

Abstract
Due to alternative splicing events in eukaryotic species, the identification of mRNA isoforms (or splicing variants) is a difficult problem. Traditional experimental methods for this purpose are time consuming and cost ineffective. The emerging RNA-Seq technology provides a possible effective method to address this problem. Although the advantages of RNA-Seq over traditional methods in transcriptome analysis have been confirmed by many studies, the inference of isoforms from millions of short sequence reads (e.g., Illumina/Solexa reads) has remained computationally challenging. In this work, we propose a method to calculate the expression levels of isoforms and infer isoforms from short RNA-Seq reads using exon-intron boundary, transcription start site (TSS) and poly-A site (PAS) information. We first formulate the relationship among exons, isoforms, and single-end reads as a convex quadratic program, and then use an efficient algorithm (called IsoInfer) to search for isoforms. IsoInfer can calculate the expression levels of isoforms accurately if all the isoforms are known and infer novel isoforms from scratch. Our experimental tests on known mouse isoforms with both simulated expression levels and reads demonstrate that IsoInfer is able to calculate the expression levels of isoforms with an accuracy comparable to the state-of-the-art statistical method and a 60 times faster speed. Moreover, our tests on both simulated and real reads show that it achieves a good precision and sensitivity in inferring isoforms when given accurate exon-intron boundary, TSS, and PAS information, especially for isoforms whose expression levels are significantly high. The software is publicly available for free at http://www.cs.ucr.edu/∼jianxing/IsoInfer.html.
Key words: alternative splicing, convex quadratic programming, deep sequencing, isoform inference, RNA-Seq
1. Introduction
Transcriptome study (or transcriptomics) aims to discover all the transcripts and their quantities in a cell or an organism under different external environmental conditions. A large amount of work has been devoted to transcriptomics, which includes the international projects EST (Boguski et al., 1994; Boguski, 1995), FANTOM (The FANTOM Consortium, 2005), and ENCODE (The ENCODE Project Consortium, 2007; Weinstock, 2007). Many technologies have been introduced in recent years, including array-based experimental methods such as tiling arrays (Bertone et al., 2004), exon arrays (Kwan et al., 2008), and exon-junction arrays (Johnson et al., 2003; Kapranov et al., 2007); and tag-based approaches such as MPSS (Brenner et al., 2000; Reinartz et al., 2002), SAGE (Velculescu et al., 1995; Harbers and Carninci, 2005), CAGE (Shiraki et al., 2003; Kodzius et al., 2005), PMAGE (Kim et al., 2007), and GIS (Ng et al., 2005). However, due to various constraints intrinsic to these technologies, the speed of advance in transcriptomics is far from being satisfactory, especially on eukaryotic species because of widespread alternative splicing events.
Applying next generation sequencing technologies to transcriptomes, the recently developed RNA-Seq technology is quickly becoming an important tool in functional genomics and transcriptomics. It can be used to identify all genes and exons and their boundaries (Nagalakshmi et al., 2008; Trapnell et al., 2009), and to study gene functions and perform transcriptome analysis (Graveley, 2008). For example, based on an unannotated genomic sequence and millions of short reads from RNA-Seq, Yassour et al. (2009) developed a general method for the discovery of a complete transcriptome, including the identification of coding regions, ends of transcripts, splice junctions, and splice site variations. Their application of the method to S.cerevisiae (yeast) showed a high degree of agreement with the existing knowledge of the yeast transcriptome. Besides yeast (Wilhelm et al., 2008; Nagalakshmi et al., 2008), RNA-Seq has been applied to the transcriptome analysis of mouse (Cloonan et al., 2008; Mortazavi et al., 2008) and human (Marioni et al., 2008; Sultan et al., 2008). These results demonstrate that RNA-Seq is a powerful quantitative method to sample a transcriptome deeply at an unprecedented resolution. Moreover, DNA sequencing technologies are under fast development. Some of them now could provide, for example, long reads, paired-end reads, and DNA-strand-sequencing of mRNA transcripts. For a comprehensive analysis of the advantages of RNA-Seq over traditional methods in genome-wide transcriptome analysis and the challenges faced by this technology, see Wang et al. (2008b).
Very recently, several methods have been proposed to characterize the expression level of each transcript (Lacroix et al., 2008; Jiang and Wong, 2009) using RNA-Seq data. In Lacroix et al. (2008), the authors showed that short (single-end or paired-end) read sequences cannot theoretically guarantee a unique solution to the transcriptome reconstruction problem (i.e., the reconstruction of all expressed isoforms and their expression levels) in general even if the reads are sampled perfectly according to the length of each transcript (without random distortions and noise). However, under the same assumption, the authors also showed that paired-end reads could help reconstruct the transcripts uniquely and determine their expression levels for most of the currently known isoforms of human, and single-end reads could allow us to determine the expression levels correctly if all the isoforms are known. However, these results are mostly of theoretical interest because sequence read data are random in nature and may contain noise in practice. Jiang and Wong (2009) presented a more practical way to estimate the expression levels of known isoforms. The method uses maximum likelihood estimation followed by importance sampling from the posterior distribution.
The availability of all the isoforms is the basis of accurate estimation of isoform expression levels (Jiang and Wong, 2009), which could then be used to calculate all the splicing variants quantitatively and qualitatively. Variations in isoform expression levels and splicing junctions could provide useful insight in many studies such as the study of diseases (Pagani and Baralle, 2004; Srebrow and Kornblihtt, 2006) and drug development (Williams, 2005).
A lot of effort has been devoted to the identification of transcripts/isoforms from the more traditional EST, cDNA data. Instead of a comprehensive review, we will just name a few results below. To enumerate all possible isoforms, a core ingredient is the splicing graph (Heber et al., 2002; Sammeth et al., 2008a). A predetermined parameter “dimension” decides how many transcripts are compared simultaneously. The parameter is usually fixed to two, but recently, Sammeth et al. (2008a) extended the method to arbitrary dimensions. There are several methods that assemble transcripts from EST data using the splicing graph and its variations (Xing et al., 2004; Bonizzoni et al., 2009). Newly proposed experimental methods in Djebali et al. (2008) and Salehi-Ashtiani et al. (2008) could be used to identify new isoforms. However, it is still unclear whether these methods can be applied in a large scale.
RNA-Seq has shown great success in transcriptome analysis, but it has not been used to infer isoforms. Although it is straightforward to infer the existence of novel isoforms from RNA-Seq data that exhibit novel transcribed regions (Mortazavi et al., 2008; Bertone et al., 2004), it is not so obvious how to use RNA-Seq data to infer the existence of novel isoforms in known transcribed regions, because the observed reads could be sampled from either known or unknown isoforms. The problem has remained challenging for two reasons. The first is that RNA-Seq reads are usually very short. The second is due to the randomness and biases of the reads sampled from all the transcripts. In fact, to our best knowledge, there has been no published work to computationally infer isoforms from (realistic) short RNA-Seq reads.
Due to the high combinatorial complexity of isoforms of genes with a (moderately) large number of exons, the inference of isoforms from short reads (and other available biological information) should be realistically divided into two sub-problems. The first is to discover all the exon-intron boundaries as well as the transcription start site (TSS) and poly-A site (PAS) of each isoform. As mentioned above, there are several effective methods for detecting exon-intron boundaries from RNA-Seq read data (Nagalakshmi et al., 2008; Trapnell et al., 2009). The identification of TSSs and PASs is an indispensable part of many large genomics projects such as The FANTOM Consortium (2005) and The ENCODE Project Consortium (2007). The technology of GIS-PET (Gene Identification Signature/Paired-End Tags) can also be used to identify TSS-PAS pairs (Ng et al., 2005; Fullwood et al., 2009). The second sub-problem is to find combinations of exons that can properly explain the RNA-Seq data, given the exon-intron boundary and TSS-PAS pair information.
In this paper, we are concerned with the second sub-problem in isoform inference. Assuming that the exon-intron boundary and TSS-PAS pair information is given, we propose a method (called IsoInfer) to infer isoforms from short RNA-Seq reads (e.g., Illumina/Solexa data). Although our method works for single-end data and data with both single-end and paired-end reads, we will use single-end reads as the primary source of data and paired-end reads as a secondary data which can be used to filter out false positives. We formulate the relationship among exons, isoforms, and single-end reads as a convex quadratic program, and design an efficient algorithm to search for isoforms. Our method can calculate the expression levels of isoforms accurately if all the isoforms are known. To demonstrate this, we have compared IsoInfer with the simple counting method in Pan et al. (2008) and Wang et al. (2008a) and the method in Jiang and Wong (2009) on simulated expression levels and reads, and found that our method is much more accurate than the simple counting method and has a comparable accuracy as the method in Jiang and Wong (2009) but is 60 times faster. Most importantly, IsoInfer can infer isoforms from scratch when they are sufficiently expressed, by trying to find a minimum set of isoforms to explain the read data. Our experimental tests on both simulated and real reads show that it is possible to infer the precise combination of exons in a sufficiently expressed isoform from RNA-Seq short read data with a reasonably good accuracy, when accurate exon-intron boundary and TSS-PAS pair information is provided. To our best knowledge, this is the first computational method to infer isoforms from short RNA-Seq reads.
2. Methods
2.1. Assumptions and terminology
Traditionally, only five types of alternating splicing (AS) events have been proposed, including exon skipping, mutually exclusive exons, intron retention, and alternative donor and acceptor sites (Breitbart et al., 1987). However, these events are not adequate to describe complex AS events as more experimental knowledge has become available (Sammeth et al., 2008b). In this work, we describe isoforms or AS events in a much general way, which is referred to as a “bit matrix” in Sammeth et al. (2008b).
The exon-intron boundaries of a gene divide the gene into disjoint segments, as shown in Figure 1. A segment is expressed if it has mapped reads. Thus, every expressed isoform consists of a subset of expressed segments. Two segments are adjacent if they are adjacent in the reference genome (i.e., they share a common boundary). For example, in Figure 1, s2 and s3 are adjacent but s1 and s2 are not. Any two segments may form a segment junction which is not necessarily an exon junction in the traditional sense. For example, s2 and s3 form a segment junction, which is not an exon junction. In the following, “junction” refers to “segment junction” unless otherwise stated.
FIG. 1.

Expressed segments. Every exon-intron boundary introduces a boundary of some segment. Every expressed segment is a part of an exon.
As stated in the introduction, we first assume that exon-intron boundaries are known. Our second assumption is that the short reads are uniformly randomly sampled from all the expressed isoforms (i.e., mRNA transcripts). We have to further assume that the short reads have been mapped to the referenced genome. The mapping of RNA-Seq reads can be done by many recent tools, for example, Bowtie (Langmead et al., 2009), Maq (Li et al., 2008a), SOAP (Li et al., 2008b), RNA-MATE (Cloonan et al., 2009), and mrFAST (Alkan et al., 2009). The mapping of multi-reads (i.e., reads that match several locations of the reference genome) is addressed in Mortazavi et al. (2008) and Hashimoto et al. (2009). We will use Bowtie in our work due to its efficiency and accuracy. The last assumption concerns paired-end reads, which will be discussed in Section 2.3.
2.2. Quadratic programming formulation
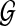 denotes the set of all the genes. Each g gene defines a set of expressed segments
denotes the set of all the genes. Each g gene defines a set of expressed segments 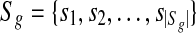 (given exon-intron boundaries), where the expressed segments are sorted according to their positions in the reference genome. The junctions on this gene are all the pairs of expressed segments (si,sj), 1 ≤ i < j ≤ |Sg|. The length of segment si is li. Denote the set of all known isoforms of this gene as Fg. Each isoform
(given exon-intron boundaries), where the expressed segments are sorted according to their positions in the reference genome. The junctions on this gene are all the pairs of expressed segments (si,sj), 1 ≤ i < j ≤ |Sg|. The length of segment si is li. Denote the set of all known isoforms of this gene as Fg. Each isoform 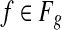 consists of a subset of expressed segments. The expression level (i.e., the number of reads per base) of isoform f is denoted by xf. The sum of the length of all transcripts, weighted by their expression levels, over all genes, is
consists of a subset of expressed segments. The expression level (i.e., the number of reads per base) of isoform f is denoted by xf. The sum of the length of all transcripts, weighted by their expression levels, over all genes, is 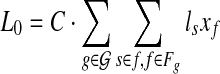 , for some constant C that defines the linear relationship between the expression level and the number of transcripts corresponding to an isoform. C can be inferred from data as shown in Mortazavi et al. (2008).
, for some constant C that defines the linear relationship between the expression level and the number of transcripts corresponding to an isoform. C can be inferred from data as shown in Mortazavi et al. (2008).
From now on, we will consider a fixed gene g and omit the subscript g when there is no ambiguity. Let M be the total number of single-end reads mapped to the reference genome and di the number of reads falling into expressed segment si. Under the uniform sampling assumption, di is the observed value of a random variable (denoted as ri) that follows the binomial distribution B(M,pi), where pi = Cyili/L0 and 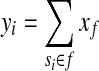 . Because M is usually very large, pi is very small and Mpi is sufficiently large in most cases, the binomial distribution can be approximated by a normal distribution
. Because M is usually very large, pi is very small and Mpi is sufficiently large in most cases, the binomial distribution can be approximated by a normal distribution 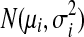 , with
, with 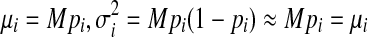 , similar to the approximation in Jiang and Wong (2009). Therefore, the random variables
, similar to the approximation in Jiang and Wong (2009). Therefore, the random variables 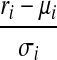 , for every expressed segment si, follow the same distribution approximately. Define
, for every expressed segment si, follow the same distribution approximately. Define 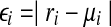 . Then, the variable
. Then, the variable 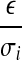 also follows the same distribution approximately for every si.
also follows the same distribution approximately for every si.
Let L1 denote the length of a single-end read. In order to map reads to junctions, we will also think of each junction (si,sj) as a segment of length 2L1 − 2, consisting of the last L1 − 1 bases of si and the first L1 − 1 bases of sj. Denote the set of the junctions as 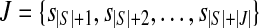 . The relationship among the expressed segments of gene g, its expressed isoforms, and the single-end reads mapped to each expressed segment and junction can be captured by the following quadratic program (QP):
. The relationship among the expressed segments of gene g, its expressed isoforms, and the single-end reads mapped to each expressed segment and junction can be captured by the following quadratic program (QP):
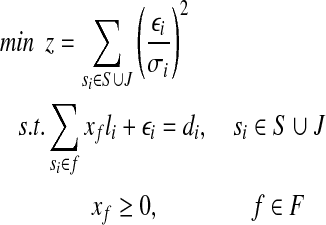 |
where σi is the standard deviation in the normal distribution 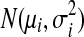 and will be empirically estimated from di.
and will be empirically estimated from di.
Note that if each ri follows the normal distribution strictly, then the random variables 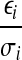 is i.i.d. and thus the solution of the above QP would correspond to the maximum likelihood estimation of the xf's if each σi is fixed (Bishop, 2007), and the objective function z is a random variable obeying the χ2 distribution with freedom |S| + |J|. This QP can be easily shown to be a convex QP by a simple transformation and solved in polynomial time by a public program QuadProg++ which implements the dual method of Goldfarb and Idnani (Goldfarb, 1983) for convex quadratic programming. Since σi is unknown, we use the empirical estimate of σi,
is i.i.d. and thus the solution of the above QP would correspond to the maximum likelihood estimation of the xf's if each σi is fixed (Bishop, 2007), and the objective function z is a random variable obeying the χ2 distribution with freedom |S| + |J|. This QP can be easily shown to be a convex QP by a simple transformation and solved in polynomial time by a public program QuadProg++ which implements the dual method of Goldfarb and Idnani (Goldfarb, 1983) for convex quadratic programming. Since σi is unknown, we use the empirical estimate of σi, 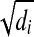 , for σi as an approximation. Let QPsolver denote the above algorithm for solving the convex QP program. Given S,F, and di's, QPsolver returns the values of xi's and z.
, for σi as an approximation. Let QPsolver denote the above algorithm for solving the convex QP program. Given S,F, and di's, QPsolver returns the values of xi's and z.
When the isoforms in F are given, minimizing the objective function means to find a combination of the expression level (xf) of each isoform in F such that the observed values (di's) can be explained the best. In this case, the value of the objective function serves as an indicator of whether the isoforms in F can explain the observed data. More specifically, p-value(z) denotes the probability of P(Z ≥ z), where Z is a random variable following the χ2 distribution with freedom |S| + |J|. We empirically choose a cutoff of 0.05. If p-value(z) is less than 0.05 we conclude that F cannot explain d.
2.3. Paired-end reads
Figure 2 (left) illustrates some concepts concerning paired-end reads. A paired-end read consists of a pair of short (single-end) reads separated by a gap. The figure also defines the read length, span, start position, center position and end position of a paired-end read. If the span of a paired-end read is a random variable following some probability distribution h(x), then three possible strategies for generating paired-end reads will be considered in this article:
Strategy (a): The start position of a paired-end read is uniformly and randomly sampled from all the expressed isoforms. Then the span of this paired-end is generated following the distribution h(x). If the end position of this paired-end read falls out of the isoform, the paired-end read is truncated such that the end position of this read is at the end of the isoform.
Strategy (b): The center position of a paired-end read is uniformly and randomly sampled from all the expressed isoforms. Then the span of this paired-end is generated following the distribution h(x). This strategy was used in Korbel et al. (2009). Again, if the start (or end) position of this paired-end read falls out of the isoform, the paired-end read is truncated such that the start (or end, respectively) position of this read is at the start (or end, respectively) of the isoform.
Strategy (c): The end position of a paired-end read is uniformly and randomly sampled from all the expressed isoforms. Then the span of this paired-end is generated following the distribution h(x). If the start position of this paired-end read falls out of the isoform, the paired-end read is truncated such that the start position of this read is at the start of the isoform.
FIG. 2.

(Left) A paired-end read consisting of two short reads of length L2 that are separated by a gap. (Right) Three consecutive intervals on an isoform.
Let w1,w2,w3 be the lengths of three consecutive intervals on an isoform as shown in Figure 2 (right). When any of the strategies (a–c) is applied to generate a certain number of paired-end reads, the following Theorem 1 gives a non-trivial upper bound on the probability of not observing any reads with start positions in the first interval and end positions in the third interval.
Theorem 1.
Suppose that the expression level of this isoform is α RPKM (i.e., reads per kilobase of exon model per million mapped reads) (Mortazavi et al., 2008), and the span of each paired-end read follows some distribution h(x). If M paired-end reads are generated by any of the strategies (a–c), the probability that there are no paired-end reads that have start positions in the first interval and end positions in the third interval is upper bounded by
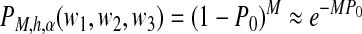 |
where 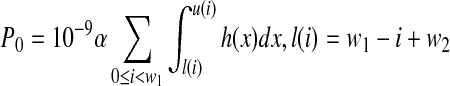 , and u(i) = w1 − i + w2 + w3.
, and u(i) = w1 − i + w2 + w3.
Proof.
For simplicity, we assume that the distributions involved in the following proof are discrete. Let q(x) be the probability that the span of a randomly generated paired-end read is x, and p(x) the probability of a uniformly randomly selected position from all isoforms being at position x on the given isoform. Every paired-end read can be represented by its start (or center or end) position and span uniquely. Denote the set of all possible start positions as Ψ and the set of all possible spans as Ω. Let V ⊂ Ψ × Ω defines the set of paired-end reads that have start positions in the first interval and end positions in the third interval. Under strategy (a), the probability of a uniformly randomly generated paired-end read being in V is:
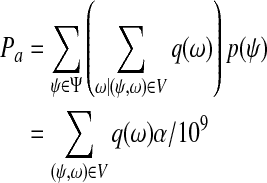 |
Similarly, we define the set of possible center positions of paired-end reads as Ψ′. Let V′ ⊂ Ψ′ × Ω define the set of paired end reads that have start positions in the first interval and end positions in the third interval. Under strategy (b), the probability of a uniformly randomly generated paired-end read being in V′ is:
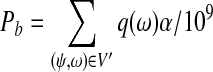 |
Because 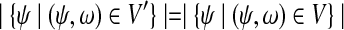 for
for 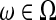 , we have Pa = Pb. The argument is also applicable to case when strategy (c) is applied.
, we have Pa = Pb. The argument is also applicable to case when strategy (c) is applied.
When strategy (a) is applied and the end position of the third interval is not the end position of the given isoform, if the start position of a uniformly randomly generated paired-end read is i, 0 ≤ i < w1 in the first interval, then the probability of the end position of this paired-end read being in the third interval is
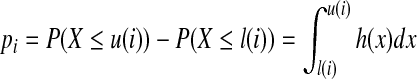 |
where l(i) = w1 − i + w2, u(i) = w1 − i + w2 + w3. When the end position of the third interval is the end position of the given isoform and strategy (a) is applied, we have
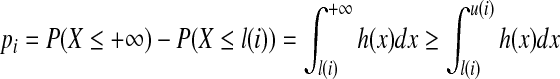 |
Because the start position of a paired-end read is uniformly randomly selected,
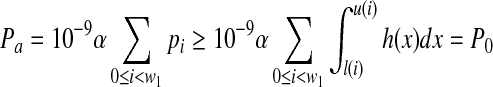 |
Because M paired-end reads are generated, the probability that none of the reads have start positions in the first interval and end positions in the third interval is 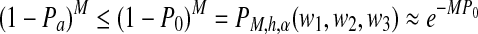 .
.
Similar arguments hold when strategies (b) and (c) are applied to generate the reads. ▪
2.4. Valid isoforms
For a gene with expressed segments 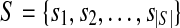 , an isoform f of this gene can be expressed as a binary vector with length |S|. The ith element f[i] of f is 1 if and only if expressed segment si is contained in f. Denote the set of all possible binary vectors with n elements as B(n). Similarly, a single-end or paired-end short read that is mapped to a subset S′ ⊆ S of expressed segments can be represented as a binary vector
, an isoform f of this gene can be expressed as a binary vector with length |S|. The ith element f[i] of f is 1 if and only if expressed segment si is contained in f. Denote the set of all possible binary vectors with n elements as B(n). Similarly, a single-end or paired-end short read that is mapped to a subset S′ ⊆ S of expressed segments can be represented as a binary vector 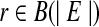 such that r[i] = 1 if and only if
such that r[i] = 1 if and only if 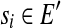 . A subset E′ of expressed segments is supported by single-end or paired-end reads if there is at least one single-end or paired-end read r such that
. A subset E′ of expressed segments is supported by single-end or paired-end reads if there is at least one single-end or paired-end read r such that 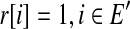 .
.
Although single-end reads, paired-end reads, and TSS-PAS information data do not provide exact combinations of expressed segments of isoforms, they can be used to eliminate many isoforms from consideration. Each of these types of data provides some information that can be used to define a condition which will be satisfied by all isoforms inferred by our algorithm (to be described in the next subsection).
Junction information. A junction (si,sj) is on an isoform f if f[i] = f[j] = 1 and f[k] = 0,i < k < j. If si and sj are adjacent, then junction (si,sj) is an adjacent junction. An isoform satisfies condition I if all the non-adjacent junctions on this isoform are supported by single-end short reads. In practice, most sufficiently expressed isoforms satisfy this condition. For example, when 40 millions single-end reads with length 30bps are mapped, the probability of an isoform with expression level 6 RPKM satisfying condition I is 99.3% and 92.8%, if this isoform contains 10 and 100 exons, respectively. See Theorem 2 below for the details.
Start-end segment pair information. For an isoform f, expressed segment si is the start expressed segment of f if f[i] = 1 and f[j] = 0, 1 ≤ j < i. Expressed segment si is the end expressed segment of f if f[i] = 1 and f[j] = 0,i < j ≤ |S|. The TSS-PAS pair information describes the start and end expressed segments of each isoform and will be referred to as the start-end segment pair data. An isoform satisfies condition II if the start-end segment pair of this isoform appears in the given set of start-end segment pairs. If the TSS-PAS pair information is missing, then any expressed segment can theoretically be the start or end expressed segment. However, in this case, many short (and thus unrealistic) isoforms could be introduced, which will make isoform inference difficult. Therefore, when the TSS-PAS pair information is missing, we allow an expressed segment si to be the start (or end) expressed segment of any isoform if there is no expressed segment sj with j < i (or i < j) such that junction (sj,si) (or (si,sj), respectively) is adjacent or supported by some read.
Paired-end read data. A pair of expressed segments (si,sj),i < j on an isoform f is an informative pair if f[i] = f[j] = 1 and PM,h,α(li + L2 − 1,gi,j,lj + L2 − 1) < 0.05, assuming that the span of a paired-end read follows some probability distribution h(x), the expression level of this isoform is α RPKM and M paired-end reads have been mapped. Here, L2 is the read length of a paired-end read,
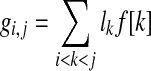 , and PM,h,α is defined in Theorem 1. According to the theorem, if (si,sj) is informative, then the probability that there are no paired-end reads with start positions in segment si and end positions in segment sj is less than 0.05. A triple of expressed segments (si,si + 1,sj),i + 1 < j is an informative triple if f[i] = f[i + 1] = f[j] = 1 and PM,h,α(L2 − 1,gi,j,lj + L2 − 1) < 0.05. Similarly, (si,si + 1,sj),j < i is an informative triple if PM,h,α(L2 − 1,gj,i + 1,lj + L2 − 1) < 0.05. An isoform satisfies condition III if every informative pair or triple on this isoform is supported by paired-end reads. A larger α makes this condition more stringent. Because in many cases, two isoforms can only be distinguished by a pair or triple of segments, it is necessary to require that every informative pairs or triple (instead of some of them) are supported by paired-end reads.
, and PM,h,α is defined in Theorem 1. According to the theorem, if (si,sj) is informative, then the probability that there are no paired-end reads with start positions in segment si and end positions in segment sj is less than 0.05. A triple of expressed segments (si,si + 1,sj),i + 1 < j is an informative triple if f[i] = f[i + 1] = f[j] = 1 and PM,h,α(L2 − 1,gi,j,lj + L2 − 1) < 0.05. Similarly, (si,si + 1,sj),j < i is an informative triple if PM,h,α(L2 − 1,gj,i + 1,lj + L2 − 1) < 0.05. An isoform satisfies condition III if every informative pair or triple on this isoform is supported by paired-end reads. A larger α makes this condition more stringent. Because in many cases, two isoforms can only be distinguished by a pair or triple of segments, it is necessary to require that every informative pairs or triple (instead of some of them) are supported by paired-end reads.
Note that while the junction information is always available given the single-end read data and exon-intron boundary information, the start-end segment pair information and paired-end read data are not necessarily always available. We define an isoform as valid if it satisfies conditions I, II, and/or III whenever the corresponding types of data are provided. The following theorem gives a lower bound on the probability that type I condition is satisfied by an isoform.
Theorem 2
Under the uniform sampling assumption, the probability that an isoform f consisting of t exons with expression level x RPKM satisfies type I condition is at least 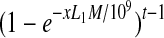 , where e is the base of natural logarithm, M the number of single-end reads mapped, and L1 the length of single-end reads.
, where e is the base of natural logarithm, M the number of single-end reads mapped, and L1 the length of single-end reads.
Proof.
If expression level of y RPKM of the isoform f corresponds to one transcript of f, the total number of the expressed transcripts of f is x/y. Based on the definition of RPKM, y = (106 · 103)/L0 = 109/L 0, where L0 is the total length of all the expressed transcripts with duplications. For any junction, the probability of a read falling into this junction is xL/yL0. So, the probability that none of the reads fall into this junction is 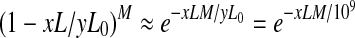 . In order for this isoform to be valid, each of the t − 1 junctions contains at least one read. Therefore, the probability of this isoform being valid is
. In order for this isoform to be valid, each of the t − 1 junctions contains at least one read. Therefore, the probability of this isoform being valid is 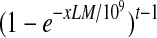 . Note that the sequencing noise does not decrease the above probability although it may provide some spurious junction reads. ▪
. Note that the sequencing noise does not decrease the above probability although it may provide some spurious junction reads. ▪
2.5. Isoform inference algorithm
We now describe our algorithm, IsoInfer, for inferring isoforms. The algorithm uses the following types of data: the reference genome, single-end short reads, exon-intron boundaries, TSS-PAS pairs, gene boundary information from the reference genome annotation, and paired-end short reads. The first three pieces of information (i.e., the reference genome, exon-intron boundaries, and single-end short reads) are required in the algorithm. If TSS-PAS pairs are not provided, gene boundaries would be required. The flow of data processing in IsoInfer is illustrated in Figure 3. The third step of the algorithm requires an external tool, e.g., Bowtie (Langmead et al., 2009), to map the short reads to the reference genome and junction sequences. In the fifth step, any two segments that are adjacent or supported by a junction read will be clustered together. Note that, such a cluster may contain expressed segments from more than one gene or contain only a subset of expressed segments from a single gene, but these cases do not happen very often. Furthermore, in each cluster, if there is a sequence of consecutive expressed segments such that every internal segment has no non-adjacent junction with any other expressed segment other than its left or right neighbor in the sequence, then we will combine the expressed segments into a single segment. This compression will be important because it reduces the problem size drastically for some isoforms containing a very large number of expressed segments. The details of the clustering and compression step are straightforward and omitted.
FIG. 3.

The flow of data processing in algorithm IsoInfer.
In the following, we give more details of the last step in IsoInfer, i.e., inferring isoforms. Each cluster of expressed segments defines an instance of the isoform inference problem. Denote such an instance as I(S,R,T,d), where 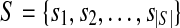 is the set of expressed segments in the cluster, R the set of short (single-end and paired-end) reads mapped to the segments in the cluster, T the set of start-end segment pairs, and d a function such that
is the set of expressed segments in the cluster, R the set of short (single-end and paired-end) reads mapped to the segments in the cluster, T the set of start-end segment pairs, and d a function such that 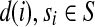 , denotes the number of single-end reads mapped to segment si and d(i,j), 1 ≤ i < j ≤ |S|, denotes the number of single-end reads mapped to junction (si,sj).
, denotes the number of single-end reads mapped to segment si and d(i,j), 1 ≤ i < j ≤ |S|, denotes the number of single-end reads mapped to junction (si,sj).
The inference procedure is summarized in Algorithm 1. It first enumerates all the valid isoforms in step 1. However, for a cluster with a large number of expressed segments and isoforms, the number of valid isoforms could be too large to be enumerated efficiently even though conditions I, II and/or III could be used to filter out many invalid isoforms. Therefore, the algorithm enumerates valid isoforms with high expression levels first, where the expression level of an isoform is defined by the least number of single-end reads on any junction of the isoform. The enumeration terminates when a preset number (denoted as γ) of valid isoforms are enumerated. The parameter γ is used to avoid the rare cases that the number of valid isoforms is too large to be handled by subsequent steps of IsoInfer. We set γ = 1000 by default based on our empirical knowledge of the real data considered in Section 4. For example, over 97.5%, 98.5%, and 99% cases, the number of valid isoforms is no more than 1000 in the tests on mouse brain, liver and muscle tissues, respectively, when the exact boundary and TSS-PAS information is extracted from the UCSC knownGene table. The impact of the omitted isoforms is minimized because highly expressed isoforms are enumerated first.
Algorithm 1.
IsoformInference. Given an instance I(S,R,T,d), the algorithm infers a set of isoforms to explain the read data
| 1: Among all segment junctions of an isoform f, denote m(f) as the minimum number of single-end reads mapped to any of these junctions. Enumerate all the valid isoforms f in the descending order of m(f) until a preset number (γ) of valid isoforms is obtained. Denote the set of all the enumerated valid isoforms as F. |
| 2: Remove all the short reads and start-end segment pairs that are not validated by F. |
| 3: for 5 ≤ u ≤ β do |
4: w(f) ← 0 for 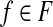 . . |
| 5: for 0 ≤ m ≤ |S|− udo |
| 6: n ← m + u. |
| 7: V(m,n) ← BestCombination(I(m,n)). |
8: For each 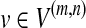 , define , define 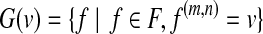 and for each and for each 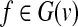 , let w(f) = w(f) + 1/|G(v)|. , let w(f) = w(f) + 1/|G(v)|. |
| 9: endfor |
| 10: Sort F by w in increasing order. |
11: for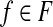 do do
|
| 12: ifw(f) < 1 and F − {f} is a feasible solution of Ithen |
| 13: F ← F − {f}. |
| 14: endif |
| 15: endfor |
| 16: endfor |
17: w′(f) ← 1/w(f) for 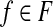 . . |
18: Solve the weighted set cover instance (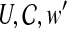 ), where ), where 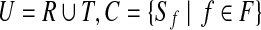 , and , and 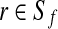 if r is validated by f for if r is validated by f for 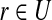 for each for each 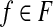 by the branch-and-bound method implemented in GNU package GLPK. Return the set of the valid isoforms corresponding to the optimal solution of set cover. by the branch-and-bound method implemented in GNU package GLPK. Return the set of the valid isoforms corresponding to the optimal solution of set cover. |
A short read r is validated by a set of isoforms if the set contains an isoform f such that f[i] = 1 when r[i] = 1. A start-end segment pair is validated by a set of isoforms if this pair is the start-end segment pair of some f in the set. A set of isoforms is a feasible solution of I(S,R,T,d) if every read in R and start-end segment pair in T are validated by the set. Due to possible noise in sequencing and the incompleteness of the enumeration of valid isoforms in step 1, it may happen that some reads or start-end segment pairs are not supported by the set of isoforms F enumerated in step 1. Step 2 of the algorithm removes such invalidated reads and start-end segment pairs to make F feasible.
To find a subset of valid isoforms to explain the data, a simple idea is to try all possible combinations of the valid isoforms in F and find a minimum combination that can explain all the short reads, as done in procedure BestCombination (Algorithm 2). The procedure BestCombination gradually increases the number of valid isoforms considered and enumerates all possible combinations of such a number of isoforms until a preset condition is met.
Algorithm 2.
BestCombination. Given an instance I(S,R,F,d), find a “best” subset of F such that the read data can be explained by enumerating all possible subsets of F
| 1: for 1 ≤ i ≤ |S|do |
| 2: p ← 0 and F′ ← ∅. |
| 3: for each F″ ⊂ F where |F″| = i and F″ is a feasible solution of Ido |
| 4: {z,x} ← QPsolver(I(S,F″,d)). |
| 5: ifp < p-value(z) then |
| 6: p ← p-value(z) and F′ ← F″. |
| 7: end if |
| 8: end for |
| 9: ifp ≥ 0.05 then |
| 10: Return F′. |
| 11: end if |
| 12: end for |
However, it is often infeasible to enumerate all possible combinations of the valid isoforms of a given size. When this happens, we decompose an the instance into some sub-instances. In each sub-instance, only a subset of expressed segments are considered. More specifically, for an instance I(S,R,F,d), where F is the set of valid isoforms enumerated, a sub-instance I(m,n) = I(S(m,n), R(m,n), d(m,n), F(m,n)), 0 ≤ m < n ≤ |S|, is defined concerning the subset 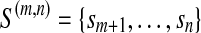 of expressed segments of S. It is formally defined as follows. For each
of expressed segments of S. It is formally defined as follows. For each 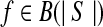 , define
, define 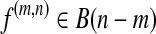 and f(m,n)[i] = f[i + m], 1 ≤ i ≤ n − m. In other words, f(m,n) denotes the sub-vector of f spanning the interval [m + 1,n]. Let
and f(m,n)[i] = f[i + m], 1 ≤ i ≤ n − m. In other words, f(m,n) denotes the sub-vector of f spanning the interval [m + 1,n]. Let 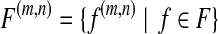 ,
, 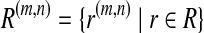 d(m,n)(i) = d(i + m), 1 ≤ i ≤ n − m, and d(m,n)(i,j) = d(i + m, j + m), 1 ≤ i < j ≤ n − m. Note that the start-end segment information is not needed in sub-instances.
d(m,n)(i) = d(i + m), 1 ≤ i ≤ n − m, and d(m,n)(i,j) = d(i + m, j + m), 1 ≤ i < j ≤ n − m. Note that the start-end segment information is not needed in sub-instances.
The parameter β appearing in step 3 controls the maximum size of a sub-instance. Larger sub-instances make the results of procedure BestCombination more reliable. However, the execution time of BestCombination increases exponentially with the number of valid isoforms which grows with the size of the sub-instance. Therefore, instead of a fixed size, a set of sub-instance sizes from the interval [5,β] are attempted. For a fixed sub-instance size, BestCombination is executed on each sub-instance of the size in step 7. According to the results of BestCombination, each valid isoform is assigned a weight in Step 8 which roughly indicates the frequency that the isoform appears in the combinations found by BestCombination. A subset of valid isoforms with weights less than 1 are removed in steps 11–15 without making F infeasible.
In steps 17 and 18 of the algorithm, a weighted set cover instance is constructed such that an optimal solution implies a subset of valid isoforms with a minimum total weight such that all the short reads and start-end segments can be explained. The set cover problem can be solved by using the branch-and-bound method implemented in GNU package GLPK, since it involves only small instances.
3. Simulation Test Results
We test IsoInfer on mouse genes. The reference genomic sequence and known isoforms of all mouse genes are downloaded from UCSC (mm9, NCBI Build 37) (Karolchik et al., 2008). All exon-intron boundaries of the known isoforms are extracted. This dataset contains 26,989 genes and 49,409 isoforms. 16,392 (60.7%) of the genes have only one isoform and 59 (0.2%) of the genes have more than 10 isoforms. Of the genes, 5830 (21.6%) have only one exon and 384 (1.4%) have more than 40 exon-intron boundaries. For the simulation study, only genes with at least two known isoforms are used, which result in 10,595 genes. We further extract all the start-end segments and randomly generate relative expression levels of every isoform. Although it would be natural to assume that expression levels follow a uniform distribution, it has been reported previously (Alter et al., 2008; Konishi, 2004; Wijaya et al., 2008) that the expression levels of isoforms tend to obey a log-normal distribution. Therefore, we consider three types of distributions.
Base10: For each isoform, a random number r following the standard normal distribution is generated and then 10r is assigned as the relative expression level of this isoform.
Base2: For each isoform, a random number r following the standard normal distribution is generated and then 2r is assigned as the relative expression level of this isoform.
Uniform: For each isoform, a random number r uniformly generated from [0,1] is assigned as the relative expression level of this isoform.
Then 40M single-end and 10M paired-end short reads are randomly generated according to the relative expression levels of the isoforms. In the simulation, we assume that the span of a paired-end read is a random variable obeying the normal distribution N(μ,σ2) (Richter et al., 2008) so we could evaluate the impact of the mean and deviation of the spans of paired-end reads on the performance of IsoInfer. Note that IsoInfer does not depend on this assumption and works for paired-end reads drawn from any distribution.
Finally, IsoInfer is used to recover all the known isoforms using the start-end segments and single-end and paired-end reads. In the simulation, the read lengths of single-end and paired-end reads are 25 and 20 bps, respectively. The parameter α is set to 1 RPKM, β = 7, and γ = 1000. We consider three measures of the performance: sensitivity, effective sensitivity, and precision. A known isoform is recovered if it is in the output of IsoInfer. Sensitivity is defined as the number of recovered isoforms divided by the number of all known isoforms. Precision is defined as the number of recovered isoforms divided by the number of isoforms inferred. Since IsoInfer only intends to infer isoforms that are sufficiently expressed, it is useful to consider how many sufficiently expressed isoforms are recovered by the algorithm. Since Theorem 2 shows that an isoform with a sufficiently high expression level is likely to satisfy condition I (i.e., all its exon-intron junctions are supported by the read data) with high probability, we define effective sensitivity as the number of recovered isoforms divided by the number of known isoforms whose exon-intron junctions are supported by the read data.
3.1. Calculation of expression levels
To estimate the effectiveness of our QP formulation, we randomly generate Base10 expression levels and single-end short reads on the known mouse isoforms and check whether it can recover the correct expression levels of the known isoforms. For an isoform f with expression level xf and calculated expression level 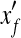 , the relative difference
, the relative difference 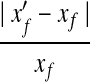 is used to measure the accuracy of calculation. A simple and widely used method of calculating expression levels of isoforms is based on counting reads mapped to its unique exons and exon junctions (Wang et al., 2008a; Pan et al., 2008). Clearly, this simple strategy fails if the isoform does not have any unique exons or exon junctions. We compare our method with the simple method (termed Uniq in this article) and the method based on maximum likelihood estimation (MLE) and importance sampling (IS) (Jiang and Wong, 2009). The comparison is depicted in Figure 4.
is used to measure the accuracy of calculation. A simple and widely used method of calculating expression levels of isoforms is based on counting reads mapped to its unique exons and exon junctions (Wang et al., 2008a; Pan et al., 2008). Clearly, this simple strategy fails if the isoform does not have any unique exons or exon junctions. We compare our method with the simple method (termed Uniq in this article) and the method based on maximum likelihood estimation (MLE) and importance sampling (IS) (Jiang and Wong, 2009). The comparison is depicted in Figure 4.
FIG. 4.

Comparison of the accuracies of different methods in estimating isoform expression levels. The y-axis shows the percentage of isoforms whose estimated/calculated expression levels are within a certain relative difference range from the truth. 10 million reads (left) and 80 million reads (right) are sampled in each of the figures.
The comparison shows that MLE followed by IS (MLE + IS) is the most accurate and Uniq is the worst. IsoInfer achieves a performance comparable to that of MLE + IS. An advantage of MLE + IS is that it also provides a 95% confidence interval for each expression level estimation. On the other hand, IsoInfer calculates the expression levels much faster than MLE + IS does (3 minutes vs. 3 hours for all mouse genes on a standard desktop PC). The efficiency of IsoInfer makes the search for novel isoforms possible.
3.2. The influence of the distribution of expression levels
In this section, we analyze the influence of the distribution of expression levels on the performance of IsoInfer in inferring isoforms. The distribution of the span of paired-end reads are fixed as the normal distribution N(300, 302). The sensitivities and precisions grouped by number of known isoforms per gene are depicted in Figure 5.
FIG. 5.

The sensitivity (top left), effective sensitivity (top right) and precision (bottom left) of IsoInfer on genes with a certain number of isoforms when different distributions of expression levels are generated. (Bottom right) Sensitivity of IsoInfer on different expression levels when different distributions of expression level are applied. In the graph, the expression levels are log2 transformed. Expression level x corresponds to 25 · 2x RPKM. The vertical line corresponds to expression level 1/8 = 3.125 RPKM.
The overall sensitivities and precisions of IsoInfer on (Base10, Base2, Uniform) expression levels are (39.7%, 75.0%, 72.5%) and (79.3%, 82.1%, 81.3%), respectively. The sensitivities for Base10 expression levels are much lower than those for Base2 and Uniform expression levels, because a large faction of the isoforms are not significant expressed. The effective sensitivity of three cases are 83.5%, 77.4%, and 77.4%, respectively. Figure 5 gives detailed sensitivity, effective sensitivity and precision of IsoInfer on genes with a certain number of isoforms. The high effective sensitivity shown in the figure is also confirmed by the sensitivity results on different expression levels, also given in Figure 5 which shows that isoforms with high expression levels are identified with high sensitivities. For example, for Base10 expression levels, isoforms with expression level above 3 (or 6) RPKM are identified with sensitivity above 56.0% (or 81.0%, respectively).
3.3. The importance of start-end expressed segment pairs
As mentioned before, single-end short reads are necessary for our algorithm but start-end segment pairs and paired-end reads are optional. To estimate the importance of the last two pieces of information, we compare the results when different types of data are available. Four combinations are possible—denoted as I, I + II, I + III, and I + II + III—where I, II, and III correspond to single-end reads (which provide the junction information), start-end segment pairs and paired-end data, respectively. The combination I + III means that the single-end and paired-end read data are available but not the start-end segment pairs. In the simulation, Base10 expression levels are generated and the span distribution of paired-end reads is fixed as N(300, 302). Figure 6 shows that start-end segment pairs are much more important than paired-end reads for our algorithm. For example, the sensitivities and precisions for combinations I + II and I + III are (38.9%, 78.5%) and (29.5%, 16.5%), respectively.
FIG. 6.

The sensitivity (top left), effective sensitivity (top right) and precision (bottom left) of IsoInfer on genes with a certain number of isoforms when different combinations of type I, II, and III data are provided. (Bottom right) Sensitivity of IsoInfer on different expression levels when different combinations of type I, II, and III data are used. Again, the expression levels are log2 transformed. Expression level x corresponds to 25 · 2x RPKM. The vertical line corresponds to expression level 1/8 = 3.125 RPKM.
3.4. The influence of span distribution
The span of paired-end reads follows the normal distribution N(μ,σ2). We run IsoInfer on different combinations of μ and σ. On each combination, 10 million pair-end reads are randomly generated. Since start-end segment pairs are much more important than paired-end reads, as shown in the above subsection, the span distribution should not have a significant influence on the inference results when start-end segment pairs are available. This is confirmed by Tables 1 and 2. The precision and sensitivity of IsoInfer vary by at most 1.5% when different span distributions are applied.
Table 1.
Sensitivities for Various Span Distributions Grouped by the Number of Isoforms per Gene
| No. of isoforms per gene | 2 | 3 | 4 | 5 | 6 | 7 | ≥8 |
|---|---|---|---|---|---|---|---|
| No PE reads | 0.392 | 0.402 | 0.392 | 0.383 | 0.374 | 0.346 | 0.391 |
| 300, 10 | 0.393 | 0.406 | 0.402 | 0.391 | 0.385 | 0.357 | 0.402 |
| 300, 30 | 0.393 | 0.407 | 0.404 | 0.392 | 0.386 | 0.362 | 0.402 |
| 300, 50 | 0.393 | 0.407 | 0.402 | 0.393 | 0.385 | 0.366 | 0.402 |
| 300, 100 | 0.393 | 0.408 | 0.404 | 0.395 | 0.385 | 0.359 | 0.405 |
| 1100, 110 | 0.387 | 0.401 | 0.399 | 0.395 | 0.392 | 0.363 | 0.403 |
| 3000, 300 | 0.392 | 0.404 | 0.403 | 0.400 | 0.390 | 0.366 | 0.413 |
Here, “No PE reads” means that no paired-end reads are applied. The first column lists various combinations of the mean and standard deviation in the span (normal) distributions considered. The corresponding effective sensitivities range from 63.4% to 97.4%.
Table 2.
Specificities for Various Span Distributions Grouped by the Number of Isoforms per Gene
| No. of isoforms per gene | 2 | 3 | 4 | 5 | 6 | 7 | ≥8 |
|---|---|---|---|---|---|---|---|
| No PE reads | 0.893 | 0.824 | 0.774 | 0.732 | 0.704 | 0.638 | 0.733 |
| 300, 10 | 0.897 | 0.830 | 0.784 | 0.738 | 0.717 | 0.648 | 0.740 |
| 300, 30 | 0.897 | 0.831 | 0.786 | 0.739 | 0.718 | 0.657 | 0.740 |
| 300, 50 | 0.897 | 0.830 | 0.784 | 0.740 | 0.714 | 0.663 | 0.737 |
| 300, 100 | 0.896 | 0.830 | 0.786 | 0.743 | 0.713 | 0.649 | 0.740 |
| 1100, 110 | 0.896 | 0.829 | 0.782 | 0.739 | 0.720 | 0.657 | 0.729 |
| 3000, 300 | 0.896 | 0.828 | 0.776 | 0.741 | 0.709 | 0.648 | 0.736 |
The first column lists various combinations of the mean and standard deviation in the span (normal) distributions considered.
The above small effect of paired-end read data on the performance of IsoInfer is because the parameter α is set to 1. When a large α is applied, IsoInfer trades sensitivity for precision. For example, when the span distribution of paired-end read is fixed as N(300, 302), if α is set to 1, the sensitivity and precision on genes with at least 8 isoforms are 40.2% and 74.0%, respectively. The two measures will change to 35.4% and 78.1%, respectively, when α is set to 20. The performance of IsoInfer when α is set to different values is shown in Figure 7.
FIG. 7.

The sensitivity and precision of IsoInfer when α is set to different values.
3.5. The influence of noise
To further evaluate the performance of IsoInfer in a real scenario, we introduce noise in the simulation test. For simplicity, we will only consider noisy reads that are sampled from the reference genome directly instead of expressed isoforms. Such a noise may generate sampled reads unproportional to the expression levels of expressed isoforms. In the RNA-Seq experiments conducted by Mortazavi et al. (2008), about 7% of the short reads are mapped to introns and intergenic regions, while introns and intergenic regions comprise more than 98% of the entire mouse genome. By taking into account the fact that the size of the mouse genome is about 2.5G, the noise level in the reads analyzed in Mortazavi et al. (2008) could be estimated as less than 0.03 RPKM. Therefore, we will consider three different noise levels: 0.03, 0.05, and 0.1 RPKM. Again, the simulation generates 40M single-end reads of length 25 bps. Table 3 shows the overall precision, sensitivity, and effective sensitivity of IsoInfer when different noise levels are adopted. The results demonstrate that the noise only has a slight (no more than 2.5%) influence on the accuracy of IsoInfer when noise levels are in the given range.
Table 3.
Performance of IsoInfer on Simulated Reads When Different Noise Levels Are Adopted
| Noise level (RPKM) | 0 | 0.03 | 0.05 | 0.1 |
|---|---|---|---|---|
| Precision | 0.794 | 0.786 | 0.782 | 0.769 |
| Sensitivity | 0.396 | 0.396 | 0.395 | 0.397 |
| Effective sensitivity | 0.811 | 0.810 | 0.808 | 0.802 |
4. Recovery of Known Isoforms From Real Reads
The evaluation uses the following four data sets: (1) known mouse isoforms downloaded from UCSC Karolchik et al. (2008), which contains 49,409 transcripts; (2) mouse mRNAs expressed in various tissues downloaded from UCSC containing 228,779 mRNAs; (3) RNA-Seq data from brain, liver, and skeletal muscle tissues of mouse (Mortazavi et al., 2008), which contains 47,781,892, 44,279,807, and 38,210,358 single-end reads for brain, liver, and muscle, respectively; and (4) 104,710 exon junctions that were predicted by TopHat from the above RNA-Seq data for mouse brain tissue (Trapnell et al., 2009).
As in the simulation tests, on a specific tissue, one can only expect that isoforms with expression levels above a certain threshold can be detected by RNA-Seq experiments, so as to be inferred by IsoInfer. Given a set of mapped reads, an isoform is said to be theoretically expressed if each exon except for the first and last one of this isoform has expression level at least 1 RPKM and every exon junction on this isoform is supported by short reads. (Note that this does not really guarantee that the isoform is actually expressed.) The expression levels of the first and last exons are ignored here because of the possible 3′ and 5′ sampling biases in RNA-Seq (Wang et al., 2008b; Mortazavi et al., 2008). The theoretically expressed isoforms among known mouse isoforms and mRNAs are used as benchmarks. Note that the benchmarks change when different tissues are considered, because the expression levels of isoforms change from tissue to tissue.
We have done two groups of tests. The first one is to use the TSS-PAS pair and exon-intron boundary information from the known mouse isoforms and/or mRNAs from UCSC and RNA-Seq short reads to infer isoforms. The predicted isoforms are compared with the theoretically expressed isoforms in the corresponding benchmark. An isoform is recovered by IsoInfer if one of isoforms inferred by IsoInfer matches this isoform precisely (i.e., the two isoforms contain exactly the same set of exons with exactly the same boundaries). The inference results are shown in Table 4. These results demonstrate that when accurate exon-intron boundary and TSS-PAS pair information is provided, IsoInfer achieves a reasonably good precision, and the precision increases as the size of the benchmark increases. When known mouse isoforms are used, IsoInfer achieves decent effective sensitivities (i.e., 72.9% for brain, 82.2% for liver and 83.0% for muscle). Because mRNAs were collected from different sources and tissues, a large fraction of them may not really be expressed in a specific tissue. Therefore, effective sensitivity of IsoInfer drops when mRNAs are used as the benchmark.
Table 4.
Performance of IsoInfer When Different Exon-Intron Boundary and TSS-PAS Pair Information and Corresponding Benchmarks Are Used
| |
Known isoforms |
mRNAs |
Union |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Tissue | Brain | Liver | Muscle | Brain | Liver | Muscle | Brain | Liver | Muscle |
| No. of theoretically expressed | 18521 | 12411 | 11723 | 87178 | 72594 | 69086 | 101392 | 82199 | 78298 |
| Precision | 0.493 | 0.592 | 0.627 | 0.572 | 0.670 | 0.712 | 0.591 | 0.697 | 0.737 |
| Effective sensitivity | 0.729 | 0.822 | 0.830 | 0.328 | 0.352 | 0.366 | 0.335 | 0.365 | 0.381 |
Here, “Union” means that the exon-intron boundary and TSS-PAS pair information is extracted from both known mouse isoforms and mRNAs, and the benchmark is the union of the known mouse isoforms and mRNAs.
The second test measures the performance of IsoInfer when the exact exon-intron boundary information is unavailable. The test uses exon-intron boundaries predicted by TopHat from the RNA-Seq read data on the mouse brain tissue and the TSS-PAS pair information extracted from the known mouse isoforms and/or mRNAs. The test results are shown in Table 5. Although it is reported in Trapnell et al. (2009) that over 80% of the exon junctions predicted by TopHat are also exon junctions in the UCSC known mouse isoforms, the inference result on the known mouse isoforms is much worse than the result when exact exon-intron boundary information is provided. On the other hand, when mRNA is used as the benchmark, the exon-intron boundaries provided by TopHat lead IsoInfer to a more aggressive prediction (and thus achieving a better effective sensitivity).
Table 5.
Performance of IsoInfer When the Exon-Intron Boundary Information Is Extracted from the Exon Junctions Predicted by TopHat
| Known isoforms | mRNAs | Union | |
|---|---|---|---|
| Precision | 0.240 | 0.362 | 0.378 |
| Effective sensitivity | 0.496 | 0.532 | 0.508 |
These results are all on the mouse brain tissue. The TSS-PAS pair information is extracted from the known mouse isoforms and/or mRNAs, depending on the benchmark. Again, “Union” means that the TSS-PAS pair information is extracted from both known mouse isoforms and mRNAs, and the benchmark is the union of the known mouse isoforms and mRNAs.
Although the test results in Tables 4 and 5 demonstrate the importance of having accurate exon-intron boundary information, we should take these results with a grain of salt because we do not know what isoforms are really expressed in each of the tissues. Hence, we think that the simulation results in Figures 4–6 and Table 3 might represent a better characterization of the true performance of IsoInfer under various conditions. On the other hand, we do not know how well the simulated data capture real RNA-Seq data from practice.
In each of the above tests, the last three steps of IsoInfer shown in Figure 3 took less than 80 minutes on an Intel P8600 processor.
5. Conclusion
We have proposed a novel method to infer isoforms from single-end and paired-end short RNA-Seq reads and information concerning exon-intron boundaries and TSS-PAS pairs. While the single-end data is necessary for our algorithm, the TSS-PAS pairs could greatly improve the performance. Our experimental results on simulated and real read data demonstrate that our algorithm is possible to infer isoforms with reasonably good accuracy and speed. The software is available publicly at http://www.cs.ucr.edu/∼jianxing/IsoInfer.html
IsoInfer is based on a critical assumption: the reads are uniformly sampled from all the transcripts. However, because of sequencing biases and errors and the issue of multi-reads, this assumption may not always hold. Therefore, it would be interesting to study more robust isoform inference methods that could correct sequencing errors and biases and deal with multi-reads.
Acknowledgments
We thank Pirola Yuri for useful discussions. The research is supported in part by a CSC scholarship, NSF grant IIS-0711129, and NIH grants 2R01LM008991 and AI078885.
Disclosure Statement
No competing financial interests exist.
References
- Alkan C. Kidd J.M. Marques-Bonet T., et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter M.D. Rubin D.B. Ramsey K., et al. Variation in the large-scale organization of gene expression levels in the hippocampus relates to stable epigenetic variability in behavior. PLoS ONE. 2008;3:e3344. doi: 10.1371/journal.pone.0003344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertone P. Stolc V. Royce T.E., et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- Bishop C.M. Pattern Recognition and Machine Learning. Springer; New York: 2007. [Google Scholar]
- Boguski M.S. The turning point in genome research. Trends Biochem. Sci. 1995;20:295–296. doi: 10.1016/s0968-0004(00)89051-9. [DOI] [PubMed] [Google Scholar]
- Boguski M.S. Tolstosher C.M. Bassett D.E., Jr Gene discovery in dbEST. Science. 1994;265:1993–1994. doi: 10.1126/science.8091218. [DOI] [PubMed] [Google Scholar]
- Bonizzoni P., et al. Detecting alternative gene structures from spliced ESTs: a computational approach. J. Comput. Biol. 2009;16:43–66. doi: 10.1089/cmb.2008.0028. [DOI] [PubMed] [Google Scholar]
- Breitbart R.E., et al. Alternative splicing: a ubiquitous mechanism for the generation of multiple protein isoforms from single genes. Annu. Rev. Biochem. 1987;56:467–495. doi: 10.1146/annurev.bi.56.070187.002343. [DOI] [PubMed] [Google Scholar]
- Brenner S., et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol. 2000;18:630–634. doi: 10.1038/76469. [DOI] [PubMed] [Google Scholar]
- Cloonan N. Forrest A.R. Kolle G., et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods. 2008;5:613–619. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- Cloonan N. Xu Q. Faulkner G.J., et al. RNA-MATE: a recursive mapping strategy for high-throughput RNA-sequencing data. Bioinformatics. 2009;25:2615–2616. doi: 10.1093/bioinformatics/btp459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S., et al. Efficient targeted transcript discovery via array-based normalization of RACE libraries. Nat. Methods. 2008;5:629–635. doi: 10.1038/nmeth.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood M.J., et al. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res. 2009;19:521–532. doi: 10.1101/gr.074906.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb D.I.A. A numerically stable dual method for solving strictly convex quadratic programs. Math Program. 1983;27:1–33. [Google Scholar]
- Graveley B.R. Molecular biology: power sequencing. Nature. 2008;453:1197–1198. doi: 10.1038/4531197b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbers M. Carninci P. Tag-based approaches for transcriptome research and genome annotation. Nat. Methods. 2005;2:495–502. doi: 10.1038/nmeth768. [DOI] [PubMed] [Google Scholar]
- Hashimoto T. de Hoon M.J. Grimmoud S.M., et al. Probabilistic resolution of multi-mapping reads in massively parallel sequencing data using MuMRescueLite. Bioinformatics. 2009;25:2613–2614. doi: 10.1093/bioinformatics/btp438. [DOI] [PubMed] [Google Scholar]
- Heber S., et al. Splicing graphs and EST assembly problem. Bioinformatics. 2002;18:S181–S188. doi: 10.1093/bioinformatics/18.suppl_1.s181. [DOI] [PubMed] [Google Scholar]
- Jiang H. Wong W.H. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009;25:1026–1032. doi: 10.1093/bioinformatics/btp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.M. Castle J. Garrett-Engele P., et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- Kapranov P. Cherg J. Dike S., et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- Karolchik D. Kuhn R.M. Baertsch R., et al. The UCSC genome browser database: 2008 update. Nucleic Acids Res. 2008;36:D773–D779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.B., et al. Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science. 2007;316:1481–1484. doi: 10.1126/science.1137325. [DOI] [PubMed] [Google Scholar]
- Kodzius R., et al. CAGE: cap analysis of gene expression. Nat. Methods. 2005;3:211–222. doi: 10.1038/nmeth0306-211. [DOI] [PubMed] [Google Scholar]
- Konishi T. Three-parameter lognormal distribution ubiquitously found in cdna microarray data and its application to parametric data treatment. BMC Bioinformatics. 2004;5:5. doi: 10.1186/1471-2105-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel J., et al. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009;10:R23. doi: 10.1186/gb-2009-10-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan T. Benovoy D. Dias C., et al. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008;40:225–231. doi: 10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- Lacroix V., et al. Exact transcriptome reconstruction from short sequence reads. Proc. WABI '08. 2008:50–63. [Google Scholar]
- Langmead B., et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008a;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., et al. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008b;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- Marioni J., et al. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P., et al. Gene identification signature (GIS) analysis for transcriptome characterization and genome annotation. Nat. Methods. 2005;2:105–111. doi: 10.1038/nmeth733. [DOI] [PubMed] [Google Scholar]
- Pagani F. Baralle F.E. Genomic variants in exons and introns: identifying the splicing spoilers. Nat. Rev. Genet. 2004;5:389–396. doi: 10.1038/nrg1327. [DOI] [PubMed] [Google Scholar]
- Pan Q., et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Reinartz J., et al. Massively parallel signature sequencing (MPSS) as a tool for in-depth quantitative gene expression profiling in all organisms. Brief Funct. Genomic Proteomic. 2002;1:95–104. doi: 10.1093/bfgp/1.1.95. [DOI] [PubMed] [Google Scholar]
- Richter D.C., et al. MetaSima sequencing simulator for genomics and metagenomics. PLoS ONE. 2008;3:e3373. doi: 10.1371/journal.pone.0003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi-Ashtiani K. Yang X. Derti A., et al. Isoform discovery by targeted cloning, “deep-well” pooling and parallel sequencing. Nat. Methods. 2008;5:597–600. doi: 10.1038/nmeth.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammeth M., et al. Bubbles: alternative splicing events of arbitrary dimension in splicing graphs. Lect. Notes Comput. Sci. 2008a;4955:372–395. [Google Scholar]
- Sammeth M., et al. A general definition and nomenclature for alternative splicing events. PLoS Comput. Biol. 2008b;4:e1000147. doi: 10.1371/journal.pcbi.1000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki T., et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA. 2003;100:15776–15781. doi: 10.1073/pnas.2136655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srebrow A. Kornblihtt A.R. The connection between splicing and cancer. J. Cell Sci. 2006;119:2635–2641. doi: 10.1242/jcs.03053. [DOI] [PubMed] [Google Scholar]
- Sultan M., et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The FANTOM Consortium. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Trapnell C., et al. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velculescu V.E., et al. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Wang E.T. Sandberg R. Luo S., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008a;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. Gestein M. Snyder M., et al. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2008b;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock G.M. ENCODE: more genomic empowerment. Genome Res. 2007;176:667–668. doi: 10.1101/gr.6534207. [DOI] [PubMed] [Google Scholar]
- Wijaya E., et al. Modeling the marginal distribution of gene expression with mixture models. Proc FGCN '08. 2008:84–89. [Google Scholar]
- Wilhelm B.T., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–43. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- Williams W.V. Editorial hot topic: transcriptome analysis in drug development. Curr. Mol. Med. 2005;5:1–2. doi: 10.2174/1566524053152915. [DOI] [PubMed] [Google Scholar]
- Xing Y., et al. The multiassembly problem: reconstructing multiple transcript isoforms from EST fragment mixtures. Genome Res. 2004;14:426–441. doi: 10.1101/gr.1304504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassour M., et al. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc. Natl. Acad. Sci. USA. 2009;106:3264–3269. doi: 10.1073/pnas.0812841106. [DOI] [PMC free article] [PubMed] [Google Scholar]