

Abstract
Intracerebral hemorrhage (ICH) is an often fatal type of stroke which kills about 30,000 people annually in the USA. If the patient survives the ictus, the resulting hematoma within brain parenchyma triggers a series of adverse events causing secondary insults and severe neurological deficits. This article discusses selected aspects of secondary brain injury after ICH and outlines key mechanisms associated with hematoma toxicity, oxidative stress, and inflammation. Finally, this review discusses the relevance of hematoma resolution processes as a target for ICH therapy, and presents potential clinically relevant molecular targets that could be harnessed to treat secondary injury associated with ICH injury.
Keywords: Intracerebral hemorrhage, hematoma toxicity, oxidative stress, hematoma clearance, Nrf2, PPARγ, NF-κB
During intracerebral hemorrhage (ICH), rapid accumulation of blood within brain parenchyma leads to disruption of normal anatomy and increased local pressure. Depending on the dynamic of hematoma expansion (growth), the primary damage occurs within minutes to hours from the onset of bleeding and is primarily the result of mechanical damage associated with the mass effect.1 Secondary damage is for the most part due to the presence of intraparenchymal blood may be dependent upon the initial hematoma volume, age or ventricular volume1, and it may occur through many parallel pathological pathways including: (1) cytotoxicity of blood,2-3 (2) hypermetabolism,4 (3) excitotoxicity,5 (4) spreading depression,6 (5) oxidative stress and inflammation.3,7-13 Ultimately, this pathogenesis leads to irreversible disruption of the components of the neurovascular unit, constituting gray and white matter, and is followed by blood-brain-barrier disruption and deadly brain edema with massive brain cell death.2-3, 5, 7, 14-16 While inflammatory mediators generated locally in response to brain death/injury have the capacity to augment damage caused by ICH (secondary injury), the involvement of inflammatory cells e.g. microglia/macrophages is vital for removal/cleanup of cellular debris from hematoma, the source of ongoing inflammation.17 The timely removal of damaged tissue is essential for reducing the length of deleterious pathological process and thereby allowing for faster and more efficient recovery.
Blood cytotoxicity and oxidative stress as mediators of cell death after ICH
After ICH the extravasated blood components (primarily erythrocytes and plasma proteins) and the “damage”-associated molecular patterns (DAMPs), including nucleic acids, extracellular matrix components, proteins, lipid mediators, ATP and uric acid released from necrotic and damaged tissue, impose a strong cytotoxic, pro-oxidative and pro-inflammatory insult toward adjacent viable brain cells, and could be seen as early as minutes after onset of ICH. At this early stage the toxicity of extravasated blood plasma components including blood derived coagulation factors, complement components, immunoglobulins and other bioactive molecules are proposed to act as contributors to ICH-affected tissue damage.2-3, 11 Subsequently, red blood cell (RBC) lysis starts at approximately 24h and continues for the next several days, leading to release of cytotoxic hemoglobin (Hb) with further deterioration of the pathological status quo.2 Hb and its degradation products, heme and iron, directly compromise the well-being of neighboring brain cells.2, 18-20 Hb and heme are potent cytotoxic chemicals capable of causing death to many brain cells. Prominently, the mechanism of Hb toxicity is via generating free radicals (mainly through Fenton-type mechanism) and massive oxidative damage to proteins, nucleic acids, carbohydrates and lipids. 2, 18, 21-22 Therefore, finding means of controlling hemolysis, and detoxification, and absorption of Hb, heme and iron (Fig 1) may have important clinical implications.
Fig 1.
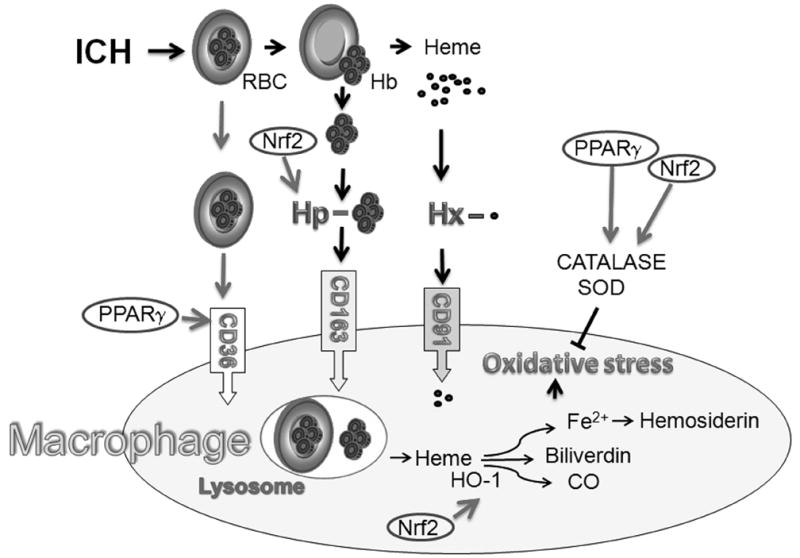
During ICH blood is released into the brain matter. Erythrocytes (RBC) are cleared from the parenchyma by microglia/macrophages through cell-surface scavenger receptor CD36-mediated phagocytosis. Timely clearance of the extravasated RBCs and irreversibly injured cells prevents them from undergoing lyses and subsequent spillage of toxic contents into the brain parenchyma. In the case of hemolysis, what unavoidably occurs after ICH, hemoglobin (Hb) needs to be removed quickly from the extracellular space to avoid its cytotoxic effects. Haptoglobin (Hp), a protein arrived to the brain from blood and synthesized locally by oligodendroglia, tightly binds Hb forming less toxic Hb-Hp complexes which are endocytosed by microglia/macrophages through scavenger receptor, CD163. The toxic extracellular free heme generated from Hb can be neutralized by binding to hemopexin (Hx). The heme-Hx complexes are subsequently removed by phagocytes via CD91 scavenger receptor-mediated endocytosis. In phagocyte heme is metabolized by heme oxygenase (HO) (primarily HO-1) to biliverdin, carbon monoxide (CO) and pro-oxidative iron. To prevent oxidative cell damage iron is sequestrated within phagocyte by iron binding proteins such as hemosiderin or ferritin. Excessive production of iron may saturate storing capacity of hemosiderin, leading to oxidative injury from the free irons. CD36 is expressed under control of PPARγ and Hp expression is increased with Nrf2. Both Nrf2 and PPARγ increase expression of antioxidant proteins (e.g. catalase or superoxide dismutase, SOD). Thus activation of these transcription factors may represent new targets for ICH treatment.
We will now review the role of selected pathways involved in blood detoxification process, including transcriptional regulation of selected molecules with a role in hematoma clearance.
Haptoglobin and hemopexin acts to combat hemoglobin/heme toxicity after ICH
Haptoglobin (Hp), an acute phase protein, is an abundant blood plasma component that is normally synthesized and released into blood circulation primarily by hepatocytes but to a lesser extent by lungs, kidneys, skin, and adipose tissue.20, 23 Hp is a heteromeric enzyme composed of alpha and beta chains. The primary function of Hp in blood is to bind and neutralize nephrotoxic free Hb in case of intravascular hemolysis.23 Hp-Hb complexes are consequently removed from the circulation by a specialized subclass of macrophages expressing CD163, a scavenger receptor and a member of the group B scavenger receptor cysteine-rich superfamily.23 Under normal circumstances, Hp represents an effective mechanism by which our body is protected from Hb toxicity. However, because Hp synthesis is not increased by low Hp levels and Hp is not recycled by macrophages, it may take 5-7 days for Hp level to recover if completely sequestered by hemoglobin. Thus massive hemolysis may lead to persistent hypohaptoglobinemia. Interestingly, we have recently demonstrated that Hp is also produced locally in rat brain after ICH and its expression is significantly increased around the hematoma within hours from the onset of ICH.20 The brain derived Hp appeared to be synthesized and released by oligodendrocytes. Because oligodendroglia are abundant in white matter and are present throughout the gray matter, local production of Hp by these cells likely represents an important endogenous mechanism protecting brain against the extravascular Hb toxicity. Indirect support for such a claim comes from: (1)-primary oligodendrocytes protect neurons in culture from Hb toxicity via Hp release, (2)-animals made hypohaptoglobinemic with repetitive hemoglobin administration, prior to ICH, suffer more extensive brain damage, (3)-mice genetically engineered to overexpress Hp are less susceptible to ICH injury, while (4) Hp-deficient mice are more vulnerable to ICH injury.20 In the context of therapeutic relevance, we have determined that pharmacological intervention with sulforaphane, a naturally occurring agent that acts as NF-E2-related factor-2 (Nrf2) transcription factor activator, increases Hp in blood plasma and brain20 and notably reduces brain damage in animal models of ICH.24
In humans, Hp gene exists in two major allelic forms designated as Hp1 and Hp2. Hp2 is produced via intragenic duplication of a 1.7-kb DNA fragment of Hp1 gene. Consequently, three major Hp genotypes are formed, Hp1-1, Hp1-2 and Hp2-223. Hp2-2, the largest of the three Hp genotypes, has the lowest Hb-binding-activity (Hp1-1>Hp1-2>Hp2-2) and due to its size represents potentially less bioavailable moieties. This may lead to a more defective hemoglobin clearing system.23 Interestingly, according to epidemiological studies, patients carrying Hp2-2 genotype demonstrate more severe vasospasm after subarachnoid hemorrhage, positive association with idiopathic seizures (and possibly posttraumatic epilepsy), as well as higher incidence of carotid atherosclerosis in diabetic patients.23, 25-26 The relevance of Hp genotype as a factor modifying the outcome after ICH has not been studied to date.
It could also be relevant for this review to indicate that in addition to haptoglobin-hemoglobin-Hb//CD163 scavenging system, an independent system exists to help remove hemoglobin breakdown products, heme and iron.27 Hemopexin (Hx) is a blood plasma glycoprotein synthesized primarily by hepatocytes. Hx has been shown to bind to heme with a high affinity and forms stable Hx-heme complexs.2, 27 The heme-Hx complexes are readily endocytosed by macrophages expressing CD91 (α2-macroglobulin receptor, also known as low-density lipoprotein receptor-related protein – LRP1). While under physiological conditions CD91 plays a role in recycling iron, in response to extravascular hemolysis in hematoma affected tissue, the Hx-heme/CD91 system may facilitate removal of pro-oxidative heme by microglia/macrophages.27 Recent studies28 and ongoing research in this lab support this notion and suggest that Hx-deficient mice experience augmented ICH injury. More studies on the role of Hx in ICH are warranted.
Heme oxygenase in ICH
Phagocytosis of intact erythrocytes, as well endocytosis of hemoglobin and heme by macrophages/microglia is instrumental for removing the pro-oxidative heme from the extracellular space of brain parenchyma. Phagocytic cells involved in this process express heme oxygenase (HO), a rate-limiting enzyme involved in heme catabolism which converts heme to biliverdin, carbon monoxide, and iron (II).29-30 There are two isoenzymes of HO; HO1 (also called HSP32) is an inducible form and HO2 is constitutively expressed.29-30 While HO2 is moderately abundant in most cell types, including neurons29, HO1 expression after ICH is primarily induced in endothelial cells and microglia/macrophages.29 Based on studies using genetically engineered mice, it was determined that HO2-null mice are more susceptible29 while HO1-null mice are more resistant30 to ICH-mediated damage, as compared to wild-type mice. The improved outcome in the HO1-null mice after ICH is indeed a surprise, as HO1 deficiency in many other brain injury models including ischemic stroke are associated with augmented brain damage. One potential explanation for such discrepancy is that HO1 deficiency could reduce the excessive liberation of free iron from erythrocytes in hematoma (feature that is unique to ICH) and consequently limit the iron-mediated oxidative stress. While the above scenario may provide logistics for reduced ICH injury in HO1-deficient mice, the presence of increased levels of Hp and Hx in HO1-deficiency animals31 needs to be acknowledged, as these factors may contribute to cytoprotection independently of HO-1. Additional limitation regarding interpretation of the data on HO1 null mice susceptibility to ICH injury is that the outcome was characterized only up to 3 days and it did not address the role HO1 could play at later stages when a majority of hemolysis and cleanup takes place. This timing issue is of particular importance because HO1-deficiency makes phagocytic cells, the main executors of brain cleanup process, more prone to self-injury upon processing of engulfed heme-containing RBC31. After ICH in mice, the cleanup process may least 3-4 weeks.32
Another intriguing finding is that a robust HO1 upregulation in ICH affected brain in response to treatment with sulforaphane coincides with a significant reduction of brain damage24. Sulforaphane promotes HO1 expression via Nrf2 acting through ARE, an antioxidant responsive element. Interestingly, porphyrin derivatives which can be used to inhibit HO1, in contrast to sulforaphane were showed reduced ICH damage.2, 33 Thus, more studies are required to clarify the role of HO1 in ICH.
Oxidative stress and ICH injury
As indicated above, oxidative stress appears to play a prominent role in ICH pathogenesis. A direct evidence for the causal relationship between free radicals and ICH injury was by demonstrating the efficacy of anti-oxidants as therapeutic agents. Specifically, the free radical scavengers such as dimethylthiourea, α-phenyl-N-tert-butyl nitrone (PBN), NXY-059 (a sulfonyl derivative of PBN) or deferoxamine, a drug chelating pro-oxidative iron, significantly reduced brain injury in animal models of ICH. 7, 19, 34-35 In agreement with this pharmacologic experiments, mouse with generically deleted NADPH-oxidase, an key enzyme involved in reactive oxygen species generating, showed reduced damage after ICH. 13 It has recently been demonstrated that estrogen reduces ferrous iron toxicity in vivo and in vitro, indicating that gender difference in susceptibility to ICH may in part be associated with differences in handling iron toxicity.36
Although, pre-clinical evidence exists for efficacy of therapy based on free radical neutralization, clinical trial with NXY-059 was surprisingly disappointing. NXY-059 was not only ineffective in ischemic stroke, but also did not benefit ICH patients. 37 The reason for these negative outcomes is unclear; however factors such as pharmacokinetics of the drug (no blood brain barrier permeability) and inability to stoichiometrically neutralize high level of free radicals could likely contribute to these neutral results.
Deferoxamine and iron detoxification
Deferoxamine is another anti-oxidant currently at early promising stages of clinical trial.34 As indicated earlier, toxicity of free iron originated from extravascular hemolysis and hem oxygenase-mediated catabolism is well documented.3 Iron (FeII), by reacting with H2O2 generates hydroxyl radical; deferoxamine via forming stable complexes with iron prevents its engagement in oxidative reactions. One caveat exists that deferoxamine may act not only through preventing iron-mediated pro-oxidative catalyses, but also via inhibiting prolyl hydroxylase activity (enzyme using iron as cofactor), which leads to HIF1α availability, a pathway with neuroprotective effect. Regardless of its mechanism, pre-clinical studies in a rat and pig ICH model showed promise for deferoxamine as therapy.34, 38 Recently, a phase-I study assessing the feasibility, safety, and maximum tolerated dose of deferoxamine in ICH patients was successfully completed (Magdy Selin; personal communications).
Role of Nrf2 in oxidative damage
In addition to increased free radical generation, damage to brain tissue may result from the impairment of endogenous anti-oxidative enzyme system in response to ICH. Hua et.al reported a significant reduction in the levels of manganese superoxide dismutase (Mn-SOD) and copper/zinc SOD (Cu-Zn-SOD), the key enzymes of antioxidant defense system in brains, following intracerebral injection of lysed erythrocytes.39 Thus, since the anti-oxidant capacity of ICH-affected brain may be defective, strategies based on stimulating endogenous antioxidant capacity of the brain could offer an effective therapeutic approach. To test this hypothesis, we and others took advantage of ubiquitous transcription factor, Nrf2, a key player in anti-oxidative homeostasis12, 24. By binding to the antioxidant response element (ARE), Nrf2 regulates the expression of many detoxification and antioxidant enzymes, including SOD, catalase, glutathione-S-transferase (GST), glutathione peroxidase, heme oxygenase-1, NAD(P)H quinone oxidoreductase-1, peroxiredoxin or thioredoxin.40 To activate Nrf2 in animals after ICH, we again utilized a naturally occurring organosulfur compound, sulforaphane. As expected, treatment with sulforaphane effectively increased the expression of Nrf2-regulated antioxidant genes including catalase, SOD and GST, in brain tissue after ICH.24 Notably, this expression of anti-oxidants corresponded to reduced oxidative damage to proteins and lipids within the ICH-affected brain, and importantly with less severe neurological deficits.24 To produce ultimate evidence for the beneficial involvement of Nrf2 in ICH pathogenesis, we established that mice deficient in Nrf2 (knockout) displayed more severe neurological deficit when subjected to ICH, implying that Nrf2 plays critical safeguard function in defending brain against oxidative stress associated with ICH pathogenesis.24
Cytoprotective effect of PPARγ
PPARγ is a ligand-dependent transcription factor that regulates target gene expression by binding to conserved DNA sequences termed peroxisome-proliferator response elements (PPREs), as heterodimers with another nuclear receptor, the retinoic acid receptor (RXR).41 In addition to a well-characterized regulation of expression of genes involved in lipid and glucose metabolism and energy storage, activation of PPARγ induces expression of antioxidant catalase and Cu-Zn-SOD, two ubiquitous enzymes capable of alleviating oxidative stress by decomposing hyperactive H2O2 into molecular oxygen and water molecules, and dismutation of superoxide into oxygen and H2O2, respectively.17, 42 The ligands for PPARγ include fatty acids, non-steroidal anti-inflammatory drugs, 15d-Δ12,14-PGJ2 (15d-PGJ2, a reactive membrane lipid metabolite and downstream product, prostaglandin D2, proposed to act as a physiologic PPARγ agonist), and the thiazolidinediones (TZDs, specifically pioglitazone and rosiglitazone), a class of compounds commonly used to treat type II diabetes.41 Using rat model of ICH based on intrastriatal autologous blood injection, we recently reported that intra-hemorrhage injection of 15d-PGJ2 indeed activates PPARγ in the brain tissue and increases expression of catalase in ICH-affected brain, primarily in neurons and microglia.17, 42 These biochemical responses were associated with decreased neuronal death, as demonstrated by the incidence of DNA scission in neurons, and reduced neurological dysfunction.42 The above findings are consistent with studies demonstrating that PPARγ plays an important role in protecting neurons and other brain cells from injury caused by oxidative stress and ischemia.17, 43
Inflammation, blood toxicity and hematoma removal as target for ICH treatment
The brain-resident phagocytes, microglia, are highly abundant (10-15% of total glial cells) in brain and become readily activated within minutes after ICH. 7, 11, 44 The activated microglia release pro-inflammatory cytokines and chemotactic factors, which help to recruit hemotogeneous inflammatory cells to the ICH injury sites. This is characterized first by the transient (18h-4d) infiltration of neutrophils and then a long term (1d-months) presence of hematogenous macrophages.7-8, 44 The inflammatory signaling involves a coordinated effort of different molecules and cells types and is largely coordinated by a ubiquitous transcription factor nuclear factor kappaB, NF-κB.9, 45 The gene targets of NF-κB include various adhesion molecules (including these involved in immune cells extravasation; e.g. ICAM-1), cytokines and chemokines (involved in pro-inflammatory signaling and NF-κB activation; e.g. IL-1β and TNFα), metalloproteinases (e.g. MMP-9), immune receptors, acute phase proteins, cell surface receptors and inflammatory enzymes (e.g. iNOS, COX-2 and PLA2). It is important to mention that free radical acts as important signaling molecules in NF-κB activation. This property of NF-κB may in part explain how oxidative stress could enhance inflammation after ICH. Experimental studies demonstrate that NF-κB is activated in ICH-affected hemisphere as early as 15 min after the onset of ICH, reaches maximum between 1 and 3 days and remains elevated for weeks.45 It is pertinent to indicate that NF-κB target genes including IL-1β, TNFα, and MMP-9 are involved in ICH-mediated brain injury.11, 46-49
Albeit some inflammatory responses generated by microglia/macrophages after ICH may aggravate brain injury, microglia/macrophages-mediated phagocytosis is instrumental in conducting brain cleanup, the process which must occur to allow for tissue repair and functional recovery.17 A fast and efficient removal of apoptotic, dislocated (e.g. extravascular erythrocytes) and damaged cells, before the discharge of injurious and pro-inflammatory cell contents (DAMPs) occur, may help to reduce secondary damage.
Microglia and macrophages express various cell surface receptors, including scavenger receptors (SR) (e.g. CD91, SR-A, MARCO, SR-BI and CD36) that assist in phagocytosis/endocytosis-mediated removal of cellular debris following tissue injury, including brain injury after ICH.50 One specific study evaluated CD36, a class II scavenger receptor which is transcriptionally regulated by PPARγ. This study used in vitro and in vivo models and demonstrated that (1) microglia/macrophages utilize CD36 to promote phagocytosis of RBC and that (2) treating animals with PPARγ agonists (e.g. rosiglitazone, pioglitazone or 15d-PGJ2), which increased CD36 expression results in faster hematoma resolution and improved functional recovery after ICH.17 The most intriguing observation in this study was that the activation of phagocytosis with PPARγ agonist in both in vitro and in vivo experiments was associated with the reduced pro-inflammatory responses (e.g. reduced IL-1β, TNFα, and MMP-9 mRNA expression).17 Since one of the mechanisms of PPARγ is to trans-repress DNA binding of NFκB, it is likely that the anti-inflammatory phenotype of PPARγ is mediated through inhibition of NFκB.41 Based on these promising pre-clinical data with PPARγ agonists, a randomized clinical trial evaluating safety of Pioglitazone for hematoma resolution in intracerebral hemorrhage was initiated in 2009 (http://clinicaltrials.gov).
In conclusion, existing experimental data suggest that interventions interfering with pro-inflammatory and pro-oxidative blood toxicity through either direct neutralization of Hb, heme and iron or improvement of its removal from extracellular space by phagocytes (e.g. through modulation of PPARγ, NF-κB and Nrf2) represent potential therapeutic targets for ICH (Fig 2).
Fig 2.
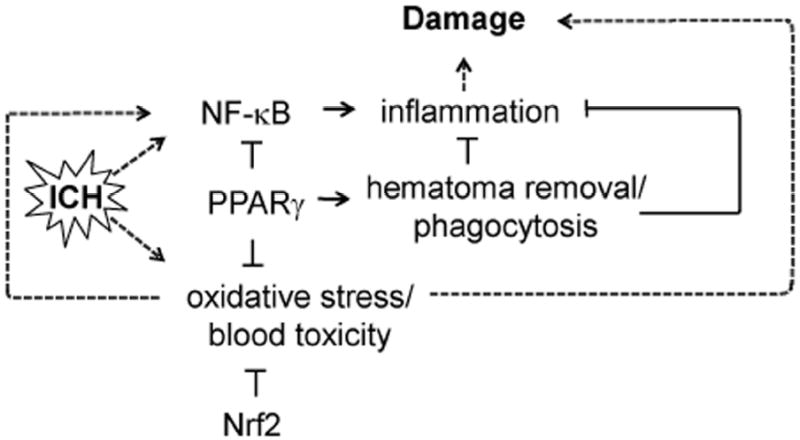
Intracerebral hemorrhage activates transcription factor NF-κB., which then perpetuates inflammation that along with oxidative stress leads to secondary brain damage. Transcription factors PPARγ via inhibition of NF-κB and induction of anti-oxidative defense components reduces inflammation and oxidative stress and protects ICH-affected brain. Transcription factor Nrf2 acts as an effective regulator of oxidative stress and blood detoxification components. In addition, PPARγ stimulates phagocytosis-mediated hematoma cleanup, thus facilitating removal of hematoma, the source of toxicity and inflammation.
Acknowledgments
Funding Sources: Supported in part by National Institute of Health, National Institute of Neurological Disorders and Stroke of RO1NS060768, R21NS057284 and R01NS064109.
Footnotes
Conflict of Interest: NONE
References
- 1.Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373:1632–1644. doi: 10.1016/S0140-6736(09)60371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: Role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–652. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- 3.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 4.Ardizzone TD, Lu A, Wagner KR, Tang Y, Ran R, Sharp FR. Glutamate receptor blockade attenuates glucose hypermetabolism in perihematomal brain after experimental intracerebral hemorrhage in rat. Stroke. 2004;35:2587–2591. doi: 10.1161/01.STR.0000143451.14228.ff. [DOI] [PubMed] [Google Scholar]
- 5.Qureshi AI, Ali Z, Suri MF, Shuaib A, Baker G, Todd K, et al. Extracellular glutamate and other amino acids in experimental intracerebral hemorrhage: An in vivo microdialysis study. Crit Care Med. 2003;31:1482–1489. doi: 10.1097/01.CCM.0000063047.63862.99. [DOI] [PubMed] [Google Scholar]
- 6.Mun-Bryce S, Wilkerson AC, Papuashvili N, Okada YC. Recurring episodes of spreading depression are spontaneously elicited by an intracerebral hemorrhage in the swine. Brain Res. 2001;888:248–255. doi: 10.1016/s0006-8993(00)03068-7. [DOI] [PubMed] [Google Scholar]
- 7.Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: Experience from preclinical studies. Neurol Res. 2005;27:268–279. doi: 10.1179/016164105X25225. [DOI] [PubMed] [Google Scholar]
- 8.Gong C, Hoff JT, Keep RF. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res. 2000;871:57–65. doi: 10.1016/s0006-8993(00)02427-6. [DOI] [PubMed] [Google Scholar]
- 9.Hickenbottom SL, Grotta JC, Strong R, Denner LA, Aronowski J. Nuclear factor-kappab and cell death after experimental intracerebral hemorrhage in rats. Stroke. 1999;30:2472–2477. doi: 10.1161/01.str.30.11.2472. discussion 2477-2478. [DOI] [PubMed] [Google Scholar]
- 10.Xue M, Del Bigio MR. Intracerebral injection of autologous whole blood in rats: Time course of inflammation and cell death. Neurosci Lett. 2000;283:230–232. doi: 10.1016/s0304-3940(00)00971-x. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, et al. Role of nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang J, Liu J, Zhou C, Ostanin D, Grisham MB, Neil Granger D, et al. Role of nadph oxidase in the brain injury of intracerebral hemorrhage. J Neurochem. 2005;94:1342–1350. doi: 10.1111/j.1471-4159.2005.03292.x. [DOI] [PubMed] [Google Scholar]
- 14.Felberg RA, Grotta JC, Shirzadi AL, Strong R, Narayana P, Hill-Felberg SJ, et al. Cell death in experimental intracerebral hemorrhage: The “Black hole” Model of hemorrhagic damage. Ann Neurol. 2002;51:517–524. doi: 10.1002/ana.10160. [DOI] [PubMed] [Google Scholar]
- 15.Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: Role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Mori T, Sumii T, Lo EH. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: Caspase activation and oxidative stress. Stroke. 2002;33:1882–1888. doi: 10.1161/01.str.0000020121.41527.5d. [DOI] [PubMed] [Google Scholar]
- 17.Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: Role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol. 2007;61:352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
- 18.Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–2969. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]
- 19.Peeling J, Yan HJ, Chen SG, Campbell M, Del Bigio MR. Protective effects of free radical inhibitors in intracerebral hemorrhage in rat. Brain Res. 1998;795:63–70. doi: 10.1016/s0006-8993(98)00253-4. [DOI] [PubMed] [Google Scholar]
- 20.Zhao X, Song S, Sun G, Strong R, Zhang J, Grotta JC, et al. Neuroprotective role of haptoglobin after intracerebral hemorrhage. J Neurosci. 2009;29:15819–15827. doi: 10.1523/JNEUROSCI.3776-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner KR, Packard BA, Hall CL, Smulian AG, Linke MJ, De Courten-Myers GM, et al. Protein oxidation and heme oxygenase-1 induction in porcine white matter following intracerebral infusions of whole blood or plasma. Dev Neurosci. 2002;24:154–160. doi: 10.1159/000065703. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura T, Keep RF, Hua Y, Hoff JT, Xi G. Oxidative DNA injury after experimental intracerebral hemorrhage. Brain Res. 2005;1039:30–36. doi: 10.1016/j.brainres.2005.01.036. [DOI] [PubMed] [Google Scholar]
- 23.Zuwala-Jagiello J. Haemoglobin scavenger receptor: Function in relation to disease. Acta Biochim Pol. 2006;53:257–268. [PubMed] [Google Scholar]
- 24.Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, et al. Transcription factor nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke. 2007;38:3280–3286. doi: 10.1161/STROKEAHA.107.486506. [DOI] [PubMed] [Google Scholar]
- 25.Borsody M, Burke A, Coplin W, Miller-Lotan R, Levy A. Haptoglobin and the development of cerebral artery vasospasm after subarachnoid hemorrhage. Neurology. 2006;66:634–640. doi: 10.1212/01.wnl.0000200781.62172.1d. [DOI] [PubMed] [Google Scholar]
- 26.Panter SS, Sadrzadeh SM, Hallaway PE, Haines JL, Anderson VE, Eaton JW. Hypohaptoglobinemia associated with familial epilepsy. J Exp Med. 1985;161:748–754. doi: 10.1084/jem.161.4.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. 2005;106:2572–2579. doi: 10.1182/blood-2005-03-1185. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, Zhang X, Chen-Roetling J, Regan RF. Increased striatal injury and behavioral deficits after intracerebral hemorrhage in hemopexin knockout mice. J Neurosurg. 2010 doi: 10.3171/2010.10.JNS10861. published ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Dore S. Heme oxygenase 2 deficiency increases brain swelling and inflammation after intracerebral hemorrhage. Neuroscience. 2008;155:1133–1141. doi: 10.1016/j.neuroscience.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Dore S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007;130:1643–1652. doi: 10.1093/brain/awm095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: Effects on macrophage viability and tissue iron distribution. Blood. 2010;116:6054–6062. doi: 10.1182/blood-2010-03-272138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao X, Sun G, Strong R, Song W, Gonzales N, Grotta JC, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: Role for pparg in microglia/macrophage. Ann Neurol. 2007;61:352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
- 33.Wagner KR, Dwyer BE. Hematoma removal, heme, and heme oxygenase following hemorrhagic stroke. Ann N Y Acad Sci. 2004;1012:237–251. doi: 10.1196/annals.1306.020. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. 2004;100:672–678. doi: 10.3171/jns.2004.100.4.0672. [DOI] [PubMed] [Google Scholar]
- 35.Peeling J, Del Bigio MR, Corbett D, Green AR, Jackson DM. Efficacy of disodium 4-[(tert-butylimino)methyl]benzene-1,3-disulfonate n-oxide (nxy-059), a free radical trapping agent, in a rat model of hemorrhagic stroke. Neuropharmacology. 2001;40:433–439. doi: 10.1016/s0028-3908(00)00170-2. [DOI] [PubMed] [Google Scholar]
- 36.Gu Y, Xi G, Liu W, Keep RF, Hua Y. Estrogen reduces iron-mediated brain edema and neuronal death. Acta Neurochir Suppl. 2010;106:159–162. doi: 10.1007/978-3-211-98811-4_29. [DOI] [PubMed] [Google Scholar]
- 37.Lyden PD, Shuaib A, Lees KR, Davalos A, Davis SM, Diener HC, Svensson HH, Rodichok L, Wasiewski WW, Ahlberg G, et al. Safety and tolerability of nxy-059 for acute intracerebral hemorrhage: The chant trial. Stroke. 2007;38:2262–2269. doi: 10.1161/STROKEAHA.106.472746. [DOI] [PubMed] [Google Scholar]
- 38.Gu Y, Hua Y, Keep RF, Morgenstern LB, Xi G. Deferoxamine reduces intracerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke. 2009;40:2241–2243. doi: 10.1161/STROKEAHA.108.539536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu J, Hua Y, Keep RF, Schallert T, Hoff JT, Xi G. Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res. 2002;953:45–52. doi: 10.1016/s0006-8993(02)03268-7. [DOI] [PubMed] [Google Scholar]
- 40.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the keap1-nrf2-are pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 41.Sundararajan S, Jiang Q, Heneka M, Landreth G. Ppargamma as a therapeutic target in central nervous system diseases. Neurochem Int. 2006;49:136–144. doi: 10.1016/j.neuint.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 42.Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J. 15d-prostaglandin j2 activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab. 2006;26:811–820. doi: 10.1038/sj.jcbfm.9600233. [DOI] [PubMed] [Google Scholar]
- 43.Zhao X, Strong R, Zhang J, Sun G, Tsien JZ, Cui Z, et al. Neuronal ppargamma deficiency increases susceptibility to brain damage after cerebral ischemia. J Neurosci. 2009;29:6186–6195. doi: 10.1523/JNEUROSCI.5857-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Rogove AD, Tsirka AE, Tsirka SE. Protective role of tuftsin fragment 1-3 in an animal model of intracerebral hemorrhage. Ann Neurol. 2003;54:655–664. doi: 10.1002/ana.10750. [DOI] [PubMed] [Google Scholar]
- 45.Zhao X, Zhang Y, Strong R, Zhang J, Grotta JC, Aronowski J. Distinct patterns of intracerebral hemorrhage-induced alterations in nf-kappab subunit, inos, and cox-2 expression. J Neurochem. 2007;101:652–663. doi: 10.1111/j.1471-4159.2006.04414.x. [DOI] [PubMed] [Google Scholar]
- 46.Mayne M, Ni W, Yan HJ, Xue M, Johnston JB, Del Bigio MR, et al. Antisense oligodeoxynucleotide inhibition of tumor necrosis factor- alpha expression is neuroprotective after intracerebral hemorrhage. Stroke. 2001;32:240–248. doi: 10.1161/01.str.32.1.240. [DOI] [PubMed] [Google Scholar]
- 47.Masada T, Hua Y, Xi G, Yang GY, Hoff JT, Keep RF. Attenuation of intracerebral hemorrhage and thrombin-induced brain edema by overexpression of interleukin-1 receptor antagonist. J Neurosurg. 2001;95:680–686. doi: 10.3171/jns.2001.95.4.0680. [DOI] [PubMed] [Google Scholar]
- 48.Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- 49.Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, et al. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–1145. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- 50.Maderna P, Godson C. Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim Biophys Acta. 2003;1639:141–151. doi: 10.1016/j.bbadis.2003.09.004. [DOI] [PubMed] [Google Scholar]