

Abstract
Intestinal fibrosis is a serious complication of Crohn's disease (CD) that can lead to stricture formation, which requires surgery. Mechanisms underlying intestinal fibrosis remain elusive because of a lack of suitable mouse models. Herein, we describe a spontaneous mouse model of intestinal inflammation with fibrosis and the profibrotic role of arginase I. The Src homology 2 domain–containing inositol polyphosphate 5′-phosphatase–deficient (SHIP–/–) mice developed spontaneous discontinuous intestinal inflammation restricted to the distal ileum starting at the age of 4 weeks. Mice developed several key features resembling CD, including inflammation and fibrosis. Inflammation was characterized by abundant infiltrating Gr-1–positive immune cells, granuloma-like immune cell aggregates that contained multinucleated giant cells, and a mixed type 2 and type 17 helper T-cell cytokine profile. Fibrosis was characterized by a thickened ileal muscle layer, collagen deposition, and increased fibroblasts at the sites of collagen deposition. SHIP–/– ilea had increased arginase activity and arginase I expression that was inversely proportional to nitrotyrosine staining. SHIP–/– mice were treated with the arginase inhibitor S-(2-boronoethyl)-l-cysteine, and changes in the disease phenotype were measured. Arginase inhibition did not affect the number of immune cell infiltrates in the SHIP–/– mouse ilea; rather, it reduced collagen deposition and muscle hyperplasia. These findings suggest that arginase activity is a potential target to limit intestinal fibrosis in patients with CD.
Inflammatory bowel disease includes two major presentations of intestinal disease: Crohn's disease (CD) and ulcerative colitis. Both are characterized by chronic, relapsing and remitting, or progressive inflammation of the intestine. Patients experience severe intestinal pathological conditions that cause physical symptoms, including pain, nausea, and diarrhea.1 Ulcerative colitis is restricted to the colon, whereas CD can occur in any part of the gastrointestinal tract.2
Intestinal fibrosis is a serious complication of CD. It can lead to strictures and stenoses that narrow the lumen of the intestine and can cause dangerous obstructions. Approximately one in three people with CD develops strictures within 10 years of diagnosis and requires surgery to remove the diseased bowel. The most common site of surgical resection in patients with CD is the distal ileum.3,4 Furthermore, patients who have undergone surgery for fibrosis frequently relapse, developing intestinal inflammation and fibrotic strictures at the same location as the previous resection.5 Anti–tumor necrosis factor-α therapy is effective at reducing inflammation in active CD, but the current thinking is that it is not effective at reducing fibrosis or fibrogenic gene expression.6–9 To our knowledge, there are no antifibrotic drugs available for use in patients with CD.10
Fibrosis can be considered a pathological consequence of an excessive healing response. Normally, in response to tissue injury, the healing process includes the deposition of extracellular matrix proteins, the most abundant of which are members of the collagen family.11 Thus, some degree of mild subclinical fibrosis may occur in all patients with CD.12,13 The cell types that contribute to scar formation are mesenchymal cells, including myofibroblasts, smooth muscle cells, and fibroblasts.4 Patients with CD and fibrosis experience accumulation of fibroblasts through the gut wall.14
Despite the urgent need to prevent or treat intestinal stricturing in patients with CD, little is known about the mechanisms underlying this process, in part because of a lack of suitable animal models for the study of intestinal fibrosis. There are many mouse models of intestinal inflammation,15,16 but studies17–22 of intestinal fibrosis have been limited to six colitis models. There are two genetic mouse models of spontaneous intestinal inflammation in the distal ileum: transgenic tumor necrosis factorΔARE and SAMP1/YitFc mice.23,24 (SAMP1/YitFc mice are a subline of the senescence-accelerated mouse line.) Both of these models recapitulate some features of CD and have been used to study the development of chronic ileitis. The SAMP1/YitFc mouse is reported to develop fibrostenotic strictures at the age of 40 weeks24 but has not been used as a model for the treatment of intestinal fibrosis.
The Src homology 2 domain–containing inositol polyphosphate 5′-phosphatase (SHIP) is a hematopoietic-specific negative regulator of the phosphatidylinositol-3-kinase (PI3K) pathway. It removes the 5′ phosphate group from class IA PI3K-generated phosphatidylinositol 3,4,5-triphosphate, removing this important second messenger from the cell membrane, thereby discontinuing PI3K activity. SHIP–/– mice were generated in 1998 by the laboratories of Krystal, Humphries, and colleagues.25 SHIP–/– mice have a myeloproliferative disorder, with a twofold to threefold increase in myeloid cells at different sites, lung pathological features, and a shortened life span.25 Myeloid cells from these mice are hyperresponsive to various stimuli,26 and in vivo differentiated SHIP–/– macrophages are alternatively activated and constitutively express arginase I (ArgI).27 Recently, SHIP–/– mice were reported to develop spontaneous intestinal CD-like inflammation.28
Herein, we confirm and extend these findings describing the inflammatory infiltrates, cytokine responses, and specific features that define ileal fibrosis. SHIP–/– mice develop thickened muscle layers, have increased collagen deposition, and accumulate fibroblasts in the affected ileum. In addition, the SHIP–/– mouse ilea have increased ArgI expression and arginase activity. More importantly, treating SHIP–/– mice with the arginase inhibitor, S-(2-boronoethyl)-l-cysteine (BEC), reduced ileal muscle hyperplasia and collagen deposition. These findings identify arginase activity as a novel therapeutic target for the treatment of intestinal fibrosis in CD.
Materials and Methods
Mice
SHIP heterozygotes, an F2 generation of C57BL/6 × 129Sv mice, were bred to generate SHIP+/+ and SHIP–/– littermates. Mice were maintained in sterilized filter-top cages and fed autoclaved food and water under specific pathogen-free conditions at the Animal Care Facility at the Child & Family Research Institute (Vancouver, BC). Sentinel mice were routinely screened for pathogens using a comprehensive serological profile service (Radil, Columbia, MO). All mice used were between the ages of 4 and 16 weeks. Experimentation was performed in accordance with institutional and Canadian Council on Animal Care guidelines.
Histological Characteristics
Mouse tissues were fixed in 10% neutral-buffered formalin at 4°C for 24 hours before paraffin processing. Paraffin sections (5-μm thick) were stained with H&E for histological examination. Masson's trichrome staining was performed according to the manufacturer's instructions (Sigma, St Louis, MO). Images were acquired and analyzed using a Zeiss Axiovert 200 microscope, a Zeiss AxiocamHR camera, and the Zeiss Axiovision 4.0 software imaging system.
Histological Analyses
Crypt depth and villus length were determined by counting epithelial cell nuclei from the base of crypts to the crypt-villus junction and from the crypt-villus junction to the villus tip, respectively, on uniform horizontal cross sections of crypts and villi. Goblet cells per crypt-villus were counted from the base of the crypt to the tip of the villus. Representative crypts-villi (10 per mouse) were counted in 5 to 10 H&E-stained sections of distal ileum for six 8-week-old SHIP+/+ and SHIP–/– mice by two individuals blinded to genotype (K.W.M. and A.E.C.). To count immune cell infiltrates, four representative fields of H&E-stained cross sections of seven untreated and seven BEC-treated SHIP–/– mouse ilea were obtained at ×20 magnification; infiltrates were counted according to nuclear morphological features in the circular muscularis externa and submucosa. The thickness of the muscularis externa from the serosa to the muscularis mucosa was measured at six points in 10 cross sections of distal ileum from seven untreated and seven BEC-treated SHIP–/– mice. In all cases, sections were nonconsecutive, separated by at least 50 μm, and counted at ×20 magnification.
IHC Procedures
For detection of CD3, F4/80, Gr-1/Ly6, ArgI, and nitrotyrosine slide-mounted 5-μm sections of FFPE tissues were deparaffinized and rehydrated. For CD3 and ArgI detection, heat-induced epitope retrieval was performed by immersing the slides in 1 mmol/L EDTA (pH 8.0) at 95°C for 20 minutes and allowing them to cool to room temperature. For F4/80, Gr-1, and nitrotyrosine detection, enzyme-induced epitope retrieval was performed by incubating samples with 20 μg/mL proteinase K in PBS for 15 minutes at room temperature. All slides were rinsed thoroughly in Tris-buffered saline with 0.1% Tween 20. Endogenous peroxidase activity was blocked with 1.5% H2O2 in PBS for 10 minutes. Endogenous avidin and biotin were blocked with an avidin-biotin blocking kit, according to the manufacturer's instructions (Vector Laboratories, Burlingame, CA). The primary antibodies used were rabbit anti-CD3 (Abcam, Cambridge, MA), rat anti–Gr-1 (Acris Antibodies, Herford, Germany), rat anti-F4/80 (AbD Serotec, Oxford, UK), mouse anti-ArgI (BD Biosciences, Mississauga, ON), and mouse anti-nitrotyrosine (Millipore, Billerica, MA). Blocking buffers, biotinylated secondary antibodies, and avidin–biotin–horseradish peroxidase or alkaline phosphatase complexes were prepared and used from rabbit IgG, rat IgG, or mouse-on-mouse immunohistochemistry (IHC) detection kits, according to the manufacturer's instructions (Vector Laboratories). Signals were detected with a diaminobenzidine chromogen system (Dako, Carpinteria, CA) or Vector Red (Vector Laboratories), and developed sections were counterstained with Harris' hematoxylin (Sigma). For vimentin staining, fresh tissue was flash frozen in dry ice-isopentane and embedded in optimal cutting temperature compound (Sakura Finetech; VWR Canada, Mississauga). Thaw-mounted sections were fixed in ice-cold acetone and blocked with the Mouse-on-Mouse detection system. Sections were incubated with mouse anti-vimentin antibody (BD Biosciences), followed by secondary Alexa 568–conjugated goat anti-mouse IgG antibody (Molecular Probes/Invitrogen, Eugene, OR). Sections were mounted in Vectashield mounting medium with DAPI (Vector Laboratories). Negative controls omitted primary antibodies and were performed for all antibodies.
Mouse Cytokine Arrays
Mouse-specific cytokine levels were determined in 150-mg samples of ilea from SHIP+/+ mice and inflamed ilea from SHIP–/– mice (n = 4 per group) using the Mouse Cytokine Array Panel A kit from R&D Systems (Minneapolis, MN), according to the manufacturer's instructions. Protein (200 μg) was used for each array. Spot densities were quantified using ImageJ software version 1.43 (http://rsbweb.nih.gov/ij/index.html) and exported to GraphPad Prism 5 version 5 (GraphPad Software Inc., San Diego, CA). Spot densities were corrected for individual background to diminish interarray variations.
Sircol Assays
Sections of fresh mouse distal ileum (30 to 100 mg) were minced with surgical scissors in 500 μL of 0.5 mol/L acetic acid with 10 mg/mL pepsin and agitated (at 1000 rpm with a VWR microplate shaker) overnight at room temperature. Tissue debris was removed and collagen was measured by the Sircol soluble collagen assay (Biocolor Ltd, Carrickfergus, UK), according to the manufacturer's instructions.
Arginase Assays
Fresh tissue samples were collected and homogenized in 1-mL arginase lysis buffer using a Polytron MR2100 bench-top homogenizer. Homogenates were cleared by centrifugation at 16,000 × g for 10 minutes at 4°C. Arginase activity was determined indirectly by measuring the concentration of urea generated by the arginase-dependent hydrolysis of l-arginine, as previously described.29
Mouse Treatment with BEC
BEC was synthesized by Dr Jean-Luc Boucher (Université Paris Descartes, Paris, France), as previously described.30 BEC was dissolved in deionized distilled water, adjusted to pH 7, and filter sterilized. BEC (100 μL of a 0.2% solution) was administered daily by oral gavage to 4-week-old SHIP–/– mice for 2 weeks.
Statistical Analyses
Unpaired two-tailed Student's t-tests were performed when indicated using GraphPad Prism version 5 (GraphPad Software Inc.). For multiple comparisons made during cytokine assays, the Bonferroni correction was applied. Differences were considered significant at P < 0.05.
Results
SHIP–/– Mice Have Spontaneous Intestinal Inflammation Resembling CD
In our work with SHIP–/– mice, we found that adult SHIP–/– mice consistently showed spontaneous intestinal inflammation. In situ intestines were thick and opaque (Figure 1A). This phenotype was restricted to the distal third of the ileum and usually within 10 cm of the ileocecal junction (Figure 1B). Discontinuous patches of active inflammation were visible as areas of redness on the gut wall (Figure 1, A and B).
Figure 1.
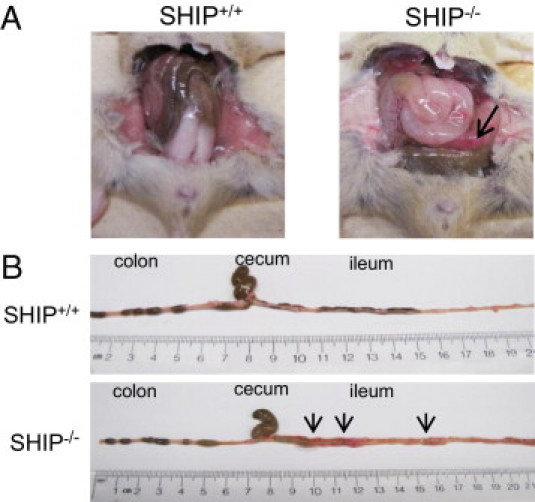
SHIP–/– mice develop spontaneous intestinal inflammation. A: The ilea of 6-week-old SHIP+/+ and SHIP–/– mice were compared in situ. SHIP–/– mice have thickened intestines with patchy inflammation (arrow). B: On extraction and extension of each ileum, it was determined that inflammation in the SHIP–/– mice was restricted to the distal ileum. Arrows indicate sites of inflammation.
The onset of the spontaneous intestinal inflammation was determined by H&E staining to occur between the ages of 4 and 8 weeks. At the age of 4 weeks, SHIP–/– mouse ilea showed no histopathological features (Figure 2A). However, inflammation was present in all SHIP–/– mice from the age of 6 weeks onward (data not shown). By 8 weeks, the ilea of the SHIP–/– mice showed thickened muscularis externa, especially the circular muscle layer, goblet cell hypertrophy, and immune cell infiltrates and aggregates throughout the gut wall (Figure 2A, bottom right). Immune cell aggregates were identified as poorly or partially formed granuloma-like structures (Figure 2B, left) and contained multinucleated giant cells (Figure 2B, middle). There was also evidence of bleeding into the lumen of the gut (Figure 2B, right). In addition, SHIP–/– mouse ilea showed epithelial cell hyperplasia (see Supplemental Figure S1 at http://ajp.amjpathol.org). SHIP–/– mouse histopathological features were restricted to the ileum because colons and ceca appeared normal (see Supplemental Figure S2 at http://ajp.amjpathol.org).
Figure 2.
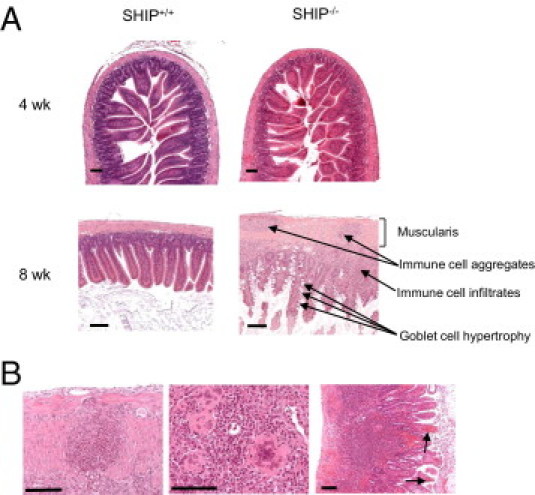
Intestinal inflammation in SHIP–/– mice develops between the ages of 4 and 8 weeks (wk) and is histologically similar to CD. A: H&E analysis of SHIP+/+ and SHIP–/– ilea at 4 wk (top) revealed no signs of pathological features in either mouse group. At the age of 8 wk (bottom), the ilea of SHIP–/– mice revealed thickening of the muscularis, immune cell aggregates and infiltrates, and goblet cell hypertrophy. Scale bar = 100 μm. B: SHIP–/– mice (aged 8 wk) shared additional histopathological features with CD, including immune cell aggregates that are organized into poorly formed granuloma-like structures in the muscularis (left) and contain multinucleated giant cells (middle); the ilea revealed signs of bleeding into the lumen (arrows, right). Sections are representative of 12 individual mice examined at the age of 6 to 8 wk. Scale bars: 100 μm (left and middle); 500 μm (right).
Villi and Lymphoid Aggregates Contain Abundant Gr-1/Ly6+ Granulocytes
Having established that SHIP–/– mice have increased ileal inflammation, we analyzed the immune cell infiltrates and immune cell aggregates in the SHIP–/– ilea. Fixed sections were stained by IHC to detect granulocytes/monocytes (Gr1+), macrophages (F4/80+), and T cells (CD3+). Gr-1+ cells were present through the entire thickness of the gut wall and were abundant in immune cell infiltrates and immune cell aggregates (Figure 3). Gr-1+ cells had multilobed nuclei, suggesting that they are neutrophils (Figure 3). There were a few F4/80+ macrophages within immune cell aggregates (see Supplemental Figure S3 at http://ajp.amjpathol.org). There were a few CD3+ T cells within crypts and villi (see Supplemental Figure S4 at http://ajp.amjpathol.org). The absence of abundant T cells within the SHIP–/– ilea was unexpected but is consistent with a recent report.28
Figure 3.
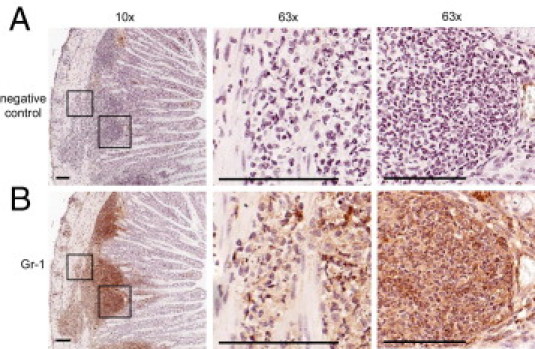
Immune cell aggregates and infiltrates from 8-week-old SHIP–/– mice are predominantly Gr-1+. A: IHC anti–Gr-1 antibody–negative control. B: Gr-1–stained serial section (brown). Gr-1+ cells were present through the entire thickness of the gut wall (hematoxylin, blue). IHC staining is shown at ×10 magnification (left). Insets (defined): ×63 magnification of immune cell infiltrates in the muscularis (middle) and in immune cell aggregates (right). Multilobed nuclear morphological features are visible at ×63 magnification. Scale bars = 50 μm. Sections are representative of 12 mice.
SHIP–/– Mouse Inflamed Ilea Have Increased Type 2 and Type 17 Helper T-Cell Cytokines
To describe the type of inflammation in the SHIP–/– mouse ileum, relative cytokine levels present in ileal tissue homogenates from 8-week-old SHIP–/– mice were compared with those in SHIP+/+ littermate controls. SHIP–/– mouse ileal homogenates had significantly increased levels of type 2 helper T-cell (Th2) cytokines, IL-4, and IL-13, but there was no difference in IL-5 (Figure 4A). SHIP–/– ilea had increased IL-23 (Th17) but no difference in IL-17 (Figure 4B). The levels of Th1 cytokines, IL-12p70, and interferon-γ were also the same between the two groups (Figure 4C).
Figure 4.
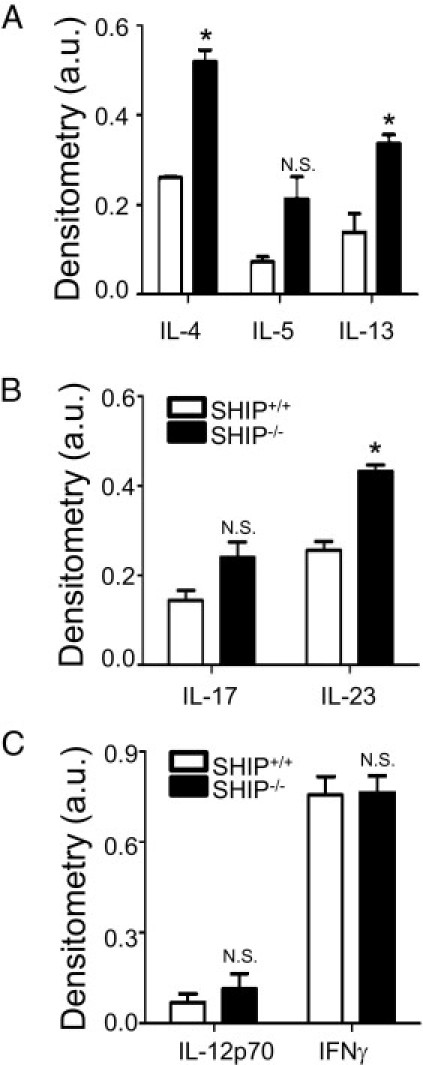
SHIP–/– mouse ileal cytokine expression shows a mixed Th2-Th17 profile. Cytokine levels were measured in 8-week-old SHIP+/+ and SHIP–/– mice ileal homogenates. Protein (200 μg) was analyzed for IL-4, IL-5, IL-13, IL-17, IL-23, IL-12p70, and interferon (IFN)-γ expression levels. Graphs show tissue expression levels of representative Th2 (A), Th17 (B), and Th1 (C) cytokines, expressed as mean ± SD signal intensity for four mice per group. IL-4, IL-13, and IL-23 were significantly increased in SHIP–/– mice ilea compared with SHIP+/+ littermate controls. No significant (N.S.) differences were observed for IL-5, IL-17, IL-12p70, or IFN-γ. *P < 0.05 comparing SHIP+/+ with SHIP–/– mice ilea using a Student's t-test with Bonferroni correction. a.u. indicates arbitrary unit.
SHIP–/– Mice Ilea Have More Collagen
Because SHIP–/– mouse ilea were thick and inflexible, we examined them for evidence of fibrosis. H&E staining revealed a thickened muscle layer in 8-week-old SHIP–/– mice compared with SHIP+/+ littermate controls (Figure 5A). Masson's trichrome stain for fibrosis in adjacent sections showed abundant collagen (blue) present in the submucosa and between muscle layers in the SHIP–/– mouse ilea (Figure 5B). However, given the larger overall size of the SHIP–/– mouse ilea, we measured the collagen per milligram of tissue. Sircol assays for soluble collagen were performed comparing SHIP–/– with SHIP+/+ distal ilea. SHIP–/– mouse ilea had 2.1-fold more collagen than age-matched SHIP+/+ control mouse ilea, with significant differences at the ages of 6 to 8 and 10 to 12 weeks (Figure 5C).
Figure 5.
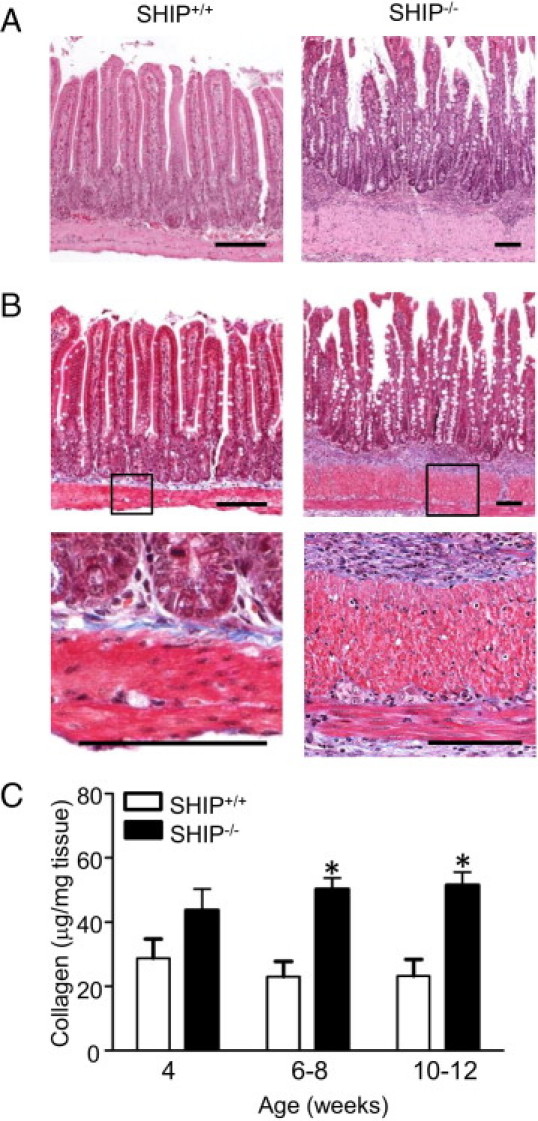
SHIP–/– mice have elevated collagen deposition in their distal ilea, indicative of fibrosis. A: H&E-stained horizontal sections of the distal ileum of 8-week-old SHIP+/+ (left) and SHIP–/– (right) mice revealed a thickened muscle layer in SHIP–/– mice. B: Masson's trichrome stain (blue) of adjacent sections from the same mice revealed increased collagen deposition. Insets: Higher magnifications show muscularis externa and muscularis mucosa and the presence of collagen between muscle layers (bottom). Scale bars = 100 μm. C: Sircol assays comparing collagen levels between SHIP+/+ and SHIP–/– ilea. Bars represent the mean ± SD for 6 to 12 mice at each age (*P < 0.05).
SHIP–/– Mice Ilea Have More Vimentin+ Fibroblasts, Increased ArgI, and Arginase Activity
Because fibroblasts are key players in intestinal fibrosis, we compared the numbers of fibroblasts present in the ilea of SHIP–/– mice with those in the ilea of SHIP+/+ mice by immunocytochemistry for vimentin. Masson's trichrome staining of SHIP+/+ and SHIP–/– mouse ilea were performed at the same magnification used for vimentin immunocytochemistry to show the thickness of the muscle layers (Figure 6A). SHIP+/+ mouse ilea have few vimentin+ cells within the muscle layer, whereas SHIP–/– mouse ilea have abundant spindle-shaped vimentin+ cells within the muscle layer and in the submucosa (Figure 6A).
Figure 6.
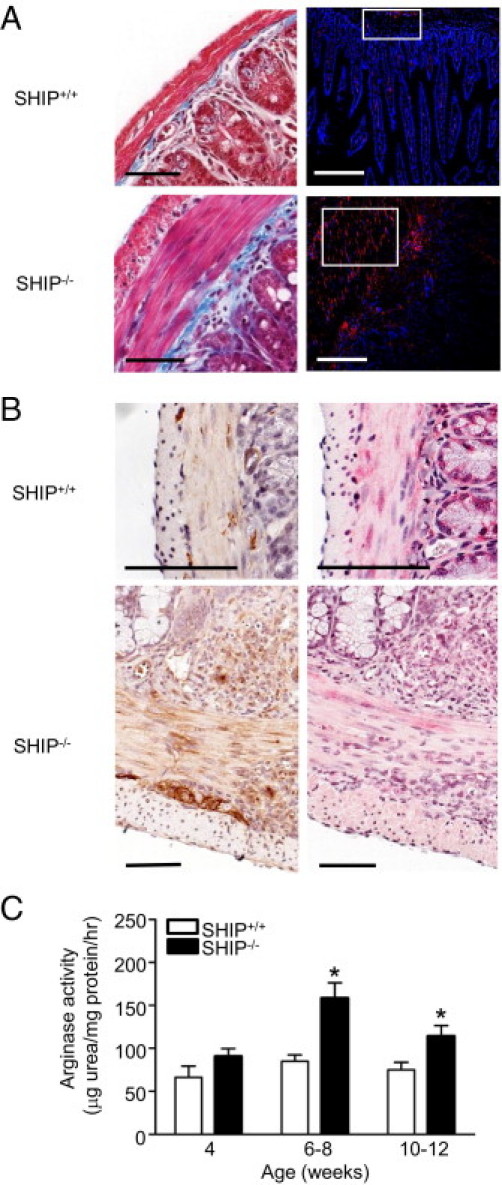
Increased collagen deposition correlates with increased fibroblasts, increased ArgI expression and activity, and decreased NO production in SHIP–/– mouse distal ilea. A: Masson's trichrome staining (blue, left) and anti-vimentin labeling (red, right; DAPI, blue) in ileal cross sections from SHIP+/+ and SHIP–/– mice. Scale bars = 50 μm. B: IHC analysis of ArgI (brown, left) and nitrotyrosine (pink/red, right) levels in serial sections from SHIP+/+ and SHIP–/– mouse ilea (hematoxylin, blue). Scale bars = 100 μm. C: ArgI activity in SHIP+/+ and SHIP–/– mouse ilea. Bars represent the mean ± SD for 6 to 12 mice at each age (*P < 0.05).
Given that ArgI is up-regulated in vivo in SHIP–/– mouse macrophages and is upstream of the l-proline required for collagen biosynthesis, we investigated ArgI protein expression levels and arginase activity in the SHIP–/– mouse ilea. The SHIP–/– mouse ilea have increased ArgI expression, including cells within immune cell aggregates, the submucosa, and the muscularis (Figure 6B). Arginases compete with NO synthases for their common substrate, l-arginine; therefore, increased ArgI is predicted to reduce NO production in tissues. Indeed, in the SHIP–/– mouse ilea, staining of nitrotyrosine (which is produced downstream of NO production) of serial sections was reduced compared with that of SHIP+/+ littermate controls (Figure 6B). Arginase activity was also measured in ileal tissue homogenates to determine whether it was increased. SHIP–/– mouse ilea had a 1.85-fold increased arginase activity compared with SHIP+/+ littermate controls at the age of 6 to 8 weeks, and arginase activity was significantly higher at the age of 10 to 12 weeks (Figure 6C).
Arginase Inhibition Reduced Collagen Deposition and Muscle Hyperplasia in SHIP–/– Ilea
Increased arginase activity in the SHIP–/– mouse ilea correlated with increased collagen deposition; therefore, SHIP–/– mice were treated with the arginase inhibitor, BEC, an arginine analogue that was previously used in other models of intestinal inflammation.31,32 Because the onset of increased arginase activity and collagen deposition in SHIP–/– ilea occurred between the ages of 4 and 6 weeks, we treated SHIP–/– mice daily with 100 μL of 0.2% BEC in water by oral gavage during this period. BEC treatment caused a 50% reduction in arginase activity compared with untreated 6-week-old SHIP–/– mice (Figure 7A), and ArgI protein expression was also decreased (Figure 7B). Immune cell infiltrates were still present in the BEC-treated mice and were not significantly reduced relative to untreated SHIP–/– controls (Figure 7C). BEC-treated SHIP–/– mice had decreased collagen deposition, evident from reduced Masson's trichrome staining (Figure 7D), and ileal collagen was also reduced by 25% compared with untreated SHIP–/– mice in Sircol assays (Figure 7E). The thickness of the muscle layers was reduced in BEC-treated SHIP–/– mice relative to untreated SHIP–/– mice (Figure 7E).
Figure 7.
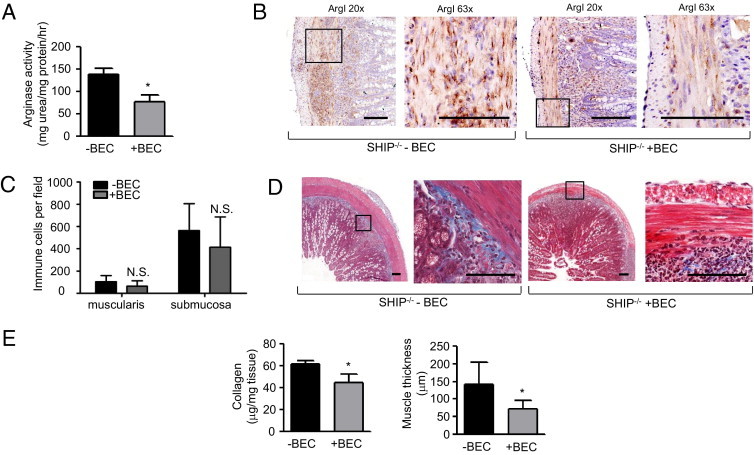
Inhibition of ArgI activity reduces collagen deposition and muscle hyperplasia in SHIP–/– ilea. SHIP–/– mice were administered 0.2% BEC in distilled water by oral gavage daily from the age of 4 to 6 weeks. A: Arginase activity from ilea of untreated (-BEC) and BEC-treated (+BEC) SHIP–/– mice (*P < 0.02). B: IHC labeling of ArgI in ileal cross sections of 6-week-old -BEC and +BEC SHIP–/– mice. Insets: Regions of magnification (×63). Scale bar = 100 μm. C: Assessment of immune cell infiltrates within the muscularis and submucosa of -BEC and +BEC SHIP–/– mice. The nuclei of infiltrating immune cells were counted. Bars represent the mean ± SD of four representative fields, counted at ×20 magnification in ileal cross sections from six mice from each group. N.S. indicates not significant. D: Masson's trichrome stain (blue) of ileal sections of -BEC and +BEC SHIP–/– mice. Scale bar = 100 μm. E: Sircol assays for soluble collagen (left) and thickness of muscle layer (right) in SHIP–/– mouse ilea, for -BEC or +BEC. Bars represent the mean ± SD (n = 7) performed in three independent experiments (*P < 0.001).
Discussion
The current treatment of CD includes therapies to reduce intestinal inflammation, improving quality of life for patients.33 Despite improvements in therapies reducing inflammation, fibrosis remains a serious complication of disease.34 Research studying intestinal fibrosis has been challenging, in part, because of a lack of suitable animal models. Herein, we report that the SHIP–/– mouse is a model of spontaneous intestinal inflammation that shares key features with CD. More importantly, these mice develop fibrosis, characterized by thickening of the muscle layers, collagen deposition, and fibroblast accumulation in the distal ileum (Figures 5 and 6).
There are several inducible murine models of intestinal fibrosis. Intestinal fibrosis can be induced in mouse colon and cecum by chronic Salmonella enterica infection19 and in the colon by overexpression of transforming growth factor-β120 or monocyte chemoattractant protein-121 or by repeated induction of colitis by trinitrobenzene sulfonic acid.22 These models do not cause fibrosis in the ileum, where it is most frequently found in patients with CD, and do not provide insight into spontaneous development of intestinal fibrosis. There are two genetic mouse models of spontaneous ileal inflammation. The tumor necrosis factorΔARE mouse develops ileal inflammation beginning before the age of 4 weeks, with inflammatory infiltrates in the submucosa containing scattered neutrophils and collections of histiocytes but do not display muscle thickening or collagen deposition.23 The SAMP1/YitFc mouse develops ileal inflammation with thickened circular muscle and collagen deposition in the submucosa and has perianal fistulae.24 Fibrosis develops slowly over 40 weeks in these mice, and the multigenic changes that cause this phenotype remain undefined.
Features reminiscent of CD in the SHIP–/– mouse include both inflammatory and fibrotic components. CD is characterized by discontinuous inflammation primarily in the distal ileum,35 and SHIP–/– mouse inflammation is discontinuous and restricted to the distal third of the ileum (Figure 1B). Sites of inflammation in patients with CD are dominated by neutrophils,36 and abundant neutrophils are present in the SHIP–/– ileum (Figure 3). An additional feature of CD is the formation of rudimentary granulomata in the ileum,37 and a diagnostic feature of the disease is the presence of multinucleated giant cells in the inflamed ileum.38,39 Similarly, SHIP–/– mice also have poorly formed granuloma containing multinucleated giant cells within the ileum (Figure 2B). Fibrotic features of CD, such as muscle hyperplasia and collagen deposition,40 are also present in our model (Figure 5). Transmural fibroblast accumulation that occurs during intestinal fibrosis in patients with CD is also apparent in our model as an increase in vimentin+ cells (Figure 6A).
CD has traditionally been considered to be a Th1- and Th17-mediated disease based on increased production of interferon-γ, IL-12p70, and IL-23 in the early phases of inflammation.2,41-43 Recently, the SHIP–/– mouse was reported to be Th2 skewed,44 which was attributed to hyperactive basophils producing IL–4.45 Unlike the inflammatory response characterized during active CD, fibrosis is a Th2-driven process.46 IL-4 and IL-13 stimulate type I collagen gene expression (Col1a2)47 and collagen production by fibroblasts.47,48 The inflamed and fibrotic SHIP–/– mouse ilea express a mixed Th2 and Th17 cytokine profile, with significantly increased production of IL-4, IL-13, and IL-23 (Figure 4). Alternatively activated macrophages, fibroblasts, and dysregulated Th2 and Th17 cytokine production have recently been suggested as the critical players in the development of an exaggerated healing response that leads to fibrosis.49 The SHIP–/– mouse has a myeloproliferative disorder and alternatively activated macrophages, abundant fibroblasts, and increased Th2 and Th17 cytokine responses that may cooperate to cause the dramatic ileal fibrosis in the SHIP–/– mouse.
We recently found that PI3K, and specifically PI3Kp110δ, activity is required for robust expression of ArgI in response to IL-4.50 Increased ArgI expression and arginase activity are features of murine alternatively activated macrophages26; in vivo SHIP–/– mouse macrophages, including intestinal macrophages, are alternatively activated and constitutively express ArgI.27,51 On examination, ArgI expression and arginase activity were up-regulated in the distal ilea of SHIP–/– mice (Figure 6). Arginase could play an indirect role in the SHIP–/– mouse ileal fibrosis by affecting inflammation. Alternatively, arginase could play a direct role in promoting fibrosis by increasing proline production, which is required for collagen biosynthesis, and/or by increasing ornithine production, which is metabolized by ornithine decarboxylase to produce polyamines that promote cell cycle progression and could lead to muscle hyperplasia.52 Based on this, we hypothesized that arginase inhibition could reduce intestinal fibrosis. Blocking arginase activity in the intestine with BEC has exacerbated inflammation, including immune cell infiltration, during Citrobacter rodentium–induced colitis,31 but limits disease during Helicobacter pylori infection by increasing NO-mediated killing of the pathogen.32 In our SHIP–/– mouse genetic model of intestinal inflammation, BEC treatment reduced arginase activity and expression in the SHIP–/– mouse ilea (Figure 7A) but had no impact on the number of immune cell infiltrates or immune cell aggregates present in the SHIP–/– mouse ilea (Figure 7D). More importantly, BEC treatment reduced fibrosis, collagen deposition, and muscle hyperplasia in the SHIP–/– mouse ilea (Figure 7E).
SHIP acts by negatively regulating signaling through the PI3K pathway, which is reported to be an important signaling pathway in inflammatory bowel disease.53 Interestingly, human SHIP is encoded in the 2q37 region of human chromosome 2, where a polymorphism that is associated with early-onset inflammatory bowel disease has recently been described.54 Our study identifies SHIP deficiency as a genetic cause of murine intestinal inflammation with CD-like features, highlighting the importance of the SHIP and PI3K pathway as regulators of intestinal homeostasis. The power of this model of CD is that it accurately and consistently recapitulates both the ileal localization of inflammation and the ileal fibrosis that occurs in some patients with CD. ArgI activity correlates with fibrosis in two murine models of lung fibrosis,55,56 and arginase expression is significantly higher in resected gut tissues from patients with CD.57 Arginase inhibition reduced the fibrosis that we observed in the SHIP–/– mouse ileum, identifying arginase as a potential target for the treatment of intestinal fibrosis.
Footnotes
Supported by a Canadian Association of Gastroenterology (CAG) summer studentship (A.E.C.), a Crohn's and Colitis Foundation of Canada grant in aid of research (L.M.S.), and a CAG/Canadian Institutes of Health Research/Crohn's and Colitis Foundation of Canada new investigator award (L.M.S.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.03.018.
Supplementary data
SHIP–/– mouse ilea have epithelial cell hyperplasia. Ileal crypt lengths, villus lengths, and goblet cells per crypt-villus in SHIP+/+ and SHIP–/– mice. Bars represent the mean ± SD (from 10 crypts and villi counted, in five sections each from 6- to 8-week-old mice; *P < 0.001).
SHIP–/– ileitis does not extend to the colon or cecum. H&E-stained cross sections of colon or cecum from 8-week-old SHIP+/+ and SHIP–/– mice. Scale bars = 100 μm.
Few F4/80+ macrophages are present in immune cell aggregates of 8-week-old SHIP–/– ilea. A: F4/80 IHC-negative controls. B: Ileal tissue sections of 8-week-old SHIP–/– mice were labeled with anti-F4/80 antibody (brown; hematoxylin, blue). Scale bars = 50 μm.
Few CD3+ T cells are present within villi of 8-week-old SHIP–/– ilea. A: CD3 IHC-negative controls. B: Ileal tissue sections of 8-week-old SHIP–/– mice were labeled with anti-CD3 antibody (brown; hematoxylin, blue). Scale bars = 50 μm.
References
- 1.Bernstein C.N., Wajda A., Svenson L.W., MacKenzie A., Koehoorn M., Jackson M., Fedorak R., Israel D., Blanchard J.F. The epidemiology of inflammatory bowel disease in Canada: a population-based study. Am J Gastroenterol. 2006;101:1559–1568. doi: 10.1111/j.1572-0241.2006.00603.x. [DOI] [PubMed] [Google Scholar]
- 2.Brand S. Crohn's disease: Th1, Th17 or both? the change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn's disease. Gut. 2009;58:1152–1167. doi: 10.1136/gut.2008.163667. [DOI] [PubMed] [Google Scholar]
- 3.Louis E., Collard A., Oger A.F., Degroote E., Aboul Nasr El Yafi F.A., Belaiche J. Behaviour of Crohn's disease according to the Vienna classification: changing pattern over the course of the disease. Gut. 2001;49:777–782. doi: 10.1136/gut.49.6.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke J.P., Mulsow J.J., O'Keane C., Docherty N.G., Watson R.W., O'Connell P.R. Fibrogenesis in Crohn's disease. Am J Gastroenterol. 2007;102:439–448. doi: 10.1111/j.1572-0241.2006.01010.x. [DOI] [PubMed] [Google Scholar]
- 5.Van Assche G., Geboes K., Rutgeerts P. Medical therapy for Crohn's disease strictures. Inflamm Bowel Dis. 2004;10:55–60. doi: 10.1097/00054725-200401000-00009. [DOI] [PubMed] [Google Scholar]
- 6.Sorrentino D., Terrosu G., Avellini C. Infliximab in Crohn's disease: a look at the (not so distant) future. Dig Liver Dis. 2008;40(Suppl 2):S229–S235. doi: 10.1016/S1590-8658(08)60531-9. [DOI] [PubMed] [Google Scholar]
- 7.Sorrentino D. Role of biologics and other therapies in stricturing Crohn's disease: what have we learnt so far? Digestion. 2008;77:38–47. doi: 10.1159/000117306. [DOI] [PubMed] [Google Scholar]
- 8.Burke J.P., Ferrante M., Dejaegher K., Watson R.W., Docherty N.G., De Hertogh G., Vermeire S., Rutgeerts P., D'Hoore A., Penninckx F., Geboes K., Van Assche G., O'Connell P.R. Transcriptomic analysis of intestinal fibrosis-associated gene expression in response to medical therapy in Crohn's disease. Inflamm Bowel Dis. 2008;14:1197–1204. doi: 10.1002/ibd.20482. [DOI] [PubMed] [Google Scholar]
- 9.Rieder F., Fiocchi C. First international summit on fibrosis in intestinal inflammation: mechanisms and biological therapies. Fibrogenesis Tissue Repair. 2010;3:22. doi: 10.1186/1755-1536-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szabò H., Fiorino G., Spinelli A., Rovida S., Repici A., Malesci A.C., Danese S. Review article: anti-fibrotic agents for the treatment of Crohn's disease - lessons learnt from other diseases. Aliment Pharmacol Ther. 2010;31:189–201. doi: 10.1111/j.1365-2036.2009.04171.x. [DOI] [PubMed] [Google Scholar]
- 11.Aumailley M., Gayraud B. Structure and biological activity of the extracellular matrix. J Mol Med. 1998;76:253–265. doi: 10.1007/s001090050215. [DOI] [PubMed] [Google Scholar]
- 12.Rieder F., Fiocchi C. Intestinal fibrosis in IBD: a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. 2009;6:228–235. doi: 10.1038/nrgastro.2009.31. [DOI] [PubMed] [Google Scholar]
- 13.Bhogal R.K., Stoica C.M., McGaha T.L., Bona C.A. Molecular aspects of regulation of collagen gene expression in fibrosis. J Clin Immunol. 2005;25:592–603. doi: 10.1007/s10875-005-7827-3. [DOI] [PubMed] [Google Scholar]
- 14.Regan M.C., Flavin B.M., Fitzpatrick J.M., O'Connell P.R. Stricture formation in Crohn's disease: the role of intestinal fibroblasts. Ann Surg. 2000;231:46–50. doi: 10.1097/00000658-200001000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizoguchi A., Mizoguchi E. Animal models of IBD: linkage to human disease. Curr Opin Pharmacol. 2010;10:578–587. doi: 10.1016/j.coph.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nell S., Suerbaum S., Josenhans C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nat Rev Microbiol. 2010;8:564–577. doi: 10.1038/nrmicro2403. [DOI] [PubMed] [Google Scholar]
- 17.Flier S.N., Tanjore H., Kokkotou E.G., Sugimoto H., Zeisberg M., Kalluri R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J Biol Chem. 2010;285:20202–20212. doi: 10.1074/jbc.M110.102012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rigby R.J., Hunt M.R., Scull B.P., Simmons J.G., Speck K.E., Helmrath M.A., Lund P.K. A new animal model of postsurgical bowel inflammation and fibrosis: the effect of commensal microflora. Gut. 2009;58:1104–1112. doi: 10.1136/gut.2008.157636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grassl G.A., Valdez Y., Bergstrom K.S., Vallance B.A., Finlay B.B. Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology. 2008;134:768–780. doi: 10.1053/j.gastro.2007.12.043. [DOI] [PubMed] [Google Scholar]
- 20.Vallance B.A., Gunawan M.I., Hewlett B., Bercik P., Van Kampen C., Galeazzi F., Sime P.J., Gauldie J., Collins S.M. TGF-beta1 gene transfer to the mouse colon leads to intestinal fibrosis. Am J Physiol Gastrointest Liver Physiol. 2005;289:G116–G128. doi: 10.1152/ajpgi.00051.2005. [DOI] [PubMed] [Google Scholar]
- 21.Motomura Y., Khan W.I., El-Sharkawy R.T., Verma-Gandhu M., Verdu E.F., Gauldie J., Collins S.M. Induction of a fibrogenic response in mouse colon by overexpression of monocyte chemoattractant protein 1. Gut. 2006;55:662–670. doi: 10.1136/gut.2005.068429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawrance I.C., Wu F., Leite A.Z., Willis J., West G.A., Fiocchi C., Chakravarti S. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology. 2003;125:1750–1761. doi: 10.1053/j.gastro.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 23.Kontoyiannis D., Boulougouris G., Manoloukos M., Armaka M., Apostolaki M., Pizarro T., Kotlyarov A., Forster I., Flavell R., Gaestel M., Tsichlis P., Cominelli F., Kollias G. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn's-like inflammatory bowel disease. J Exp Med. 2002;196:1563–1574. doi: 10.1084/jem.20020281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rivera-Nieves J., Bamias G., Vidrich A., Marini M., Pizarro T.T., McDuffie M.J., Moskaluk C.A., Cohn S.M., Cominelli F. Emergence of perianal fistulizing disease in the SAMP1/YitFc mouse, a spontaneous model of chronic ileitis. Gastroenterology. 2003;124:972–982. doi: 10.1053/gast.2003.50148. [DOI] [PubMed] [Google Scholar]
- 25.Helgason C.D., Damen J.E., Rosten P., Grewal R., Sorensen P., Chappel S.M., Borowski A., Jirik F., Krystal G., Humphries R.K. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sly L.M., Ho V., Antignano F., Ruschmann J., Hamilton M., Lam V., Rauh M.J., Krystal G. The role of SHIP in macrophages. Front Biosci. 2007;12:2836–2848. doi: 10.2741/2276. [DOI] [PubMed] [Google Scholar]
- 27.Rauh M.J., Ho V., Pereira C., Sham A., Sly L.M., Lam V., Huxham L., Minchinton A.I., Mui A., Krystal G. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–374. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Kerr W.G., Park M.Y., Maubert M., Engelman R.W. SHIP deficiency causes Crohn's disease-like ileitis. Gut. 2011;60:177–188. doi: 10.1136/gut.2009.202283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho V.W., Sly L.M. Derivation and characterization of murine alternatively activated (M2) macrophages. Methods Mol Biol. 2009;531:173–185. doi: 10.1007/978-1-59745-396-7_12. [DOI] [PubMed] [Google Scholar]
- 30.Kim N.N., Cox J.D., Baggio R.F., Emig F.A., Mistry S.K., Harper S.L., Speicher D.W., Morris S.M., Jr, Ash D.E., Traish A., Christianson D.W. Probing erectile function: s-(2-boronoethyl)-l-cysteine binds to arginase as a transition state analogue and enhances smooth muscle relaxation in human penile corpus cavernosum. Biochemistry. 2001;40:2678–2688. doi: 10.1021/bi002317h. [DOI] [PubMed] [Google Scholar]
- 31.Gobert A.P., Cheng Y., Akhtar M., Mersey B.D., Blumberg D.R., Cross R.K., Chaturvedi R., Drachenberg C.B., Boucher J.L., Hacker A., Casero R.A., Jr, Wilson K.T. Protective role of arginase in a mouse model of colitis. J Immunol. 2004;173:2109–2117. doi: 10.4049/jimmunol.173.3.2109. [DOI] [PubMed] [Google Scholar]
- 32.Lewis N.D., Asim M., Barry D.P., Singh K., de Sablet T., Boucher J.L., Gobert A.P., Chaturvedi R., Wilson K.T. Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. J Immunol. 2010;184:2572–2582. doi: 10.4049/jimmunol.0902436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Podolsky D.K. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 34.Cosnes J., Nion-Larmurier I., Beaugerie L., Afchain P., Tiret E., Gendre J.P. Impact of the increasing use of immunosuppressants in Crohn's disease on the need for intestinal surgery. Gut. 2005;54:237–241. doi: 10.1136/gut.2004.045294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lennard-Jones J.E. Classification of inflammatory bowel disease. Scand J Gastroenterol. 1989;170(Suppl):2–6. doi: 10.3109/00365528909091339. [DOI] [PubMed] [Google Scholar]; discussion, 16–19
- 36.Hanauer S.B. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12(Suppl 1):S3–S9. doi: 10.1097/01.mib.0000195385.19268.68. [DOI] [PubMed] [Google Scholar]
- 37.Xavier R.J., Podolsky D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 38.Adams D.O. The granulomatous inflammatory response: a review. Am J Pathol. 1976;84:164–192. [PMC free article] [PubMed] [Google Scholar]
- 39.Fais S., Pallone F. Inability of normal human intestinal macrophages to form multinucleated giant cells in response to cytokines. Gut. 1995;37:798–801. doi: 10.1136/gut.37.6.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graham M.F., Diegelmann R.F., Elson C.O., Lindblad W.J., Gotschalk N., Gay S., Gay R. Collagen content and types in the intestinal strictures of Crohn's disease. Gastroenterology. 1988;94:257–265. doi: 10.1016/0016-5085(88)90411-8. [DOI] [PubMed] [Google Scholar]
- 41.Kugathasan S., Saubermann L.J., Smith L., Kou D., Itoh J., Binion D.G., Levine A.D., Blumberg R.S., Fiocchi C. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut. 2007;56:1696–1705. doi: 10.1136/gut.2006.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuss I.J., Becker C., Yang Z., Groden C., Hornung R.L., Heller F., Neurath M.F., Strober W., Mannon P.J. Both IL-12p70 and IL-23 are synthesized during active Crohn's disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm Bowel Dis. 2006;12:9–15. doi: 10.1097/01.mib.0000194183.92671.b6. [DOI] [PubMed] [Google Scholar]
- 43.Monteleone G., Trapasso F., Parrello T., Biancone L., Stella A., Iuliano R., Luzza F., Fusco A., Pallone F. Bioactive IL-18 expression is up-regulated in Crohn's disease. J Immunol. 1999;163:143–147. [PubMed] [Google Scholar]
- 44.Kuroda E., Antignano F., Ho V.W., Hughes M.R., Ruschmann J., Lam V., Kawakami T., Kerr W.G., McNagny K.M., Sly L.M., Krystal G. SHIP represses Th2 skewing by inhibiting IL-4 production from basophils. J Immunol. 2011;186:323–332. doi: 10.4049/jimmunol.1002778. [DOI] [PubMed] [Google Scholar]
- 45.Kuroda E., Ho V., Ruschmann J., Antignano F., Hamilton M., Rauh M.J., Antov A., Flavell R.A., Sly L.M., Krystal G. SHIP represses the generation of IL-3-induced M2 macrophages by inhibiting IL-4 production from basophils. J Immunol. 2009;183:3652–3660. doi: 10.4049/jimmunol.0900864. [DOI] [PubMed] [Google Scholar]
- 46.Wynn T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4:583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gillery P., Fertin C., Nicolas J.F., Chastang F., Kalis B., Banchereau J., Maquart F.X. Interleukin-4 stimulates collagen gene expression in human fibroblast monolayer cultures: potential role in fibrosis. FEBS Lett. 1992;302:231–234. doi: 10.1016/0014-5793(92)80448-p. [DOI] [PubMed] [Google Scholar]
- 48.Postlethwaite A.E., Holness M.A., Katai H., Raghow R. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4. J Clin Invest. 1992;90:1479–1485. doi: 10.1172/JCI116015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barron L., Wynn T.A. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am J Physiol Gastrointest Liver Physiol. 2011;300:G723–G728. doi: 10.1152/ajpgi.00414.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weisser S.B., McLarren K.W., Voglmaier N., van Netten-Thomas C.J., Antov A., Flavell R.A., LM Sly. Alternative activation of macrophages by IL-4 requires SHIP degradation. Eur J Immunol. 2011 Mar 21 doi: 10.1002/eji.201041105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bishop J.L., Sly L.M., Krystal G., Finlay B.B. The inositol phosphatase SHIP controls Salmonella enterica serovar Typhimurium infection in vivo. Infect Immun. 2008;76:2913–2922. doi: 10.1128/IAI.01596-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Casero R.A., Jr, Marton L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 53.Wei J., Feng J. Signaling pathways associated with inflammatory bowel disease. Recent Pat Inflamm Allergy Drug Discov. 2010;4:105–117. doi: 10.2174/187221310791163071. [DOI] [PubMed] [Google Scholar]
- 54.Imielinski M., Baldassano R.N., Griffiths A., Russell R.K., Annese V., Dubinsky M. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. 2009;41:1335–1340. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Endo M., Oyadomari S., Terasaki Y., Takeya M., Suga M., Mori M., Gotoh T. Induction of arginase I and II in bleomycin-induced fibrosis of mouse lung. Am J Physiol Lung Cell Mol Physiol. 2003;285:L313–L321. doi: 10.1152/ajplung.00434.2002. [DOI] [PubMed] [Google Scholar]
- 56.Mora A.L., Torres-Gonzalez E., Rojas M., Corredor C., Ritzenthaler J., Xu J., Roman J., Brigham K., Stecenko A. Activation of alveolar macrophages via the alternative pathway in herpesvirus-induced lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:466–473. doi: 10.1165/rcmb.2006-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Horowitz S., Binion D.G., Nelson V.M., Kanaa Y., Javadi P., Lazarova Z., Andrekopoulos C., Kalyanaraman B., Otterson M.F., Rafiee P. Increased arginase activity and endothelial dysfunction in human inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1323–G1336. doi: 10.1152/ajpgi.00499.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SHIP–/– mouse ilea have epithelial cell hyperplasia. Ileal crypt lengths, villus lengths, and goblet cells per crypt-villus in SHIP+/+ and SHIP–/– mice. Bars represent the mean ± SD (from 10 crypts and villi counted, in five sections each from 6- to 8-week-old mice; *P < 0.001).
SHIP–/– ileitis does not extend to the colon or cecum. H&E-stained cross sections of colon or cecum from 8-week-old SHIP+/+ and SHIP–/– mice. Scale bars = 100 μm.
Few F4/80+ macrophages are present in immune cell aggregates of 8-week-old SHIP–/– ilea. A: F4/80 IHC-negative controls. B: Ileal tissue sections of 8-week-old SHIP–/– mice were labeled with anti-F4/80 antibody (brown; hematoxylin, blue). Scale bars = 50 μm.
Few CD3+ T cells are present within villi of 8-week-old SHIP–/– ilea. A: CD3 IHC-negative controls. B: Ileal tissue sections of 8-week-old SHIP–/– mice were labeled with anti-CD3 antibody (brown; hematoxylin, blue). Scale bars = 50 μm.