

Abstract
Acute lung inflammation can be caused by a variety of respirable agents, including cigarette smoke. Long-term cigarette smoke exposure can cause chronic obstructive pulmonary disease (COPD), a serious illness that affects >10 million Americans. Cigarette smoke is a known inducer of inflammation and is responsible for approximately 90% of all COPD cases. RelB, a member of the NF-κB family, attenuates cigarette smoke–induced inflammatory mediator production in mouse lung fibroblasts in vitro. We hypothesized that overexpression of RelB in the airways of mice would dampen acute smoke-induced pulmonary inflammation. Mice received a recombinant adenovirus encoding RelB by intranasal aspiration to induce transient RelB overexpression in the lungs and were subsequently exposed to mainstream cigarette smoke. Markers of inflammation were analyzed after smoke exposure. Neutrophil infiltration, normally increased by smoke exposure, was significantly and potently decreased after RelB overexpression. Cigarette smoke–induced proinflammatory cytokine and chemokine production, cyclooxygenase-2 expression, and prostaglandin E2 production were also significantly decreased in the context of RelB overexpression. The expression of intercellular adhesion molecule 1, an NF-κB–dependent protein, was decreased, indicating a potential mechanism through which RelB can regulate inflammatory cell migration. Therefore, increased expression and/or activation of RelB could be a novel therapeutic strategy against acute lung inflammation caused by respirable agents and possibly against chronic injury, such as COPD.
Chronic obstructive pulmonary disease (COPD), a disease characterized by airflow obstruction and destruction of the alveolar walls,1,2 is the fourth leading cause of death in the United States,3 with >10 million Americans being diagnosed as having COPD in 2007.4 Despite its high prevalence, there is no cure and current therapies neither prevent nor reverse the associated pathological changes.2 Available anti-inflammatory agents are suboptimal. Almost 25% of Americans smoke, and cigarette smoking is responsible for roughly 90% of all COPD cases.1–3 Cigarette smoke contains nearly 5000 chemicals, many of which are known carcinogens5,6 and induce injury and inflammation.7,8
Inflammation is the normal physiological response to tissue damage and is typified by increased leukocyte, particularly neutrophil and macrophage, infiltration.9,10 Cellular inflammation is typically associated with increased cytokine production,11,12 cyclooxygenase-2 (Cox-2) expression, and Cox-2–derived prostaglandins, particularly prostaglandin E2 (PGE2).7,8,13 More important, sustained inflammation is a risk factor in the development of COPD and lung cancer.2,8,14 In humans with COPD, neutrophils, macrophages, and other inflammatory cells accumulate around airways. In mouse models, cigarette smoke increases neutrophil infiltration into the bronchoalveolar space and around airways10,11 and increases production of several proinflammatory cytokines, such as IL-6, macrophage inflammatory protein 2 (MIP-2), and keratinocyte-derived chemokine (KC).11,12 In vitro, cigarette smoke stimulates Cox-2 expression and PGE2 production in lung epithelial cells and fibroblasts.7,8 Little is known about the mechanisms surrounding cigarette smoke–induced pulmonary inflammation, but evidence implicates involvement of the NF-κB pathway.
NF-κB is a complex signaling pathway involved in numerous cellular functions, including inflammatory mediator production, adhesion molecule expression, and lymphocyte activation.15,16 Although many family members are proinflammatory,15–19 recent evidence7,20–23 has shown that the NF-κB family member, RelB, has anti-inflammatory activity. RelB, which is normally bound to p100 in the cytosol, translocates to the nucleus and stimulates its target genes on activation.15 More important, NF-κB signaling is dysregulated in the lungs of some patients with COPD24; and selective NF-κB inhibition is an emerging therapeutic strategy against COPD and other inflammation-associated diseases.18,25–29
RelB is an understudied protein with significant translational potential as an attenuator of inflammation. Loss of RelB expression promotes production of inflammatory mediators both in vitro and in vivo, and reconstitution of RelB dampens inflammation.7,11 Furthermore, RelB-deficient mice exhibit severe inflammation in tissues in which RelB is normally expressed.22 RelB regulates chemokine expression in human macrophages and mouse kidney fibroblasts.20,21,23 Together, these data suggest that RelB may be a critical dampener of inflammation.
We hypothesized that lung-targeted overexpression of RelB would protect against cigarette smoke–induced inflammation by reducing inflammatory mediator production. To our knowledge, no published data exist showing that RelB overexpression dampens inflammation in any system. The experiments detailed herein demonstrate that RelB is a novel therapeutic target in the treatment of inflammation-associated lung diseases induced by cigarette smoke.
Materials and Methods
Mice
Wild-type female C57BL/6 mice were purchased at the age of 7 to 8 weeks from The Jackson Laboratory (Bar Harbor, ME) and were housed at the University of Rochester (Rochester, NY). All animal procedures were performed with permission from the University Committee on Animal Research.
Adenovirus Gene Delivery
To overexpress RelB in the airways of mice, a replication-deficient (E1- and E3-deleted) recombinant human type 5 adenovirus containing the human RelB gene (Cell Biolabs, San Diego, CA) was administered to age- and sex-matched C57BL/6 mice by intranasal aspiration. To administer the virus, isoflurane (2% vapor in air) was given to lightly anesthetize the mice; and 35 μL of sterile saline containing 5 × 108 plaque-forming units of the virus was inhaled into the lungs through the nose of each mouse. Two doses of this virus were administered to each mouse 24 hours apart. Control mice were treated with a DL-70 control recombinant adenovirus containing an empty vector.30
Cigarette Smoke Exposure
Three days after the second dose of adenovirus, mice were exposed to cigarette smoke, as previously described.11 Mice were placed in individual compartments within a wire cage, which was sealed in a Plexiglas box connected to the smoke source. Mainstream smoke was generated from research cigarettes (2R4F; University of Kentucky, Lexington) with a Baumgartner-Jaeger CSM2072i cigarette smoking machine (CH Technologies, Westwood, NJ), according to the Federal Trade Commission protocol (1 puff per minute of cigarette, 2-second duration, in a 35-mL volume). The concentration of the smoke (total particulate matter per cubic meter of air) was monitored with a MicroDust Pro aerosol monitor (Casella CEL, Bedford, UK) and confirmed by gravimetric sampling. The smoke was diluted with filtered air to a nominal value of 300 mg total particulate matter/m3 air. The average true exposure for these experiments was 335 ± 56 mg total particulate matter/m3 air. (The true CS exposure is represented as mean ± SD.) Mice received two 1-hour exposures, spaced 1 hour apart, for 3 days and were euthanized on the fourth day. Control mice were exposed to filtered air.
Tissue Harvest and Bronchoalveolar Lavage
Mice were anesthetized with Avertin (2,2,2-tribromoethanol, 250 mg/kg i.p.; Sigma-Aldrich, St. Louis, MO) and euthanized by exsanguination. The lungs were excised and lavaged twice with 0.5 mL of PBS. The bronchoalveolar lavage (BAL) fluids were centrifuged, and the supernatants were frozen at −80°C for later analysis. The BAL cell pellets were resuspended in PBS, and the total cell number was determined by counting on a hemocytometer. Differential cell counts were performed after Cytospin slide preparation (Thermo Shandon, Pittsburg, PA) and staining with Three Step Stain (Thermo Scientific, Waltham, MA). The left and right lungs were frozen immediately in liquid nitrogen and stored at −80°C for later analysis.
BAL Fluid Analysis
Proinflammatory cytokines IL-6, MIP-2, KC, JE (also known as MCP-1 or monocyte chemoattractant protein-1), and tumor necrosis factor (TNF)-α were detected in the BAL supernatants with DuoSet enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, MN). Granulocyte-macrophage colony-stimulating factor, LPS-induced CXC chemokine (LIX), interferon (IFN)-γ, and IL-10 were measured by cytokine multiplex analysis (Milliplex MAP kit MPXMCYTO-70K; Millipore, Billerica, MA) and analyzed with a Luminex 100 (Luminex, Austin, TX). PGE2 was detected by competitive enzyme immunoassay, as previously described.31
Western Blot Analysis
Mouse lungs were homogenized in an NP40 lysis buffer containing a protease inhibitor cocktail (Sigma) and were centrifuged to remove debris. The total protein concentration was determined with a bicinchoninic acid detection assay (Pierce, Rockford, IL). Total protein, 5 to 30 μg, was separated via 8% SDS-PAGE, transferred onto Immobilon-P (Millipore), and blocked with 10% nonfat dry milk in 0.1% Tween 20 in PBS. Antibodies targeting RelB (sc-226; Santa Cruz Biotechnologies, Santa Cruz, CA), Cox-2 (160112; Cayman Chemical, Ann Arbor, MI), intercellular adhesion molecule 1 (ICAM-1; AF796; R&D Systems), β-tubulin (2146; Cell Signaling Technology, Beverly, MA), and total actin (CP01; Calbiochem, San Diego) were diluted according to the manufacturers' instructions. Protein was visualized using Immobilon Western chemiluminescent horseradish peroxidase substrate (Millipore) and developed on Classic blue-sensitive X-ray film (Laboratory Product Sales, Rochester) with an X-OMAT 1000A film developer (Eastman Kodak, Rochester). Densitometric analysis was performed with Kodak 1D Imaging Software (Eastman Kodak).
Histological Analysis and Immunohistochemistry
Mouse lungs that were not lavaged were fixed with 10% neutral-buffered formalin, embedded in paraffin, and sectioned. RelB was detected with an antibody specific to the human RelB isoform (ab33917; Abcam, Cambridge, MA). Neutrophils were detected with an anti–mouse neutrophil antibody (MCA771GA; Serotec, Oxford, UK). Both were developed with NovaRed (Vector Laboratories, Burlingame, CA) and counterstained with hematoxylin. ICAM-1 was targeted with an ICAM-1–specific antibody (AF796; R&D Systems) and detected with a Texas Red–conjugated secondary antibody. Slides were coverslipped, viewed, and photographed, as previously described.7 All pictures were taken at ×400 magnification.
Myeloperoxidase Assay
Frozen mouse lungs were homogenized in 0.5 mL of 50 mmol/L potassium phosphate buffer, pH 6.0, containing 0.5% hexadecyltrimethylammonium bromide and a protease inhibitor cocktail (Sigma), were subjected to one freeze-thaw cycle in a dry ice–ethanol bath, and were centrifuged at 10,000 × g for 10 minutes. Myeloperoxidase (MPO) activity was detected in the supernatants by monitoring the oxidation of o-dianisidine dihydrochloride (Sigma) at 460 nm with a UV-Vis diode array spectrophotometer (8453; Hewlett Packard, Palo Alto, CA), as previously described.11,32
Statistical Analysis
Statistical analysis was performed with GraphPad Prism v4.0 (GraphPad Software, San Diego). All results are presented as the mean ± SEM. Statistically significant differences were assessed with either a one-way analysis of variance with a Tukey posttest or a two-way analysis of variance with a Bonferroni posttest.
Results
RelB Recombinant Adenovirus Delivery by Intranasal Aspiration Increases RelB Expression in Mouse Lungs
We first established that targeted RelB overexpression in mouse lungs could be accomplished by adenoviral gene delivery. Mice were treated with the RelB virus, and the left lungs were harvested 3, 5, and 7 days after the initial infection to confirm overexpression of RelB protein using Western blot (Figure 1A). Control mice were treated with either sterile saline or the control virus. At no time did the control adenovirus induce RelB expression. Densitometric analysis in Figure 1B revealed sevenfold, ninefold, and fivefold increases at 3, 5, and 7 days, respectively, after the first adenovirus dose compared with control levels. Furthermore, RelB was significantly overexpressed 5 days after initial infection, confirming that RelB overexpression in mouse airways can be accomplished with this technique and that it is maintained for the duration of the experiment. Overexpression of RelB was also confirmed 7 days after initial treatment by immunohistochemistry (IHC) (Figure 1, C and D).
Figure 1.
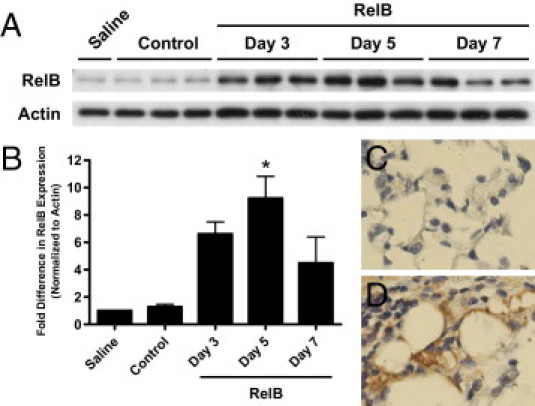
RelB recombinant adenovirus delivery by intranasal aspiration induces RelB overexpression in mouse lungs. A: Overexpression of human RelB was induced with an RelB recombinant adenovirus delivered by intranasal aspiration [two doses of 5 × 108 plaque-forming units (pfu) per mouse delivered 24 hours apart], and RelB expression was detected in whole mouse left lung homogenates using Western blot 3, 5, and 7 days after the first adenovirus dose. Saline indicates saline treated; Control, control virus treated (two doses of 5 × 108 pfu per mouse delivered 24 hours apart, both 7 days after initial treatment). B: Densitometric analysis revealed sevenfold, ninefold, and fivefold increases in RelB expression 3, 5, and 7 days, respectively, after the first treatment. RelB overexpression was confirmed 7 days after the initial treatment by IHC with an antibody specific to the human RelB isoform. C: Control virus. D: RelB virus. n = 3 for each group except saline treated, for which n = 1. *P < 0.05 for statistical significance relative to the control-treated mice (one-way analysis of variance with a Tukey posttest).
Targeted RelB Overexpression in Mouse Lungs before Cigarette Smoke Exposure Significantly Reduces Neutrophilic Inflammation
To determine whether RelB overexpression dampens inflammation in mouse lungs, we first looked at the effects of RelB on neutrophil and macrophage infiltration into the bronchoalveolar space. Mice received two doses of the RelB virus by intranasal aspiration, as previously described (days 0 and 1); 3 days later, mice were exposed to cigarette smoke twice a day for 3 days (days 4–6) and were euthanized 24 hours after the final exposure (day 7). Days 4 through 6 were chosen for smoke exposure because they corresponded to peak RelB expression (Figure 1). Control mice were treated with the control virus and filtered air. No differences in the number of neutrophils or macrophages were seen between control virus–treated and untreated mice (data not shown). Cells present in the bronchoalveolar space were detected in BAL fluids collected during the harvest by Cytospin analysis. Cigarette smoke exposure induced a significant increase in the total BAL cells (Figure 2A) that could be entirely attributed to an increase in the number of neutrophils. Overexpression of RelB in the airways of mice significantly reduced the number of neutrophils recovered by BAL (P < 0.001; Figure 2, B and C). Interestingly, significantly fewer macrophages were recovered by BAL in the smoke-treated, compared with the air-treated, control mice, but RelB overexpression before smoke exposure significantly increased the number of recovered macrophages (Figure 2D). Furthermore, no significant differences were seen between either air- and smoke-treated mice in which RelB was overexpressed or control- or RelB-treated mice exposed to filtered air. Cytospin images representative of these observations are shown in Figure 2E.
Figure 2.

RelB overexpression before cigarette smoke exposure significantly affects bronchoalveolar cell populations. Mice received either the RelB or control virus (two doses of 5 × 108 plaque-forming units per mouse delivered 24 hours apart) and either cigarette smoke or filtered air (two 1-hour exposures per day for 3 days). Cells present in the bronchoalveolar space were collected by lavage, counted, and analyzed after Cytospin slide preparation. Cigarette smoke exposure significantly increased the total number of cells (combined neutrophils and macrophages) present in the bronchoalveolar space (A). Significant increases in the total number and percentage of neutrophils (B and C, respectively) accounted for this increase, but RelB overexpression before smoke exposure damped these increases by more than threefold. RelB overexpression before smoke exposure also significantly increased the total number of macrophages present compared with the smoke-treated control group (D). No significant differences were seen between air-treated mice. *P < 0.05, **P < 0.01, and ***P < 0.001 (two-way analysis of variance with a Bonferroni posttest; n = 5 to 6 per group). E: Images representative of these observations are shown (black arrows indicate neutrophils; white arrows, macrophages).
To confirm that neutrophil infiltration was decreased in RelB-treated mice, we assessed neutrophils in unlavaged mouse lung sections by IHC. As seen in Figure 3, neutrophil infiltration into the alveolar space was increased after smoke exposure, but RelB overexpression abated this increase. We also examined MPO activity in whole mouse lungs that had not been lavaged. MPO is an enzyme present in neutrophils that is integral to the oxidative burst and is a marker of neutrophilic inflammation.32 MPO activity was increased in mouse lungs on exposure to cigarette smoke, but RelB overexpression before smoke exposure significantly reduced MPO activity (Figure 4). The mean basal MPO activity was also lowered on RelB overexpression, but this did not achieve statistical significance.
Figure 3.
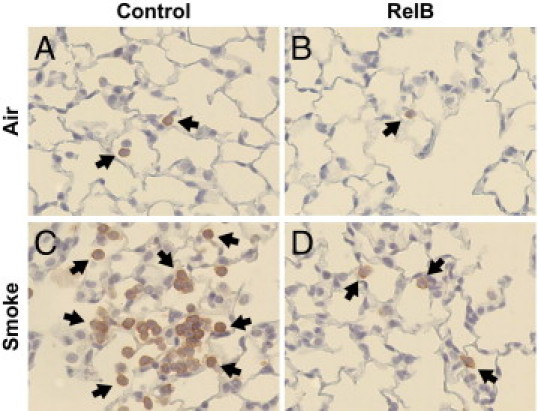
RelB overexpression reduces cigarette smoke–induced neutrophilic inflammation. Mice were treated with either the RelB or control virus (two doses of 5 × 108 plaque-forming units per mouse delivered 24 hours apart) and were exposed to either cigarette smoke or filtered air (two 1-hour exposures per day for 3 days). Unlavaged left mouse lungs were formalin fixed, sectioned, stained for neutrophils (red), and counterstained with hematoxylin. Few neutrophils were present in the lungs of air-treated mice (A and B), but neutrophil infiltration dramatically increased after cigarette smoke exposure (C). However, this increase was dramatically reduced after RelB overexpression (D). Black arrows indicate neutrophils.
Figure 4.
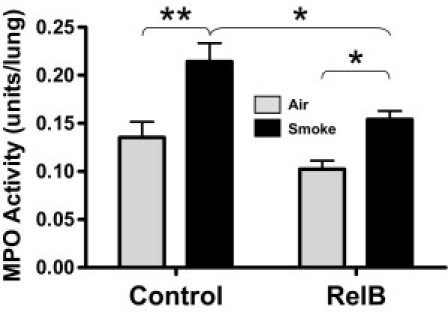
RelB overexpression reduces cigarette smoke–induced MPO activity. Mice were treated with either the RelB or control virus (two doses of 5 × 108 plaque-forming units per mouse delivered 24 hours apart) and with either smoke or filtered air (two 1-hour exposures per day for 3 days). MPO activity was detected in whole right lung homogenates, as described. MPO activity was significantly increased in smoke-treated mice. This was blocked by RelB overexpression. *P < 0.05 and **P < 0.01 (two-way analysis of variance with a Bonferroni posttest; n = 4 per group).
Targeted RelB Overexpression Significantly Reduces Cigarette Smoke–Induced Proinflammatory Cytokine and Chemokine Production
The levels of proinflammatory cytokines and chemokines were determined in BAL fluids by either enzyme-linked immunosorbent assay or cytokine multiplex analysis. As seen in Figure 5, RelB overexpression dampened the smoke-induced increase in neutrophil (MIP-2, KC, and LIX) and macrophage (JE and granulocyte-macrophage colony-stimulating factor) chemokines12,33–37 and cytokines that were otherwise involved in leukocyte recruitment and activation (IL-6, TNF-α, and IFN-γ),34,35,38–40 by roughly 50%. IL-6 production (Figure 5A) was reduced by almost sixfold. Only the reductions in MIP-2 and LIX (Figure 5, B and G, respectively) did not achieve statistical significance. Interestingly, the mean basal production of IL-6, MIP-2, KC, JE, and TNF-α were lowered on RelB overexpression (Figure 5, A–E, respectively), but only basal JE production was significantly reduced (Figure 5D). Production of IL-10 (Figure 5I) was not significantly affected by either cigarette smoke exposure or RelB overexpression.
Figure 5.
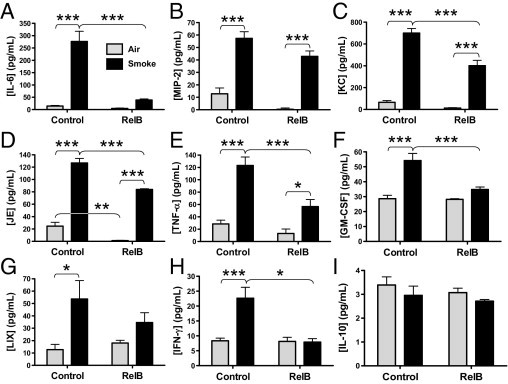
RelB overexpression reduces cigarette smoke–induced proinflammatory cytokine production in mouse lungs. Mice received either the control or RelB virus (two doses of 5 × 108 plaque-forming units per mouse delivered 24 hours apart) and either air or smoke (two 1-hour exposures per day for 3 days). Cytokine production was detected in the BAL fluids by either enzyme-linked immunosorbent assay (A–E) or cytokine multiplex assay (F–H). Cigarette smoke exposure significantly increased production of each cytokine, whereas RelB overexpression reduced the production of IL-6 (A), MIP-2 (B), KC (C), JE (D), TNF-α (E), granulocyte-macrophage colony-stimulating factor (F), LIX (G), IFN-γ (H), and IL-10 (I). Only MIP-2 (B) and LIX (G) did not achieve statistical significance. Basal production of JE was also significantly reduced on RelB overexpression (D). *P < 0.05, **P < 0.01, and ***P < 0.001 (two-way analysis of variance with a Bonferroni posttest; n = 5 to 6 per group).
Targeted RelB Overexpression Significantly Reduces Cigarette Smoke–Induced Cox-2 Expression and PGE2 Production
Next, we looked at whole-lung expression of Cox-2, the NF-κB–dependent proinflammatory enzyme involved in prostaglandin, particularly PGE2, production.7,8,19 Increased Cox-2 activity can also promote tumorigenesis.41 Left and right whole mouse lungs were homogenized after lavage, and Cox-2 expression was detected using Western blot (Figure 6A). Densitometric analysis confirmed a 4.5-fold decrease in smoke-induced Cox-2 expression after RelB overexpression (Figure 6B). Mean basal Cox-2 expression was also lowered after RelB overexpression, but this difference did not achieve statistical significance. Furthermore, significant decreases in both basal and smoke-induced production of PGE2, a potent proinflammatory lipid mediator that regulates cytokine production, vasodilation, and angiogenesis,7,8 were detected in the BAL fluids after RelB overexpression (Figure 6C).
Figure 6.
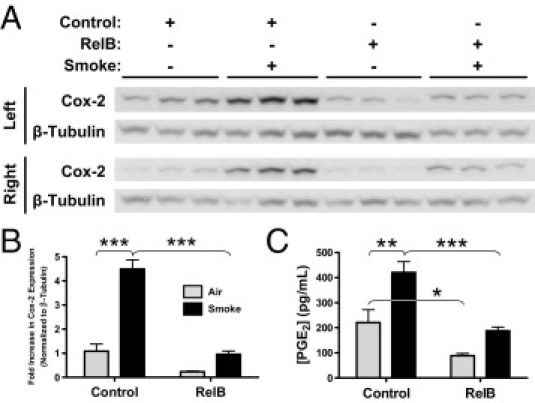
RelB overexpression reduces cigarette smoke–induced Cox-2 expression and PGE2 production. Mice were treated with either the control or RelB virus (two doses of 5 × 108 plaque-forming units per mouse delivered 24 hours apart) and were exposed to either smoke or air (two 1-hour exposures per day for 3 days). A: Cox-2 expression was detected in homogenates of both the left and right lungs using Western blot (n = 3 mice per group). B: Densitometric analysis of both blots confirmed that cigarette smoke significantly increased Cox-2 expression by 4.5-fold and that RelB overexpression before smoke exposure completely dampened this increase (n = 6 per group). C: PGE2 production, detected in the BAL fluids of the same mice, was significantly increased on smoke exposure, but RelB overexpression dampened this increase. *P < 0.05, **P < 0.01, and ***P < 0.001, two-way analysis of variance with a Bonferroni posttest (n = 5 to 6 per group).
Targeted RelB Overexpression in Mouse Lungs Reduces Cigarette Smoke–Induced ICAM-1 Expression
Next, we looked for changes in expression of ICAM-1, the predominant cell adhesion protein on both the endothelium and epithelium of the lung.42–45 The expression of ICAM-1, an NF-κB–dependent protein,45–47 is regulated by IL-6, TNF-α, IFN-γ, and PGE248–54 and is increased in vitro on exposure to cigarette smoke extract.55 ICAM-1 is also a marker of inflammation and malignancy in the lung.56 Both immunofluorescent detection and Western blot analysis (Figure 7, A and B, respectively) demonstrated that RelB overexpression dampened the cigarette smoke–induced increase in ICAM-1 expression. Densitometric analysis of the Western blot confirmed a significant sixfold reduction in smoke-induced ICAM-1 expression in RelB-treated mice compared with control groups (Figure 7C).
Figure 7.
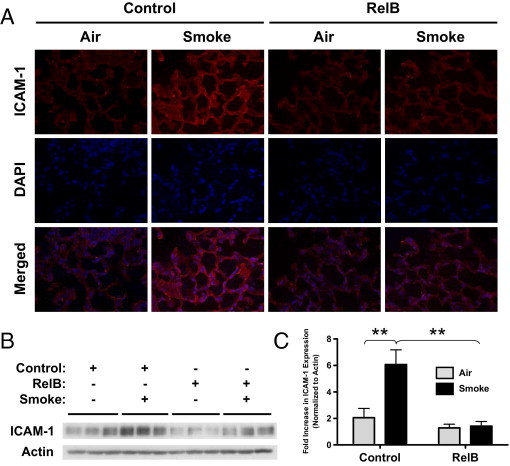
RelB overexpression reduces cigarette smoke–induced ICAM-1 expression in mouse lungs. Mice were treated with either the RelB or control virus (two doses of 5 × 108 plaque-forming units per mouse delivered 24 hours apart) and exposed to filtered air or smoke (two 1-hour exposures per day for 3 days). A: Unlavaged left lungs were formalin fixed, sectioned, and probed for ICAM-1 expression. Cigarette smoke increased ICAM-1 expression (red), but RelB overexpression before smoke exposure dampened this increase. Nuclei were detected by DAPI staining (blue). B: ICAM-1 was also detected in whole left lung homogenates using Western blot. C: Densitometric analysis confirmed that smoke exposure significantly increased ICAM-1 expression and that RelB overexpression significantly dampened this increase. RelB overexpression did not significantly affect basal ICAM-1 expression. **P < 0.01, two-way analysis of variance with a Bonferroni posttest (n = 3 per group).
Discussion
Acute lung inflammation results from exposure to toxicants, infections, and a variety of other injurious processes, including cigarette smoking.7,8 Long-term exposure to cigarette smoke promotes the development of various inflammation-associated lung diseases, such as COPD,1–3 that affect >10 million people in the United States alone.4 No cure exists for these diseases, and current therapies are limited to symptomatic treatments.57–59 The available anti-inflammatory agents have limited efficacy.
NF-κB activation, which is induced by cigarette smoke,60,61 is generally thought to be proinflammatory,15–18 but researchers7,20–22 have shown that RelB has anti-inflammatory properties. We hypothesized that transient overexpression of the NF-κB family member, RelB, in the airways of mice would dampen the proinflammatory effects of cigarette smoke and offer a potential new therapy against lung inflammation. To accomplish this, we administered a recombinant adenovirus containing the RelB gene to age- and sex-matched wild-type C57BL/6 mice by intranasal aspiration and measured RelB expression 3, 5, and 7 days after initial infection (Figure 1). The human RelB gene was used in these experiments because it interacts faithfully with mouse NF-κB and can be distinguished from mouse RelB. We found that administering two half doses of the virus 24 hours apart minimized variability between animals and between the left and right lungs in individual mice (data not shown).
Cigarette smoke is a known inducer of neutrophilic inflammation. We determined that RelB overexpression significantly dampened cigarette smoke–induced neutrophil accumulation in the lungs of mice by approximately 70% (Figures 2–4). Inhaled corticosteroids, by comparison, have varying effects on neutrophil accumulation in the lungs of smokers and patients with COPD. Therefore, current treatment strategies against neutrophilic lung inflammation are limited in their efficacy; and more effective treatment strategies are needed. However, recent research25,26,29 suggests that lung-targeted inhibition of NF-κB may dampen neutrophil accumulation. These findings highlight the significant therapeutic potential of RelB overexpression in dampening the effects of lung inflammation.
Interestingly, RelB overexpression prevented the cigarette smoke–induced reduction in the number of BAL macrophages (Figure 2D). Resident macrophages in the bronchoalveolar space may migrate to the interstitium in response to smoke exposure, and RelB overexpression may reduce this phenomenon. RelB overexpression may also decrease the expression of adhesion molecules on macrophages, which may be increased by cigarette smoke and could decrease the ease of their recovery by lavage. Therefore, cigarette smoke may not truly affect the total number of macrophages within the bronchoalveolar space. If these numbers are changing, then changes in macrophage-derived cytokines would also likely be observed, but it is unclear if the cytokines collected by BAL are derived from macrophages or other cell types. Other cell types present in the lung may compensate for the decrease in macrophage-derived cytokines. RelB overexpression may also promote the recruitment of certain macrophage subtypes, such as the M2 population, which has anti-inflammatory properties.62,63
Numerous inflammatory mediators are involved in inflammatory cell accumulation in the lung. We demonstrated that RelB overexpression almost completely ablated production of IL-6, TNF-α, and IFN-γ (Figure 5, A, E, and H, respectively), proinflammatory mediators linked to neutrophil recruitment and activation. The production of neutrophil chemokines, MIP-2, KC, and LIX was also dampened by approximately one third, but of these, only the reduction in KC achieved statistical significance (Figure 5, B, C, and G, respectively). RelB overexpression also dampened the cigarette smoke–induced increases in both Cox-2 expression and PGE2 production (Figure 6). PGE2 regulates cytokine production, vasodilation, and cell motility8,13,64; therefore, decreasing the production of this mediator should dampen inflammatory cell migration. Thus, it is likely that RelB overexpression dampens neutrophilic inflammation through a combination of mechanisms involving, at least in part, the regulation of these inflammatory mediators. Corticosteroids reduce production of the neutrophil chemokine IL-8 in the lungs of both long-term smokers and patients with COPD by as much as 40%, but these compounds have limited effects on neutrophil accumulation in the lungs in these studies.65–67 However, inhaled corticosteroids reduce IL-6 production in the lungs of patients with COPD by 26%,68 but they do not significantly affect PGE2 production69; their effects on Cox-2 expression have not yet been established. These findings further underline the limitations of current therapies for inflammatory diseases and highlight the need for newer approaches. In addition to dampening neutrophilic inflammation, selective NF-κB inhibitors have decreased proinflammatory cytokine production in the lungs26,29 and dampen cigarette smoke–induced Cox-2 expression and PGE2 production in human tracheal smooth muscle cells in vitro.26 Collectively, these data indicate that modulation of NF-κB, particularly RelB, expression may be advantageous in treating or preventing lung inflammation.
The expression of ICAM-1, the predominant cell adhesion protein in the lung epithelium and endothelium,42–46 was examined in addition to the previously mentioned inflammatory mediators. ICAM-1, an NF-κB–dependent protein,45–47 is typically up-regulated during inflammation to promote leukocyte infiltration into affected tissues.42,70 However, as seen in Figure 7, RelB overexpression dampened the cigarette smoke–induced increase in ICAM-1 expression, highlighting another mechanism through which RelB influences inflammatory cell migration in the lung. Because ICAM-1 is also regulated by IL-6, TNF-α, IFN-γ, and PGE2,48–54 all of which are significantly decreased on RelB overexpression, RelB overexpression is likely dampening cigarette smoke–induced inflammation in the lungs through multiple complex mechanisms. By contrast, inhaled corticosteroids do not significantly affect ICAM-1 expression in the lungs,67 although the oral corticosteroid, prednisone, did reduce ICAM-1 expression by approximately 30%.71 ICAM-1 expression has also been reduced in human tracheal smooth muscle cells in vitro using NF-κB inhibitors,26 indicating that modulation of key NF-κB family members, including RelB, would be a novel therapeutic strategy against acute lung inflammation.
New and efficacious therapeutic options for lung inflammation are needed. Current therapeutic strategies target symptoms and are limited in their efficacy.57,59 Alternative approaches, including the use of pharmacological inhibitors, remain largely in development.25 Our findings indicate that transient overexpression of RelB in the lungs of mice significantly reduces cigarette smoke–induced inflammation. Conversely, abnormally low RelB expression could predispose humans to a proinflammatory phenotype. Although we induced RelB overexpression with a recombinant adenoviral vector, such vectors are controversial in human gene therapy. Nevertheless, RelB could be targeted by next-generation gene therapy vectors, microRNAs, small-interfering RNAs, or small molecule activators and inhibitors. The up-regulation of RelB via these techniques could be a novel and powerful therapeutic strategy against acute and chronic lung inflammation and inflammatory diseases, such as COPD.
Acknowledgment
We thank Michael Anne, Shikha Gupta, Keith Olsen, M.S., Ramil Sapinoro, Ph.D., and Fabeha Fazal, Ph.D., for their technical expertise.
Footnotes
Supported by grants HL088325, HL088325-02S1, ES01247, T32ES07026, T32H066988, and UL1RR024160 from the NIH; a research grant from the American Lung Association; and a Parker B. Francis Fellowship (C.J.B.).
References
- 1.Baglole C.J., Bushinsky S.M., Garcia T.M., Kode A., Rahman I., Sime P.J., Phipps R.P. Differential induction of apoptosis by cigarette smoke extract in primary human lung fibroblast strains: implications for emphysema. Am J Physiol Lung Cell Mol Physiol. 2006;291:L19–L29. doi: 10.1152/ajplung.00306.2005. [DOI] [PubMed] [Google Scholar]
- 2.Snider G.L. Chronic obstructive pulmonary disease: risk factors, pathophysiology and pathogenesis. Annu Rev Med. 1989;40:411–429. doi: 10.1146/annurev.me.40.020189.002211. [DOI] [PubMed] [Google Scholar]
- 3.Pauwels R.A., Buist A.S., Calverley P.M., Jenkins C.R., Hurd S.S. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163:1256–1276. doi: 10.1164/ajrccm.163.5.2101039. [DOI] [PubMed] [Google Scholar]
- 4.National Health Interview Survey: research for the 1995–2004 redesign. Vital Health Stat 2. 1999;126:1–119. [PubMed] [Google Scholar]
- 5.Stampfli M.R., Anderson G.P. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol. 2009;9:377–384. doi: 10.1038/nri2530. [DOI] [PubMed] [Google Scholar]
- 6.Swauger J.E., Steichen T.J., Murphy P.A., Kinsler S. An analysis of the mainstream smoke chemistry of samples of the U.S. cigarette market acquired between 1995 and 2000. Regul Toxicol Pharmacol. 2002;35(Pt 1):142–156. doi: 10.1006/rtph.2001.1521. [DOI] [PubMed] [Google Scholar]
- 7.Baglole C.J., Maggirwar S.B., Gasiewicz T.A., Thatcher T.H., Phipps R.P., Sime P.J. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB. J Biol Chem. 2008;283:28944–28957. doi: 10.1074/jbc.M800685200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martey C.A., Pollock S.J., Turner C.K., O'Reilly K.M., Baglole C.J., Phipps R.P., Sime P.J. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol. 2004;287:L981–L991. doi: 10.1152/ajplung.00239.2003. [DOI] [PubMed] [Google Scholar]
- 9.Choi E.Y., Santoso S., Chavakis T. Mechanisms of neutrophil transendothelial migration. Front Biosci. 2009;14:1596–1605. doi: 10.2741/3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Profita M., Sala A., Bonanno A., Riccobono L., Ferraro M., La Grutta S., Albano G.D., Montalbano A.M., Gjomarkaj M. Chronic obstructive pulmonary disease and neutrophil infiltration: role of cigarette smoke and cyclooxygenase products. Am J Physiol Lung Cell Mol Physiol. 2010;298:L261–L269. doi: 10.1152/ajplung.90593.2008. [DOI] [PubMed] [Google Scholar]
- 11.Thatcher T.H., Maggirwar S.B., Baglole C.J., Lakatos H.F., Gasiewicz T.A., Phipps R.P., Sime P.J. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am J Pathol. 2007;170:855–864. doi: 10.2353/ajpath.2007.060391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thatcher T.H., McHugh N.A., Egan R.W., Chapman R.W., Hey J.A., Turner C.K., Redonnet M.R., Seweryniak K.E., Sime P.J., Phipps R.P. Role of CXCR2 in cigarette smoke-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2005;289:L322–L328. doi: 10.1152/ajplung.00039.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baglole C.J., Ray D.M., Bernstein S.H., Feldon S.E., Smith T.J., Sime P.J., Phipps R.P. More than structural cells, fibroblasts create and orchestrate the tumor microenvironment. Immunol Invest. 2006;35:297–325. doi: 10.1080/08820130600754960. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H., Cai B. The impact of tobacco on lung health in China. Respirology. 2003;8:17–21. doi: 10.1046/j.1440-1843.2003.00433.x. [DOI] [PubMed] [Google Scholar]
- 15.Beinke S., Ley S.C. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J. 2004;382(Pt 2):393–409. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grossmann M., Nakamura Y., Grumont R., Gerondakis S. New insights into the roles of ReL/NF-kappa B transcription factors in immune function, hemopoiesis and human disease. Int J Biochem Cell Biol. 1999;31:1209–1219. doi: 10.1016/s1357-2725(99)00068-0. [DOI] [PubMed] [Google Scholar]
- 17.Båge T., Lindberg J., Lundeberg J., Modéer T., Yucel-Lindberg T. Signal pathways JNK and NF-kappaB, identified by global gene expression profiling, are involved in regulation of TNFalpha-induced mPGES-1 and COX-2 expression in gingival fibroblasts. BMC Genomics. 2010;11:241. doi: 10.1186/1471-2164-11-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnes P.J., Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 19.Catley M.C., Chivers J.E., Cambridge L.M., Holden N., Slater D.M., Staples K.J., Bergmann M.W., Loser P., Barnes P.J., Newton R. IL-1beta-dependent activation of NF-kappaB mediates PGE2 release via the expression of cyclooxygenase-2 and microsomal prostaglandin E synthase. FEBS Lett. 2003;547:75–79. doi: 10.1016/s0014-5793(03)00672-0. [DOI] [PubMed] [Google Scholar]
- 20.Vogel C.F., Sciullo E., Li W., Wong P., Lazennec G., Matsumura F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol. 2007;21:2941–2955. doi: 10.1210/me.2007-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogel C.F., Sciullo E., Matsumura F. Involvement of RelB in aryl hydrocarbon receptor-mediated induction of chemokines. Biochem Biophys Res Commun. 2007;363:722–726. doi: 10.1016/j.bbrc.2007.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weih F., Carrasco D., Durham S.K., Barton D.S., Rizzo C.A., Ryseck R.P., Lira S.A., Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 23.Xia Y., Pauza M.E., Feng L., Lo D. RelB regulation of chemokine expression modulates local inflammation. Am J Pathol. 1997;151:375–387. [PMC free article] [PubMed] [Google Scholar]
- 24.Brown V., Elborn J.S., Bradley J., Ennis M. Dysregulated apoptosis and NFkappaB expression in COPD subjects. Respir Res. 2009;10:24. doi: 10.1186/1465-9921-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barnes P.J. Future advances in COPD therapy. Respiration. 2001;68:441–448. doi: 10.1159/000050547. [DOI] [PubMed] [Google Scholar]
- 26.Catley M.C., Sukkar M.B., Chung K.F., Jaffee B., Liao S.M., Coyle A.J., Haddad el- B., Barnes P.J., Newton R. Validation of the anti-inflammatory properties of small-molecule IkappaB kinase (IKK)-2 inhibitors by comparison with adenoviral-mediated delivery of dominant-negative IKK1 and IKK2 in human airways smooth muscle. Mol Pharmacol. 2006;70:697–705. doi: 10.1124/mol.106.023150. [DOI] [PubMed] [Google Scholar]
- 27.Birrell M.A., Wong S., Hele D.J., McCluskie K., Hardaker E., Belvisi M.G. Steroid-resistant inflammation in a rat model of chronic obstructive pulmonary disease is associated with a lack of nuclear factor-kappaB pathway activation. Am J Respir Crit Care Med. 2005;172:74–84. doi: 10.1164/rccm.200409-1257OC. [DOI] [PubMed] [Google Scholar]
- 28.Newton R., Holden N.S., Catley M.C., Oyelusi W., Leigh R., Proud D., Barnes P.J. Repression of inflammatory gene expression in human pulmonary epithelial cells by small-molecule IkappaB kinase inhibitors. J Pharmacol Exp Ther. 2007;321:734–742. doi: 10.1124/jpet.106.118125. [DOI] [PubMed] [Google Scholar]
- 29.Rajendrasozhan S., Hwang J.W., Yao H., Kishore N., Rahman I. Anti-inflammatory effect of a selective IkappaB kinase-beta inhibitor in rat lung in response to LPS and cigarette smoke. Pulm Pharmacol Ther. 2010;23:172–181. doi: 10.1016/j.pupt.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sime P.J., Xing Z., Graham F.L., Csaky K.G., Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100:768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koumas L., Phipps R.P. Differential COX localization and PG release in Thy-1(+) and Thy-1(−) human female reproductive tract fibroblasts. Am J Physiol Cell Physiol. 2002;283:C599–C608. doi: 10.1152/ajpcell.00065.2002. [DOI] [PubMed] [Google Scholar]
- 32.Bradley P.P., Priebat D.A., Christensen R.D., Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 33.Mehrad B., Strieter R.M., Moore T.A., Tsai W.C., Lira S.A., Standiford T.J. CXC chemokine receptor-2 ligands are necessary components of neutrophil-mediated host defense in invasive pulmonary aspergillosis. J Immunol. 1999;163:6086–6094. [PubMed] [Google Scholar]
- 34.Vieira S.M., Lemos H.P., Grespan R., Napimoga M.H., Dal-Secco D., Freitas A., Cunha T.M., Verri W.A., Jr, Souza-Junior D.A., Jamur M.C., Fernandes K.S., Oliver C., Silva J.S., Teixeira M.M., Cunha F.Q. A crucial role for TNF-alpha in mediating neutrophil influx induced by endogenously generated or exogenous chemokines, KC/CXCL1 and LIX/CXCL5. Br J Pharmacol. 2009;158:779–789. doi: 10.1111/j.1476-5381.2009.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith R.S., Smith T.J., Blieden T.M., Phipps R.P. Fibroblasts as sentinel cells: synthesis of chemokines and regulation of inflammation. Am J Pathol. 1997;151:317–322. [PMC free article] [PubMed] [Google Scholar]
- 36.Gregory J.L., Morand E.F., McKeown S.J., Ralph J.A., Hall P., Yang Y.H., McColl S.R., Hickey M.J. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol. 2006;177:8072–8079. doi: 10.4049/jimmunol.177.11.8072. [DOI] [PubMed] [Google Scholar]
- 37.Subramaniyam D., Steele C., Köhnlein T., Welte T., Grip O., Matalon S., Janciauskiene S. Effects of alpha 1-antitrypsin on endotoxin-induced lung inflammation in vivo. Inflamm Res. 2010;59:571–578. doi: 10.1007/s00011-010-0164-x. [DOI] [PubMed] [Google Scholar]
- 38.Li L., Huang L., Vergis A.L., Ye H., Bajwa A., Narayan V., Strieter R.M., Rosin D.L., Okusa M.D. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. 2010;120:331–342. doi: 10.1172/JCI38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sethu S., Pushparaj P.N., Melendez A.J. Phospholipase D1 mediates TNFalpha-induced inflammation in a murine model of TNFalpha-induced peritonitis. PLoS One. 2010;5:e10506. doi: 10.1371/journal.pone.0010506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fielding C.A., McLoughlin R.M., McLeod L., Colmont C.S., Najdovska M., Grail D., Ernst M., Jones S.A., Topley N., Jenkins B.J. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol. 2008;181:2189–2195. doi: 10.4049/jimmunol.181.3.2189. [DOI] [PubMed] [Google Scholar]
- 41.Subbaramaiah K., Dannenberg A.J. Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends Pharmacol Sci. 2003;24:96–102. doi: 10.1016/S0165-6147(02)00043-3. [DOI] [PubMed] [Google Scholar]
- 42.Porter J.C., Hall A. Epithelial ICAM-1 and ICAM-2 regulate the egression of human T cells across the bronchial epithelium. FASEB J. 2009;23:492–502. doi: 10.1096/fj.08-115899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu X., Zhang Y., Cheng D., Ding Y., Yang D., Jiang F., Zhou C., Ying B., Wen F. Mechanical stress upregulates intercellular adhesion molecule-1 in pulmonary epithelial cells. Respiration. 2008;76:344–350. doi: 10.1159/000137509. [DOI] [PubMed] [Google Scholar]
- 44.Noguera A., Busquets X., Sauleda J., Villaverde J.M., MacNee W., Agusti A.G. Expression of adhesion molecules and G proteins in circulating neutrophils in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;158(Pt 1):1664–1668. doi: 10.1164/ajrccm.158.5.9712092. [DOI] [PubMed] [Google Scholar]
- 45.Holden N.S., Catley M.C., Cambridge L.M., Barnes P.J., Newton R. ICAM-1 expression is highly NF-kappaB-dependent in A549 cells: no role for ERK and p38 MAPK. Eur J Biochem. 2004;271:785–791. doi: 10.1111/j.1432-1033.2004.03982.x. [DOI] [PubMed] [Google Scholar]
- 46.Textor S., Accardi R., Havlova T., Hussain I., Sylla B.S., Gissmann L., Cerwenka A. NF-κ-B-dependent upregulation of ICAM-1 by HPV16-E6/E7 facilitates NK cell/target cell interaction. Int J Cancer. 2011;128:1104–1113. doi: 10.1002/ijc.25442. [DOI] [PubMed] [Google Scholar]
- 47.Yang C.M., Luo S.F., Hsieh H.L., Chi P.L., Lin C.C., Wu C.C., Hsiao L.D. Interleukin-1beta induces ICAM-1 expression enhancing leukocyte adhesion in human rheumatoid arthritis synovial fibroblasts: involvement of ERK, JNK, AP-1, and NF-kappaB. J Cell Physiol. 2010;224:516–526. doi: 10.1002/jcp.22153. [DOI] [PubMed] [Google Scholar]
- 48.Kim H., Hwang J.S., Woo C.H., Kim E.Y., Kim T.H., Cho K.J., Kim J.H., Seo J.M., Lee S.S. TNF-alpha-induced up-regulation of intercellular adhesion molecule-1 is regulated by a Rac-ROS-dependent cascade in human airway epithelial cells. Exp Mol Med. 2008;40:167–175. doi: 10.3858/emm.2008.40.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su X., Ao L., Zou N., Song Y., Yang X., Cai G.Y., Fullerton D.A., Meng X. Post-transcriptional regulation of TNF-induced expression of ICAM-1 and IL-8 in human lung microvascular endothelial cells: an obligatory role for the p38 MAPK-MK2 pathway dissociated with HSP27. Biochim Biophys Acta. 2008;1783:1623–1631. doi: 10.1016/j.bbamcr.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wung B.S., Hsu M.C., Wu C.C., Hsieh C.W. Resveratrol suppresses IL-6-induced ICAM-1 gene expression in endothelial cells: effects on the inhibition of STAT3 phosphorylation. Life Sci. 2005;78:389–397. doi: 10.1016/j.lfs.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 51.Marino M., Scuderi F., Provenzano C., Scheller J., Rose-John S., Bartoccioni E. IL-6 regulates MCP-1, ICAM-1 and IL-6 expression in human myoblasts. J Neuroimmunol. 2008;196:41–48. doi: 10.1016/j.jneuroim.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 52.Whiteman S.C., Spiteri M.A. IFN-gamma regulation of ICAM-1 receptors in bronchial epithelial cells: soluble ICAM-1 release inhibits human rhinovirus infection. J Inflamm (Lond) 2008;5:8. doi: 10.1186/1476-9255-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spoelstra F.M., Postma D.S., Hovenga H., Noordhoek J.A., Kauffman H.F. Interferon-gamma and interleukin-4 differentially regulate ICAM-1 and VCAM-1 expression on human lung fibroblasts. Eur Respir J. 1999;14:759–766. doi: 10.1034/j.1399-3003.1999.14d06.x. [DOI] [PubMed] [Google Scholar]
- 54.Noguchi K., Iwasaki K., Shitashige M., Endo H., Kondo H., Ishikawa I. Cyclooxygenase-2-dependent prostaglandin E2 down-regulates intercellular adhesion molecule-1 expression via EP2/EP4 receptors in interleukin-1beta-stimulated human gingival fibroblasts. J Dent Res. 2000;79:1955–1961. doi: 10.1177/00220345000790120601. [DOI] [PubMed] [Google Scholar]
- 55.Chen H.W., Chien M.L., Chaung Y.H., Lii C.K., Wang T.S. Extracts from cigarette smoke induce DNA damage and cell adhesion molecule expression through different pathways. Chem Biol Interact. 2004;150:233–241. doi: 10.1016/j.cbi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 56.Gogali A., Charalabopoulos K., Zampira I., Konstantinidis A.K., Tachmazoglou F., Daskalopoulos G., Constantopoulos S.H., Dalavanga Y. Soluble adhesion molecules E-cadherin, intercellular adhesion molecule-1, and E-selectin as lung cancer biomarkers. Chest. 2010;138:1173–1179. doi: 10.1378/chest.10-0157. [DOI] [PubMed] [Google Scholar]
- 57.Black P.N., McDonald C.F. Interventions to reduce the frequency of exacerbations of chronic obstructive pulmonary disease. Postgrad Med J. 2009;85:141–147. doi: 10.1136/pgmj.2008.072439. [DOI] [PubMed] [Google Scholar]
- 58.Celli B.R. What is the best pharmacological treatment to prevent exacerbation of chronic obstructive pulmonary disease? Pol Arch Med Wewn. 2008;118:172–174. [PubMed] [Google Scholar]
- 59.Evensen A.E. Management of COPD exacerbations. Am Fam Physician. 2010;81:607–613. [PubMed] [Google Scholar]
- 60.Cheng S.E., Luo S.F., Jou M.J., Lin C.C., Kou Y.R., Lee I.T., Hsieh H.L., Yang C.M. Cigarette smoke extract induces cytosolic phospholipase A2 expression via NADPH oxidase, MAPKs, AP-1, and NF-kappaB in human tracheal smooth muscle cells. Free Radic Biol Med. 2009;46:948–960. doi: 10.1016/j.freeradbiomed.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 61.Lin C.C., Lee I.T., Yang Y.L., Lee C.W., Kou Y.R., Yang C.M. Induction of COX-2/PGE(2)/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: role of TLR4-dependent NADPH oxidase activation. Free Radic Biol Med. 2010;48:240–254. doi: 10.1016/j.freeradbiomed.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 62.van Dongen M., Savage N.D., Jordanova E.S., Briaire-de Bruijn I.H., Walburg K.V., Ottenhoff T.H., Hogendoorn P.C., van der Burg S.H., Gelderblom H., van Hall T. Anti-inflammatory M2 type macrophages characterize metastasized and tyrosine kinase inhibitor-treated gastrointestinal stromal tumors. Int J Cancer. 2010;127:899–909. doi: 10.1002/ijc.25113. [DOI] [PubMed] [Google Scholar]
- 63.Choi K.M., Kashyap P.C., Dutta N., Stoltz G.J., Ordog T., Shea Donohue T., Bauer A.J., Linden D.R., Szurszewski J.H., Gibbons S.J., Farrugia G. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology. 2010;138:2399–2409. doi: 10.1053/j.gastro.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan S.C., Kim J.W., Henderson W.R., Jr, Hanifin J.M. Altered prostaglandin E2 regulation of cytokine production in atopic dermatitis. J Immunol. 1993;151:3345–3352. [PubMed] [Google Scholar]
- 65.Ozol D., Aysan T., Solak Z.A., Mogulkoc N., Veral A., Sebik F. The effect of inhaled corticosteroids on bronchoalveolar lavage cells and IL-8 levels in stable COPD patients. Respir Med. 2005;99:1494–1500. doi: 10.1016/j.rmed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 66.Perng D.W., Tao C.W., Su K.C., Tsai C.C., Liu L.Y., Lee Y.C. Anti-inflammatory effects of salmeterol/fluticasone, tiotropium/fluticasone or tiotropium in COPD. Eur Respir J. 2009;33:778–784. doi: 10.1183/09031936.00115308. [DOI] [PubMed] [Google Scholar]
- 67.Van Overveld F.J., Demkow U., Górecka D., De Backer W.A., Zieliński J. Differences in responses upon corticosteroid therapy between smoking and non-smoking patients with COPD. J Physiol Pharmacol. 2006;57(Suppl 4):273–282. [PubMed] [Google Scholar]
- 68.Sin D.D., Lacy P., York E., Man S.F. Effects of fluticasone on systemic markers of inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170:760–765. doi: 10.1164/rccm.200404-543OC. [DOI] [PubMed] [Google Scholar]
- 69.Montuschi P., Kharitonov S.A., Ciabattoni G., Barnes P.J. Exhaled leukotrienes and prostaglandins in COPD. Thorax. 2003;58:585–588. doi: 10.1136/thorax.58.7.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horgan M.J., Ge M., Gu J., Rothlein R., Malik A.B. Role of ICAM-1 in neutrophil-mediated lung vascular injury after occlusion and reperfusion. Am J Physiol. 1991;261(Pt 2):H1578–H1584. doi: 10.1152/ajpheart.1991.261.5.H1578. [DOI] [PubMed] [Google Scholar]
- 71.Man S.F., Xuekui Z., Vessey R., Walker T., Lee K., Park D., Sin D.D. The effects of inhaled and oral corticosteroids on serum inflammatory biomarkers in COPD: an exploratory study. Ther Adv Respir Dis. 2009;3:73–80. doi: 10.1177/1753465809336697. [DOI] [PubMed] [Google Scholar]