

Abstract
We and others have shown that calcium-independent phospholipase A2 (iPLA2) is involved in epithelial ovarian cancer (EOC). Hence, we propose that iPLA2 is a potential effective and novel target for EOC. We tested this concept and found that bromoenol lactone (BEL), a selective inhibitor of iPLA2, significantly inhibited EOC metastatic tumor growth in mouse xenograft models using human SKOV3 and HEY ovarian cancer cells. Moreover, the combination of BEL with paclitaxel (PTX), one of the most commonly used therapeutic agents in EOC, almost completely blocked tumor development in the xenograft mouse model. BEL showed no detectable cytotoxic effects in mice. Another iPLA2 inhibitor, FKGK11, also inhibited tumor development in the xenograft mouse model, supporting that the major target of action was iPLA2. The additional effects of BEL with PTX in vivo likely stem from their distinct cellular effects. BEL and FKGK11 reduced adhesion, migration, and invasion of EOC cells in vitro; the reduced ability to adhere, migrate, and invade seems to increase the vulnerability of tumor cells to PTX. These results provide an important basis for the development of new treatment modalities for EOC.
Epithelial ovarian cancer (EOC) caused an estimated 13,850 deaths in 2010 in the United States.1 More effective treatment regimens are urgently required for this deadly disease. Lysophosphatidic acid (LPA) is a potent bioactive lipid with multiple roles in normal cellular function and pathogenesis. We and others have obtained ample evidence from clinical investigations, as well as experimental animal and cell culture studies, that LPA plays an important role in every stage of EOC development, including initiation, primary tumor growth, and metastasis.2–12 LPA can be produced via the action of phospholipase A1 (PLA1) or PLA213,14 and lysophospholipase D/autotaxin (ATX)/Enpp2. LPA production and degradation have been investigated as potential strategies to control cancer development. Lately, we have concentrated our efforts on calcium-independent PLA2 (iPLA2) as a potential effective and novel target for reducing the effects of LPA in EOC.10,11,15–18 We revealed a laminin–β1-integrin–caspase 3 pathway that operates to activate iPLA2β in nonapoptotic EOC cells in vitro.15 Activated iPLA2 increases extracellular LPA and arachidonic acid from EOC cells, with the former being important for chemotaxis and the latter for chemokinetics.15 The iPLA2 selective inhibitor bromoenol lactone (BEL) reduces extracellular LPA from human EOC cells17 and human peritoneal mesothelial cells.16 Using iPLA2β knockout mice and iPLA2β knockdown mouse EOC cells, we recently demonstrated that iPLA2β in EOC cells and the surrounding microenvironment interactively regulate the concentrations of several bioactive lipids (including LPA) in response to the tumor, affecting EOC development.18 LPA, but not lysophosphatidylcholine, the substrate of ATX, enhances in vivo ascites formation and metastatic tumor growth in iPLA2β−/− mice.18 Our results are highly consistent with those of another study conducted by Song et al19 showing that inhibition of iPLA2β using short hairpin RNA suppresses tumorigenicity of EOC cells.
The key to the formation of solid secondary tumors (metastases) in EOC is the ability to migrate through, attach to, and invade into the organs of the peritoneal cavity. Inhibition of these three critical steps involved in tumor metastasis will block progression of the cancer.20–22 Evidence supports that attachment of disseminating EOC cells to the organs of the peritoneum is the rate-limiting step of EOC metastasis.21 Several studies suggest that this is where the LPA-iPLA2 axis plays a key role.10,11,17 Differential patterns of invasion in iPLA2β wild-type versus knockout mice, in vector– versus iPLA2β–down-regulated mouse EOC cells, and in LPA- versus vehicle-treated EOC in vivo models have been observed.18 In addition, we have shown that iPLA2 is involved in migration and invasion in all human EOC cell lines tested and in a mouse EOC cell line using BEL and/or small-interfering/short hairpin RNA approaches.10–12,15–18
Because of the interactive effect on bioactive lipid levels, targeting iPLA2β in both host and tumor cells (with a small molecule inhibitor) is likely to be beneficial.18 Moreover, targeting host cells is also likely to be safe as suggested by the fact that iPLA2β−/− mice are grossly normal and that only male mice have reproductive deficiency.23 This is in contrast to targeting ATX, the important enzyme directly involved in LPA production, because ATX has roles in many developmental and physiologic functions and Enpp2−/− mice are embryonic lethal.24
In this work, we report the results of an experimental metastatic EOC mouse model (i.p. injection of human ovarian cancer cells in NOD/SCID mice) with two iPLA2 selective inhibitors, BEL and FKGK11. In addition, the potential additional effects of BEL and the two most commonly used therapeutic reagents for EOC, cisplatin (CDDP), and paclitaxel (PTX), were examined. In vivo toxic effects were assessed, and the molecular mechanisms by which BEL and PTX differentially affect cellular activities were investigated.
Materials and Methods
Materials
Human collagen I was obtained from Chemicon International (Temecula, CA), 18:1 LPA was from Avanti Polar Lipids (Birmingham, AL), BEL was from Calbiochem (San Diego, CA), and FKGK11 was from Cayman Chemical (Ann Arbor, MI) or was synthesized following the literature,17 with slight modification (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Cell Culture
The human EOC cells SKOV3 and HEY were cultured in RPMI 1640 medium with glutamine (2 mmol/L) and fetal bovine serum (5%). Human hepatoma cells HepG2 were obtained from Mary Maluccio (Indiana University School of Medicine) and were cultured in Dulbecco's modified Eagle's medium with glutamine and fetal bovine serum (10%). Primary human peritoneal mesothelial cells LP-9 were obtained from the Coriell Cell Repositories (Camden, NJ) and were cultured in the medium specified by the company.16
Cell Toxicity Assays
Cells were seeded into 96-well plates at 5000 cells per well in growth medium and were allowed to attach and reach 25% to 30% confluence. Then the medium was replaced by serial drug dilutions in medium containing 2% fetal bovine serum, and cells were incubated for 72 hours. MTT (5 mg/mL) dye reduction assays were used to detect living cells, as described previously.25,26 To test whether floating SKOV3 cells were more sensitive to PTX compared with attached cells, PTX was used in suspended cells with the same dilutions for 4 hours before plating.
Cell Migration, Invasion, and Adhesion
For migration assays, the 8-μm pore size membranes of a Costar Transwell assay plate (Corning Inc., Corning, NY) were coated with collagen I (10 μL, 10 μg/mL). For invasion assays, BD BioCoat Matrigel invasion chambers (BD Biosciences, Bedford, MA) were used following the manufacturer's instructions. The assays were performed as previously described.14 Data are presented as mean ± SD of cells/image/membrane, normalized to control. For adhesion assays, each well of a 96-well plate was coated with collagen I (5 μL, 10 μg/mL). Cells were allowed to attach for 4 hours. After removing unattached cells with PBS, the attached cells were fixed with methanol for 30 minutes and were stained with crystal violet, and the absorbance of the solubilized dye was read at 555 nm using a Victor3 V plate reader (PerkinElmer, Waltham, MA).17
Reagents Tested in Vivo
For BEL, the stock solution (25 mg/mL; 78.8 mmol/L in dimethyl sulfoxide) was diluted in PBS for injection (the final dimethyl sulfoxide concentration was <0.2%). The stock of FKGK11 was 5.5 mol/L and was freshly made to 2 mmol/L in PBS before use. The CDDP stock solution (25 mg/mL in H2O) was diluted in PBS for injection. PTX was dissolved in a 50:50 (v/v) mixture of Cremophor EL (Sigma-Aldrich, St. Louis, MO) and dehydrated ethanol at 50 mg/mL and was diluted in PBS for injection. For all the experiments, i.p. injections of 200 μL per mouse were given three times per week for 3 to 4 weeks starting 8 days (for HEY cells) or 10 days (for SKOV3 cells) after tumor cell injection.
Ovarian Cancer Xenograft Models
Female NOD/SCID mice were obtained from the In Vivo Therapeutics Core, Indiana University School of Medicine (Indianapolis, IN) at 6 to 8 weeks of age. SKOV3-luciferase cells were a gift from Dr. Melissa Fishel at the Indiana University Cancer Center and express both green fluorescent protein and luciferase. First, cells (107 in 500 μL of PBS) were i.p. injected into mice. Starting 10 days after tumor cell injection, the mice were i.p. injected with drugs or vehicle three times per week for 3 to 4 weeks. Mouse body weights were measured regularly. Tumors were monitored in living mice by in vivo bioluminescence imaging 2 and 4 weeks after treatment initiation as described previously.12 Thirty-eight to 40 days after tumor cell injections, mice were sacrificed and tumor development was analyzed. Tumors were counted at each metastatic location, and tumor diameters were measured. Ascites or peritoneal washings (peritoneal washings were collected in mice that did not develop ascites using 3 mL of PBS) were collected. After centrifugation, the pelleted cells were seeded and cultured to detect living tumor cells (expressing green fluorescent protein). For the HEY cell model, 8 × 106 HEY cells were i.p. injected into each mouse. Eight days later, treatment was initiated, and mice were sacrificed 32 to 33 days after tumor cell injection. All the animal protocols were approved by the Indiana University School of Medicine Animal Care and Use Committee.
IHC Analysis
Tissue preparation, staining, and immunohistochemistry (IHC) analyses were performed as previously described.14 Antibody to proliferating cell nuclear antigen (PCNA) was from Santa Cruz Biotechnology (Santa Cruz, CA) and was used at 1:100 dilution. Quantitation of PCNA staining was performed using MetaMorph software (Molecular Devices Inc., Sunnyvale, CA). The percentages of the positively stained brown cells per total cells (both brown and blue cells) were obtained from three independent tumor sections from each group of mice.
Toxicity Studies
Blood samples were collected from the facial veins of the mice using EDTA as anticoagulant and were analyzed using a Hemavet 950 analyzer (Drew Scientific Inc., Oxford, CT). Lymphocyte, neutrophil, and monocyte counts from mice in the various treatment groups were compared with the reference ranges supplied by the manufacturer. Fixed paraffin-imbedded tissue slices from kidney, liver, spleen, small intestine, lung, and brain were subjected to H&E staining and pathologic examination.
Results
BEL Inhibits EOC Development in Vivo
Down-regulation of iPLA2β expression with lentivirus-mediated RNA interference inhibited tumorigenicity of EOC cell lines in nude mice.19 However, it is more clinically practical and relevant to use small molecule inhibitors. BEL has been identified as a potent, irreversible, and mechanism-based inhibitor of iPLA2.27 There are few studies published using BEL in vivo (in mice), and these are limited to evaluation of brain inflammation.28 A long-term effect of BEL on cancer development in vivo has not been reported. We conducted a series of experiments to test whether BEL could be used for the treatment of EOC in an SKOV3 experimental metastatic mouse model. In two sets of independent experiments (five mice in each group), BEL greatly inhibited metastasis of EOC (mean ± SD total tumor number per mouse, 44.5 ± 13.7 versus 8.8 ± 2.6, P = 0.0027). To determine whether the effect of BEL is limited to SKOV3 cells, we conducted a similar experiment using another human EOC cell line, HEY (three mice in each group). Similar to what was seen with SKOV3 cells, BEL substantively inhibited HEY cell tumor development in vivo (mean ± SD total tumor number per mouse, 18.3 ± 3.2 versus 6.3 ± 2.3, P = 0.0094). Images of peritoneal tumors are shown in Figure 1A, and a summary of tumor number per mouse broken down by tumor size is given in Figure 1B. It is clear in Figure 1B that small tumors predominate in the SKOV3 model and that BEL particularly affects small tumor numbers. However, it should be noted that in the HEY cell model, all three vehicle-treated mice developed a 2-mm-thick tumor layer on the peritoneal wall (Figure 1A). This skews the count of individual small tumors (<1 mm) shown in Figure 1B and underrepresents the difference in tumor development produced by BEL treatment in the HEY cell model as shown in Figure 1B. This work was repeated in additional independent experiments, and the results were consistent (see later herein).
Figure 1.
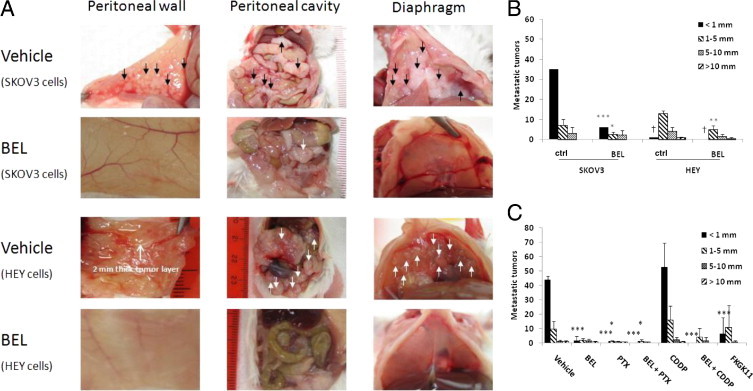
Treatment with BEL compared with vehicle decreased tumor formation in a mouse xenograft model of EOC. A: Human EOC cells SKOV3 or HEY were injected i.p. into NOD/SCID mice, and after 10 days (8 days for HEY cells), i.p. injections of BEL or vehicle were initiated. Mice were sacrificed 32 to 40 days after tumor cell injection and were examined for tumor formation. Tumors are noted with arrows on images of the peritoneal wall, the organs of the abdominal cavity, and the diaphragm of representative mice (n = 5 for each group). B: Summary of results from A showing metastatic tumors per mouse broken down by size. There are more small tumors than large tumors, and these are reduced in number by BEL (n = 5 for each group). C: Treatment with iPLA2β inhibitors alone or combined with traditional chemotherapeutic agents reduced tumor numbers in a mouse xenograft model of EOC. Human EOC cells SKOV3 were injected i.p. into NOD/SCID mice; after 10 days, i.p. injections of drugs or vehicle were initiated. The numbers of mice used are listed in Table 1. All the mice were sacrificed 38 to 40 days after tumor cell injection and were examined for tumors. Data are mean ± SD. Two-sided Student's t-test was used to obtain the P values for pair comparisons. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with vehicle. †The numbers of tumors <1 mm are underrepresented here owing to fusion of small tumors on the peritoneal wall (see Results).
BEL Has an Additional Effect with PTX in Inhibiting Metastasis of EOC
CDDP and PTX are the two most commonly used drugs for EOC treatment. However, they are cytotoxic and associated with severe adverse effects that affect quality of life. In addition, patients with EOC undergoing chemotherapy develop drug resistance. We have shown that iPLA2 inhibition reduces migration and invasion of EOC cells,2,11,15–18 which differs in mechanism from the actions of CDDP and PTX. We tested whether administration of low-dose CDDP or PTX (at which there were few or nondetectable toxic adverse effects) with BEL had an additional effect. Six groups of mice in three independent sets of experiments were treated with vehicle (solvents used), BEL (0.19 mg/kg), CDDP (0.25 mg/kg), PTX (5 mg/kg), CDDP + BEL, and PTX + BEL. All the drugs were delivered 10 days after tumor cell injection three times per week for 4 weeks. Mice were sacrificed 38 to 40 days after tumor cell injection. The use of SKOV3 cells stably expressing green fluorescent protein–luciferase allowed for real-time imaging. Before drug treatment (10 days after injection of tumor cells), the luciferase intensities were increased compared with day 1 of injection (data not shown). This finding suggests that tumors were established and growing, validating that the model mimics late-stage and disseminating human EOC diseases. Representative images taken 2 weeks after drug treatment initiation and showing the effects of the drugs are displayed in Supplemental Figure S2 (available at http://ajp.amjpathol.org). The imaging results from all the mice tested are quantitated in Supplemental Table S1 (available at http://ajp.amjpathol.org). All the drugs tested had inhibitory effects on tumor development (Table 1 and Figure 1C). As seen previously herein (Figure 1B), BEL dramatically reduced the number of tumors developed compared with vehicle (5.3 ± 1.2 versus 54.8 ± 6.2, P < 0.001), and small tumors in particular were reduced (1.7 ± 2.9 versus 43.8 ± 2.5, P < 0.001) (Figure 1C). The combination of BEL with PTX showed additional inhibitory effects (0.8 ± 1.8 versus 5.3 ± 1.2, P = 0.008) (Figure 1C), as five of seven mice did not develop any detectable tumors and the other two mice had smaller tumors compared with the other groups (Table 1 and Figure 1C). Ascites did not develop in any of the mice in the BEL plus PTX group compared with ascites development in other groups (Table 1). Although the combination of BEL with CDDP did not further reduce the number of tumors compared with the BEL-only group (5.3 ± 8.6 versus 5.3 ± 1.2, P = 0.988), BEL plus CDDP was more effective than either drug alone, as shown by reduced tumor incidence (three of six mice did not develop tumors) (Table 1 and Figure 1C).
Table 1.
Effect of BEL, FKGK11, and Combinations in Mouse Xenograft Models of EOC
| Injected cells | Treatment | No. of mice | Ascites incidence | Invaded organs⁎ | Tumor incidence | Floating cells/peritoneum |
|---|---|---|---|---|---|---|
| SKOV3 | Vehicle | 10 | 4 of 10 | W, D, M, O, L, SI | 10 of 10 | 10 of 10 |
| BEL | 11 | 4 of 11 | W, D (45%), M (27%), O (45%) | 11 of 11 | 11 of 11 | |
| PTX | 8 | 2 of 8 | W, O (25%), M (25%) | 8 of 8 | 0 of 8 | |
| PTX + BEL | 7 | 0 of 7 | O (29%) | 2 of 7 | 0 of 7 | |
| CDDP | 8 | 2 of 8 | W, D, M (75%), O (75%), L (75%), SI | 8 of 8 | 8 of 8 | |
| CDDP + BEL | 6 | 1 of 6 | W (33%), D (33%), M (33%), O (33%), L (33%), SI (33%) | 3 of 6 | 3 of 6 | |
| FKGK11 | 7 | 4 of 7 | W (43%), D (43%), M (43%), O (57%), SI (14%) | 4 of 7 | 4 of 7 | |
| HEY | Vehicle | 3 | 3 of 3 | W (100%), D (100%), M (100%), O (100%) | 3 of 3 | 3 of 3 |
| BEL | 3 | 2 of 3 | W (33%), D (33%), M, O (67%) | 3 of 3 | 3 of 3 |
D, diaphragm; L, liver; M, mesentery; O, omentum; SI, small intestine; W, peritoneal wall.
If <100%, the percentage of mice is indicated.
No Apparent Cytotoxicity Is Observed in Mice Treated with BEL or BEL Combined with Low Doses of CDDP or PTX
The potential cytotoxicity of the reagents tested was assessed. The body weights of mice from the different treatment groups did not change with time over the 32 to 40 days of treatment. Also, no systematic changes in body weight between groups were seen (Figure 2A). White blood cell counts were within the reference range in all the groups except the solvent control group, where slightly higher counts were found (Table 2). Lymphocyte counts were normal in all six groups. Neutrophil and monocyte counts were increased above the reference ranges in several groups (Table 2). However, neutrophil counts were normal in the BEL and BEL with PTX groups, and monocyte counts were normal (or close to normal) in the PTX, BEL with PTX, and BEL- and CDDP-only groups (Table 2). These data suggest that BEL and the low doses of CDDP or PTX used do not generate blood cell toxic effects. Rather, reduced tumor loads were associated with normalized blood cell counts. Finally, the major organs of mice from the six treatment groups were examined for evidence of drug-induced abnormalities. H&E-stained tissue sections from the kidney, liver, spleen, small intestine, lung, and brain were compared (Figure 2B). No evidence of drug-induced pathologic or toxic effects was seen in any tissues as judged by a professional pathologist (R.E.).
Figure 2.
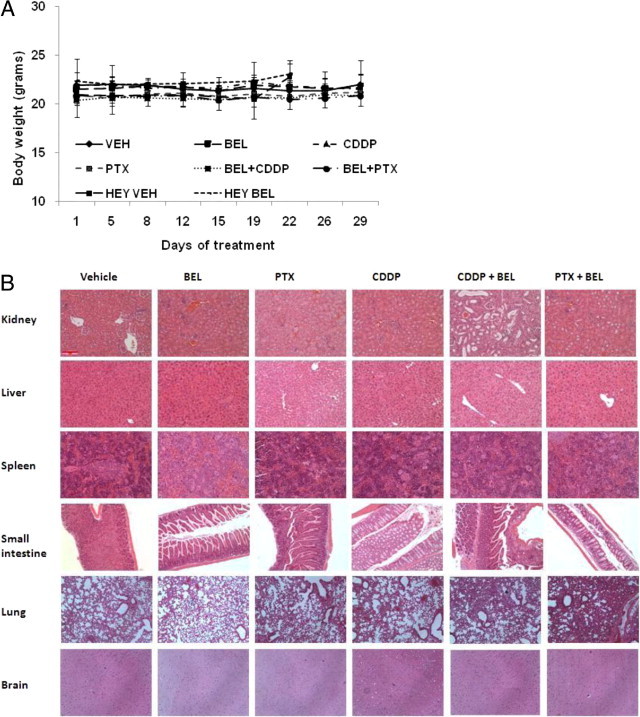
Treatment with iPLA2β inhibitors alone or combined with traditional chemotherapeutic agents did not cause toxic effects in NOD/SCID mice. A: The mean ± SD body weights of mice in the treatment groups starting on the day of tumor injection (day 1). VEH, vehicle. B: Representative H&E-stained tissue sections from mice in the treatment groups. Original magnification, ×200.
Table 2.
Blood Cell Counts at Sacrifice from NOC/SCID Mice i.p. Injected with Human EOC Tumor Cells and Treated with Various Drugs as Described in Materials and Methods
| Group | Counts, mean ± SD (103/μL) |
|||
|---|---|---|---|---|
| White blood cells | Neutrophils | Lymphocytes | Monocytes | |
| Vehicle | 7.83 ± 5.10 | 4.86 ± 4.28⁎ | 2.32 ± 1.90 | 0.60 ± 0.51⁎ |
| BEL | 3.71 ± 1.21 | 1.66 ± 0.60 | 1.41 ± 0.48 | 0.64 ± 0.26⁎ |
| PTX | 4.56 ± 3.97 | 2.80 ± 2.99⁎ | 1.48 ± 1.06 | 0.26 ± 0.13 |
| CDDP | 7.29 ± 2.40 | 2.66 ± 0.66⁎ | 3.72 ± 1.72 | 0.82 ± 0.006⁎ |
| PTX + BEL | 4.15 ± 4.07 | 1.23 ± 1.23 | 2.5 ± 2.77 | 0.35 ± 0.11 |
| CDDP + BEL | 4.52 ± 1.86 | 2.73 ± 1.46⁎ | 1.55 ± 0.94 | 0.17 ± 0.08 |
| Reference range | 1.8 to 10.7 | 0.1 to 2.4 | 0.9 to 9.3 | 0 to 0.4 |
Values are outside the reference range.
Determination of Optimal Dosages for BEL and PTX
We tested several doses of BEL and PTX in the SKOV3 xenograft mouse model. BEL was tested in a range of 0.05 to 0.475 mg/kg. Even at 0.475 mg/kg, BEL did not show detectable toxic effects using the assays described previously herein (data not shown). Although concentrations of BEL <0.19 mg/kg showed a dose-dependent antitumor effect, the effect was not further improved at >0.19 mg/kg (detailed data not shown). Thus, the optimal dose for the in vivo effects of BEL on metastasis was approximately 0.19 mg/kg (equivalent to 200 μL of 60 μmol/L BEL for each i.p. injection, assuming a 20-g mouse). Although apparent toxic effects were not observed when 5 mg/kg of PTX was used (see previously herein), 10 mg/kg of PTX caused liver toxicity presented as many white spots on the liver surface (see Supplemental Figure S3 at http://ajp.amjpathol.org). H&E staining revealed that many leukocytes had infiltrated the liver (see Supplemental Figure S3 at http://ajp.amjpathol.org). These results further support the advantage of using combined BEL with PTX at relatively lower doses of each drug.
FKGK11 Has Similar Effects as BEL in Inhibiting EOC Development
It has been reported that BEL has off-target effects.29 Recently, a more structurally based iPLA2 inhibitor, FKGK11, was synthesized and tested in vivo in two mouse models of brain inflammation. In these studies, FKGK11 was used at 2 mmol/L in 200 μL for each i.p injection.30–32 The SKOV3 xenograft model was used to test whether FKGK11 has similar effects as BEL on EOC development. FKGK11 (5.6 mg/kg, equivalent to 2 mmol/L in 200 μL of solvent per injection) was i.p injected into each mouse three times per week for 4 weeks. Although the control mice all grew tumors, three of seven FKGK11-treated mice developed neither tumors nor ascites, and the other four had reduced tumor volume (Figure 1C and Table 1). The similar effects of these two structurally and mechanistically distinct iPLA2 selective inhibitors in vivo support that iPLA2 is likely to be at least a major target for both these drugs.
BEL, FKGK11, CDDP, and PTX Have Different Dose-Dependent Cytotoxic Effects
We compared the dose-dependent cytotoxic effects of BEL, FKGK11, CDDP, and PTX in human EOC cells (SKOV3 and HEY), primary human mesothelial cells (LP-9) as representative cells for peritoneal host cell toxicity, and human hepatoma cells (HepG2) for evaluation of liver toxicity. We found that human EOC cell lines were sensitive to PTX (Figure 3B). The inhibition curves were biphasic: in the first phase between 1 nmol/L and 10 μmol/L, PTX inhibited 50% to 60% of cell proliferation; in the second phase, PTX further inhibited cell proliferation dose dependently (cytotoxic). LP-9 and HepG2 are relatively slower-growing cells and seemed to be modestly more resistant to the same concentrations of PTX. CDDP dose dependently inhibited cell proliferation in all four cell lines tested, with estimated inhibitory concentration of 50% values of 1.7, 3.5, 1.7, and 1.0 μmol/L for SKOV3, HEY, LP-9, and HepG2 cells, respectively (Figure 3A). Low concentrations of BEL (<3 μmol/L) did not affect proliferation of any of the four cell lines tested (Figure 3C). At a higher concentration, BEL inhibited proliferation, with estimated inhibitory concentration of 50% values of 23, 34, 7.8, and 55 μmol/L for SKOV3, HEY, LP-9, and HepG2 cells, respectively. FKGK11 is a potentially more specific but less potent inhibitor than BEL for iPLA2.30,31 The inhibitory concentration of 50% values for the effect of FKGK11 on cell proliferation were estimated to be 180, 180, 300, and 380 μmol/L for SKOV3, HEY, LP-9, and HepG2 cells, respectively (Figure 3D).
Figure 3.
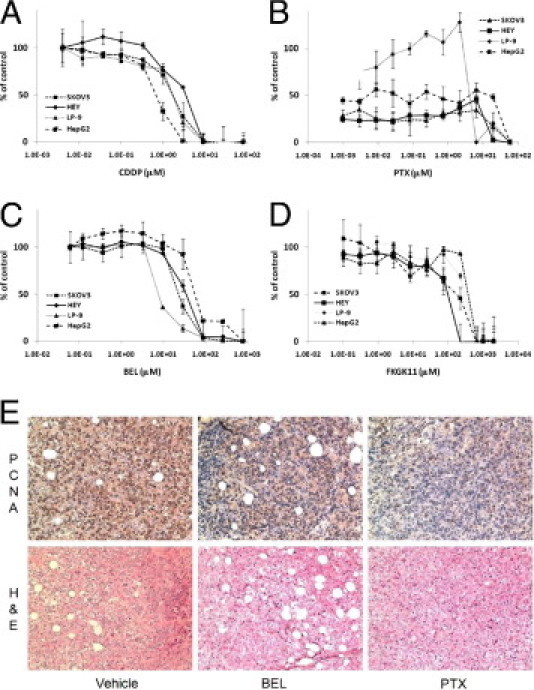
Proliferation inhibition/cytotoxicity profiles for drugs used in this study. Ovarian cancer SKOV3 and HEY cells, peritoneal mesothelial LP-9 cells, and hepatoma HepG2 cells were incubated with the indicated concentrations of CDDP (A), PTX (B), BEL (C), and FKGK11 (D) for 72 hours, and viability was assessed using the MTT assay as described in Materials and Methods. Absorbance was normalized to the control. Data are mean ± SD of triplicate determinations. E: Tumor sections from mice treated with vehicle, BEL, or PTX and processed for IHC detection of PCNA or stained with H&E. Original magnification, ×200.
Although none of these reagents has a highly selective power against tumor cells, the effective concentration ranges are different. We compared the concentration-dependent in vitro and in vivo effects of PTX, CDDP, BEL, and FKGK11. In vivo, PTX, CDDP, BEL, and FKGK11 were injected in a volume of 200 μL at concentrations of 585, 167, 60, and 2000 μmol/L (equivalent to approximately 5.0, 0.5, 0.19, and 5.6 mg/kg), respectively. To estimate the initial peritoneal concentrations of the drugs in vivo, we assumed that they would be diluted approximately 15-fold based on the total volume of a mouse peritoneal cavity (3 mL) divided by the original injection volume (0.2 mL); hence, i.p. concentrations of PTX, CDDP, BEL, FKGK11 were 39, 11, 4, and 66.7 μmol/L, respectively. At these in vivo concentrations, PTX and CDDP inhibited 90% to 100% of cell proliferation in vitro, whereas BEL and FKGK11 had no and approximately 30% proliferation inhibitory effects, respectively (Figure 3). We recognize that the estimated concentrations are not the actual and local concentrations of these drugs and that metabolism is not taken into consideration. However, applying the same rule to all of the reagents is a fair comparison and supports the notion that the major actions of BEL and FKGK11 at the concentrations used in vivo are not their cytotoxic effects. This was supported by the IHC study using the proliferation marker PCNA. Tumors from the control mice had the strongest PCNA staining (mean ± SD: 94% ± 9% cells were positive). Although BEL decreased the mean ± SD percentage of positively stained cells (73% ± 15%), it was not significantly different from that of the control group (P = 0.099) (Figure 3E). In contrast, PCNA-positive cells were significantly fewer in PTX-treated tumors (mean ± SD: 52% ± 12%; P = 0.03) (representative images are shown in Figure 3E).
BEL and FKGK11 Inhibit Adhesion, Migration, and Invasion of EOC Cells in Vitro, Critical Processes for the Formation of Tumors/Metastases in Vivo
We observed an intriguing phenomenon: similar to human EOC diseases, large numbers of floating tumor cells were present in ascites and/or peritoneal washings (not all mice developed ascites) at sacrifice from all vehicle- and BEL-treated mice injected with either SKOV3 or HEY cells (Table 1). We found that the floating tumor cells were alive and tumorigenic, as confirmed when we cultured them and reinjected them into mice (data not shown). A similar and complementary phenomenon was observed when we injected syngeneic mouse EOC cells (ID8) into iPLA2β−/− mice.18 Even in mice where no visible solid tumors were observed as we reported, large numbers of floating tumor cells were present in the peritoneal cavities. The floating ID8 cells were also viable and could form tumors when cultured and reinjected into wild-type mice. This phenomenon led us to hypothesize that the effect of BEL (or FKGK11) was primarily to prevent cell migration, followed by attachment of the floating cells and invasion to form tumors. This hypothesis is consistent with the observation that BEL particularly reduced the number of small tumors.
To support this idea further, we determined the ability of BEL and FKGK11 to reduce EOC cell adhesion, migration, and invasion in vitro. Cell adhesion to collagen I33,34 was dose dependently inhibited by BEL and FKGK11 (Figure 4A). As with their different potencies in inhibition of cell proliferation, FKGK11 was approximately 10 to 15 times less potent than BEL in inhibition of adhesion. At approximately 5 and 100 μmol/L, where cell proliferation was <20% inhibited, these compounds almost completely blocked cell migration (Figure 4B) and invasion through Matrigel (Figure 4C).
Figure 4.
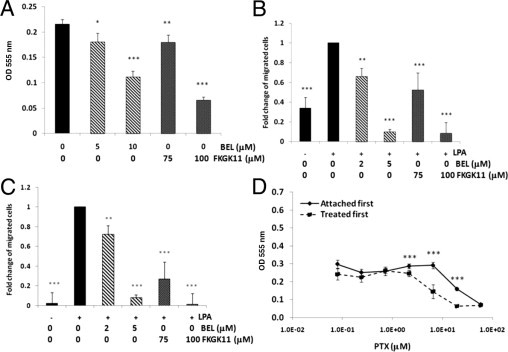
Effect of iPLA2 inhibitors on adhesion, migration, and invasion of SKOV3 cells. A: Adhesion to collagen I. Cells were treated with the indicated concentrations of drug for 30 minutes and then were tested for the ability to attach to 96-well plates coated with collagen I as described in Materials and Methods. Data are mean ± SD of at least three replicates. B: Haptotactic migration to collagen I. Cells were treated with the indicated concentrations of drug for 30 minutes and then were assayed for migration with LPA (1 μmol/L) as chemoattractant as described in Materials and Methods. C: Invasion through Matrigel. Cells were treated with the indicated concentrations of drug for 30 minutes and then were assayed for invasion with LPA (5 μmol/L) as chemoattractant as described in Materials and Methods. Data are mean ± SD of at least three experiments. D: SKOV3 cells in suspension are more sensitive to PTX compared with cells that are attached. SKOV3 cells were plated first and allowed to attach before treatment with PTX for 72 hours or were treated with PTX while suspended for 4 hours at 37°C and then counted and added to 96-well plates in the same media for 72 hours. MTT assay was performed as described in Materials and Methods, with at least three replicates per data point. The experiment was repeated twice with similar results. *P < 0.05, **P < 0.01, and ***P < 0.001.
In contrast to BEL-treated mice, mice treated with PTX alone or combined with BEL showed complete eradication of all floating tumor cells, suggesting that PTX is highly effective in killing unattached cells. The effect of BEL in preventing cell adhesion, migration, and/or invasion may, therefore, facilitate the action of PTX, making low-dose PTX more potent. We tested this concept in vitro by comparing the dose response to PTX of SKOV3 cells pretreated in suspension for 4 hours before plating with that of cells plated and attached for 4 hours before treatment. Viability was assessed after 72 hours by MTT assays (Figure 4D). Cells treated before plating realized the toxic effects of PTX at an approximately threefold lower concentration, consistent with greater vulnerability. Collectively, these data suggest that the complementary effects of BEL and PTX may represent a new and lower-toxicity therapeutic regimen for EOC.
Discussion
The data presented herein support the hypothesis that targeting iPLA2 with small molecule inhibitors, especially combined with low doses of current chemotherapeutic agents, is a promising new strategy against late-stage and metastatic EOC. Although many enzymes, including different PLA2s, ATX, and lipid phosphate phosphohydrolases, are involved in controlling LPA production and degradation, the ideal therapeutic target should be rate limiting and not cause excessive toxic effects. ATX, due to its direct role in LPA production, has been the major focus.13,35,36 However, ATX is involved in many normal physiologic processes, including blood vessel formation during development. ATX-deficient mice are embryonic lethal with profound vascular defects.24 Thus, targeting ATX requires overcoming the specific tumor cell targeting issues. In contrast, previous and current in vivo and in vitro works10–12,15–18 suggest that controlling iPLA2 may be effective and safe. LPA and several other bioactive lipids in the tumor environment of EOC are regulated by iPLA2β expressed in host and tumor cells.18 These effects can be directly or indirectly related to iPLA2 enzymatic activity, which has been substantially discussed previously.18 Although tumor-specific targeting is a major challenge for drug development, it may not be such a concern for iPLA2β because it is not an essential gene in adult female mice.23 Rather, the present data suggest that targeting both host and tumor cells is an advantage because iPLA2β in both cell types is functionally involved in EOC development.18 The inhibitors used may also target iPLA2γ and other new group GVI PLA2 members recently identified.29 However, the toxicity studies suggest that mice tolerate these inhibitors well. Hence, small molecule inhibition of iPLA2 without specific tumor cell targeting could be a promising option for reducing the oncogenic effects of bioactive lipids.
LPA is involved in many physiologic processes, but we found that iPLA2 deficiency did not affect LPA levels in mice, suggesting redundancy for normal physiologic functions. However, after tumor cells were injected, the local (in the peritoneum, but not in the blood) concentrations of LPA were increased in an iPLA2-dependent manner.18 These data suggest that iPLA2 is dispensable under normal physiologic conditions (the iPLA2 knockout female mice are grossly normal as reported by John Turk's group23) but is involved in tumor-induced LPA elevation in the tumor microenvironment. This is the foundational basis for the development of iPLA2-targeted therapy.
BEL has been used as a selective, potent, irreversible, mechanism-based inhibitor of iPLA2 since 1991.27 However, BEL was originally recognized as an inhibitor of chymotrypsin,37,38 and it also inhibits the lipid phosphatase phosphatidate phosphohydrolase 1.39 iPLA2 and chymotrypsin are inhibited by BEL with Ki values of 60 to 180 nmol/L and 635 nmol/L, respectively, suggesting that chymotrypsin may not be blocked at concentrations effective for inhibiting iPLA2. In addition, the similar biological effects of FKGK11, which is structurally and mechanistically different from BEL as we reported herein, support the notion that iPLA2 is the major target. Chymotrypsin is a serine protease and iPLA2 is a serine lipase.40 BEL inactivates these enzymes via these serine-dependent activities to generate a diffusible bromomethyl keto acid that alkylates cysteine thiols.40 Although FKGK11 is approximately 10 times less potent than BEL (Figure 4), it is presumably more specific for iPLA2. This is because FKGK11 is a product analogue of PLA2, which does not have the nonspecific cysteine inactivation mechanism.31 Although more detailed and in-depth studies are warranted to further address the targets of BEL and FKGK11 in vivo, it is important that toxic effects in blood and major organs were not detected, suggesting that the off-target effects, if any, are harmless or even beneficial. Moreover, although short hairpin RNA down-regulation–based iPLA2β studies (potentially more specific) support this target for EOC treatment,19 small molecules are more practical in the development of clinically useful therapies.
Another important advancement of this work is the observed additional effects of BEL with PTX, one of the most commonly used chemotherapeutic agents for treating patients with EOC. Although PTX was highly effective when used alone in the mouse models, it did not completely inhibit EOC development when low-toxicity doses were used (a higher dose caused liver toxic effects; see Supplemental Figure S3 at http://ajp.amjpathol.org. This is consistent with clinical experience, where most patients with EOC who undergo chemotherapy experience adverse effects with reduced quality of life.41 We show that in combination, BEL with low-dose PTX effectively inhibited tumor growth, providing a new direction for the development of novel therapeutics. The additional effects of BEL with CDDP were less optimal at the doses we used, which may be related to the lower effectiveness of CDDP in eradicating floating tumor cells (Table 1). Although we did not directly address the issue of drug resistance in this work, whether BEL and FKGK11 have the ability to overcome PTX and/or CDDP drug resistance warrants further testing in appropriate models.
The differential effects of BEL and PTX on floating/living tumor cells in the peritoneal cavity is the key observation allowing us to speculate on the mechanism underlying these effects. The present in vitro data (Figures 3 and 4) support the complementary actions of these drugs used together. The major in vivo effect of BEL is unlikely to be on cell proliferation directly based on the PCNA data (Figure 3E). In addition, BEL preferentially affected small tumor numbers, presumably related to its ability to interfere with cell attachment, because bigger tumors are probably derived from tumor cells attached before drug treatment. We previously conducted rather extensive signaling and mechanistic iPLA2 studies in human and mouse EOC cells, including using more specific genetic tools (such as small-interfering/short hairpin RNA approaches and iPLA2β−/− mice) and identifying signaling molecules, such as PI3K and p38 MAK, that are involved.11,16–18 Hence, these studies are not repeated here, and the present work focuses on preclinical evaluation of reagents for EOC treatment. In summary, these results provide a basis for development of new regimens for efficient treatment of late-stage EOC by interfering with the critical steps of cell adhesion/migration/invasion through inhibition of the iPLA2-LPA axis combined with a more traditional cytotoxic eradication of floating tumor cells.
Footnotes
This work was supported in part by NIH grant R01CA95042 and the Mary Fendrich Hulman Charitable Trust (Y.X.) and by NIH grant CA126937 (Z.-Y.Z.).
H.L., Z.Z., and C.A. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.03.027.
Current address of H.L. and G.W., Jilin Province Cancer Hospital, Changchun, People's Republic of China.
Supplementary data
All the reagents were purchased from Sigma-Aldrich (St. Louis, MO). 1H and 13C NMR spectra were obtained using a Bruker Avance II 500-MHz NMR spectrometer (Billerica, MA). Flash column chromatography was performed using 230- to 400 mesh silica gel (Dynamic Adsorbents Inc., Norcross, GA), with a mixture of ethyl acetate and hexane as the mobile phase. FKGK11 was synthesized as described,31 with slight modification. Under N2 protection, dimethyl formamide (50 μL) was added to a solution of 5-phenylvaleric acid (1.78 g, 10 mmol) in anhydrous CH2Cl2 (150 mL). Oxalylchloride (2.5 mL, 30 mmol) was added dropwise. The resulting solution was stirred at room temperature for 3 hours. CH2Cl2 and excess oxalylchloride were evaporated under vacuum using a Rotavapor R-210 (Buchi Corp., New Castle, DE). The residue was dissolved in dry CH2Cl2 (80 mL) under N2 protection. The solution was cooled to 0°C with an ice bath. Pentafluoropropionic acid anhydride (12 mL, 60 mmol) was added slowly at 0°C. Then pyridine (6.4 mL, 80 mmol) was added dropwise to this solution at 0°C under N2 protection. The resulting solution was stirred for 30 minutes at 0°C and then for 2 hours at room temperature. The reaction solution was cooled to 0°C again with an ice bath, and water (2 mL) was added dropwise to quench the reaction. After the addition of water, the reaction mixture was stirred for 30 minutes at 0°C and then for 30 minutes at room temperature. CH2Cl2 (200 mL) was added to the reaction mixture, and the organic phase was washed with brine (50 mL, three times). The organic phase was dried over Na2SO4 for 20 minutes. The solvent was evaporated under vacuum. The residual was purified by flash column with ethyl acetate-hexane (1:9) as the eluting phase, and pure FKGK11 (1.2 g, 43% yield) was obtained. The structure was confirmed by NMR. 1H NMR (CDCl3 with TMS as internal standard): d 7.33-7.14 (m, 5 H), 2.75 (2 H, t, J = 6.8 Hz), 2.64 (2 H, t, J = 7.3 Hz), 1.79-1.60 (4 H, m); 13C NMR (CDCl3): 194.2 (t, J = 26.5 Hz), 141.60, 128.4, 128.3, 126.0, 117.8 (qt, J = 286.4, J = 34.1 Hz), 106.9 (tq, J = 266.8 Hz, J = 38.1 Hz), 37.2, 35.5, 30.4, 21.9.
NOD/SCID mice were i.p. injected with SKOV3-luciferase cells. Two weeks after treatment initiation, mice were imaged for bioluminescence. Two representative mice from each treatment group are shown. Quantitated luciferase signal intensities are given in Supplemental Table S1.
A: Image of a liver from a mouse treated with 10 mg/kg of PTX. Arrows point to white spots on the surface of the liver. B: Tissue section of a liver stained with H&E. Arrows point to areas of leukocyte infiltration.
References
- 1.Jemal A., Siegel R., Xu J., Ward E. Cancer Statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Sengupta S., Wang Z., Tipps R., Xu Y. Biology of LPA in health and disease. Semin Cell Dev Biol. 2004;15:503–512. doi: 10.1016/j.semcdb.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Xu Y., Wang D., Wang Z. Lipid generation and signaling in ovarian cancer. Cancer Treat Res. 2009;149:241–267. doi: 10.1007/978-0-387-98094-2_12. [DOI] [PubMed] [Google Scholar]
- 4.Xu Y., Sengupta S., Singh S., Steinmetz R. Novel lipid signaling pathways in ovarian cancer cells. Cell Sci Rev. 2006;3:168–197. [Google Scholar]
- 5.Mills G.B., Moolenaar W.H. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 6.Do T.V., Symowicz J.C., Berman D.M., Liotta L.A., Petricoin E.F., III, Stack M.S., Fishman D.A. Lysophosphatidic acid down-regulates stress fibers and up-regulates pro-matrix metalloproteinase-2 activation in ovarian cancer cells. Mol Cancer Res. 2007;5:121–131. doi: 10.1158/1541-7786.MCR-06-0319. [DOI] [PubMed] [Google Scholar]
- 7.Fishman D.A., Liu Y., Ellerbroek S.M., Stack M.S. Lysophosphatidic acid promotes matrix metalloproteinase (MMP) activation and MMP-dependent invasion in ovarian cancer cells. Cancer Res. 2001;61:3194–3199. [PubMed] [Google Scholar]
- 8.Mills G.B., Eder A., Fang X., Hasegawa Y., Mao M., Lu Y., Tanyi J., Tabassam F.H., Wiener J., Lapushin R., Yu S., Parrott J.A., Compton T., Tribley W., Fishman D., Stack M.S., Gaudette D., Jaffe R., Furui T., Aoki J., Erickson J.R. Critical role of lysophospholipids in the pathophysiology, diagnosis, and management of ovarian cancer. Cancer Treat Res. 2002;107:259–283. doi: 10.1007/978-1-4757-3587-1_12. [DOI] [PubMed] [Google Scholar]
- 9.Symowicz J., Adley B.P., Woo M.M., Auersperg N., Hudson L.G., Stack M.S. Cyclooxygenase-2 functions as a downstream mediator of lysophosphatidic acid to promote aggressive behavior in ovarian carcinoma cells. Cancer Res. 2005;65:2234–2242. doi: 10.1158/0008.5472.CAN-04-2781. [DOI] [PubMed] [Google Scholar]
- 10.Kim K.S., Sengupta S., Berk M., Kwak Y.G., Escobar P.F., Belinson J., Mok S.C., Xu Y. Hypoxia enhances lysophosphatidic acid responsiveness in ovarian cancer cells and lysophosphatidic acid induces ovarian tumor metastasis in vivo. Cancer Res. 2006;66:7983–7990. doi: 10.1158/0008-5472.CAN-05-4381. [DOI] [PubMed] [Google Scholar]
- 11.Sengupta S., Kim K.S., Berk M.P., Oates R., Escobar P., Belinson J., Li W., Lindner D.J., Williams B., Xu Y. Lysophosphatidic acid downregulates tissue inhibitor of metalloproteinases, which are negatively involved in lysophosphatidic acid-induced cell invasion. Oncogene. 2007;26:2894–2901. doi: 10.1038/sj.onc.1210093. [DOI] [PubMed] [Google Scholar]
- 12.Li H., Wang D., Zhang H., Kirmani K., Zhao Z., Steinmetz R., Xu Y. Lysophosphatidic acid stimulates cell migration, invasion, and colony formation as well as tumorigenesis/metastasis of mouse ovarian cancer in immunocompetent mice. Mol Cancer Ther. 2009;8:1692–1701. doi: 10.1158/1535-7163.MCT-08-1106. [DOI] [PubMed] [Google Scholar]
- 13.Aoki J., Inoue A., Okudaira S. Two pathways for lysophosphatidic acid production. Biochim Biophys Acta. 2008;1781:513–518. doi: 10.1016/j.bbalip.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Okudaira S., Yukiura H., Aoki J. Biological roles of lysophosphatidic acid signaling through its production by autotaxin. Biochimie. 2010;92:698–706. doi: 10.1016/j.biochi.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 15.Zhao X., Wang D., Zhao Z., Xiao Y., Sengupta S., Xiao Y., Zhang R., Lauber K., Wesselborg S., Feng L., Rose T.M., Shen Y., Zhang J., Prestwich G., Xu Y. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J Biol Chem. 2006;281:29357–29368. doi: 10.1074/jbc.M513105200. [DOI] [PubMed] [Google Scholar]
- 16.Ren J., Xiao Y.J., Singh L.S., Zhao X., Zhao Z., Feng L., Rose T.M., Prestwich G.D., Xu Y. Lysophosphatidic acid is constitutively produced by human peritoneal mesothelial cells and enhances adhesion, migration, and invasion of ovarian cancer cells. Cancer Res. 2006;66:3006–3014. doi: 10.1158/0008-5472.CAN-05-1292. [DOI] [PubMed] [Google Scholar]
- 17.Sengupta S., Xiao Y.J., Xu Y. A novel laminin-induced LPA autocrine loop in the migration of ovarian cancer cells. FASEB J. 2003;17:1570–1572. doi: 10.1096/fj.02-1145fje. [DOI] [PubMed] [Google Scholar]
- 18.Li H., Zhao Z., Wei G., Yan L., Wang D., Zhang H., Sandusky G.E., Turk J., Xu Y. Group VIA phospholipase A2 in both host and tumor cells is involved in ovarian cancer development. FASEB J. 2010;24:4103–4116. doi: 10.1096/fj.10-161356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song Y., Wilkins P., Hu W., Murthy K.S., Chen J., Lee Z., Oyesanya R., Wu J., Barbour S.E., Fang X. Inhibition of calcium-independent phospholipase A2 suppresses proliferation and tumorigenicity of ovarian carcinoma cells. Biochem J. 2007;406:427–436. doi: 10.1042/BJ20070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steeg P.S., Theodorescu D. Metastasis: a therapeutic target for cancer. Nat Clin Pract Oncol. 2008;5:206–219. doi: 10.1038/ncponc1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kenny H.A., Krausz T., Yamada S.D., Lengyel E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int J Cancer. 2007;121:1463–1472. doi: 10.1002/ijc.22874. [DOI] [PubMed] [Google Scholar]
- 22.Jatoi A., Podratz K.C., Gill P., Hartmann L.C. Pathophysiology and palliation of inoperable bowel obstruction in patients with ovarian cancer. J Support Oncol. 2004;2:323–327. [PubMed] [Google Scholar]
- 23.Bao S., Miller D.J., Ma Z., Wohltmann M., Eng G., Ramanadham S., Moley K., Turk J. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Meeteren L.A., Ruurs P., Stortelers C., Bouwman P., van Rooijen M.A., Pradere J.P., Pettit T.R., Wakelam M.J., Saulnier-Blache J.S., Mummery C.L., Moolenaar W.H., Jonkers J. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol. 2006;26:5015–5022. doi: 10.1128/MCB.02419-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y., Fang X.J., Casey G., Mills G.B. Lysophospholipids activate ovarian and breast cancer cells. Biochem J. 1995;309(pt 3):933–940. doi: 10.1042/bj3090933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Y., Gaudette D.C., Boynton J.D., Frankel A., Fang X.J., Sharma A., Hurteau J., Casey G., Goodbody A., Mellors A. Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin Cancer Res. 1995;1:1223–1232. [PubMed] [Google Scholar]
- 27.Hazen S.L., Zupan L.A., Weiss R.H., Getman D.P., Gross R.W. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2: mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 28.Yeo J.F., Ong W.Y., Ling S.F., Farooqui A.A. Intracerebroventricular injection of phospholipases A2 inhibitors modulates allodynia after facial carrageenan injection in mice. Pain. 2004;112:148–155. doi: 10.1016/j.pain.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 29.Burke J.E., Dennis E.A. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009;50(Suppl):S237–S242. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez-Vales R., Navarro X., Shimizu T., Baskakis C., Kokotos G., Constantinou-Kokotou V., Stephens D., Dennis E.A., David S. Intracellular phospholipase A(2) group IVA and group VIA play important roles in Wallerian degeneration and axon regeneration after peripheral nerve injury. Brain. 2008;131:2620–2631. doi: 10.1093/brain/awn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baskakis C., Magrioti V., Cotton N., Stephens D., Constantinou-Kokotou V., Dennis E.A., Kokotos G. Synthesis of polyfluoro ketones for selective inhibition of human phospholipase A2 enzymes. J Med Chem. 2008;51:8027–8037. doi: 10.1021/jm800649q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalyvas A., Baskakis C., Magrioti V., Constantinou-Kokotou V., Stephens D., Lopez-Vales R., Lu J.Q., Yong V.W., Dennis E.A., Kokotos G., David S. Differing roles for members of the phospholipase A2 superfamily in experimental autoimmune encephalomyelitis. Brain. 2009;132:1221–1235. doi: 10.1093/brain/awp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sodek K.L., Brown T.J., Ringuette M.J. Collagen I but not Matrigel matrices provide an MMP-dependent barrier to ovarian cancer cell penetration. BMC Cancer. 2008;8:223. doi: 10.1186/1471-2407-8-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fishman D.A., Kearns A., Chilukuri K., Bafetti L.M., O'Toole E.A., Georgacopoulos J., Ravosa M.J., Stack M.S. Metastatic dissemination of human ovarian epithelial carcinoma is promoted by α2β1-integrin-mediated interaction with type I collagen. Invasion Metastasis. 1998;18:15–26. doi: 10.1159/000024495. [DOI] [PubMed] [Google Scholar]
- 35.Tokumura A., Majima E., Kariya Y., Tominaga K., Kogure K., Yasuda K., Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem. 2002;277:39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 36.Umezu-Goto M., Kishi Y., Taira A., Hama K., Dohmae N., Takio K., Yamori T., Mills G.B., Inoue K., Aoki J., Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daniels S.B., Cooney E., Sofia M.J., Chakravarty P.K., Katzenellenbogen J.A. Haloenol lactones: potent enzyme-activated irreversible inhibitors for α-chymotrypsin. J Biol Chem. 1983;258:15046–15053. [PubMed] [Google Scholar]
- 38.Daniels S.B., Katzenellenbogen J.A. Halo enol lactones: studies on the mechanism of inactivation of α-chymotrypsin. Biochemistry. 1986;25:1436–1444. doi: 10.1021/bi00354a037. [DOI] [PubMed] [Google Scholar]
- 39.Balsinde J., Dennis E.A. Bromoenol lactone inhibits magnesium-dependent phosphatidate phosphohydrolase and blocks triacylglycerol biosynthesis in mouse P388D1 macrophages. J Biol Chem. 1996;271:31937–31941. doi: 10.1074/jbc.271.50.31937. [DOI] [PubMed] [Google Scholar]
- 40.Song H., Ramanadham S., Bao S., Hsu F.F., Turk J. A bromoenol lactone suicide substrate inactivates group VIA phospholipase A2 by generating a diffusible bromomethyl keto acid that alkylates cysteine thiols. Biochemistry. 2006;45:1061–1073. doi: 10.1021/bi052065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McWhinney S.R., Goldberg R.M., McLeod H.L. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009;8:10–16. doi: 10.1158/1535-7163.MCT-08-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All the reagents were purchased from Sigma-Aldrich (St. Louis, MO). 1H and 13C NMR spectra were obtained using a Bruker Avance II 500-MHz NMR spectrometer (Billerica, MA). Flash column chromatography was performed using 230- to 400 mesh silica gel (Dynamic Adsorbents Inc., Norcross, GA), with a mixture of ethyl acetate and hexane as the mobile phase. FKGK11 was synthesized as described,31 with slight modification. Under N2 protection, dimethyl formamide (50 μL) was added to a solution of 5-phenylvaleric acid (1.78 g, 10 mmol) in anhydrous CH2Cl2 (150 mL). Oxalylchloride (2.5 mL, 30 mmol) was added dropwise. The resulting solution was stirred at room temperature for 3 hours. CH2Cl2 and excess oxalylchloride were evaporated under vacuum using a Rotavapor R-210 (Buchi Corp., New Castle, DE). The residue was dissolved in dry CH2Cl2 (80 mL) under N2 protection. The solution was cooled to 0°C with an ice bath. Pentafluoropropionic acid anhydride (12 mL, 60 mmol) was added slowly at 0°C. Then pyridine (6.4 mL, 80 mmol) was added dropwise to this solution at 0°C under N2 protection. The resulting solution was stirred for 30 minutes at 0°C and then for 2 hours at room temperature. The reaction solution was cooled to 0°C again with an ice bath, and water (2 mL) was added dropwise to quench the reaction. After the addition of water, the reaction mixture was stirred for 30 minutes at 0°C and then for 30 minutes at room temperature. CH2Cl2 (200 mL) was added to the reaction mixture, and the organic phase was washed with brine (50 mL, three times). The organic phase was dried over Na2SO4 for 20 minutes. The solvent was evaporated under vacuum. The residual was purified by flash column with ethyl acetate-hexane (1:9) as the eluting phase, and pure FKGK11 (1.2 g, 43% yield) was obtained. The structure was confirmed by NMR. 1H NMR (CDCl3 with TMS as internal standard): d 7.33-7.14 (m, 5 H), 2.75 (2 H, t, J = 6.8 Hz), 2.64 (2 H, t, J = 7.3 Hz), 1.79-1.60 (4 H, m); 13C NMR (CDCl3): 194.2 (t, J = 26.5 Hz), 141.60, 128.4, 128.3, 126.0, 117.8 (qt, J = 286.4, J = 34.1 Hz), 106.9 (tq, J = 266.8 Hz, J = 38.1 Hz), 37.2, 35.5, 30.4, 21.9.
NOD/SCID mice were i.p. injected with SKOV3-luciferase cells. Two weeks after treatment initiation, mice were imaged for bioluminescence. Two representative mice from each treatment group are shown. Quantitated luciferase signal intensities are given in Supplemental Table S1.
A: Image of a liver from a mouse treated with 10 mg/kg of PTX. Arrows point to white spots on the surface of the liver. B: Tissue section of a liver stained with H&E. Arrows point to areas of leukocyte infiltration.