

Abstract
Increased glutamatergic input in the paraventricular nucleus (PVN) is important for high sympathetic outflow in hypertension, but the associated molecular mechanisms remain unclear. Here, we determined the role of protein kinase CK2 (formerly casein kinase II) in increased N-methyl-d-aspartate receptor (NMDAR) activity in spinally projecting PVN neurons and sympathetic vasomotor tone in spontaneously hypertensive rats (SHRs). The selective CK2 inhibitors 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) or 4,5,6,7-tetrabromobenzotriazole (TBB) significantly decreased the frequency of miniature EPSCs (mEPSCs) of labeled PVN neurons in SHRs but not in Wistar-Kyoto (WKY) normotensive rats. Also, DRB abolished the inhibitory effect of the NMDAR antagonist AP5 on the frequency of mEPSCs in SHRs. Treatment with DRB or TBB significantly reduced the amplitude of evoked NMDA-EPSCs but not AMPA-EPSCs in SHRs. Furthermore, DRB significantly decreased the firing activity of PVN neurons in SHRs but not in WKY rats. The membrane protein level of CK2α in the PVN, but not brainstem and prefrontal cortex, was significantly higher in SHRs than in WKY rats. Lowering blood pressure with celiac ganglionectomy in SHRs did not alter the increased CK2α level and the effects of DRB on mEPSCs and NMDA-EPSCs. In addition, intracerebroventricular injection of DRB not only significantly reduced blood pressure and lumbar sympathetic nerve discharges but also eliminated the inhibitory effect of AP5 microinjected into the PVN on sympathetic nerve activity in SHRs. Our findings suggest that augmented CK2 activity critically contributes to increased presynaptic and postsynaptic NMDAR activity in the PVN and elevated sympathetic vasomotor tone in essential hypertension.
Introduction
Uncontrolled high blood pressure can lead to stroke, coronary artery disease, or kidney failure. Essential (primary) hypertension is the most prevalent type of hypertension affecting 90–95% of hypertensive patients. However, the etiology of essential hypertension is poorly understood. The pathogenesis of and mechanisms involved in essential hypertension are complex and likely multifactorial. Considerable evidence shows that essential hypertension is initiated primarily in the CNS and is later reinforced by non-neural factors. For example, increased sympathetic outflow is well recognized in animal models of hypertension, including spontaneously hypertensive rats (SHRs) (Judy et al., 1976; Allen, 2002; Li and Pan, 2007) and in patients with essential hypertension (Anderson et al., 1989; Grassi, 1998; Greenwood et al., 1999; Mancia et al., 1999). However, the cellular and molecular mechanisms responsible for the sustained elevation of sympathetic vasomotor tone in essential hypertension are not clear.
The paraventricular nucleus (PVN) of the hypothalamus is an important source of the excitatory drive for sympathetic vasomotor tone in essential hypertension (Swanson and Sawchenko, 1983; Kannan et al., 1989; Martin and Haywood, 1993; Allen, 2002; Li and Pan, 2007). The PVN neurons that project to sympathetic preganglionic neurons in the intermediolateral cell column of the spinal cord and vasomotor neurons in the rostral ventrolateral medulla in the brainstem play a crucial role in the regulation of sympathetic outflow and arterial blood pressure (ABP) (Yamashita et al., 1984; Kannan et al., 1989; Martin and Haywood, 1993; Ranson et al., 1998; Li et al., 2003). We have shown that the glutamatergic tone is increased in the PVN in SHRs, which is maintained by increased presynaptic glutamate release and postsynaptic N-methyl-d-aspartate receptor (NMDAR) activity (Li and Pan, 2007). Nevertheless, the molecular mechanisms underlying increased glutamatergic input and NMDAR activity in the PVN in hypertension remain unclear.
Casein kinase II (CK2) is a ubiquitous and essential serine/threonine protein kinase that modulates intracellular signaling, protein expression, and ion channel activity through phosphorylation (Bolanos-Garcia et al., 2006; Bachhuber et al., 2008; Lou et al., 2008). CK2 is a heterotetrameric holoenzyme composed of two catalytic α or α′ subunits and two regulatory β subunits (Niefind et al., 2001; Litchfield, 2003) and is widely expressed in synaptosomes and the postsynaptic density in the brain, including the PVN (Soto et al., 2004). Previous studies have shown that CK2 plays an important role in regulating NMDAR function in the hippocampus (Lieberman and Mody, 1999; Kimura and Matsuki, 2008). However, the role of CK2 in the increased glutamatergic input to the PVN in hypertension has yet to be determined.
In the present study, we determined the contribution of CK2 to increased glutamatergic input to spinally projecting PVN neurons in SHRs. Furthermore, we measured the protein level of CK2 in the PVN and determined its role in controlling sympathetic vasomotor tone in SHRs, a widely used animal model of essential hypertension. Our findings strongly suggest that CK2 is critically involved in potentiation of presynaptic and postsynaptic NMDAR activity of PVN presympathetic neurons and contributes to the increased sympathetic outflow in hypertension.
Materials and Methods
Male Wistar-Kyoto (WKY) rats and SHRs (11–13 weeks old, Harlan) were used in this study. We selected SHRs for this study because this is the most commonly used and best-characterized animal model of essential hypertension. The surgical procedures and experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center (Houston, TX) and conformed to the National Institutes of Health Guidelines on the ethical use of animals. Blood pressure was measured daily for at least 1 week before the electrophysiological experiments by using a noninvasive tail–cuff system (IITC Life Science) in a subset of animals. The systolic ABP in 13-week-old SHRs (210.43 ± 3.86 mmHg, n = 25) was significantly higher than that of the age-matched WKY rats (126.39 ± 2.16 mmHg, n = 20 rats).
Retrograde labeling of PVN presympathetic neurons.
We identified the PVN presympathetic neurons via retrograde labeling of spinally projecting neurons as we described previously (Li et al., 2003, 2008). Briefly, rats were anesthetized by intraperitoneal injection of a mixture of ketamine (70 mg/kg) and xylazine (6 mg/kg), and a limited dorsal laminectomy between T2 and T4 was performed to expose the spinal cord. FluoSpheres microspheres (0.04 μm; Invitrogen) were pressure ejected (Nanojector II, Drummond Scientific) via a glass pipette bilaterally into intermediolateral cell column of the spinal cord at T2-T4 level (∼500 μm from the midline and ∼500 μm below the dorsolateral sulcus) in 3–4 separate 50 nl injections. The rat was returned to its cage for 3–5 d to permit the FluoSpheres microspheres to be transported to the PVN.
Brain slice preparation.
Coronal hypothalamic slices (300 μm thick) containing the PVN were obtained from FluoSpheres microsphere-injected rats using a vibrating microtome. Briefly, the rat was rapidly decapitated under isoflurane-induced anesthesia. The brain was quickly removed and sectioned in an ice-cold artificial CSF (aCSF) solution containing (in mm): 126 NaCl, 3 KCl, 1.5 MgSO4, 2.4 CaCl2, 1.2 NaH2PO4, 10 glucose, and 26 NaHCO3 saturated with 95% O2 and 5% CO2. The slice was preincubated in the aCSF at 34°C for at least 1 h before recording. To verify the injection and diffusion sites of FluoSpheres microspheres, the spinal cord was sectioned at the injection level and viewed under a microscope immediately after the rat was killed (Li et al., 2003, 2004). Data were excluded from analysis if the injection site was not located within the intermediolateral cell column of the spinal cord.
Electrophysiological recordings in brain slices.
All the whole-cell, patch-clamp recording was performed in labeled PVN neurons visually identified under an upright microscope (BX51WI, Olympus) with epifluorescent and infrared differential interference contrast optics. All recordings were obtained at 34°C using borosilicate glass microelectrodes (resistance, 4–6 MΩ) pulled using a micropipette puller (Sutter Instruments). The pipette solution contained (in mm): 110 Cs2SO4, 2.0 MgCl2, 0.5 CaCl2, 5.0 EGTA, 5.0 MgATP, 0.5 Na2GTP, and 10 HEPES. The pH was adjusted to 7.3 with CsOH (280–300 mOsm). Signals were processed using Axopatch 700B amplifier (Molecular Devices), filtered at 1–2 kHz, digitized at 20 kHz using DigiData 1440 (Molecular Devices), and saved to a computer hard drive.
The miniature EPSCs (mEPSCs) of labeled PVN neurons were recorded at a holding potential of −60 mV in the presence of 10 μm bicuculline and 1 μm tetrodotoxin (TTX). The evoked EPSCs were elicited by electrical stimulation (0.1 ms, 0.8 mA, and 0.2 Hz) through a bipolar tungsten electrode connected to a stimulator (Grass Instruments). The tip of the stimulation electrode was placed on the ventral side ∼150 μm away from the recorded neuron within the PVN (Li et al., 2003, 2005). Evoked AMPA-EPSCs were recorded at a holding potential of −60 mV in the presence of 10 μm bicuculline, and evoked NMDA-EPSCs were recorded at a holding potential of +40 mV in the presence of 10 μm bicuculline and 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX). A sodium channel blocker, lidocaine N-ethyl bromide (10.0 mm), was included in the pipette solution to suppress the firing activity of the recorded neuron.
We recorded the NMDA-induced current using puff application of NMDA to the recorded neuron at a holding potential of −60 mV. The puff pipette (∼10 μm tip diameter) was placed about 80 μm away from the recorded cell. Positive pressure (4 psi; Toohey Company) was applied to eject the agonist onto the recorded cell over 15 ms. Because the NMDA channel is blocked by Mg2+ at a negative holding potential (Nowak et al., 1984) and coactivated by glycine (Kleckner and Dingledine, 1988), the NMDA current was recorded at a low concentration of Mg2+ (0.1 mm), 10 μm glycine, and 1 μm TTX.
The spontaneous firing activity of labeled PVN neurons was recorded using the whole-cell, current-clamp technique (Li et al., 2003). The recording procedures were similar to those used for EPSC recordings described above, except that Cs2SO4 in the pipette solution was replaced with potassium gluconate and TTX and QX-314 were not used. The spontaneous firing activity was recorded when it reached a steady state.
All drugs were freshly prepared in aCSF before the experiments and delivered via syringe pumps at their final concentrations. 5,6-Dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), chelerythrine, 4,5,6,7-tetrabromobenzotriazole (TBB), glycine, GABA, and NMDA were purchased from Sigma-Aldrich. d-2-Amino-5-phosphonopentanoate (AP5), CNQX, bicuculline, and TTX were obtained from Ascent Scientific. Lidocaine N-ethyl bromide was purchased from Alomone Labs.
Western immunoblotting.
Rats were anesthetized with isoflurane and decapitated. Using the third ventricle as a reference, the PVN tissue (∼0.5 mm in diameter) spanning from −1.08 bregma to −2.12 bregma was micro-punched bilaterally on a cold plate under the microscope. Total and membrane proteins were extracted with radioimmunoprecipitation assay buffer in the presence of the inhibitor mixture of proteases (serine proteases, cysteine proteases, aspartic proteases, and metalloproteases). Membrane protein fraction (PVN tissues from three rats were pooled) was obtained using ProteoExtract subcellular proteome extraction kit (Calbiochem) according to the manufacturer's instructions. The protein concentrations were determined using the Bradford protein assay. Samples were subjected to 12% SDS-PAGE gels and transferred to a polyvinylidene difluoride membrane (Millipore). The blot was probed with an anti-CK2α (1:1000, Santa Cruz Biotechnology) or anti-CK2β antibody (1:1000, Santa Cruz Biotechnology) and then reprobed with anti-β-actin or anti-GAPDH antibody (1:2000, Sigma-Aldrich) for protein loading controls. ECL kit (GE Healthcare) was used to detect the protein band, and the band intensities were quantified by ImageJ software. The specificity of the CK2 antibodies has been reported previously (Kang et al., 2009; Pallares et al., 2009). The amounts of total and membrane CK2 subunits were quantified by normalizing the optical density of their protein band to that of β-actin and GAPDH, respectively. The mean values of CK2 subunits in WKY rats were considered to be 1.
In vivo recording of lumbar sympathetic nerve activity, ABP, and heart rate.
The SHRs were initially anesthetized using 2% isoflurane in O2 before intraperitoneal injection of a mixture of α-chloralose (60–75 mg/kg) and urethane (800 mg/kg). The depth of anesthesia level was confirmed before surgery by the absence of both corneal reflexes and paw withdrawal responses to a noxious pinch. The trachea was cannulated for mechanical ventilation using a rodent ventilator with 100% O2. The ABP was monitored with a pressure transducer through a catheter placed into the left femoral artery. Heart rate (HR) was counted by triggering from the pulsatile blood pressure. The right femoral vein was cannulated for intravenous administration of drugs. Supplemental doses of α-chloralose and urethane were administered as necessary to maintain an adequate depth of anesthesia. A small branch of the left lumbar postganglionic sympathetic nerve was isolated under an operating microscope through a retroperitoneal incision. The lumbar sympathetic nerve was cut distally to ensure that afferent activity was not recorded. The nerve was then immersed in mineral oil and placed on a stainless steel recording electrode (Li and Pan, 2007). The nerve signal was amplified and bandpass filtered (100–3000 Hz) by an alternating current amplifier (model P511; Grass Instruments), and the lumbar sympathetic nerve activity (LSNA) was monitored using an audio amplifier (Grass Instruments). The LSNA and ABP were recorded using a 1401-PLUS analog-to-digital converter and Spike2 system (Cambridge Electronic Design). The level of background electrical noise was determined by a suppressing the LSNA via i.v. injection of phenylephrine (20 μg/kg i.v.) before euthanasia and 5 min after the rats were killed by an overdose of pentobarbital sodium (200 mg/kg i.v.) at the end of each experiment. The respective electrical noise levels measured using these two methods were similar and were subtracted from the integrated LSNA values, and the percentage change in LSNA from baseline was calculated.
Intracerebroventricular injection and PVN microinjection.
A guide cannula was placed through a 2 mm burr hole drilled in the skull over the lateral ventricle in the following coordinates: 1.5 mm lateral to the midline, 1.0 mm caudal to the bregma, and 3.0 mm ventral to the dura. Injections were performed by using a Hamilton syringe connected to an injection cannula, which was advanced 3.5 mm ventral from the surface of the dura. The tip of the injection cannula protruded 0.5 mm beyond the tip of the guide cannula. Each injection consisted of 10 μl of solution delivered over 1 min. The DRB was initially dissolved in DMSO and diluted with aCSF (0.5% DMSO) in a concentration of 4 mm. To ensure that the entire injection was delivered, the injection cannula was allowed to remain in place for an additional minute before being removed. Intracerebroventricular (ICV) injections were randomized to the right and left hemispheres, whereas each PVN microinjection was performed on the contralateral site.
For PVN microinjections, rats were placed in a stereotactic frame and the brain was exposed at the level of the hypothalamus. A glass microinjection pipette (tip diameter, 20–30 μm) was advanced into the PVN using the following stereotactic coordinates: 1.6–2.0 mm caudal to the bregma, 0.5 mm lateral to the midline, and 7.0–7.5 mm ventral to the dura. The injection sites of the PVN were first confirmed by the depressor responses to microinjection of 5.0 nmol GABA (20 nl, 250 mm) (Li and Pan, 2007). The microinjection was done by using a calibrated microinjection system (Nanojector II; Drumond Scientific) and monitored using an operating microscope. GABA microinjections were separated by 10–15 min intervals to allow for recovery of the depressor response. The PVN vasomotor site was considered to have been located when the GABA injection decreased the mean ABP by at least 10 mmHg, and the stereotactic coordinates at which the prior GABA microinjection elicited the greatest depressor responses were used in the same rat for the subsequent microinjection of AP5. After microinjection of the drugs, the glass pipette was left in place for 1–2 min to ensure adequate delivery of the drug to the injection site. The location of the pipette tip and diffusion of the drugs in the PVN were determined by including 5% FluoSpheres microspheres (0.04 μm; Invitrogen) in the injection solution. The rat brain was removed rapidly at the end of the experiment and fixed in 10% buffered formalin solution overnight. Frozen coronal sections (40 μm thick) were cut on a freezing microtome and mounted on slides. Rhodamine-labeled fluorescent regions were identified using an epifluorescent microscope and plotted on standardized sections from the atlas of Paxinos and Watson (1997). Rats were not included for data analysis if they had one misplaced microinjection outside the PVN.
Celiac ganglionectomy and blood pressure measurement with telemetry.
Surgery was performed aseptically in SHRs anesthetized with 2% isofluorane. The celiac ganglion area was exposed through a midline laparotomy. The bifurcations of the superior mesenteric artery and celiac plexus were identified, and the celiac ganglion was located within the area near the superior mesenteric artery and celiac artery. For rats undergoing celiac ganglionectomy (CGx), the celiac plexus and all visible nerves were dissected under a surgical microscope and removed as completely as possible by stripping. In sham control rats, the celiac ganglion plexus was exposed but not disturbed.
To measure blood pressure in SHRs, the Millar catheter of the telemetry system (Telemetry Research) was inserted into the abdominal aorta below the bifurcation of the renal arteries. The transmitter was implanted in the abdominal cavity and attached with a suture to the peritoneum. The abdomen was closed in two layers with interrupted sutures. The rats were housed singly, and buprenorphine (0.3 mg/kg, s.c.) and penicillin were given for 3 d after the surgery. The blood pressure signal was monitored in conscious rats through the receiver, and the data were collected every 2 d and analyzed with a data acquisition system (LabChart; AD Instruments). Two weeks after CGx or sham surgery, rats were used for brain slice recordings or Western blotting.
Data analysis.
Data are presented as the mean ± SEM. The action potential and mEPSCs were analyzed off-line using a peak detection program (MiniAnalysis, Synaptosoft). The junction potential was corrected offline on the basis of the composition of the internal and external solutions used for recordings. The peak amplitude of evoked EPSCs was determined and analyzed using pClamp 10 (Molecular Devices). In all the slice experiments, only one neuron was recorded in each slice and at least four rats were used in each group. The LSNA, ABP, and HR were analyzed using the Spike2 software program. LSNA was rectified and integrated offline after subtracting the background noise. Control values were obtained by averaging the signal over a 60 s period immediately before each treatment. Response values after each intervention were averaged over 30 s when the maximal responses occurred. For comparisons of two datasets, statistical significance was tested using the Student's t test. For comparisons of more than two datasets, the repeated-measures ANOVA with Dunnett's test or two-way ANOVA with Bonferroni's post hoc test was performed to compare responses within or between experimental groups, respectively. p < 0.05 was considered statistically significant.
Results
Effects of CK2 inhibitors on mEPSCs of labeled PVN neurons
The frequency but not the amplitude of mEPSCs was significantly higher in SHRs than in WKY rats (Fig. 1). The mEPSCs were completely abolished by bath application of the non-NMDAR antagonist CNQX (20 μm; data not shown). To determine the role of CK2 in presynaptic glutamate release in the PVN, the mEPSCs were recorded in hypothalamic slices from WKY rats and SHRs incubated with the specific CK2 inhibitor DRB (100 μm) for 2–3 h. The concentration of DRB has been shown to inhibit CK2 activity in rat hippocampal slices (Lieberman and Mody, 1999). Treatment with DRB significantly reduced the frequency of mEPSCs in SHRs but not in WKY rats (n = 10 neurons in each group) (Fig. 1A,B). The amplitude of mEPSCs was not significantly different in slices treated with a vehicle or DRB in either WKY rats or SHRs (Fig. 1A,B). Furthermore, pretreatment of the slices with another structurally dissimilar CK2 inhibitor, TBB (2 μm) (Sarno et al., 2001), also normalized the frequency of mEPSCs in SHRs to the level of that in WKY rats without changing the amplitude of mEPSCs (n = 8 neurons in each group) (Fig. 1B).
Figure 1.

Effects of CK2 selective inhibitors on glutamatergic mEPSCs in labeled PVN neurons in WKY rats and SHRs. A, Representative current traces show mEPSCs of the labeled PVN neuron treated with vehicle or DRB (100 μm) in WKY rats and SHRs. B, Summary data show the frequency and amplitude of mEPSCs of PVN neurons treated with vehicle (n = 10), DRB (n = 10), or TBB (2 μm, n = 8) in WKY rats and SHRs. *p < 0.05, compared with the WKY rat vehicle control; #p < 0.05, compared with the SHR vehicle control.
Presynaptic NMDARs can regulate synaptic glutamate release in the neocortex and spinal cord (Sjöström et al., 2003; Zeng et al., 2006). To determine whether increased CK2 activity plays a role in increased synaptic glutamate release through presynaptic NMDARs in SHRs, we blocked postsynaptic NMDARs by adding the specific NMDA channel blocker MK-801 (1 mm) to the recording pipette solution (Zhou et al., 2010). Bath application of the NMDAR antagonist AP5 (50 μm) significantly reduced the frequency of mEPSCs in labeled PVN neurons in SHRs (n = 7 neurons) but not in WKY rats (n = 8 neurons) (Fig. 2A–C). However, AP5 failed to reduce the frequency of mEPSCs in brain slices pretreated with DRB in SHRs (n = 8 neurons) (Fig. 2A–C).
Figure 2.
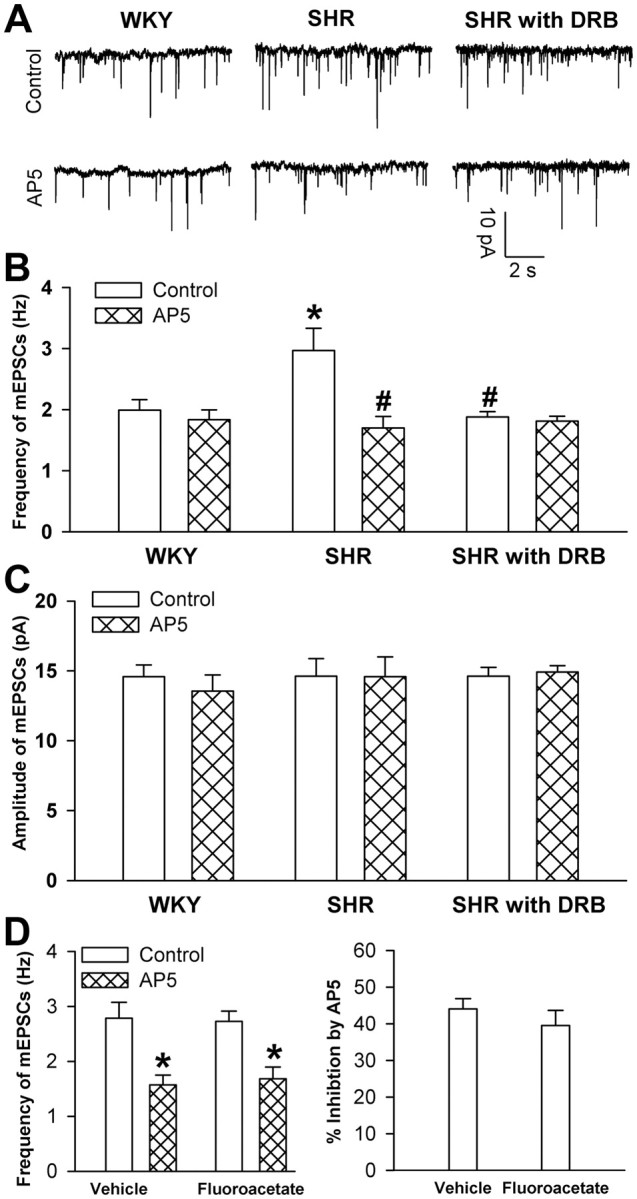
Effects of AP5 on mEPSCs in labeled PVN neurons in WKY rats and SHRs. A, Representative recordings show mEPSCs of the labeled PVN neuron on a vehicle-treated brain slice from a WKY rat (left) and an SHR (middle panel) and a DRB-treated brain slice from an SHR (right panel) before and after bath application of AP5 (50 μm). The NMDAR antagonist MK-801 (1 mm) was added to the intracellular pipette solution to block postsynaptic NMDARs. B, C, Summary data show the effects of AP5 on the frequency (B) and amplitude (C) of mEPSCs of PVN neurons (n = 7 in each group) in SHRs and WKY rats. D, Group data show the effect of AP5 on the frequency of mEPSCs of PVN neurons (n = 7 in each group) in vehicle-treated and fluoroacetate-treated brain slices from SHRs. *p < 0.05, compared with the control value in the WKY rat group; #p < 0.05, compared with the control value in the SHR group.
To assess the possible contribution of presynaptic NMDARs present on glia to increased frequency of mEPSCs of PVN neurons in SHRs, we next examined the effect of AP5 on mEPSCs of PVN neurons in brain slices treated with fluoroacetate, a selective inhibitor of glial metabolism (Hülsmann et al., 2000). Incubation of the brain slices with 5 mm fluoroacetate for 30–50 min did not significantly change mEPSCs and the inhibitory effect of AP5 on the mEPSC frequency of PVN neurons in SHRs (Fig. 2D).
Effects of CK2 inhibitors on postsynaptic NMDAR activity in WKY rats and SHRs
To determine the role of CK2 in increased postsynaptic NMDAR activity in SHRs, we examined the effect of DRB and TBB on NMDA-EPSCs and AMPA-EPSCs evoked by focal stimulation in the hypothalamic slices of both WKY rats and SHRs. The amplitude of electrically evoked AMPA-EPSCs of labeled PVN neurons was similar in SHRs and WKY rats (Fig. 3A,B). However, the amplitude of evoked NMDA-EPSCs was about twofold larger in SHRs than in WKY rats (Fig. 3A,B). Incubation of the brain slices with DRB (100 μm) for 2–3 h did not significantly affect evoked AMPA-EPSCs in either SHRs or WKY rats (Fig. 3A,B). DRB significantly reduced the amplitude of evoked NMDA-EPSCs and the ratio of NMDA-EPSCs to AMPA-EPSCs in SHRs but not in WKY rats (Fig. 3A,B). Furthermore, another CK2 inhibitor, TBB (2 μm), also significantly reduced the amplitude of evoked NMDA-EPSCs but not AMPA-EPSCs in SHRs (Fig. 3C). TBB did not significantly affect either NMDA-EPSCs or AMPA-EPSCs in WKY rats.
Figure 3.

Effects of CK2 inhibitors on evoked AMPA-EPSCs and NMDA-EPSCs in labeled PVN neurons in WKY rats and SHRs. A, Examples of electrically evoked AMPA-EPSCs and NMDA-EPSCs recorded in the same labeled PVN neuron treated with vehicle or DRB (100 μm) in one WKY rat and one SHR. AMPA-EPSCs were record at a holding potential of −60 mV in the presence of 10 μm bicuculline and NMDA-EPSCs at +40 mV in the presence of 20 μm CNQX and 10 μm bicuculline. B, Summary data show the evoked AMPA-EPSCs, NMDA-EPSCs, and the ratio of NMDA-EPSCs to AMPA-EPSCs in labeled PVN neurons of WKY rats and SHRs treated with vehicle (n = 8), DRB (n = 7), or TBB (n = 8 in each group). C, Original recordings showing NMDA puff application-induced NMDA currents of a PVN neuron treated with vehicle or DRB (100 μm) in one WKY rat and one SHR. Bath application of 50 μm AP5 abolished NMDA-induced currents. D, Summary data show the amplitude and current density of NMDA-induced currents of the labeled PVN neurons treated with vehicle or DRB in WKY rats and SHRs (n = 7 in each group). *p < 0.05, compared with the WKY rat vehicle control; #p < 0.05, compared with SHRs vehicle control.
We also determined the effects of DRB on NMDA currents induced by puff application of 100 μm NMDA to labeled PVN neurons in WKY rats and SHRs. The amplitude of NMDA-induced currents was significantly greater in SHRs (n = 7 neurons) than in WKY rats (n = 8 neurons). Although DRB treatment had no significant effects on the NMDA currents in WKY rats, it significantly decreased NMDA currents in SHRs to the level seen in WKY rats (Fig. 3C,D).
Additionally, we examined the effect of the inhibitor of protein kinase C (PKC), another protein kinase using ATP as a phosphate donor (Gardoni et al., 2001), on the mEPSCs and evoked EPSCs of labeled PVN neurons in SHRs. The PKC inhibitor chelerythrine (10 μm) had no significant effect on the frequency (vehicle control, 3.04 ± 0.32; chelerythrine treatment, 3.08 ± 0.49) and amplitude (vehicle control, 14.25 ± 0.71; chelerythrine treatment, 14.27 ± 1.21) of mEPSCs of PVN neurons (n = 7 in each group). Chelerythrine also failed to significantly change the amplitude of evoked NMDA-EPSCs (vehicle control, 142.68 ± 6.14; chelerythrine treatment, 148.96 ± 11.87) and AMPA-EPSCs (vehicle control, 210.34 ± 9.86; chelerythrine treatment, 217.71 ± 14.58) of separate groups of labeled PVN neurons (n = 7 in each group) in SHRs.
Effect of CK2 inhibition on the firing activity of labeled PVN neurons in SHRs
To determine the role of CK2 in the increased firing activity of spinally projecting PVN neurons in essential hypertension, we compared the firing rate of spinally projecting PVN neurons in slices treated with and without DRB in SHRs and WKY rats. Inhibition of CK2 with DRB significantly reduced the firing rate of PVN neurons in SHRs but not in WKY rats (n = 8 neurons in each group) (Fig. 4). Furthermore, blocking NMDARs with AP5 (50 μm) significantly decreased the firing rate of labeled PVN neurons in vehicle-treated but not DRB-treated slices from SHRs (Fig. 4A,B).
Figure 4.
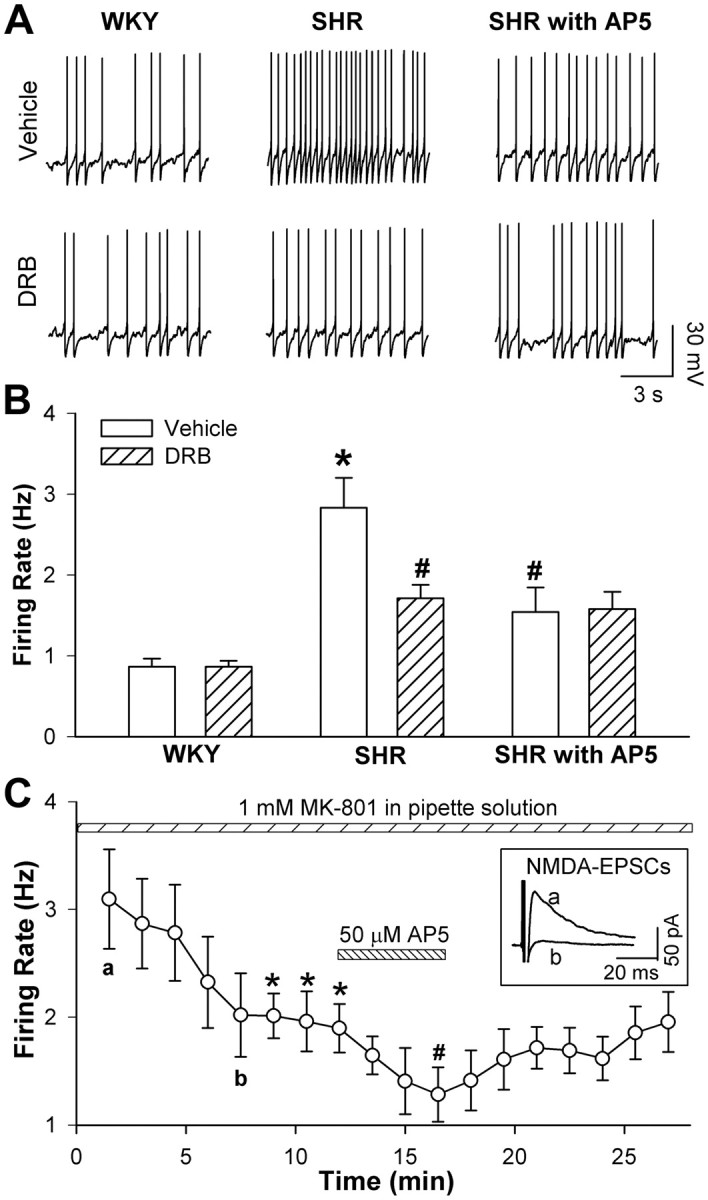
Effects of DRB on the firing activity of labeled PVN neurons in WKY rats and SHRs. A, Representative recordings show the firing activity of the labeled PVN neuron on the vehicle- and DRB-treated brain slices from a WKY rat (left) and a SHR in the absence (middle) or presence (right) of 50 μm AP5. B, Summary data show the effect of DRB on the firing activity of labeled PVN neurons in WKY rats and SHRs (n = 8 in each group). *p < 0.05, compared with WKY rat vehicle control; #p < 0.05, compared with the SHRs vehicle control. C, Group data show the effect of intracellular dialysis of 1 mm MK-801 and the subsequent effect of bath application of 50 μm AP5 on the firing activity of labeled PVN neurons (n = 7) in SHRs. Inset, original current traces of evoked NMDA-EPSCs recorded at the time point shown in C. *p < 0.05, compared with the baseline control; #p < 0.05, compared with the effect of intracellular dialysis of MK-801 at 10 min.
To determine the relative contribution of presynaptic and postsynaptic NMDARs to increased firing activity of PVN neurons in SHRs, we also examined the effect of AP5 on the spontaneous firing activity of spinally projecting PVN neurons by including MK-801 (1 mm), an NMDAR channel blocker (Zhou et al., 2010), in the pipette solution. Intracellular dialysis with MK-801 abolished NMDA-EPSCs within 10 min and significantly inhibited the firing activity of 7 PVN neurons in SHRs. Moreover, subsequent bath application of 50 μm AP5 further significantly reduced the firing activity of these PVN neurons in SHRs (Fig. 4C).
Responses of LSNA, ABP, and HR to ICV injection of DRB and bilateral microinjection of AP5 into the PVN of SHRs
To determine the role of CK2 in controlling the sympathetic vasomotor tone in SHRs, the CK2 inhibitor DRB (30 nmol, 10 μl) was injected ICV (3 times at an interval of 30 min) in 10 SHRs. DRB significantly decreased the LSNA, ABP, and HR by 28.6 ± 4.6%, 27.8 ± 3.5 mmHg, and 38.5 ± 5.4 beats per minute (bpm), respectively (Fig. 5). ICV injection of the vehicle (0.5% DMSO) did not significantly affect the ABP, HR, or LSNA in another nine SHRs. To further determine whether inhibition of CK2 activity suppresses the ability of NMDARs in the PVN to support elevated sympathetic vasomotor tone in SHRs, AP5 was microinjected bilaterally into the PVN following ICV DRB administration. Microinjection of AP5 (1.0 nmol, 50 nl) significantly decreased the ABP, HR, and LSNA in SHRs treated with ICV vehicle. However, bilateral microinjection of AP5 into the PVN failed to further decrease the LSNA, ABP, and HR in SHRs treated with ICV injection of DRB (Fig. 5).
Figure 5.

Response of ABP, HR, and LSNA to ICV injection of DRB and microinjection of AP5 into the PVN in SHRs. A, B, Representative recordings show the effect of ICV injection of DRB (30 nmol, 10 μl) and bilateral microinjection of AP5 (1.0 nmol, 50 nl) into the PVN on the ABP, HR, and LSNA in SHRs. C, D, Summary data show the changes in the mean ABP and LSNA in response to microinjection of AP5 into the PVN after ICV injection of DRB (n = 10 rats) or vehicle (Veh, n = 9 rats); BL denotes baseline. E, Schematic drawings show the locations of AP5 microinjection sites in SHRs with ICV injection of a vehicle (○) or DRB (•); AH denotes anterior hypothalamus. *p < 0.05, compared with the vehicle control.
CK2 protein levels in the PVN of SHRs and WKY rats
CK2 activity is correlated with its expression and subunit composition (Guerra et al., 1999; Filhol et al., 2004; Bibby and Litchfield, 2005). To determine the CK2 protein levels in the PVN in WKY rats and SHRs, we used Western immunoblotting to measure the total and membrane-fraction proteins of CK2 in micro-punched PVN tissues. There was only a single protein band present on the gel using the CK2α or CK2β primary antibody. The total protein level of CK2α and CK2β subunits in the PVN did not differ significantly between SHRs and WKY rats (Fig. 6A,C). However, the membrane protein level of the CK2α, but not CK2β, subunit in the PVN was significantly higher in the SHRs than in WKY rats (Fig. 6B,D).
Figure 6.
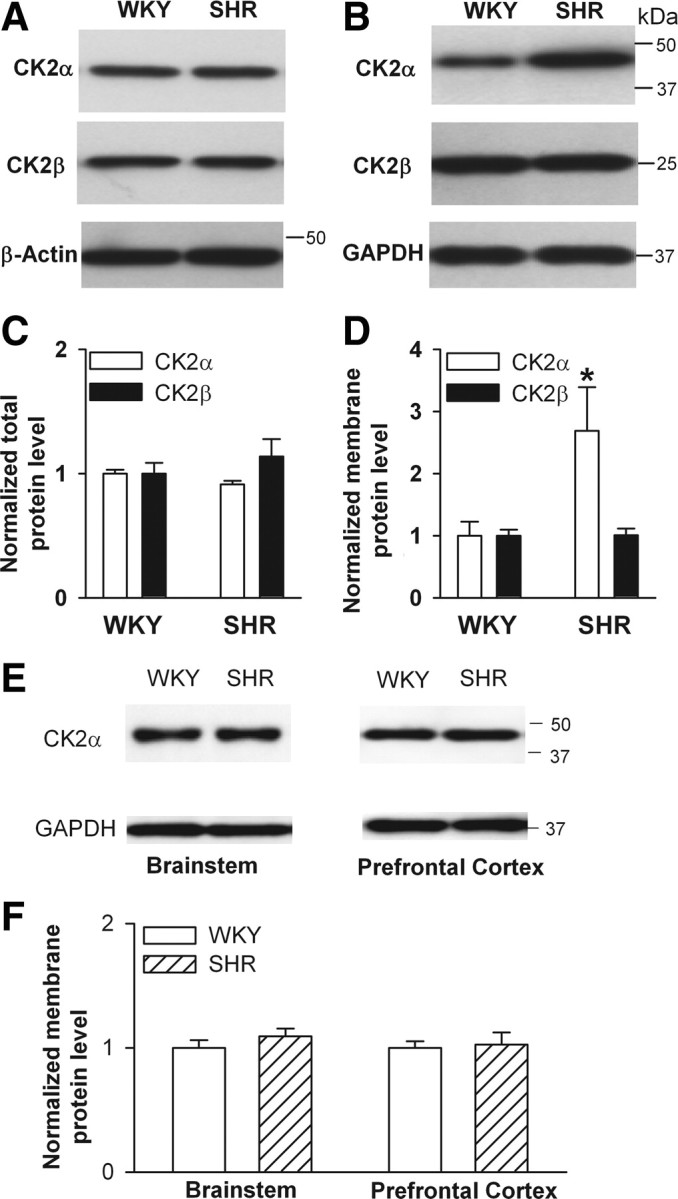
CK2 protein levels in the PVN, brainstem, and prefrontal cortex of SHRs and WKY rats. A, B, Representative blots show the total (A, 15 μg/lane) and membrane (B, 20 μg/lane) protein levels of CK2α and CK2β subunits in the PVN in SHRs and WKY rats. C, D, Changes in the total (C) and membrane (D) protein levels of CK2α and CK2β subunits in the PVN in SHRs and WKY rats. E, Representative gel images show the membrane (20 μg/lane) protein levels of CK2α subunit in the brainstem and prefrontal cortex in SHRs and WKY rats. F, Membrane protein levels of CK2α in the brainstem and prefrontal cortex in SHRs and WKY rats. The molecular weight was indicated on the right side of the gel. In all the protein assays, four samples (four different pools of three rats) were used in each group. *p < 0.05, compared with the WKY rat group.
To determine whether there is a general increase in CK2α protein level in the brain of SHRs, we also measured the CK2α protein levels in the brainstem and prefrontal cortex of WKY rats and SHRs. There were no significant differences in the membrane CK2α protein levels in the brainstem and prefrontal cortex between WKY rats and SHRs (Fig. 6E,F).
CK2 protein levels in the PVN and effects of CK2 inhibition on NMDAR activity in SHRs subjected to CGx
To further determine whether increased CK2 activity in the PVN is the cause or result of hypertension in SHRs, we examined the CK2α level and the effects of CK2 inhibition on presynaptic and postsynaptic NMDAR activity in the PVN in SHRs subjected to CGx. CGx, but not sham surgery, significantly lowered the mean ABP in SHRs for at least 2 weeks (Fig. 7A). However, the increased CK2α protein level in the PVN and the inhibitory effects of DRB on the frequency of mEPSCs and the amplitude of evoked NMDA-EPSCs of spinally projecting PVN neurons observed in untreated SHRs were not significantly altered after lowering ABP with CGx in SHRs (Fig. 7B–D).
Figure 7.

CK2α protein level in the PVN and effects of CK2 inhibition on presynaptic and postsynaptic NMDAR activity in SHRs subjected to CGx. A, Original traces and summary data show the effect of CGx and sham surgery on the mean ABP in SHRs (n = 6 rats in each group). B, Original gel images and group data show the membrane CK2α protein levels in the PVN in SHRs subjected to CGx or sham surgery (n = 4 samples in each group). C, Effect of DRB (100 μm) on the mEPSC frequency of labeled PVN neurons (n = 7 in each group) in SHRs subjected to CGx. D, Effect of DRB on evoked AMPA-EPSCs and NMDA-EPSCs of labeled PVN neurons (n = 7) in SHRs subjected to CGx. *p < 0.05, compared with the sham or vehicle control.
Discussion
In this study, we determined the contribution of protein kinase CK2 to increased glutamatergic input in the PVN and to elevated sympathetic outflow in SHRs, an animal model of essential hypertension. We found that inhibition of CK2 significantly reduced the frequency of glutamatergic mEPSCs, evoked NMDA-EPSCs, and the firing activity of PVN presympathetic neurons in SHRs only. Furthermore, we found that the CK2 membrane protein level in the PVN was significantly increased and that CK2 inhibition significantly decreased the LSNA, ABP, and HR and abolished the sympathoinhibitory effect of the NMDAR antagonist injected into the PVN in SHRs. In addition, lowering blood pressure with CGx did not significantly affect increased CK2 protein levels in the PVN and the effects of CK2 inhibition on mEPSCs and NMDA-EPSCs of PVN neurons in SHRs. Thus, our findings provide novel evidence showing that CK2 plays a critical role in the increased presynaptic and postsynaptic NMDAR activity and glutamatergic tone in the PVN and in the elevated sympathetic outflow in essential hypertension.
Although hypertension is a well known risk factor for stroke and heart diseases, the underlying causes in the majority of patients with hypertension are unknown. Our previous studies have demonstrated that enhanced excitatory glutamatergic input in the PVN plays a major role in increased sympathetic outflow in SHRs (Li and Pan, 2007; Li et al., 2008). In the present study, we found that treatment of brain slices with the two structurally distinct CK2 inhibitors, DRB and TBB, significantly inhibited the frequency of mEPSCs in SHRs but not in WKY rats. Our data suggest that increased CK2 activity is involved in the elevated synaptic glutamate release in the PVN of SHRs. The CK2 inhibitor DRB is considered to be highly specific, with no effect on PKC, PKA, CaMKII, or the Src tyrosine kinase (Pinna, 1990). Previous studies have shown that NMDARs exist at glutamatergic terminals in the cortex (Berretta and Jones, 1996), hippocampus (Petralia et al., 1994), and spinal cord (Liu et al., 1997). Because activation of presynaptic NMDARs could increase the frequency of mEPSCs (Sjöström et al., 2003; Zhou et al., 2010), inhibition of CK2 in the PVN may attenuate presynaptic glutamate release through its effect on presynaptic NMDARs in SHRs. To test this hypothesis, we determined whether presynaptic NMDARs contribute to increased glutamate release in the PVN in SHRs. We found that after including the NMDA channel blocker MK-801 in the intracellular pipette solution (to block postsynaptic NMDARs), bath application of the NMDAR antagonist AP5 significantly reduced the frequency of mEPSCs in SHRs but not in WKY rats. This suggests that elevated activity of presynaptic NMDARs contributes to increased glutamate release in the PVN in SHRs. Furthermore, AP5 failed to reduce the frequency of mEPSCs in brain slices from SHRs pretreated with DRB, suggesting that CK2 increases synaptic glutamate release through potentiation of presynaptic NMDAR activity in these hypertensive animals. Because the glial toxin fluoroacetate did not significantly alter mEPSCs and the effect of AP5 on the mEPSC frequency of PVN neurons in SHRs, it seems that presynaptic NMDARs present on glia do not contribute significantly to increased presynaptic NMDAR activity and glutamate release in the PVN in this animal model of hypertension.
We found that inhibiting CK2 with DRB or TBB significantly decreased the amplitude of evoked NMDA-EPSCs and the ratio of evoked NMDA-EPSCs to AMPA-EPSCs in PVN neurons in SHRs but not in WKY rats. Also, DRB significantly inhibited NMDA currents produced by NMDA puff application in SHRs but not in WKY rats. It is possible that the NMDAR activity may not be tonically controlled by CK2 in WKY rats. Alternatively, the CK2 activity in the PVN may be very low in WKY rats, which could explain why CK2 inhibitors had no significant effects on presynaptic and postsynaptic NMDAR activity in normotensive WKY rats. Similarly, we found that AP5 failed to significantly affect the frequency of mEPSCs in WKY rats. Because glutamate release (reflected by the mEPSC frequency) in the PVN was much lower in WKY rats than in SHRs, it may not be sufficient to spillover and activate presynaptic NMDARs in WKY rats. Our results suggest that the activity of CK2 is upregulated in the PVN and contributes to increased presynaptic and postsynaptic NMDAR activity in SHRs. On the other hand, neither DRB nor TBB had any significant effects on the amplitude of mEPSCs or evoked AMPA-EPSCs in PVN neurons in SHRs or WKY rats. CK2 may increase the phosphorylation level of NMDARs, but not AMPA receptors, in the PVN in SHRs. Consistent with our findings, it has been shown that CK2 increases the NMDAR activity in the hippocampus (Lieberman and Mody, 1999).
We found that the membrane protein level of CK2α, but not CK2β, was significantly increased in the PVN in SHRs compared with that in WKY rats. Interestingly, the total CK2 protein level in the PVN did not differ significantly between SHRs and WKY rats. CK2α is moderately active alone as a monomer (Allende and Allende, 1995). Free CK2α subunits, alone or associated with other proteins, have been observed in various subcellular compartments, including the nucleus, plasma membrane, and cytoplasm (Filhol et al., 2004; Bibby and Litchfield, 2005). Elevated CK2α protein level on the cell membrane in the PVN may increase the probability of its interaction with NMDARs and their binding proteins in SHRs. Although the CK2 protein level is increased in the PVN in SHRs, this change may not be unique to PVN neurons projecting to the spinal cord. Interestingly, we found that the membrane CK2α level in the brainstem and prefrontal cortex did not differ significantly between WKY rats and SHRs, suggesting that increased CK2α protein level does not occur in all brain regions in this rat model of hypertension.
The precise mechanisms by which CK2 increases NMDAR activity remain unclear. We found that inhibition of PKC had no significant effect on the frequency of mEPSCs and the amplitude of NMDA-EPSCs of PVN neurons in SHRs, suggesting that increased ATP level in the PVN may not account for increased CK2 activity in SHRs. CK2 may regulate NMDAR function indirectly through phosphorylation of PSD95/SAP90, an essential protein for the assembling of NMDARs. PSD95/SAP90 is a CK2 substrate, and phosphorylation of PSD95/SAP90 by CK2 may enhance its binding with NMDARs and subsequently increase NMDAR activity (Iwamoto et al., 2004; Soto et al., 2004). Indirect effects of CK2 on NMDARs may also include its phosphorylation of calmodulin (Sacks et al., 1992), a Ca2+-binding protein that is also a CK2 substrate (Allende and Allende, 1995). The basal activity of CK2 toward calmodulin is low under physiological conditions, because calmodulin can only interact with CK2α monomers but not α2β2 holoenzymes (Meggio et al., 1987; Quadroni et al., 1994). It has been demonstrated that calmodulin inhibits NMDAR activity through binding to the NR1 subunit in a Ca2+-dependent manner (Ehlers et al., 1996; Krupp et al., 1999). Phosphorylation of calmodulin by CK2 may weaken the calmodulin-NR1 interaction, leading to the disinhibition of NMDARs (Ehlers et al., 1996; Arrigoni et al., 2004).
Increased glutamatergic input is responsible for the elevated firing activity of PVN presympathetic neurons in SHRs (Li et al., 2008). In the present study, we found that CK2 inhibitors significantly reduced the firing rate of spinally projecting PVN neurons in SHRs but not in WKY rats. Because DRB and TBB significantly inhibited NMDAR rather than AMPA receptor activity in SHRs, we reasoned that CK2 inhibition may reduce the firing activity of PVN presympathetic neurons through its effect on presynaptic and postsynaptic NMDARs. Therefore, we tested the effects of treatment with the NMDAR antagonist AP5 on the firing activity of labeled PVN neurons in brain slices treated with DRB or a vehicle. We found that AP5 significantly inhibited the firing activity of PVN neurons in vehicle-treated slices but failed to further inhibit the firing activity of PVN neurons in DRB-treated slices from SHRs. Intracellular dialysis with MK-801 to block postsynaptic NMDARs significantly inhibited the firing activity of PVN neurons. Furthermore, bath application of AP5 significantly reduced the firing activity of these PVN neurons in SHRs. Our data suggest that CK2 increases the firing activity of PVN presympathetic neurons through potentiation of both presynaptic and postsynaptic NMDAR activity in SHRs. Importantly, we found that ICV injection of DRB largely decreased sympathetic vasomotor tone in SHRs. This treatment also abolished the sympathoinhibitory effect of AP5 microinjected into the PVN in SHRs. Our results provide further in vivo evidence that increased CK2 activity in SHRs is crucial to elevated sympathetic outflow and ABP through potentiation of NMDAR activity in the PVN.
Because the blood pressure and sympathetic nerve activity were significantly reduced after microinjection of the CK2 inhibitor into the PVN in SHRs, upregulated CK2 activity seems to be the cause of PVN neuronal hyperactivity in this animal model of hypertension. To further determine whether increased CK2 activity in the PVN is a result or the cause of hypertension in SHRs, we determined the CK2 level and the effects of CK2 inhibition in the PVN after lowering ABP with CGx in SHRs. It has been shown that the sympathetic nerve activity directed to the splanchnic vascular bed is particularly high in SHRs (Ekas and Lokhandwala, 1981; Morrison and Whitehorn, 1984). Consistent with these findings, we observed that CGx effectively lowered ABP for >2 weeks in SHRs, demonstrating a critical role of splanchnic sympathetic nerve discharges in the maintenance of elevated ABP in SHRs. We found that the membrane CK2α level and the effects of CK2 inhibition on presynaptic and postsynaptic NMDAR activity in the PVN were not significantly affected by lowering blood pressure with CGx in SHRs. Thus, our findings suggest that increased CK2 activity in the PVN contributes to the pathogenesis of hypertension in SHRs.
In summary, our study provides important new information that increased CK2 activity in the PVN is responsible for enhanced synaptic glutamate release and presynaptic and postsynaptic NMDAR activity in SHRs. Our data suggest that increased CK2 activity increases the firing activity of spinally projecting PVN neurons as well as the sympathetic outflow and ABP through potentiation of glutamatergic input in SHRs. Our findings provide new insight into the molecular mechanisms underlying the elevated sympathetic vasomotor tone in essential hypertension. Therefore, CK2 inhibition may represent a novel strategy for the treatment of neurogenic hypertension.
Footnotes
This study was supported by National Institutes of Health Grant R01HL077400, a postdoctoral fellowship from the American Heart Association South Central Affiliate (to Z.-Y.Y.), and the N. G. and Helen T. Hawkins Endowment (to H.-L.P).
References
- Allen AM. Inhibition of the hypothalamic paraventricular nucleus in spontaneously hypertensive rats dramatically reduces sympathetic vasomotor tone. Hypertension. 2002;39:275–280. doi: 10.1161/hy0202.104272. [DOI] [PubMed] [Google Scholar]
- Allende JE, Allende CC. Protein kinases. 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J. 1995;9:313–323. doi: 10.1096/fasebj.9.5.7896000. [DOI] [PubMed] [Google Scholar]
- Anderson EA, Sinkey CA, Lawton WJ, Mark AL. Elevated sympathetic nerve activity in borderline hypertensive humans. Evidence from direct intraneural recordings. Hypertension. 1989;14:177–183. doi: 10.1161/01.hyp.14.2.177. [DOI] [PubMed] [Google Scholar]
- Arrigoni G, Marin O, Pagano MA, Settimo L, Paolin B, Meggio F, Pinna LA. Phosphorylation of calmodulin fragments by protein kinase CK2. Mechanistic aspects and structural consequences. Biochemistry. 2004;43:12788–12798. doi: 10.1021/bi049365c. [DOI] [PubMed] [Google Scholar]
- Bachhuber T, Almaça J, Aldehni F, Mehta A, Amaral MD, Schreiber R, Kunzelmann K. Regulation of the epithelial Na+ channel by the protein kinase CK2. J Biol Chem. 2008;283:13225–13232. doi: 10.1074/jbc.M704532200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience. 1996;75:339–344. doi: 10.1016/0306-4522(96)00301-6. [DOI] [PubMed] [Google Scholar]
- Bibby AC, Litchfield DW. The multiple personalities of the regulatory subunit of protein kinase CK2: CK2 dependent and CK2 independent roles reveal a secret identity for CK2beta. Int J Biol Sci. 2005;1:67–79. doi: 10.7150/ijbs.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolanos-Garcia VM, Fernandez-Recio J, Allende JE, Blundell TL. Identifying interaction motifs in CK2beta–a ubiquitous kinase regulatory subunit. Trends Biochem Sci. 2006;31:654–661. doi: 10.1016/j.tibs.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Zhang S, Bernhadt JP, Huganir RL. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- Ekas RD, Jr, Lokhandwala MF. Sympathetic nerve function and vascular reactivity in spontaneously hypertensive rats. Am J Physiol. 1981;241:R379–R384. doi: 10.1152/ajpregu.1981.241.5.R379. [DOI] [PubMed] [Google Scholar]
- Filhol O, Martiel JL, Cochet C. Protein kinase CK2: a new view of an old molecular complex. EMBO Rep. 2004;5:351–355. doi: 10.1038/sj.embor.7400115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Bellone C, Cattabeni F, Di Luca M. Protein kinase C activation modulates alpha-calmodulin kinase II binding to NR2A subunit of N-methyl-d-aspartate receptor complex. J Biol Chem. 2001;276:7609–7613. doi: 10.1074/jbc.M009922200. [DOI] [PubMed] [Google Scholar]
- Grassi G. Role of the sympathetic nervous system in human hypertension. J Hypertens. 1998;16:1979–1987. doi: 10.1097/00004872-199816121-00019. [DOI] [PubMed] [Google Scholar]
- Greenwood JP, Stoker JB, Mary DA. Single-unit sympathetic discharge: quantitative assessment in human hypertensive disease. Circulation. 1999;100:1305–1310. doi: 10.1161/01.cir.100.12.1305. [DOI] [PubMed] [Google Scholar]
- Guerra B, Siemer S, Boldyreff B, Issinger OG. Protein kinase CK2: evidence for a protein kinase CK2beta subunit fraction, devoid of the catalytic CK2alpha subunit, in mouse brain and testicles. FEBS Lett. 1999;462:353–357. doi: 10.1016/s0014-5793(99)01553-7. [DOI] [PubMed] [Google Scholar]
- Hülsmann S, Oku Y, Zhang W, Richter DW. Metabolic coupling between glia and neurons is necessary for maintaining respiratory activity in transverse medullary slices of neonatal mouse. Eur J Neurosci. 2000;12:856–862. doi: 10.1046/j.1460-9568.2000.00973.x. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Yamada Y, Hori K, Watanabe Y, Sobue K, Inui M. Differential modulation of NR1-NR2A and NR1-NR2B subtypes of NMDA receptor by PDZ domain-containing proteins. J Neurochem. 2004;89:100–108. doi: 10.1046/j.1471-4159.2003.02293.x. [DOI] [PubMed] [Google Scholar]
- Judy WV, Watanabe AM, Henry DP, Besch HR, Jr, Murphy WR, Hockel GM. Sympathetic nerve activity: role in regulation of blood pressure in the spontaenously hypertensive rat. Circ Res. 1976;38:21–29. doi: 10.1161/01.res.38.6.21. [DOI] [PubMed] [Google Scholar]
- Kang H, Jung JW, Kim MK, Chung JH. CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PLoS One. 2009;4:e6611. doi: 10.1371/journal.pone.0006611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan H, Hayashida Y, Yamashita H. Increase in sympathetic outflow by paraventricular nucleus stimulation in awake rats. Am J Physiol. 1989;256:R1325–R1330. doi: 10.1152/ajpregu.1989.256.6.R1325. [DOI] [PubMed] [Google Scholar]
- Kimura R, Matsuki N. Protein kinase CK2 modulates synaptic plasticity by modification of synaptic NMDA receptors in the hippocampus. J Physiol. 2008;586:3195–3206. doi: 10.1113/jphysiol.2008.151894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science. 1988;241:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J Neurosci. 1999;19:1165–1178. doi: 10.1523/JNEUROSCI.19-04-01165.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Pan HL. Glutamatergic inputs in the hypothalamic paraventricular nucleus maintain sympathetic vasomotor tone in hypertension. Hypertension. 2007;49:916–925. doi: 10.1161/01.HYP.0000259666.99449.74. [DOI] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan HL. Angiotensin II stimulates spinally projecting paraventricular neurons through presynaptic disinhibition. J Neurosci. 2003;23:5041–5049. doi: 10.1523/JNEUROSCI.23-12-05041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan HL. VR1 receptor activation induces glutamate release and postsynaptic firing in the paraventricular nucleus. J Neurophysiol. 2004;92:1807–1816. doi: 10.1152/jn.00171.2004. [DOI] [PubMed] [Google Scholar]
- Li DP, Atnip LM, Chen SR, Pan HL. Regulation of synaptic inputs to paraventricular-spinal output neurons by alpha2 adrenergic receptors. J Neurophysiol. 2005;93:393–402. doi: 10.1152/jn.00564.2004. [DOI] [PubMed] [Google Scholar]
- Li DP, Yang Q, Pan HM, Pan HL. Pre- and postsynaptic plasticity underlying augmented glutamatergic inputs to hypothalamic presympathetic neurons in spontaneously hypertensive rats. J Physiol. 2008;586:1637–1647. doi: 10.1113/jphysiol.2007.149732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman DN, Mody I. Casein kinase-II regulates NMDA channel function in hippocampal neurons. Nat Neurosci. 1999;2:125–132. doi: 10.1038/5680. [DOI] [PubMed] [Google Scholar]
- Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Mantyh PW, Basbaum AI. NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature. 1997;386:721–724. doi: 10.1038/386721a0. [DOI] [PubMed] [Google Scholar]
- Lou DY, Dominguez I, Toselli P, Landesman-Bollag E, O'Brien C, Seldin DC. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol Cell Biol. 2008;28:131–139. doi: 10.1128/MCB.01119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancia G, Grassi G, Giannattasio C, Seravalle G. Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension. 1999;34:724–728. doi: 10.1161/01.hyp.34.4.724. [DOI] [PubMed] [Google Scholar]
- Martin DS, Haywood JR. Hemodynamic responses to paraventricular nucleus disinhibition with bicuculline in conscious rats. Am J Physiol. 1993;265:H1727–H1733. doi: 10.1152/ajpheart.1993.265.5.H1727. [DOI] [PubMed] [Google Scholar]
- Meggio F, Brunati AM, Pinna LA. Polycation-dependent, Ca2+-antagonized phosphorylation of calmodulin by casein kinase-2 and a spleen tyrosine protein kinase. FEBS Lett. 1987;215:241–246. doi: 10.1016/0014-5793(87)80154-0. [DOI] [PubMed] [Google Scholar]
- Morrison SF, Whitehorn D. Enhanced preganglionic sympathetic nerve responses in spontaneously hypertensive rats. Brain Res. 1984;296:152–155. doi: 10.1016/0006-8993(84)90522-5. [DOI] [PubMed] [Google Scholar]
- Niefind K, Guerra B, Ermakowa I, Issinger OG. Crystal structure of human protein kinase CK2: insights into basic properties of the CK2 holoenzyme. EMBO J. 2001;20:5320–5331. doi: 10.1093/emboj/20.19.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Pallares J, Llobet D, Santacana M, Eritja N, Velasco A, Cuevas D, Lopez S, Palomar-Asenjo V, Yeramian A, Dolcet X, Matias-Guiu X. CK2beta is expressed in endometrial carcinoma and has a role in apoptosis resistance and cell proliferation. Am J Pathol. 2009;174:287–296. doi: 10.2353/ajpath.2009.080552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 3. San Diego: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Wenthold RJ. The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J Neurosci. 1994;14:6102–6120. doi: 10.1523/JNEUROSCI.14-10-06102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinna LA. Casein kinase 2: an ‘eminence grise’ in cellular regulation? Biochim Biophys Acta. 1990;1054:267–284. doi: 10.1016/0167-4889(90)90098-x. [DOI] [PubMed] [Google Scholar]
- Quadroni M, James P, Carafoli E. Isolation of phosphorylated calmodulin from rat liver and identification of the in vivo phosphorylation sites. J Biol Chem. 1994;269:16116–16122. [PubMed] [Google Scholar]
- Ranson RN, Motawei K, Pyner S, Coote JH. The paraventricular nucleus of the hypothalamus sends efferents to the spinal cord of the rat that closely appose sympathetic preganglionic neurones projecting to the stellate ganglion. Exp Brain Res. 1998;120:164–172. doi: 10.1007/s002210050390. [DOI] [PubMed] [Google Scholar]
- Sacks DB, Davis HW, Williams JP, Sheehan EL, Garcia JG, McDonald JM. Phosphorylation by casein kinase II alters the biological activity of calmodulin. Biochem J. 1992;283:21–24. doi: 10.1042/bj2830021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarno S, Reddy H, Meggio F, Ruzzene M, Davies SP, Donella-Deana A, Shugar D, Pinna LA. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 ('casein kinase-2′) FEBS Lett. 2001;496:44–48. doi: 10.1016/s0014-5793(01)02404-8. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Soto D, Pancetti F, Marengo JJ, Sandoval M, Sandoval R, Orrego F, Wyneken U. Protein kinase CK2 in postsynaptic densities: phosphorylation of PSD-95/SAP90 and NMDA receptor regulation. Biochem Biophys Res Commun. 2004;322:542–550. doi: 10.1016/j.bbrc.2004.07.158. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci. 1983;6:269–324. doi: 10.1146/annurev.ne.06.030183.001413. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Inenaga K, Koizumi K. Possible projections from regions of paraventricular and supraoptic nuclei to the spinal cord: electrophysiological studies. Brain Res. 1984;296:373–378. doi: 10.1016/0006-8993(84)90077-5. [DOI] [PubMed] [Google Scholar]
- Zeng J, Thomson LM, Aicher SA, Terman GW. Primary afferent NMDA receptors increase dorsal horn excitation and mediate opiate tolerance in neonatal rats. J Neurosci. 2006;26:12033–12042. doi: 10.1523/JNEUROSCI.2530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Chen H, Pan HL. Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J Neurosci. 2010;30:4460–4466. doi: 10.1523/JNEUROSCI.5857-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]