleworth C.A., Kielty C.M. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J Cell Biol. 2007;177:489–500. doi: 10.1083/jcb.200608093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veron D., Reidy K.J., Bertuccio C., Teichman J., Villegas G., Jimenez J., Shen W., Kopp J.B., Thomas D.B., Tufro A. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 2010;77:989–999. doi: 10.1038/ki.2010.64. [DOI] [PubMed] [Google Scholar]
- 44.Sison K., Eremina V., Baelde H., Min W., Hirashima M., Fantus I.G., Quaggin S.E. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol. 2010;21:1691–1701. doi: 10.1681/ASN.2010030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vitalone M.J., O'Connell P.J., Jimenez-Vera E., Yuksel A., Wavamunno M., Fung C.L., Chapman J.R., Nankivell B.J. Epithelial-to-mesenchymal transition in early transplant tubulointerstitial damage. J Am Soc Nephrol. 2008;19:1571–1583. doi: 10.1681/ASN.2007050580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mann J.F., Schmieder R.E., McQueen M., Dyal L., Schumacher H., Pogue J., Wang X., Maggioni A., Budaj A., Chaithiraphan S., Dickstein K., Keltai M., Metsarinne K., Oto A., Parkhomenko A., Piegas L.S., Svendsen T.L., Teo K.K., Yusuf S., ONTARGET investigators Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008;372:547–553. doi: 10.1016/S0140-6736(08)61236-2. [DOI] [PubMed] [Google Scholar]
- 47.Brandes R.P., Schmitz-Winnenthal F.H., Félétou M., Gödecke A., Huang P.L., Vanhoutte P.M., Fleming I., Busse R. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proc Natl Acad Sci USA. 2000;97:9747–9752. doi: 10.1073/pnas.97.17.9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwakiri Y., Cadelina G., Sessa W.C., Groszmann R.J. Mice with targeted deletion of eNOS develop hyperdynamic circulation associated with portal hypertension. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1074–G1081. doi: 10.1152/ajpgi.00145.2002. [DOI] [PubMed] [Google Scholar]
- 49.Yang S., Wei S., Pozzi A., Capdevila J.H. The arachidonic acid epoxygenase is a component of the signaling mechanisms responsible for VEGF-stimulated angiogenesis. Arch Biochem Biophys. 2009;489:82–91. doi: 10.1016/j.abb.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Webler A.C., Michaelis U.R., Popp R., Barbosa-Sicard E., Murugan A., Falck J.R., Fisslthaler B., Fleming I. Epoxyeicosatrienoic acids are part of the VEGF-activated signaling cascade leading to angiogenesis. Am J Physiol Cell Physiol. 2008;295:C1292–C1301. doi: 10.1152/ajpcell.00230.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishimura S., Manabe I., Nagasaki M., Hosoya Y., Yamashita H., Fujita H., Ohsugi M., Tobe K., Kadowaki T., Nagai R., Sugiura S. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes. 2007;56:1517–1526. doi: 10.2337/db06-1749. [DOI] [PubMed] [Google Scholar]
- 52.Tam J., Duda D.G., Perentes J.Y., Quadri R.S., Fukumura D., Jain R.K. Blockade of VEGFR2 and not VEGFR1 can limit diet-induced fat tissue expansion: role of local versus bone marrow-derived endothelial cells. PLoS One. 2009;4:e4974. doi: 10.1371/journal.pone.0004974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pruna A., Peyri N., Berard M., Boffa M.C. Thrombomodulin is synthesized by human mesangial cells. Kidney Int. 1997;51:687–693. doi: 10.1038/ki.1997.99. [DOI] [PubMed] [Google Scholar]