

Abstract
Exposure to the excitotoxin domoic acid (DOM) has been shown to produce cardiac lesions in both clinical and animal studies. We have previously shown that DOM failed to directly affect cardiomyocyte viability and energetics, but the development of this cardiomyopathy has remained unexplained. The present study compared effects of high-level seizure induction obtained by intraperitoneal (2 mg/kg) or intrahippocampal (100 pmol) bolus administration of DOM on development of cardiac pathologies in a rat model. Assessment of cardiac pressure derivatives and coronary flow rates revealed a significant time-dependent decrease in combined left ventricular (LV) systolic and diastolic function at 1, 3, 7, and 14 days after intraperitoneal administration and at 7 and 14 days after intrahippocampal DOM administration. LV dysfunction was matched by a similar time-dependent decrease in mitochondrial respiratory control, associated with increased proton leakage, and in mitochondrial enzyme activities. Microscopic examination of the LV midplane revealed evidence of progressive multifocal ischemic damage within the subendocardial, septal, and papillary regions. Lesions ranged from reversible early damage (vacuolization) to hypercontracture and inflammatory necrosis progressing to fibrotic scarring. Plasma proinflammatory IL-1α, IL-1β, and TNF-α cytokine levels were also increased from 3 days after seizure induction. The observed cardiomyopathies did not differ between intraperitoneal and intrahippocampal groups, providing strong evidence that cardiac damage after DOM exposure is a consequence of a seizure-evoked autonomic response.
Neurological deficits and cardiomyopathy arising from domoic acid (DOM) exposure have been documented in a range of marine mammals and in one major documented outbreak of DOM poisoning in humans after the ingestion of contaminated shellfish.1,2 DOM, a potent excitotoxic structural analog of glutamic and kainic acids, is produced by algae and marine diatoms (eg, Pseudo-nitzschia spp.) and is consequently accumulated in a number of seafoods.3,4 Clinical reports of neurological symptoms including seizures, amnesia, and coma coupled with postmortem evidence of hippocampal and amygdaloid nucleus necrosis and astrocytosis were particularly noted among severe cases of DOM ingestion.1,5,6 Among these cases, the authors also reported cardiovascular disturbances, including cardiac arrhythmias and hypotension.1
As a consequence of heightened interest in the mechanism of DOM toxicity, a number of experimental studies have investigated the consequences of DOM exposure on behavioral and neurological responses in laboratory animals.4,7–13 Within neuronal tissue, DOM has been shown to activate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainic acid (KA) receptors and results in the release of glutamate.14 Overstimulation of ionotropic glutamate receptors (iGluR), namely N-methyl-D-aspartate (NMDA) receptors, provokes pathophysiological increases in intracellular Ca2+ and consequently leads to the activation of several overlapping Ca2+-dependent processes, such as the generation of mitochondrial reactive oxygen species and loss of cellular viability.15–20 DOM has been shown to be 8 to 10 times more potent than kainic acid in rodents, producing a similar reproducible pattern of behavioral toxicity culminating in seizures.8,21,22 Studies conducted in rats, mice, and cynomolgus monkeys have shown a consistent pattern of DOM-induced damage to the hippocampal pyramidal neurons, as well as to the thalamic, amygdalar, entorhinal, cortical, and septal neurons after DOM administration (0.22 to 4.4 mg/kg i.p.).9,23–25 With the use of electroencephalographic recordings, these studies showed that the hippocampal damage resulted in generalized epileptiform activity.
In addition to extensive neuronal damage, perimortem studies conducted on marine mammals after natural exposure to DOM have shown clear evidence of myocardial lesions.26,27 Cardiac lesions within a Californian sea otter population have been described to include myocardial pallor and multifocal myocardial necrosis associated with myocardial hemorrhage. Inflammatory cells were also observed in both the atrial and ventricular myocardium.26 In contrast, however, DOM toxicity studies conducted on an exposed Californian sea lion population showed myocardial degeneration limited to the ventricles.28 The link between DOM exposure and the reported cardiomyopathies has not as yet been characterized, and it remains to be determined whether the damage is the consequence of a direct (ie, cardiotoxic) or indirect [ie, central nervous system (CNS)/autonomic discharge] interaction. Gill and coworkers29,30 reported the presence of various ionotropic and metabotropic glutamate receptor subtypes within the intrinsic cardiac ganglia, nerve fibers, and specific components of the conducting system of murine, primate, and human hearts. Given the location of these glutamate subunits, it has been postulated that they may affect the regulation and conduction of impulses in the heart and may in part explain the cardiovascular effects reported after DOM intoxication. Functional evidence of receptor responsiveness to DOM exposure within the context of the cardiac pathology described, however, remains to be elucidated. Earlier work by our group demonstrated that, although DOM does traverse the membrane of rat cardiac H9c2 cells, it is not cytotoxic, nor does it compromise mitochondrial function within intact murine cardiomyocytes.31 This finding suggests that the mechanism of DOM-induced cardiac damage does not involve the same direct excitotoxic cell death processes described in neuronal tissue, but rather may represent the consequence of an indirect overactivation of glutamate receptors within the CNS.
CNS regulation of cardiovascular function has traditionally been considered to fall under the control of the rostral ventrolateral medullary pressor area. Emerging evidence, however, has indicated that the hippocampus and hypothalamic paraventricular nucleus, which project into the cardiac regulatory centers, also play a significant role in regulating cardiovascular function.32 This finding is supported by earlier studies showing that injection of kainic acid or NMDA into the paraventricular nucleus resulted in lesions of the parvocellular elements and evoked an excitotoxin-induced myocardial necrosis resembling the acute myocardial necrosis reported with systemic β-agonist administration.33–35 These studies support the hypothesis that the reported cardiotoxicity is a consequence of a catecholamine surge mediated by excitotoxin stimulation of glutamate receptors in the CNS. Other neuronal pathologies, such as stroke and epilepsy, have also been shown to result in long-term abnormal electrocardiographic activity and arrhythmias.36–38 Clinical peri-ictal studies of generalized tonic-clonic seizure patients reporting disturbances in blood pressure, heart rate and rhythm, and QT waves have offered an insight into the cause of the cardiomyopathy described as sudden unexplained death due to epileptic periods (SUDEP).39–42 The presentation of ischemic infarct and fibrotic scarring to the ventricular wall as a consequence of repeated epilepsy-induced autonomic discharges to the heart in SUDEP may represent a mechanism of DOM-induced cardiomyopathy.42
As our previous studies firmly established that domoic concentrations of ≤10 μmol/L have no direct effect on cardiomyocyte integrity or viability,31 we hypothesized that the DOM-induced cardiomyopathies occur as a consequence of seizure-evoked response. Here, we report that administration of DOM to the right ventral hippocampus results in seizure activity and prolonged cardiac dysfunction. Furthermore, we show that, although administration of DOM into the peritoneum also resulted in cardiac injury, acute exposure of isolated hearts to DOM failed to evoke any cardiac hemodynamic changes. The present study provides new insights into the cardiac dysfunction noted in DOM intoxication and offers new evidence for SUDEP-induced cardiac pathology.
Materials and Methods
Materials
Chemicals used in study protocols were obtained from BDH (Palmerston North, New Zealand), Sigma-Aldrich (Castle Hill, NSW, Australia; St. Louis, MO), and Roche Diagnostics (Mannheim, Germany), unless otherwise specified. DOM was purchased from Sapphire Bioscience (Waterloo, Australia) and was freshly dissolved in normal saline as needed. A Bio-Plex rat cytokine 9-Plex assay kit was purchased from Bio-Rad (Bio-Rad Laboratories, Albany, Auckland, NZ; Hercules, CA). DOM-specific ASP direct competitive enzyme-linked immunosorbent (cELISA) assay kits were purchased from Biosense Laboratories (Bergen, Norway).
Animal Care
Male Sprague-Dawley rats (2 to 3 months old; 280 to 275 g) were obtained from the University of Otago Animal Resource Unit. Rats were housed at 25°C under controlled 12-hour light/dark cycles and fed ad libitum on a standard rat diet before experimentation. All surgical and euthanasia procedures, including seizure induction and monitoring, were conducted in accordance with the regulations of the Otago University Committee on Ethics in the Care and Use of Laboratory Animals and the Guide for Care and Use of Laboratory Animals (NIH Publication 85-23, 1996).
Experimental Protocol
The study animals were randomly divided into two main groups, receiving DOM either through an intraperitoneal (2 mg/kg) bolus dose or through intrahippocampal (100 pmol) infusion; the two groups were matched to equivalent sham treatment vehicle control (0.9% saline) groups. The DOM doses used here have reproducibly been shown by our group to elicit similar, moderately high seizure scores.11,43 Seizure level scoring was performed for the first 2 hours immediately after drug or vehicle administration, as described below. Cumulative seizure score analysis, calculated as the sum of the highest individual scores recorded during the observation period, were used to produce excitotoxin intraperitoneally and intrahippocampally treated groups with similar seizure scores. After observation, animals within each group were randomly assigned into different groups for termination at specific endpoints. Serum samples were collected at sacrifice for analysis of circulating inflammatory cytokines and hemodynamic function assessed ex vivo. A matched naïve (untreated) control group was included, to standardize hemodynamic responses. Cardiac tissue was then processed for mitochondrial electron transport chain and citric acid cycle enzyme analysis. Midplane sections of the hearts were simultaneously retained for histological studies. Finally, a separate study measuring the same hemodynamic parameters was conducted, to examine the effects of direct DOM (1 and 5 μmol/L) administration in isolated perfused hearts.
DOM-Induced Cardiomyopathy Model
Intraperitoneal Dosing
To investigate the possibility of a direct effect of DOM on cardiac function, a single intraperitoneal bolus injection of DOM (2 mg/kg) was administered and the rats were subjected to immediate behavioral analysis.11 Animals receiving systemic DOM (intraperitoneally) were then randomly allocated into four subgroups (n = 8/group) and were sacrificed at 1, 3, 7, and 14 days after DOM administration. Two age- and weight-matched intraperitoneally saline-treated animal control groups were included (n = 5/group) and were sacrificed 1 and 14 days after saline administration.
Intrahippocampal Dosing
The potential indirect CNS-mediated effects of DOM on cardiac function were investigated by delivering DOM (100 pmol) directly into the hippocampus, as described previously.11,43 Cannula implantation surgery was performed on the intrahippocampal study group 1 week before DOM or vehicle administration. Rats were pretreated with dihydrostreptomycin (250 IU, s.c.) and carprofen (4 mg/kg, s.c.), anesthetized with ketamine (70 mg/kg, s.c.) and medetomidine hydrochloride (0.3 mg/kg, s.c.), and stereotactically (Narishige Scientific Instruments, Tokyo) implanted with a unilateral drug delivery cannula into the ventral hippocampus (using bregma as reference: 5.2 mm posterior, 5.0 mm lateral to the right, 5.2 mm depth from cranial surface). The cannula, a 24-gauge oblique needle with a 30-gauge obturator, was inserted slowly to reduce damage to the surrounding tissue and was anchored using cranioplastic cement. Body temperature was maintained at 37°C throughout the surgical procedure with a heating pad. After cannula implantation, the animals were allowed to recover and received bolus saline subcutaneously as needed to maintain their hydration status for 7 consecutive days after surgery. DOM (100 pmol) and vehicle doses were infused at rate of 1 μL/min (total volume of 1 μL) to minimize tissue injury. After intrahippocampal DOM administration and seizure analysis, animals that reached a similar seizure level as those receiving intraperitoneal DOM were randomly assigned into two groups (n = 8/group) and were sacrificed at 7 and 14 days after intrahippocampal DOM administration. A third group consisting of saline-treated controls (n = 8) received saline through the same intrahippocampal route and were sacrificed at 14 days after treatment. After termination of the studies, the brains were dissected to confirm correct cannula placement.
Behavioral Analysis after DOM Administration
In both the intraperitoneal and the intrahippocampal treatment groups, animals were habituated to an acrylic glass observation chamber for 30 minutes before DOM or vehicle administration. Experiments were conducted in a randomized, single-blind fashion. Immediately after drug administration, the animals were returned to the observation chamber and their behaviors were observed for 2 hours in a cycle of 1 minute on and 4 minutes off, with all behavioral responses during each observation period coded and logged electronically, as described previously.8,11,13 Behaviors were rated using a modified version of the seizure scale: level 0, normal resting or exploratory behaviors; level 1, discomfort behaviors; level 2, stereotypical behaviors confined to the head and neck region; level 3, moderate seizure behaviors associated with stereotypical movements of the limbs or trunk; and level 4, severe generalized seizure behaviors, with level 4 leading to level 5, clonic-tonic convulsions. Animals experiencing more than two periods of prolonged clonic-tonic convulsions were immediately sacrificed in compliance with ethical requirements and all subsequent observation periods were logged as level 5. Cumulative scores were derived as the sum of the highest scores observed during each 1-minute observation period recorded over the 2 hours after DOM or vehicle administration.8,11
DOM Levels in Serum and Myocardium after Dosing
The concentration of DOM in the heart and serum at 1 hour after administration was investigated in a separate group of animals. Male Sprague-Dawley rats (250 g, n = 5/group) were injected with DOM (2 mg/kg i.p. or 100 pmol i.h.) or intraperitoneal saline and were observed for 1 hour. Rats were decapitated without anesthesia and a trunk blood sample was taken. The cardiac ventricles were minced and washed (×5) in isolation medium A containing (in mmol/L) 225 mannitol, 75 sucrose, 10 Tris, and 0.1 EDTA (dipotassium salt) and 0.1 mmol/L phenylmethylsulfonyl fluoride (PMSF) protease inhibitor (pH 7.2, 4°C) and were homogenized in five times their volume on ice, using a glass-Teflon homogenizer. The homogenates were centrifuged (3000 × g) to produce a clear supernatant and were stored with the serum samples at −80°C until analysis. The concentration of DOM within the diluted cardiac homogenate (100-fold) and serum (1000-fold) samples was quantified using an ASP direct cELISA kit, as described previously.31,44
Cardiac Hemodynamics
Study groups were sacrificed at the specified time points. Both DOM- and vehicle-treated rats were decapitated without anesthesia and the hearts were rapidly excised into cold (4°C), filtered Krebs-Heinsleit buffer containing (in mmol/L), 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.23 KH2PO4, 24 NaHCO3, 11.1 glucose, and 1.2 CaCl2 (pH 7.4). Within 30 seconds after excision, the heart was cannulated via the aorta and perfused with oxygenated (95% O2/5% CO2) filtered Krebs-Heinsleit buffer at a constant pressure of 100 cmH2O at 37°C, using a Langendorff apparatus, as described previously.45,46 Left ventricular (LV) pressure was measured isovolumetrically using a transducer connected to a balloon inserted through the mitral valve. Analog signals were continuously recorded using a PowerLab data acquisition system with LabChart 5.2 software (ADInstruments, Sydney, Australia). The heart was allowed to equilibrate for 20 minutes and coronary flow rate (mL/min) was assessed before hemodynamic measurements were taken. Left ventricular developed pressure (LVDP) was calculated as the peak systolic pressure minus the end diastolic pressure set at 10 mmHg. Maximal and minimal first derivatives of LVDP (dP/dt maximum and dP/dt minimum, respectively) and heart rate were also assessed.
Morphological Characterization of Myocardial Injury
Perfused hearts were sectioned into 3-mm transverse sections corresponding to the middle of the ventricles and were fixed for 24 hours in 10% neutral buffered formalin and processed immediately. Paraffin-embedded sections (5 μm) were stained with Ehrlich's H&E or with picro-Sirius red (Sirius Red F3BA in aqueous saturated picric acid) to detect collagen fiber accumulation, a marker of cardiac fibrosis. The sections were examined using an Axioplan microscope with the AxioVision 3.1 image analysis system (Carl Zeiss, Göttingen, Germany). Qualitative histological evaluations were performed in a double-blind manner on five randomly selected individual fields from each ventricular section. Qualitative evaluation was made, noting formation of contractile band lesions and fragmentation of fibers (as representative of infarct damage), the presence of leukocytic inflammatory infiltrate, and subendocardial and perivascular collagen deposition (as evidence of reparative fibrosis).
Examination of Ventricular Mitochondria Function and Energetics
After sectioning, the remaining perfused ventricular tissue was minced at 4°C in isolation medium A containing the protease enzyme nagase (5 mg/g wet weight of tissue) to ensure recovery of interfibrillar as well as subsarcolemmal mitochondria. The digested tissue was repeatedly washed in medium A containing phenylmethylsulfonyl fluoride (0.1 mmol/L) and homogenized using a glass-Teflon homogenizer. A purified mitochondrial pellet was obtained using differential centrifugation, as described previously.45,46 Mitochondrial samples were aliquoted and either flash-frozen and stored at −80°C for later analysis of enzyme activities or subjected to immediate respiratory analysis. Intact mitochondrial respiratory function was measured polarographically using an oxygen electrode (World Precision Instruments, Sarasota, FL), as described previously.45,46 Oxidative phosphorylation was assessed by recording the rate of mitochondrial O2 consumption during state 4 respiration (substrate-driven rate alone) and state 3 respiration (substrate-driven rate in the presence of adenosine diphosphate). All experiments were conducted at 32°C using a standard respiratory medium saturated with oxygen, containing 100 mmol/L KCl, 0.05 mmol/L EDTA (dipotassium salt), 75 mmol/L mannitol, 25 sucrose, 10 mmol/L Tris-HCl, and 10 mmol/L KH2PO4 (pH 7.4). Mitochondrial respiration was initiated by the addition of cardiac mitochondria (0.5 mg) in the presence of essentially fat-free bovine serum albumin (0.2 mg) to make up a final chamber volume of 250 μL. After equilibration, at which point a steady endogenous rate of respiration had been reached, flavin adenine dinucleotide (FAD)-linked state 4 respiration was initiated by administration of succinate (7 mmol/L final concentration) in the presence of the NADH-dehydrogenase inhibitor rotenone (50 mmol/L). State 3 respiration was initiated by the addition of ADP (200 nmol). Mitochondrial respiratory control indices were calculated as the ratio of state 3 to state 4 respiration rates.
Mitochondrial energetic assays were conducted on lysed mitochondrial samples fractured by three rapid freeze-thawing cycles and diluted to a final concentration of 1 mg mitochondrial protein mL−1 in PMSF containing isolation medium A. Assays for mitochondrial electron transport chain enzyme kinetics: complex I (NADH-ubiquinone oxidoreductase, EC 1.6.99.3), complex II/III (succinate-ubiquinone/ubiquinol-cytochrome c reductase, EC 1.8.3.1), and complex V (ATPase, EC 3.6.1.3), as well as citrate synthase (EC 4.1.3.7; a mitochondrial matrix enzyme) and aconitase [citrate (isocitrate) hydro-lyase, EC 4.2.1.3; a marker of oxidative stress], were performed essentially as described previously.31,45,46 Mitochondrial enzyme activities were assessed using a SpectraMax-Plus 96-well spectrophotometer (Molecular Devices, Crawley, UK) at 30°C. Optical path-lengths were corrected for microplate use, and enzyme activities were expressed as nmol substrate/min/mg protein.
Circulating Inflammatory Cytokine Analysis
Cytokine levels were quantitatively assessed in 50-μL plasma samples collected at the time of sacrifice from all treatment groups using a Bio-Plex bead-array immunoassay kit (Bio-Rad Laboratories) with a Luminex 100 analyzer (Luminex, Austin, TX), as described previously.47,48 Cytokine concentrations were calculated from a standard curve (detectable range of 2 to 32,000 pg/mL) for rat interleukins IL-1α and IL-1β, tumor necrosis factor-α (TNF-α), and granular monocyte-colony stimulating factor (GM-CSF), using Bio-Plex Manager software (Bio-Rad Laboratories).
Exposure of Isolated Hearts to DOM
The acute direct effect of DOM (1 and 5 μmol/L) was assessed in the isolated heart model using the same hemodynamic and mitochondrial respiratory function analysis as described above. Hearts isolated from Sprague-Dawley rats (n = 5 per group) were perfused with oxygenated (95% O2/5% CO2) filtered Krebs-Henseleit buffer (37°C) and were allowed to equilibrate at a constant pressure of 100 cmH2O. After baseline hemodynamic recordings, hearts were perfused with DOM (final concentration of 1 or 5 μmol/L dissolved in the perfusate) delivered through a side port in the aortic cannula for 20 minutes. Cardiac function was immediately assessed at 10 mmHg LV diastolic pressure and the hearts were subjected to a 20-minute washout period before a final assessment of hemodynamic function at 40 minutes after initial DOM exposure. Results were compared with data obtained in time-matched saline-treated controls.
Statistics
Statistical analysis was performed using SigmaStat v2.03 (SPSS, Chicago, IL) by a one-way analysis of variance (ANOVA) or Student's t-test for paired comparison. Bonferroni and Tukey pairwise tests were used for post hoc comparisons between control and treatment groups. Results were expressed as means ± SEM, with a P value of <0.05 being considered significant. Inverse log correlations were performed if equal variance failed, and one-way ANOVA and post hoc pairwise analysis were repeated.
Results
Behavioral Seizure Profile after Intraperitoneal or Intrahippocampal DOM
Animals receiving saline through either intraperitoneal or intrahippocampal routes did not differ significantly from each other in their behavior and exhibited low cumulative behavioral score (Figure 1). DOM intraperitoneal administration, however, resulted in significantly elevated cumulative behavioral scores, compared with saline-treated animals (ANOVA, F = 103.446; P < 0.001). The levels of seizures experienced by all animals within this intraperitoneal DOM group were similar, with rats exhibiting stage 4 seizures, approaching clonic-tonic convulsions, within 2 hours of DOM administration. Animals receiving intrahippocampal DOM also exhibited significantly higher cumulative behavioral scores, compared with saline-treated animals (ANOVA, F = 32.498; P < 0.001). Level 4 behavior was observed earlier, at approximately 40 minutes after intrahippocampal DOM infusion. The level of seizures experienced by animals receiving either intrahippocampal or intraperitoneal DOM treatment were similar and not significantly different (one-way ANOVA, F = 1.484; P = 0.229).
Figure 1.
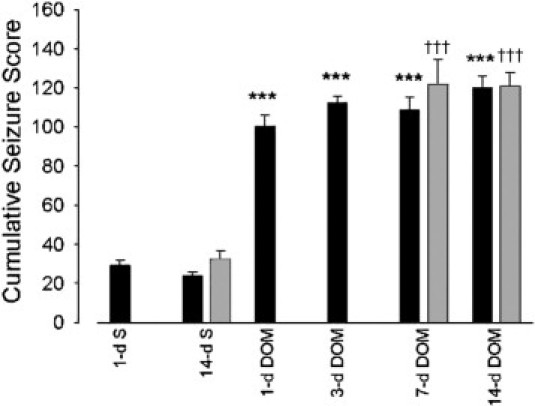
Cumulative behavioral seizure scores in rats were recorded over 2 hours after intraperitoneal (black bars; 1, 3, 7, and 14 days) or intrahippocampal (gray bars; 7 and 14 days) administration of domoic acid (DOM). Data are expressed as means ± SEM of 8 separate experiments. ***P < 0.001 versus intraperitoneal saline-treated animals (1 and 14 day); †††P < 0.001 versus intraperitoneal intrahippocampal saline-treated animals (1 and 14 day). S, saline.
DOM Levels in Serum and Heart after Intraperitoneal or Intrahippocampal Administration
Analysis of serum and cardiac samples at 1 hour after a single intraperitoneal injection of DOM confirmed the presence of DOM in serum (630.1 ± 121.4 ng/mL) and in ventricular homogenates (19.6 ± 2.7 ng/g tissue). Levels of DOM in saline-treated rats fell below the limits of detection of the immunoassay. Intrahippocampal administration of 100 pmol DOM also did not result in a detectable level of DOM in the systemic circulation.
Comparison of Cardiac Hemodynamics after Intraperitoneal and Intrahippocampal DOM-Induced Seizures
Myocardial function was evaluated by measuring LVDP at 10 mmHg diastolic pressure in perfused hearts isolated from animals at specific times after intraperitoneal or intrahippocampal DOM administration (Figures 2 and 3). Naïve control and saline-treated (1 and 14 day) animals did not significantly differ in their LVDP indices. DOM intraperitoneal administration resulted in decreased LVDP values by the first day, compared with all other controls in this group (naïve control, P < 0.01; 1-day saline, P = 0.002; 14-day saline, P = 0.019) (Figure 2A). LVDP was reduced further in a time-dependent fashion at 3 days (P < 0.01 versus untreated and saline-treated controls), 7 days (P < 0.001 versus untreated and saline-treated controls), and 14 days (ANOVA, F = 44.075; P < 0.001 compared with all three control groups) after DOM administration. Both rate derivatives of diastolic (dP/dt minimum) and systolic (dP/dt maximum) pressures were also compromised over the same 14-day time scale after intraperitoneal DOM dosing (Figure 2, B and C). Heart rate recorded in isolated hearts (at 10 mmHg diastolic pressure) after intraperitoneal dosing was not affected at any time point.
Figure 2.
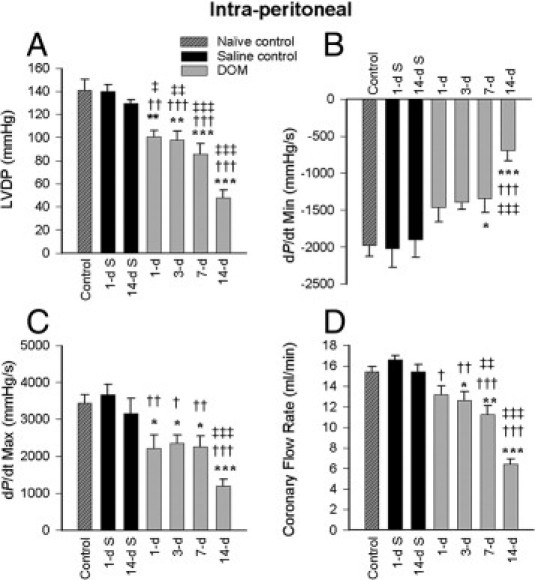
Cardiac hemodynamic function at 1, 3, 7, and 14 days after intraperitoneal DOM administration, compared with naïve control and 1- and 14-day saline-treated control rats: left ventricular developed pressure (LVDP) (A), dP/dt minimum (B), dP/dt maximum (C), and coronary flow at 10 mmHg diastolic pressure (D). Data are expressed as means ± SEM of 8 separate experiments. S, saline. *P < 0.05, **P < 0.01, and ***P < 0.001 versus naïve control; †P < 0.05, ††P < 0.01, and †††P < 0.001 versus 1-day saline; ‡P < 0.05, ‡‡P < 0.01, and ‡‡‡P < 0.001 versus 14-day saline.
Figure 3.
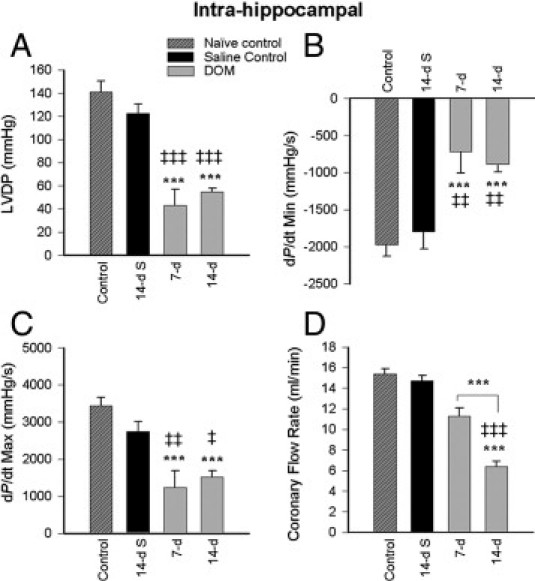
Cardiac hemodynamic function at 7 and 14 days after intrahippocampal DOM administration, compared with naïve control and 14-day saline-treated control rats: LVDP (A), dP/dt minimum (B), dP/dt maximum (C), and coronary flow at 10 mmHg diastolic pressure (D). Data are expressed as means ± SEM of 8 separate experiments. S, saline. ***P < 0.001 versus naïve control; ‡P < 0.05, ‡‡P < 0.01, and ‡‡‡P < 0.001 versus 14-day saline.
Comparison of LVDP values between naïve untreated animals and intrahippocampal saline-treated animals at 14 days (Figure 3A) confirmed that intrahippocampal cannula implantation did not compromise cardiac hemodynamic function. LVDP was significantly reduced at 7 days after DOM (intrahippocampal) treatment (P < 0.001 versus naïve control; P = 0.003 versus 14-day saline) with no further reduction seen at 14 days. Comparison of cardiac LVDP indices in hearts isolated at 7 days showed that intrahippocampal DOM dosing (Figure 3A) reduced developed pressure indices to a greater extent than intraperitoneal DOM dosing (Figure 2A) (Student's t-test, t = −2.55; P = 0.043). Both pressure-rate derivatives (dP/dt minimum and maximum) were also significantly and irreversibly impaired by 7 days after intrahippocampal DOM administration (Figure 3B and 3C). As in the intraperitoneal model, intrahippocampal dosing with DOM produced no significant residual effect on heart rate.
Systemic (intraperitoneal) dosing of DOM resulted in a time-dependent decrease in coronary flow rate over the 14-day time period (1 day versus 1-day saline, P < 0.05; 3 days versus 1-day saline, P < 0.01; ANOVA, F = 6.621; 7 days versus 1-day saline, P < 0.001; 7 days versus 14-day saline, P < 0.01) (Figure 2D). Intrahippocampal delivery of DOM also resulted in a gradual and equivalent decline in coronary flow (Figure 3D) which reached significance at 14 days (ANOVA, F = 16.795; P < 0.001 versus naïve control and 14-day saline). No significant difference was seen in coronary flow response at 14 days between either route of DOM administration.
Cardiac Mitochondrial Respiratory Damage after Intraperitoneal and Intrahippocampal DOM-Induced Seizures
To explore the mechanism behind the hemodynamic dysfunction observed, mitochondria were isolated from the hearts and respiratory function was assessed in the pooled preparation of interfibrillar and subsarcolemmal mitochondria. FAD-linked mitochondrial respiratory parameters were assessed in isolated cardiac mitochondria at each time point after intraperitoneal (Table 1) and intrahippocampal (Table 2) DOM dosing. Systemic (intraperitoneal) administration of DOM resulted in a significant impairment of mitochondrial respiratory function, as indicated by a decline in respiratory control index, evident at 1, 3, and 7 days after seizure induction. Maximal mitochondrial dysfunction was evident at 14 days after DOM-induced seizures, compared with all control groups (ANOVA, F = 15.507; P < 0.001). State 4 respiration rate was significantly increased, indicative of proton leak at 3, 7 (P < 0.05), and 14 days (ANOVA, F = 63.857; P < 0.001) after DOM, compared with 14-day saline-treated animals (Table 1). State 3 respiratory rate, however, was significantly affected only at 14 days after intraperitoneal DOM-induced seizures (P < 0.05 versus 14-day saline).
Table 1.
Temporal Assessment of Cardiac Mitochondrial Respiratory Function (FAD-Linked) at 1 to 14 Days after Systemic Administration of Domoic Acid
| Treatment | RCI | Rate (ng O2 min−1 mg protein−1) |
|
|---|---|---|---|
| State 4 | State 3 | ||
| Naïve control | 6.852 ± 0.304 | 41.549 ± 2.89 | 249.039 ± 18.241 |
| Saline | |||
| 1 day | 7.135 ± 0.348 | 37.199 ± 2.824 | 268.22 ± 25.66 |
| 14 days | 7.037 ± 0.252 | 34.785 ± 1.768 | 242.638 ± 5.557 |
| Domoic acid | |||
| 1 day | 5.487 ± 0.445†‡ | 41.382 ± 7.748 | 214.669 ± 29.596 |
| 3 days | 5.689 ± 0.399 | 43.204 ± 1.846 | 244.183 ± 18.173 |
| 7 days | 4.874 ± 0.604⁎††‡‡‡ | 61.88 ± 6.177†‡ | 245.144 ± 3.138 |
| 14 days | 4.631 ± 0.302⁎⁎††† | 54.91 ± 1.297⁎⁎†††‡‡‡ | 263.757 ± 4.521‡ |
Each value represents the mean ± SEM of eight separate experiments.
RCI, respiratory control index.
P < 0.05,
P < 0.01 versus naïve control.
P < 0.05,
P < 0.01, and
P < 0.001 versus 1-day saline.
P < 0.05,
P < 0.001 versus 14-day saline.
Table 2.
Temporal Assessment of Cardiac Mitochondrial Respiratory Function (FAD-Linked) at 7 to 14 Days after Intrahippocampal Administration of Domoic Acid
| Treatment | RCI | Rate (ng O2/min/mg protein) |
|
|---|---|---|---|
| State 4 | State 3 | ||
| Naïve control | 6.85–0.30 | 41.55–2.89 | 249.04–18.24 |
| 14 days saline | 6.69–0.24 | 39.34–3.81 | 261.91–16.01 |
| 7 days DOM | 4.64–0.44⁎⁎⁎‡‡ | 55.81–7.62 | 268.26–56.98 |
| 14 days DOM | 4.89–0.26⁎⁎⁎‡‡ | 54.61–5.31‡ | 262.42–24.79 |
Each value represents the mean ± SEM of eight separate experiments.
DOM, domoic acid; RCI, respiratory control index.
P < 0.001 versus naïve control.
P < 0.05,
P < 0.01 versus 14 day saline.
Mitochondrial respiratory control index ratios were also equally affected at 7 days after intrahippocampal DOM (Table 2) (ANOVA, F = 13.753; P < 0.001 versus control; P = 0.003 versus 14-day saline), with no further damage detected at 14 days. Comparison of the mean oxidative (state 4) rate values among the four intrahippocampal treatment groups (ANOVA, F = 2.985; P = 0.05) showed that state 4 increased in response to intrahippocampal DOM at 14 days, reaching significance (Student's t-test; P = 0.04) versus the 14-day saline control. Phosphorylative (state 3) components of the respiration control, however, were not significantly affected by this central route of DOM delivery.
Cardiac Mitochondrial Enzyme Dysfunction after Intraperitoneal and Intrahippocampal DOM-Induced Seizures
To investigate the cardiac energetic dysfunction further, the kinetic activity of individual mitochondrial respiratory complexes was examined in the isolated mitochondria. Systemic (intraperitoneal) DOM administration provoked a reduction in complex I activity at 7 days (ANOVA, F = 5.43; P < 0.05 versus 1-day and 14-day saline) and 14 days (P < 0.001, 1-day saline; P < 0.05, 14-day saline) after bolus injection (Figure 4A). Complex II-III activity also displayed significant impairment at 1, 3, 7, and 14 days after DOM (ANOVA, F = 34.051; P < 0.001) (Figure 4B). Complex V ATPase activity (Figure 4C) became significantly compromised at 3 days (P < 0.05, 1-day saline; P < 0.01, 14-day saline), and at 7 and 14 days (P < 0.01 versus 1-day saline; P < 0.001 versus 14-day saline) after DOM administration. Citrate synthase activity (Figure 4D) was reduced as early as 1 day after DOM treatment (P = 0.023 versus 1-day saline; P = 0.006 versus 14-day saline), with the maximal loss of activity occurring in mitochondria isolated 7 days after DOM treatment (ANOVA, F = 14.17; P < 0.001). Aconitase activity was used as a marker of oxidative stress; however, there appeared to be very little change in aconitase activity, with the only significant inactivation occurring at 14 days after DOM dosing (14-day saline; P = 0.022) (Figure 4E).
Figure 4.
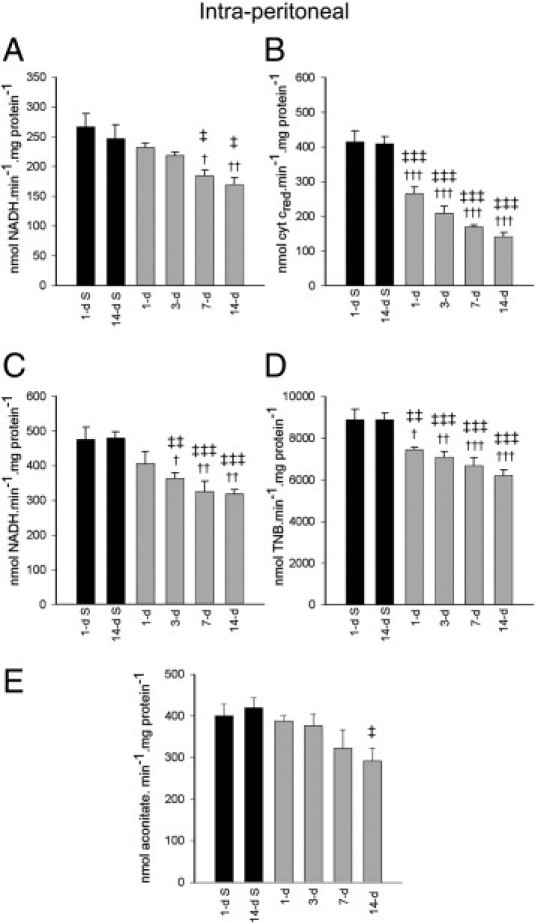
Isolated cardiac complex I (A), complex II-III (B), complex V (C), citrate synthase (D), and aconitase activities (E) at 1, 3, 7, and 14 days after intraperitoneal DOM administration (gray bars) in rats, compared with 1 and 14 days after saline administration in controls (black bars). Data are expressed as means ± SEM of 8 separate experiments. †P < 0.05, ††P < 0.01, †††P < 0.001 versus 1-day saline; ‡P < 0.05, ‡‡P < 0.01, and ‡‡‡P < 0.001 versus 14-day saline.
Infusion of DOM into the brain (intrahippocampal) also resulted in a fall in cardiac mitochondrial complex I activity, which reached significance at 14 days (P < 0.01 versus 14-day saline control) (Figure 5A). Complex II-III, complex V ATPase, citrate synthase, and aconitase activities were also significantly compromised at 7 days after intrahippocampal DOM treatment, with no further reduction observed at 14 days (Figure 5, B–E). The degree of mitochondrial enzyme impairment at 7 and 14 days after intrahippocampal administration was similar and did not differ significantly from that seen after intraperitoneal DOM administration.
Figure 5.
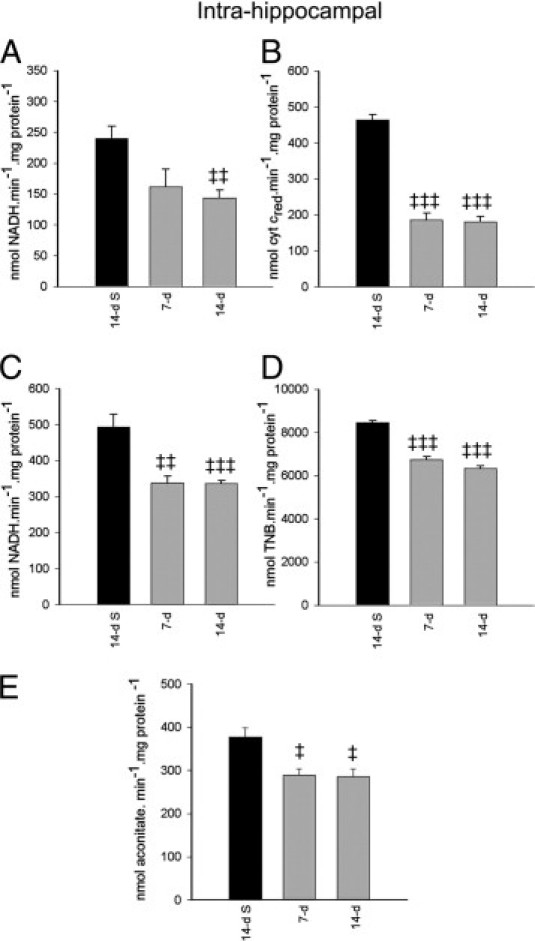
Isolated cardiac complex I (A), complex II-III (B), complex V (C), citrate synthase (D), and aconitase activities (E) at 7 and 14 days after intrahippocampal DOM administration (gray bars) in rats, compared with 14 days after saline administration in controls (black bars). Data are expressed as means ± SEM of 8 separate experiments. ‡P < 0.05, ‡‡P < 0.01, and ‡‡‡P < 0.001 versus 14-day saline.
Histopathological Findings after DOM Administration
Examination of hearts isolated from intraperitoneal and intrahippocampal saline-treated rats at day 14 showed a normal histological appearance (Figure 6, A and B), with no remarkable subendocardial fibrous deposition (Figure 7, A and D).
Figure 6.
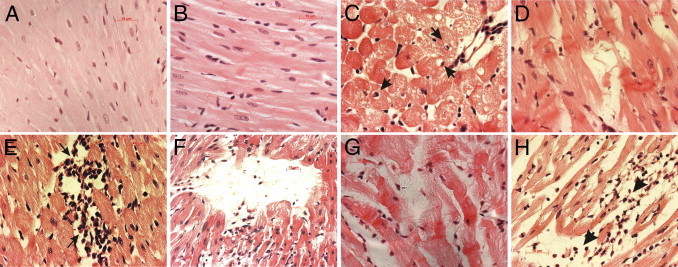
Histopathological changes in LV myocardium of saline- or DOM-treated rats, visualized using H&E stain. A: Normal histology of cardiomyocytes, 14 days after intraperitoneal saline. B: Equally intact tightly packed cardiomyocytes with cross-striations, 14 days after intrahippocampal saline. C: Reversible degenerative cardiomyopathy, evidenced by myocyte vacuolization (arrowheads indicate intracellular vacuoles displacing nuclei) and edema, 3 days after intraperitoneal DOM. D: Prominent hypercontraction bands associated with fiber derangement, 3 days after intraperitoneal DOM. E: Numerous interstitial inflammatory cells in proximity to necrotic cells typical of coagulation necrosis and evidence of interstitial edema, 7 days after intraperitoneal DOM. F: Phagocytosis of necrotic myocardium with inflammatory infiltrate and edema still present, at 14 days after intraperitoneal DOM. G: Hypercontraction bands with fiber derangement and edema, 7 days after intrahippocampal DOM. H: Muscle fibers disintegrated into eosinophilic granular material with extensive inflammatory infiltrate (arrowheads) and edema, 14 days after intrahippocampal DOM. Scale bars: 10 μm.
Figure 7.
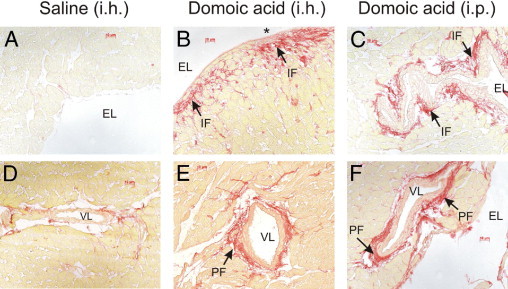
Both intraperitoneal and intrahippocampal delivery of DOM-induced cardiac fibrosis at 14 days after drug administration, as seen in representative photomicrographs of picro-Sirius-stained LV subendocardium sections. A: Saline control (intrahippocampal dosed) rats, with absence of Sirius red staining (absence of fibrosis). B and C: DOM intrahippocampal (B) and DOM intraperitoneal (C) dosed rats show evidence of interstitial fibrosis and reparative fibrosis. D–F: Evidence of increased LV perivascular fibrosis in DOM-dosed intrahippocampal (E) and intraperitoneal (F) animals, compared with saline-treated control (D) control showing modest collagen levels surrounding the coronary artery. Asterisk indicates reparative fibrosis. EL, endocardial lumen; IF, interstitial fibrosis; PF, perivascular fibrosis; VL vascular lumen. Scale bars: 10 μm.
Histological examinations of hearts from intraperitoneal DOM-treated animals using H&E stain showed evidence of diapedesis at 1 day after dosing. No myocyte alterations were detected in the heart until at 3 days after systemic DOM administration. At this time point, these alterations consisted of cardiomyocyte vacuolization, regarded as a histological marker of early reversible cardiac damage, as well as hypercontraction bands, multifocal degeneration of muscle fibers, and interstitial edema (Figure 6, C and D). Minimal inflammatory cell infiltration was observed. At 7 days, hypercontraction bands and myofiber loss (myocardial necrosis) with interstitial (Figure 6E) and perivascular inflammatory cell infiltrate were prominent mainly in the subendocardial regions of the septum and left ventricle. Evidence of mild interstitial fibrosis and perivascular fibrosis (collagen accumulation within the adventitia of coronary arteries and arterioles) was also seen in 5 out of 8 rats. These findings were still present at 14 days in all intraperitoneal DOM-dosed animals (Figure 6F), with extracellular matrix collagen accumulation seen, indicative of subendocardial reparative and interstitial fibrosis and of perivascular fibrosis (Figure 7, C and F).
Intrahippocampal DOM administration resulted in a myocardial histopathology similar to that observed after intraperitoneal DOM. However, intrahippocampal DOM-treated animals also displayed evidence at the 7-day termination point (3 out of 8 rats) and at the 14-day termination point (two out of eight rats) of erythrocyte infiltration, suggesting intramyocardial hemorrhage. Myocardial necrotic lesions consisting of hypercontraction bands and degeneration of muscle fibers coupled with interstitial edema and inflammatory cell infiltrate were also evident within the LV subendocardium, papillary muscle, and septum at 7 days after intrahippocampal DOM (Figure 6G). At 14 days after intrahippocampal dosing, necrotic myocytes with inflammatory infiltrates and dense connective tissue staining were also noted extensively throughout the same cardiac regions (Figure 6H and Figure 7, B and E).
Increases in Circulating Inflammatory Cytokines after Intraperitoneal and Intrahippocampal DOM Administration
Significant increases in circulating IL-1α levels were detected in the serum only at 14 days after DOM intraperitoneal dosing, compared with 1-day and 14-day saline-treated rats (ANOVA, F = 5.47; P < 0.05) (Figure 8A). Similar IL-1α increases were also evident in rats receiving DOM intrahippocampal (ANOVA, F = 6.426; P < 0.05) (Figure 8B). Increases in circulating IL-1β levels, however, were seen as early as 7 days after intraperitoneal DOM and continued to be elevated at 14 days (ANOVA, F = 5.49; P < 0.05) (Figure 8C). Increased IL-1β levels were also noted at 7 days after intrahippocampal DOM administration (ANOVA, F = 5.43; P < 0.05) (Figure 8D), but subsequently returned to control levels at 14 days. TNF-α levels were increased as early as 3 days after intraperitoneal DOM (ANOVA, F = 13.2; P = 0.014 versus 1-day saline; P = 0.004 versus 14-day saline) (Figure 8E), with no further increase seen at 7 or 14 days (ANOVA, F = 13.20; P < 0.05, 1-day saline; P < 0.01, 14-day saline) (Figure 8E). Significant increases in TNF-α circulating levels were evident at 7 and 14 days after intrahippocampal DOM administration (ANOVA, F = 6.81; P < 0.05) (Figure 8F), compared with 14-day saline-treated animals. GM-CSF levels were not significantly elevated, however, after either intraperitoneal (Figure 9A) or intrahippocampal (Figure 9B) DOM administration.
Figure 8.
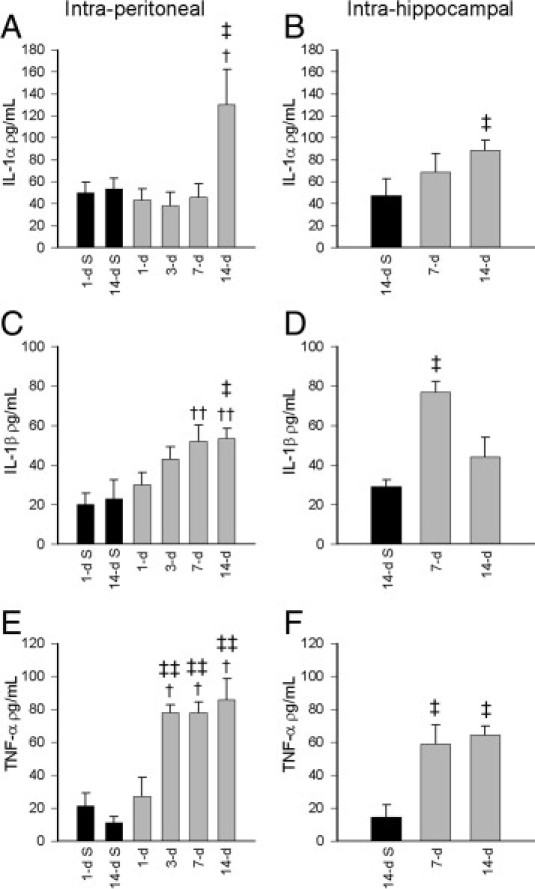
Stimulatory effect of intraperitoneal (A, C, E) and intrahippocampal (B, D, F) DOM delivery in rats on circulating proinflammatory cytokine IL-1α, IL-1β, and TNF-α levels at 1- to 14-day termination points after administration of DOM (gray bars), compared with the respective saline-treated controls (black bars). Data are expressed as means ± SEM of 8 separate experiments. †P < 0.05, ††P < 0.01 versus 1-day saline; ‡P < 0.05, ‡‡P < 0.01 versus 14-day saline.
Figure 9.
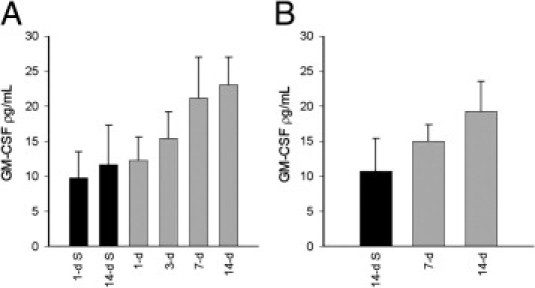
Effect of intraperitoneal (A) and intrahippocampal (B) DOM delivery in rats on circulating GM-CSF levels at termination points 1, 3, 7, and 14 days after administration of DOM (gray bars), compared with saline-treated controls at 1 and 14 days (black bars). Data are expressed as means ± SEM of 8 separate experiments.
Cardiac Function after Acute DOM Administration in Isolated Hearts
Baseline pretreatment hemodynamic indices (LVDP, dP/dt maximum and minimum, or heart rate) recorded after equilibration were not significantly different between groups. Delivery of 1 or 5 μmol/L DOM to isolated hearts failed to significantly impair cardiac hemodynamic parameters, either immediately after 20-minute exposure or at the 40-minute termination point (data not shown).
Discussion
The novel finding of the present study is that localized infusion of DOM into the hippocampus can evoke cardiac dysfunction and morphological changes resembling the cardiomyopathy obtained after systemic (intraperitoneal) administration of DOM. Previously, however, we reported that direct exposure of isolated cardiomyocytes to DOM did not affect cellular viability or energetic function, suggesting that the cardiomyopathy observed with DOM in the present study could not have resulted from a direct cardiotoxic action.31 The myocardial pathology seen in the present in vivo study was shown to be preceded in both DOM treatment groups by severe seizure activity. In each case, the functional, molecular, and microscopic presentation described was found to deteriorate with time after the initial DOM-induced insult. Examination of cardiac function in isolated hearts subjected to DOM infusion, however, failed to provoke a hemodynamic effect, confirming that DOM has no direct pathological effect in the rat heart model.
Previous descriptions of DOM toxicity in humans and animals have focused primarily on neurological sequelae culminating in seizure activity.1,5,6,8,21,22 Of direct relevance to development of our hypothesis were indications that systemic dosing with domoic and kainic acids provoke the formation of epileptiform activity originating primarily from the hippocampus.49 Using intrahippocampal dosed animals, Sawant and colleagues13 from our research group used electrocorticography to show that DOM-induced epileptiform discharges spread rapidly to surrounding extralimbic regions. As expected, animals subjected to DOM infusion into the right hippocampus in the present study exhibited consistently high level convulsive seizures (level 4) within the first 2 hours after drug administration. Similar high seizure scores were obtained in the systemically (intraperitoneal) dosed animal groups, indicating that DOM is able to traverse the blood-brain barrier. These findings are consistent with previous studies from our group, which established that intraperitoneal delivery of 2 mg/kg DOM resulted in significantly elevated levels of DOM in the brain.44 The present study has also shown that, although cardiac levels of DOM were increased within 1 hour of intraperitoneal dosing (2 mg/kg) with the excitotoxin, localized intrahippocampal infusions with 100 pmol DOM failed to produce a detectable level of DOM in the circulation. This is not surprising, because, even though the integrity of the blood-brain barrier may be compromised during seizure activity, resulting in systemic diffusion, the original 100 pmol dose applied would have been greatly diluted.11 Consequently, we discount the possibility of DOM release from the hippocampus reaching the heart.
Left ventricular hemodynamic parameters assessed in isolated hearts at specific time points after DOM administration provided clear evidence of a combined deterioration of both systolic and diastolic function. The degree of residual cardiac dysfunction exhibited at the final termination point (14 days) did not differ between the systemic and hippocampal dosing. However, the onset of damage appeared to be more rapid in the intrahippocampal animals, which sustained a comparatively greater degree of damage at day 7 than did their intraperitoneal counterparts. The impairment in LV contractile force and developed pressure indicates that ventricular compliance and cardiac output were compromised in a progressive manner suggestive of dilated cardiomyopathy.50 Although in vivo DOM administration did not appear to influence heart rate when measured in the isolated organ, suggesting a lack of residual effect of seizure on isolated cardiac automaticity, coronary flow was markedly impaired when examined at the earliest time points in both intraperitoneal and intrahippocampal groups. This reduction in coronary flow progressively increased at each time point in the present study and would have resulted in reduced oxygen delivery and hypoxia in the ventricular myocardium. Such an ischemic event is consistent with the damage seen to ventricular mitochondrial respiratory control indices in both DOM study groups. FAD-linked state 4 respiration rates were primarily affected, showing a time-dependent increase after DOM dosing, indicative of proton loss across a disrupted mitochondrial energy gradient.45,46
Mitochondrial electron transport chain enzyme activities were assessed as part of the present study and showed a similar deterioration particularly, in FAD-linked complex II, confirming a loss of energetic capability. It is well established that a large acute ischemic insult will result in damage to cardiac mitochondrial structural integrity and energetic function and generate reactive oxygen species.45,46 However, activity of cardiac mitochondrial aconitase, a superoxide labile enzyme, was not greatly impaired in the myocardium of animals until at least 7 days (intrahippocampal group) after DOM administration. This indication of low oxidative stress damage to the ventricular mitochondria suggests that the damage may have resulted from a series of small ischemic insults over time. The correlation between the loss of energetic capability and hemodynamic function seen in the present study further supports the hypothesis that DOM administration through systemic or hippocampal administration provoked a cardiac ischemic event.
The catastrophic ischemic damage to the mitochondria and hemodynamic function was reflected in the morphological appearance of the ventricular myocardium after DOM treatment. The cardiomyopathy presented by both intraperitoneal and intrahippocampal groups was distributed primarily in the subendocardial, papillary muscle, and septal regions around the left ventricle. Microscopically, the lesions manifested as multifocal discrete necrotic regions containing inflammatory cell infiltrate (most likely consisting of macrophages and leukocytes). Interspersed with this presentation of coagulation necrosis and cardiomyocyte vacuolization, evidence of catecholamine toxicity seen as hypercontraction band necrosis, was also noted.51 As the duration of time since original seizure induction progressed, the distribution of myocardial necrosis became more extensive around the subendocardial region. By day 14, cardiac tissue morphology was characterized by dead phagocytosed tissue and dense interstitial and perivascular connective tissue. A similar level of pathological change was evident in both treatment groups; however, indications of erythrocyte extravasation and intramyocardial hemorrhage were present only in animals receiving intrahippocampal DOM. This suggests that seizure induction by direct infusion of DOM into the hippocampus may have provoked a more extreme cardiac pathological event than experienced after intraperitoneal DOM administration.
The DOM-induced cardiomyopathy in this rat model was accompanied by significant increases in plasma TNF-α, IL-1α, and IL-1β levels, which were similar between the intraperitoneal and intrahippocampal paradigms. A greater increase in plasma IL-1α levels was observed in the intraperitoneal group, however, compared with the intrahippocampal group, which may have resulted from peripheral inflammatory damage provoked by systemic exposure to DOM. Similarly, although IL-1β levels fell in the intrahippocampal group at day 14, they remained significantly up-regulated at the same time point in the intraperitoneal group. IL-1α and IL-1β are structurally distinct, but have overlapping biological functions; the decrease in IL-1β at 14 days after intrahippocampal DOM administration may therefore be functionally compensated for by the increase in IL-1 α. Increased expression of proinflammatory cytokines such as IL-1β by glial cells has been shown to occur rapidly within regions of the CNS system where seizures are initiated and may exacerbate the degree of neuronal injury resulting from the seizure activity.52
The increased expression of circulating proinflammatory cytokines seen after cardiac hemodynamic and mitochondrial dysfunction in the present study, however, is more likely to be a consequence of the myocardial damage noted, rather than an indication of a neuronal inflammatory event. Indeed, proinflammatory TNF-α, IL-1β and IL-6 have been reported to be released after myocardial ischemic injury and may stimulate the expression of matrix metalloproteinases (MMPs) contributing to myocardial remodeling, and progression of heart failure.53 The delayed elevation of IL-1α at 14 days supports the scenario of perpetuated rather than initiated cardiac damage observed in the present study. IL-1 has been previously shown to stimulate adult cardiac fibroblast migration, down-regulate fibrillar collagen synthesis, increase fibronectin and nonfibrillar procollagen transcription, and increase MMP protein and activity, all characteristics of cardiac remodeling.54,55 In addition to promoting cardiac remodeling, IL-1 has also been shown to act in synergy with TNF-α to produce a delayed and prolonged phase of decreased myocardial contractility.56 These known effects of IL-1 correlate well with the fibrosis and decreased cardiac contractility observed in the present study. GM-CSF levels did not rise significantly within the time period studied after seizure induction. As both morphological integrity and hemodynamic parameters also progressively fell, these data do not support the notion of a GM-CSF/TNF-α-mediated angiogenic response over the time period studied.57
The association of the ischemic cardiomyopathy with the induction of seizures in these two models suggests that DOM has a central rather than peripheral action. Seizures originating in frontotemporal cortex, insular cortex, amygdala, and hippocampus can spread into key CNS cardiovascular control centers, including ventrolateral thalamus, paraventricular nucleus, medial parabrachial nuclei, locus ceruleus, ventrolateral pons, nucleus tractus solitarius, nucleus ambiguus, and finally the dorsal motor nucleus of the vagus nerve.58–60 Epileptiform discharges in preganglionic parasympathetic (dorsal motor nucleus of the vagus and nucleus ambiguus) and sympathetic (both caudal and rostral ventrolateral medulla) control areas are widely thought to account for supraventricular and sinus tachycardia, as well as bradycardia, atrioventricular block, and asystoles known to occur in SUDEP. Importantly, the activation of the hippocampus in temporal lobe epilepsy has been associated with neuronal discharges onto cardiac sympathetic and vagal neurons, resulting in arrhythmia.61 In the present study, we administered a scant 100 pmol of domoic acid directly into ventral hippocampus. Although we cannot rule out the spread of the neurotoxin into adjacent regions, preliminary evidence collected using telemetry confirms a rapid appearance of ictal activity in the contralateral cortex with concomitant disturbance in electrocardiography, suggesting that it is the spread of electrical discharge, and not the toxin itself, that drives autonomic disturbances. Additional proof of these cardiac sequelae to seizure-induced discharges may be obtained by repeating the intrahippocampal study component using a cardiac sympathectomy model.
The microscopic evidence collected here is strikingly similar to clinical evidence collected postmortem from patients suffering sudden death after epilepsy62 and supports the hypothesis that cardiomyopathy after DOM exposure is a consequence of seizure activity. The presentation of catecholamine-associated hypercontraction band necrosis and coagulation necrosis resulting from ischemic insult are consistent with the conclusion of a seizure-induced sympathetic storm. Catecholamine-induced coronary microvascular spasms are likely to contribute to the appearance of ischemic infarcts seen distributed throughout the subendocardium of the left ventricle. Given that this region is exposed to the highest ventricular pressure and is poorly supplied by collateral vessels, compression of the microcoronary supply by the surrounding hypercontracting myocardium renders the subendocardium exquisitely sensitive to hypoxia.63
Findings from the present study reinforce our earlier studies suggesting that domoic acid does not exert a direct effect on myocardium.31 To summarize, our novel findings of cardiomyopathy in rats infused intrahippocampally with DOM strongly suggest that the resultant ischemic structural and mitochondrial damage seen together with a combined systolic/diastolic dysfunction is a consequence of seizure-induced discharges from hippocampal and adjacent limbic structures onto cardiac sympathetic neurons. The finding that equipotent concentrations of intraperitoneal DOM also resulted in no greater degree of cardiac pathology further supports the hypothesis that the DOM-induced cardiac damage essentially resulted from a seizure-induced autonomic response. This was borne out by the absence of any significant hemodynamic effect resulting from acute exposure of isolated hearts to DOM. We have shown that the pathology resulting in the in vivo treatment studies bears a strong resemblance to clinical descriptions of cardiac injury arising after temporal lobe epilepsy and submit that this model of epileptiform induction forms an excellent basic research tool for study of the temporal arrhythmogenic and respiratory mechanisms of SUDEP. Finally, we believe that these findings strongly reinforce the call for regular cardiac monitoring in epilepsy patients.
Acknowledgments
We thank Amanda Fisher for her expertise in preparing the tissues for histology and Dr. Andrew Clarkson for his assistance with mitochondrial assays.
Footnotes
Supported by equipment grants from the National Heart Foundation of New Zealand (J.C.H., I.A.S.), from the New Zealand Lottery Health (J.C.H., I.A.S.), and by University of Otago School of Medical Sciences bequest grants (J.C.H., I.A.S., S.K.).
References
- 1.Perl T.M., Bédard L., Kosatsky T., Hockin J.C., Todd E.C.D., Remis R.S. An outbreak of toxic encephalopathy caused by eating mussels contaminated with domoic acid. N Engl J Med. 1990;322:1775–1780. doi: 10.1056/NEJM199006213222504. [DOI] [PubMed] [Google Scholar]
- 2.Teitelbaum J., Carpenter S., Cashman N.R. Neurologic sequelae after ingestion of mussels contaminated with domoic acid. N Engl J Med. 1990;323:1632–1633. doi: 10.1056/NEJM199012063232313. [DOI] [PubMed] [Google Scholar]
- 3.Lefebvre K.A., Bargu S., Kieckhefer T., Silver M.W. From sanddabs to blue whales: the pervasiveness of domoic acid. Toxicon. 2002;40:971–977. doi: 10.1016/s0041-0101(02)00093-4. [DOI] [PubMed] [Google Scholar]
- 4.Scallet A.C., Schmued L.C., Johannessen J.N. Neurohistochemical biomarkers of the marine neurotoxicant, domoic acid. Neurotoxicol Teratol. 2005;27:745–752. doi: 10.1016/j.ntt.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 5.Cendes F., Andermann F., Carpenter S., Zatorre R.J., Cashman N.R. Temporal lobe epilepsy caused by domoic acid intoxication: evidence for glutamate receptor-mediated excitotoxicity in humans. Ann Neurol. 1995;37:123–126. doi: 10.1002/ana.410370125. [DOI] [PubMed] [Google Scholar]
- 6.Jeffery B., Barlow T., Moizer K., Paul S., Boyle C. Amnesic shellfish poison. Food Chem Toxicol. 2004;42:545–557. doi: 10.1016/j.fct.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 7.Tasker R.A., Strain S.M., Drejer J. Selective reduction in domoic acid toxicity in vivo by a novel non-N-methyl-D-aspartate receptor antagonist. Can J Physiol Pharmacol. 1996;74:1047–1054. [PubMed] [Google Scholar]
- 8.Tasker R.A., Connell B.J., Strain S.M. Pharmacology of systemically administered domoic acid in mice. Can J Physiol Pharmacol. 1991;69:378–382. doi: 10.1139/y91-057. [DOI] [PubMed] [Google Scholar]
- 9.Sobotka T.J., Brown R., Quander D.Y., Jackson R., Smith M., Long S.A., Barton C.N., Rountree R.L., Hall S., Eilers P., Johannessen J.N., Scallet A.C. Domoic acid: neurobehavioral and neurohistological effects of low-dose exposure in adult rats. Neurotoxicol Teratol. 1996;18:659–670. doi: 10.1016/s0892-0362(96)00120-1. [DOI] [PubMed] [Google Scholar]
- 10.Sari P., Kerr D.S. Domoic acid-induced hippocampal CA1 hyperexcitability independent of region CA3 activity. Epilepsy Res. 2001;47:65–76. doi: 10.1016/s0920-1211(01)00295-9. [DOI] [PubMed] [Google Scholar]
- 11.Hesp B.R., Clarkson A.N., Sawant P.M., Kerr D.S. Domoic acid preconditioning and seizure induction in young and aged rats. Epilepsy Res. 2007;76:103–112. doi: 10.1016/j.eplepsyres.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Sawant P.M., Tyndall J.D., Holland P.T., Peake B.M., Mountfort D.O., Kerr D.S. In vivo seizure induction and affinity studies of domoic acid and isodomoic acids-D, -E and -F. Neuropharmacology. 2010;59:129–138. doi: 10.1016/j.neuropharm.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 13.Sawant P.M., Mountfort D.O., Kerr D.S. Spectral analysis of electrocorticographic activity during pharmacological preconditioning and seizure induction by intrahippocampal domoic acid. Hippocampus. 2010;20:994–1002. doi: 10.1002/hipo.20698. [DOI] [PubMed] [Google Scholar]
- 14.Hampson D.R., Huang X.P., Wells J.W., Walter J.A., Wright J.L. Interaction of domoic acid and several derivatives with kainic acid and AMPA binding sites in rat brain. Eur J Pharmacol. 1992;218:1–8. doi: 10.1016/0014-2999(92)90140-y. [DOI] [PubMed] [Google Scholar]
- 15.Budd S.L., Nicholls D.G. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- 16.Budd S.L., Nicholls D.G. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem. 1996;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- 17.Dykens J.A. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated Ca+ and Na+: implications for neurodegeneration. J Neurochem. 1994;63:584–591. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- 18.Luetjens C.M., Bui N.T., Sengpiel B., Münstermann G., Poppe M., Krohn A.J., Bauerbach E., Krieglstein J., Prehn J.H. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. 2000;20:5715–5723. doi: 10.1523/JNEUROSCI.20-15-05715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng T.I., Greenamyre J.T. Privileged access to mitochondria of calcium influx through N-methyl-D-aspartate receptors. Mol Pharmacol. 1998;53:974–980. [PubMed] [Google Scholar]
- 20.White R.J., Reynolds I.J. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doucette T.A., Strain S.M., Allen G.V., Ryan C.L., Tasker R.A. Comparative behavioural toxicity of domoic acid and kainic acid in neonatal rats. Neurotoxicol Teratol. 2000;22:863–869. doi: 10.1016/s0892-0362(00)00110-0. [DOI] [PubMed] [Google Scholar]
- 22.Strain S.M., Tasker R.A. Hippocampal damage produced by systemic injections of domoic acid in mice. Neuroscience. 1991;44:343–352. doi: 10.1016/0306-4522(91)90059-w. [DOI] [PubMed] [Google Scholar]
- 23.Scallet A.C., Kowalke P.K., Rountree R.L., Thorn B.T., Binienda Z.K. Electroencephalographic, behavioral, and c-fos responses to acute domoic acid exposure. Neurotoxicol Teratol. 2004;26:331–342. doi: 10.1016/j.ntt.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Peng Y.G., Taylor T.B., Finch R.E., Switzer R.C., Ramsdell J.S. Neuroexcitatory and neurotoxic actions of the amnesic shellfish poison, domoic acid. Neuroreport. 1994;5:981–985. doi: 10.1097/00001756-199404000-00032. [DOI] [PubMed] [Google Scholar]
- 25.Scallet A.C., Binienda Z., Caputo F.A., Hall S., Paule M.G., Rountree R.L., Schmued L., Sobotka T., Slikker W., Jr Domoic acid-treated cynomolgus monkeys (M. fascicularis): effects of dose on hippocampal neuronal and terminal degeneration. Brain Res. 1993;627:307–313. doi: 10.1016/0006-8993(93)90335-k. [DOI] [PubMed] [Google Scholar]
- 26.Kreuder C., Miller M.A., Lowenstine L.J., Conrad P.A., Carpenter T.E., Jessup D.A., Mazet J.A. Evaluation of cardiac lesions and risk factors associated with myocarditis and dilated cardiomyopathy in southern sea otters (Enhydra lutris nereis) Am J Vet Res. 2005;66:289–299. doi: 10.2460/ajvr.2005.66.289. [DOI] [PubMed] [Google Scholar]
- 27.Silvagni P.A., Lowenstine L.J., Spraker T., Lipscomb T.P., Gulland F.M. Pathology of domoic acid toxicity in California sea lions (Zalophus californianus) Vet Pathol. 2005;42:184–191. doi: 10.1354/vp.42-2-184. [DOI] [PubMed] [Google Scholar]
- 28.Zabka T.S., Goldstein T., Cross C., Mueller R.W., Kreuder-Johnson C., Gill S., Gulland F.M.D. Characterization of a degenerative cardiomyopathy associated with domoic acid toxicity in California sea lions (Zalophus californianus) [Erratum appeared in Vet Pathol 2009, 46:552] Vet Pathol. 2009;46:105–119. doi: 10.1354/vp.46-1-105. [DOI] [PubMed] [Google Scholar]
- 29.Gill S., Veinot J., Kavanagh M., Pulido O. Human heart glutamate receptors–implications for toxicology, food safety, and drug discovery. Toxicol Pathol. 2007;35:411–417. doi: 10.1080/01926230701230361. [DOI] [PubMed] [Google Scholar]
- 30.Gill S.S., Pulido O.M. Glutamate receptors in peripheral tissues: current knowledge, future research, and implications for toxicology. Toxicol Pathol. 2001;29:208–223. doi: 10.1080/019262301317052486. [DOI] [PubMed] [Google Scholar]
- 31.Vranyac-Tramoundanas A., Harrison J.C., Clarkson A.N., Kapoor M., Winburn I.C., Kerr D.S., Sammut I.A. Domoic acid impairment of cardiac energetics. Toxicol Sci. 2008;105:395–407. doi: 10.1093/toxsci/kfn132. [DOI] [PubMed] [Google Scholar]
- 32.Kawabe T., Chitravanshi V.C., Kawabe K., Sapru H.N. Cardiovascular function of a glutamatergic projection from the hypothalamic paraventricular nucleus to the nucleus tractus solitarius in the rat. Neuroscience. 2008;153:605–617. doi: 10.1016/j.neuroscience.2008.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rockhold R.W., Acuff C.G., Clower B.R. Excitotoxin-induced myocardial necrosis. Eur J Pharmacol. 1989;166:571–576. doi: 10.1016/0014-2999(89)90379-8. [DOI] [PubMed] [Google Scholar]
- 34.Rockhold R.W., Acuff C.G., Clower B.R. Excitotoxic lesions of the paraventricular hypothalamus: metabolic and cardiac effects. Neuropharmacology. 1990;29:663–673. doi: 10.1016/0028-3908(90)90028-p. [DOI] [PubMed] [Google Scholar]
- 35.Whitehurst V.E., Joseph X., Alleva F.R., Vick J.A., Whittaker P., Zhang J., Fry B.E., Jr, Balazs T. Enhancement of acute myocardial lesions by asthma drugs in rats. Toxicol Pathol. 1994;22:72–76. doi: 10.1177/019262339402200110. [DOI] [PubMed] [Google Scholar]
- 36.Britton M., De Faire U., Helmers C., Miah K., Ryding C., Wester P.O. Arrhythmias in patients with acute cerebrovascular disease. Acta Med Scand. 1979;205:425–428. doi: 10.1111/j.0954-6820.1979.tb06076.x. [DOI] [PubMed] [Google Scholar]
- 37.Korpelainen J.T., Sotaniemi K.A., Mäkikallio A., Huikuri H.V., Myllylä V.V. Dynamic behavior of heart rate in ischemic stroke. Stroke. 1999;30:1008–1013. doi: 10.1161/01.str.30.5.1008. [DOI] [PubMed] [Google Scholar]
- 38.Korpelainen J.T., Huikuri H.V., Sotaniemi K.A., Myllylä V.V. Abnormal heart rate variability reflecting autonomic dysfunction in brainstem infarction. Acta Neurol Scand. 1996;94:337–342. doi: 10.1111/j.1600-0404.1996.tb07076.x. [DOI] [PubMed] [Google Scholar]
- 39.Freeman R. Cardiovascular manifestations of autonomic epilepsy. Clin Auton Res. 2006;16:12–17. doi: 10.1007/s10286-006-0278-y. [DOI] [PubMed] [Google Scholar]
- 40.Surges R., Scott C.A., Walker M.C. Enhanced QT shortening and persistent tachycardia after generalized seizures. Neurology. 2010;74:421–426. doi: 10.1212/WNL.0b013e3181ccc706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naritoku D.K., Casebeer D.J., Darbin O. Effects of seizure repetition on postictal and interictal neurocardiac regulation in the rat. Epilepsia. 2003;44:912–916. doi: 10.1046/j.1528-1157.2003.48302.x. [DOI] [PubMed] [Google Scholar]
- 42.P-Codrea Tigaran S., Dalager-Pedersen S., Baandrup U., Dam M., Vesterby-Charles A. Sudden unexpected death in epilepsy: is death by seizures a cardiac disease? Am J Forensic Med Pathol. 2005;26:99–105. [PubMed] [Google Scholar]
- 43.Sawant P.M., Holland P.T., Mountfort D.O., Kerr D.S. In vivo seizure induction and pharmacological preconditioning by domoic acid and isodomoic acids A, B and C. Neuropharmacology. 2008;55:1412–1418. doi: 10.1016/j.neuropharm.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 44.Hesp B.R., Harrison J.C., Selwood A.I., Holland P.T., Kerr D.S. Detection of domoic acid in rat serum and brain by direct competitive enzyme-linked immunosorbent assay (cELISA) Anal Bioanal Chem. 2005;383:783–786. doi: 10.1007/s00216-005-0060-3. [DOI] [PubMed] [Google Scholar]
- 45.Sammut I.A., Jayakumar J., Latif N., Rothery S., Severs N.J., Smolenski R.T., Bates T.E., Yacoub M.H. Heat stress contributes to the enhancement of cardiac mitochondrial complex activity. Am J Pathol. 2001;158:1821–1831. doi: 10.1016/S0002-9440(10)64138-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adlam V.J., Harrison J.C., Porteous C.M., James A.M., Smith R.A., Murphy M.P., Sammut I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 47.Clarkson A.N., Clarkson J., Jackson D.M., Sammut I.A. Mitochondrial involvement in transhemispheric diaschisis following hypoxia-ischemia: clomethiazole-mediated amelioration. Neuroscience. 2007;144:547–561. doi: 10.1016/j.neuroscience.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 48.Hulse R.E., Kunkler P.E., Fedynyshyn J.P., Kraig R.P. Optimization of multiplexed bead-based cytokine immunoassays for rat serum and brain tissue. J Neurosci Methods. 2004;136:87–98. doi: 10.1016/j.jneumeth.2003.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujita T., Tanaka T., Yonemasu Y., Cendes F., Cashman N.R., Andermann F. Electroclinical and pathological studies after parenteral administration of domoic acid in freely moving nonanesthetized rats: an animal model of excitotoxicity. J Epilepsy. 1996;9:87–93. [Google Scholar]
- 50.Markham L.W., Michelfelder E.C., Border W.L., Khoury P.R., Spicer R.L., Wong B.L., Benson D.W., Cripe L.H. Abnormalities of diastolic function precede dilated cardiomyopathy associated with Duchenne muscular dystrophy. J Am Soc Echocardiogr. 2006;19:865–871. doi: 10.1016/j.echo.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Zhang J., Knapton A., Lipshultz S.E., Weaver J.L., Herman E.H. Isoproterenol-induced cardiotoxicity in Sprague-Dawley rats: correlation of reversible and irreversible myocardial injury with release of cardiac troponin T and roles of iNOS in myocardial injury. Toxicol Pathol. 2008;36:277–278. doi: 10.1177/0192623307313010. [DOI] [PubMed] [Google Scholar]
- 52.Vezzani A., Moneta D., Richichi C., Perego C., De Simoni M.G. Functional role of proinflammatory and anti-inflammatory cytokines in seizures. Adv Exp Med Biol. 2004;548:123–133. doi: 10.1007/978-1-4757-6376-8_10. [DOI] [PubMed] [Google Scholar]
- 53.Wang Q.D., Bohlooly-Y M., Sjöquist P.O. Murine models for the study of congestive heart failure: implications for understanding molecular mechanisms and for drug discovery. J Pharmacol Toxicol Methods. 2004;50:163–174. doi: 10.1016/j.vascn.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 54.Siwik D.A., Chang D.L., Colucci W.S. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–1265. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 55.Brown R.D., Jones G.M., Laird R.E., Hudson P., Long C.S. Cytokines regulate matrix metalloproteinases and migration in cardiac fibroblasts 1. Biochem Biophys Res Commun. 2007;362:200–205. doi: 10.1016/j.bbrc.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prabhu S.D. Cytokine-induced modulation of cardiac function 1. Circ Res. 2004;95:1140–1153. doi: 10.1161/01.RES.0000150734.79804.92. [DOI] [PubMed] [Google Scholar]
- 57.Clarkson A.N., Liu H., Schiborra F., Shaw O., Sammut I.A., Jackson D.M., Appleton I. Angiogenesis as a predictive marker of neurological outcome following hypoxia-ischemia. Brain Res. 2007;1171:111–121. doi: 10.1016/j.brainres.2007.06.100. [DOI] [PubMed] [Google Scholar]
- 58.Druschky A., Hilz M.J., Hopp P., Platsch G., Radespiel-Tröger M., Druschky K., Kuwert T., Stefan H., Neundörfer B. Interictal cardiac autonomic dysfunction in temporal lobe epilepsy demonstrated by [(123)I]metaiodobenzylguanidine-SPECT. Brain. 2001;124:2372–2382. doi: 10.1093/brain/124.12.2372. [DOI] [PubMed] [Google Scholar]
- 59.Scorza F.A., Arida R.M., Cysneiros R.M., Terra V.C., Sonoda E.Y., De Albuquerque M., Cavalheiro E.A. The brain-heart connection: implications for understanding sudden unexpected death in epilepsy. Cardiol J. 2009;16:394–399. [PubMed] [Google Scholar]
- 60.Michelini L.C., Stern J.E. Exercise-induced neuronal plasticity in central autonomic networks: role in cardiovascular control. Exp Physiol. 2009;94:947–960. doi: 10.1113/expphysiol.2009.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lathers C.M., Schraeder P.L., Bungo M.W. The mystery of sudden death: mechanisms for risks. Epilepsy Behav. 2008;12:3–24. doi: 10.1016/j.yebeh.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 62.Natelson B.H., Suarez R.V., Terrence C.F., Turizo R. Patients with epilepsy who die suddenly have cardiac disease. Arch Neurol. 1998;55:857–860. doi: 10.1001/archneur.55.6.857. [DOI] [PubMed] [Google Scholar]
- 63.Van Vleet J.F., Ferrans V.J., Herman E. Cardiovascular and skeletal muscle systems. In: Haschek W.M., Rousseaux C.G., Wallig M.A., editors. Handbook of Toxicologic Pathology. ed 2. Academic Press; San Diego: 2002. pp. 363–455. [Google Scholar]