

Abstract
In cardiac ventricular myocytes, calcium (Ca) release occurs at distinct structures (dyads) along t-tubules, where L-type Ca channels (LCCs) appose sarcoplasmic reticulum (SR) Ca release channels (RyR2s). We developed a model of the cardiac ventricular myocyte that simulates local stochastic Ca release processes. At the local Ca release level, the model reproduces Ca spark properties. At the whole-cell level, the model reproduces the action potential, Ca currents, and Ca transients. Changes in microscopic dyadic properties (e.g., during detubulation in heart failure) affect whole-cell behavior in complex ways, which we investigated by simulating changes in the dyadic volume and number of LCCs/RyR2s in the dyad, and effects of calsequestrin (CSQN) as a Ca buffer (CSQN buffer) or a luminal Ca sensor (CSQN regulator). We obtained the following results: 1), Increased dyadic volume and reduced LCCs/RyR2s decrease excitation-contraction coupling gain and cause asynchrony of SR Ca release, and interdyad coupling partially compensates for the reduced synchrony. 2), Impaired CSQN buffer depresses Ca transients without affecting the synchrony of SR Ca release. 3), When CSQN regulator function is impaired, interdyad coupling augments diastolic Ca release activity to form Ca waves and long-lasting Ca release events.
Introduction
Calcium (Ca) release in a ventricular myocyte occurs in local domains along t-tubules called dyadic spaces. In this space, ∼50–200 ryanodine receptors (RyR2s) in the terminal cisternae of the sarcoplasmic reticulum (SR), called junctional SR (JSR), closely appose five to 15 L-type Ca channels (LCCs) situated along the t-tubules (1). Ca release from RyR2s can occur in two ways: During excitation-contraction coupling (ECC), LCCs open in response to the membrane potential and cause an influx of Ca into the dyadic space from the extracellular space. This Ca influx triggers a much larger Ca release from the RyR2s through a process called calcium-induced calcium release (CICR) (2). Ca release can also occur spontaneously without an influx of Ca from LCCs in the dyad (3). A typical ventricular myocyte has ∼10,000–50,000 dyads. Global macroscopic Ca release in the whole cell is then the cumulative sum of microscopic local Ca releases from the dyads. To understand how modifications of Ca release processes at the dyadic level translate into changes in whole-cell Ca behavior, it is necessary to model Ca release processes at the local dyadic level and extend this modeling framework to include sarcolemmal ionic currents, pumps, and exchangers to form an integrated model of the cell. This is a major objective of the work presented here, motivated by the fact that various disease processes alter dyadic structure/function to cause abnormal cellular phenotypes.
In a normal ventricular myocyte, t-tubules form a regular network of invaginations mainly in the transverse direction (4). The cell-wide regular network ensures that Ca release occurs nearly synchronously during excitation due to membrane depolarization (5). There is evidence that in the failing heart, remodeling processes lead to disarray of t-tubules (6). A family of proteins known as junctophilin-2 plays a role in colocalization of t-tubules and SR, thereby ensuring proper coupling between LCCs and RyR2s in the dyad (7). Junctophilin-2 deficiency has been observed during heart failure and can lead to depressed contractility as well as hyperactive RyR2s (8). Such deficiencies may result in changes in the structural dyadic properties. ECC in a ventricular myocyte can be quite sensitive to structural changes in the dyad that affect the coupling between LCCs and RyR2s in tight restricted spaces. It is important to understand how such changes on the molecular scale alter the whole-cell ECC gain and other cellular properties.
Calsequestrin (CSQN), a native SR protein, can play an important role in the CICR process by functioning both as a Ca buffer in the SR (9) and as a luminal Ca sensor by regulating RyR2 openings via interaction with the anchoring proteins Triadin and Junctin (10). Recently, mutations in CSQN that cause changes in its Ca buffering capacity or its ability to interact with RyR2 (11,12) were implicated in hereditary cardiac arrhythmias. The mutation-induced disturbances occur at the level of the dyad but have important global consequences at the level of the whole cell. Understanding the relationships between dyadic processes and whole-cell function requires a multiscale model of Ca cycling.
Motivated by the above findings, in this work we sought to 1), develop a gating model of RyR2 in which RyR2 opening is a function of cytosolic Ca and CSQN; 2), create a model of the dyad that takes into account the stochastic nature of LCC and RyR2 openings; 3), build a multiscale model of ventricular myocyte Ca cycling and electrophysiology; and 4), investigate how changes in microscopic properties of the dyad affect whole-cell behavior. To that end, we examined the following changes in dyad properties: 1), impaired buffering capacity of CSQN; 2), impaired luminal Ca sensor function (RyR2 gating regulation by CSQN); 3), changes in the number of LCCs/RyR2s in the dyad; and 4), changes in dyadic volume.
Methods
A summary of important model properties is presented here. Detailed methods, descriptions, definitions of variables and abbreviations, and model equations are provided in the Supporting Material (including Fig. S1, Fig. S2, Fig. S3, Fig. S4, and Fig. S5). The model code can be downloaded from the research section of http://rudylab.wustl.edu. Formulation of the sarcolemmal ionic currents, pumps, and exchangers was adapted from the existing guinea-pig ventricular myocyte model (13). The following subscripts are used in abbreviations: SR, sarcoplasmic reticulum; NSR, network SR; JSR, junctional SR; ss, subsarcolemmal space (submembrane); d, dyadic space; and i, myoplasm. Superscripts cell and dyad denote whole-cell and dyadic values, respectively.
Dyad model
Each dyad (see diagram in Fig. 1 A) comprises the following Ca distribution compartments: myoplasm, NSR, JSR, dyadic space, and submembrane space. In the control myocyte model, it is assumed that 15 LCCs and 100 RyR2s are present in a dyad.
Figure 1.
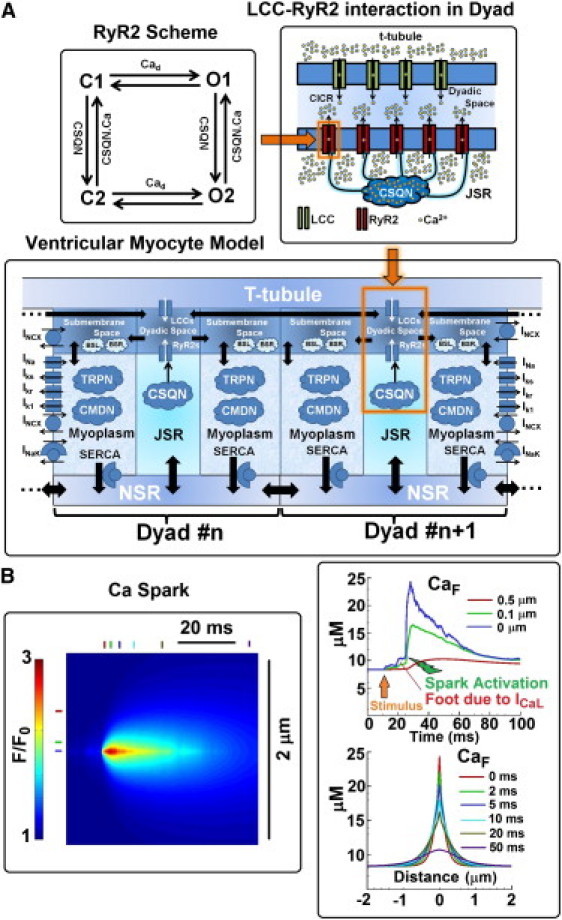
(A) Schematic diagram of the spatially distributed ventricular myocyte model and its components. The model consists of 10,000 diffusively coupled CaRUs (also called dyads). Each dyad consists of five distinct Ca compartments: NSR, JSR, dyadic space, submembrane space, and myoplasm. Ca concentration is assumed to be uniform in each compartment. The number of LCCs and SR Ca release channels (RyR2s) in a dyad is 15 and 100, respectively, and their states fluctuate stochastically. State diagram of the RyR2 model is shown in the upper-left corner. LCC-RyR2 interaction in a dyad is shown in the upper-right corner. Ca release occurs in the dyadic space. CSQN is a Ca buffer that also regulates RyR2 openings. JSR is defined as the domain of CSQN distribution, so CSQN is in all of JSR. Sarcolemmal buffers (BSL) and SR buffers (BSR) are in the submembrane space. Calmodulin (CMDN) and troponin (TRPN) are Ca buffers in the myoplasm. All Ca-dependent membrane currents, pumps, and exchangers sense Ca concentration in the compartment in which they reside. Important currents that determine membrane potential are shown. Diffusive Ca fluxes are indicated by solid thick arrows. Equations for the model are given in the Supporting Material. The model code is available to download from the research section of http://rudylab.wustl.edu. (B) Left: Simulated Ca spark (line scan); CaF indicates the concentration of Ca-bound Fluo-3 dye. Top right: Temporal profile of CaF at three different locations: the spark center (blue), 0.1 μm from spark center (green), and 0.5 μm from spark center (red). Bottom right: Spatial profile of Ca spark at several time points after initiation. It is assumed that the line scan passes through the spark origin.
Ventricular myocyte model
Fig. 1 A shows a schematic diagram of the multiscale ventricular myocyte model and its components. The model consists of 10,000 dyads in which Ca release occurs stochastically. Each dyad communicates with its neighboring dyads via Ca diffusion in the NSR and the dyadic space. Membrane ionic currents, pumps, and exchangers sense local Ca concentrations in their compartment. Dyadic concentrations are averaged to represent whole-cell macroscopic values. Dyadic currents are added to represent whole-cell values.
RyR2 model
The Markov model of RyR2 (Fig. 1 A) consists of four states and two tiers of modal gating (upper and lower). The upper tier is the activation tier, and the lower tier is the refractory tier. Km for Ca activation in the lower tier is 10 times higher than Km in the upper tier (150 μM vs. 15 μM).
CSQN regulator (luminal Ca sensor)
The transition of RyR2 channel states between the activation tier and refractory tier depends on CSQN. This dependence represents the luminal Ca sensor function of CSQN in the model. Details about the regulation of RyR2 openings by CSQN are provided in the Supporting Material.
CSQN buffer
CSQN has a total capacity of absorbing 10 mM Ca with a half constant (Kd) of 0.8 mM. It is assumed that Ca binds instantaneously to CSQN.
Results
The multiscale model of the cardiac ventricular myocyte scales processes from stochastic single-channel openings of RyR2s and LCCs to the whole-cell action potential (AP) and Ca transient (CaT). In the first half of the Results section, we present model simulations of normal physiological processes at both the local dyadic level and the whole-cell level, and show their correspondence across scales. These simulations serve to allow evaluation and validation of the model. In the second part of the Results section we discuss the macroscopic (whole-cell) consequences of changes in the microscopic properties of the dyad. In particular, we are interested in the whole-cell consequences of changes in the structural properties of the dyad, including the volume of the dyadic space and the number of LCCs/RyR2s in the dyad. Pathologically, such changes may occur during detubulation in a failing heart (6). We are also interested in the whole-cell consequences of impaired function of CSQN as a buffer of Ca or a regulator of RyR2 function (or both). Impairment of CSQN function has been implicated in genetic mutations that lead to cardiac arrhythmias (11,12). The model allows us to investigate separately the roles played by CSQN buffer and the CSQN regulator in these pathologies. In addition, we investigate the effects of interdyad coupling via Ca diffusion on whole-cell function when the above changes in dyadic properties are present.
Normal physiological function
Simulated Ca spark
Fig. 1 B shows a simulated Ca spark during myocyte pacing at 1 Hz. A temporal profile of fluorescent-dye-bound Ca (CaF) at 0, 0.1, and 0.5 μm from the spark center is shown at the top right of the figure. After application of the stimulus, a small foot of Ca increase due to ICaL is seen before the Ca spark activates, in similarity to the sparklet-triggered spark observed by Wang et al. (14). The spatial profile of the Ca spark at 0, 2, 5, 10, 20, and 50 ms after initiation of the spark is also shown (bottom right). The simulated Ca spark properties are consistent with experiments (3) and with simulated Ca sparks generated with either a square pulse of Ca release (2 pA for 10 ms) (15) or a square pulse of Ca trigger (0.5 pA for 0.5 ms) (16).
RyR2 model response to cytosolic and SR Ca
Fig. 2 A (left panel) shows simulated single-channel RyR2 records for 15 μM Cad and for 100 μM and 2 mM CaSR. Experiments performed with native RyR2s in lipid bilayers (17) for a cis Ca concentration of 1 μM, and trans Ca concentrations of 20 μM and 5 mM are shown on the right. The open probability (Po) of the channel at 15 μM Cad for different CaSR-values is shown in Fig. 2 B (left: model; right: experiment). The model behavior is similar to that seen in experiments. Quantitative differences can be attributed to the different environment of RyR2 in the cell and in lipid bilayers.
Figure 2.
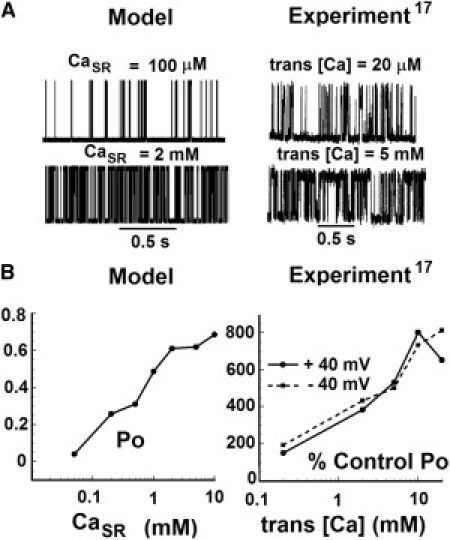
RyR2 model response. (A) Simulated (left) and experimentally measured (right) single-channel current records of RyR2 in response to Ca. In the model, the dyadic space Ca (Cad) is held at 15 μM and records are shown for two different SR Ca (CaSR) values: 100 μM and 2 mM. In experiments, cis [Ca] is held at 1 μM and traces are shown for two different trans [Ca] values: 20 μM and 5 mM (reproduced with permission from Györke and Györke (17)). (B) The Po of RyR2 as a function of CaSR at 15 μM Cad. In the simulation (left), Po represents the absolute open probability. In the experiments (right), Po is plotted relative to control conditions at 20 μM trans [Ca] and 1 μM cis [Ca]. The solid line indicates a +40 mV voltage difference between the cis and trans (cis – trans) compartments of the lipid bilayer. The dashed line indicates a voltage difference of −40 mV.
Ca currents and Ca concentrations in a single dyad during pacing
Simulated Ca currents and Ca concentrations in one of the dyads during an AP, when the cell is paced at 1 Hz, are shown in Fig. 3. During pacing at 1 Hz in response to membrane voltage, Ca influx via begins shortly after the AP upstroke. Peak reaches −0.11 pA, and SR Ca release occurs 5 ms after peak has been reached. Peak Ca release current reaches 4 pA and decays over 15 ms, consistent with reported values of an elementary SR Ca release current and decay time course (3). After the release is terminated, the release channels become refractory for the remaining duration of the AP plateau. peaks at 150 μM and decays over 20 ms, following the time course of SR Ca release. This result is consistent with the graded nature of ECC: under normal physiological conditions, a given Ca influx via LCCs triggers a much greater Ca release via RyR2s (18), and therefore is governed by SR Ca release. Local SR Ca, decays from 1.3 mM to a nadir of 1 mM and recovers in ∼250 ms, consistent with the finding that diastolic CaSR is only partially depleted during contraction (19). The results demonstrate that substantial differences in the peak concentration and time course of Ca exist among different local Ca compartments within the dyad during ECC.
Figure 3.
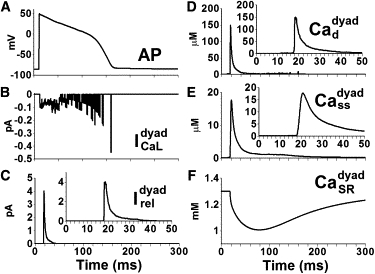
Ca currents and Ca concentrations in a single dyad in a ventricular myocyte model paced at 1 Hz. (A) AP. (B) L-type Ca current (C) Ca release current (D) Dyadic space Ca concentration (E) Submembrane Ca concentration (F) Free SR Ca concentration Insets in C–E show expanded timescale.
Whole-cell (macroscopic) Ca currents and Ca concentrations during pacing
The steady-state AP at 1 Hz, whole-cell and values, and compartmental Ca concentrations are shown in Fig. 4. The duration of the whole-cell Ca release flux, , and the average dyadic space Ca, is in the range of 20–30 ms, which is similar to the duration of local Ca release events. However, the rise time of these whole-cell parameters is slower, and the peak is lower than the local reflecting a slight temporal dispersion in the occurrence of local Ca release events. The global SR Ca depletion profile is similar to that of local SR Ca depletion ( versus ), indicating that SR Ca depletion occurs uniformly throughout the cell. The different shape of and can be attributed to the small number of LCCs in the dyad (Fig. S2).
Figure 4.
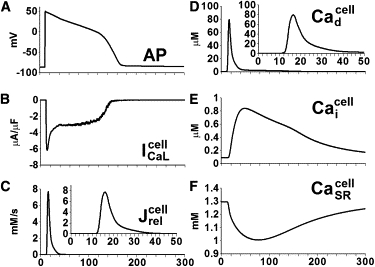
Whole-cell Ca currents and Ca concentrations in the ventricular myocyte model paced at 1 Hz. (A) AP. (B) L-type Ca current (C) Ca release flux from SR (D) Average Ca concentration in the dyadic space (E) CaT in the myoplasm (F) Average free Ca concentration in the SR Insets in C and D show expanded timescale.
Graded release and variable gain and rate dependence of myoplasmic Na and Ca
LCCs and RyR2s interact in local dyadic spaces, and stochastic openings of local LCCs can trigger openings of local RyR2s. The magnitude of Ca entry via LCCs determines the number of RyR2 openings and therefore the magnitude of SR release. This phenomenon of graded release is reproduced by the model, as demonstrated in Fig. 5 A, which shows that peak RyR2 flux is a graded function of peak LCC flux. Another property of a cardiac myocyte is the variable gain. The ratio of Ca released from the SR to Ca entry into the myocyte, also known as gain, is a function of the transmembrane potential. The model reproduces this property. For example, in Fig. 5 B, for a similar magnitude of peak LCC flux at transmembrane voltages of +50 mV and −20 mV, the peak RyR2 flux is quite different. The gain computed in the model as the ratio of peak RyR2 flux to peak LCC flux is a decreasing function of voltage (Fig. 5 C). The gain at 0 mV is 15.8, which is similar to the value of 16 previously measured in rats (20).
Figure 5.
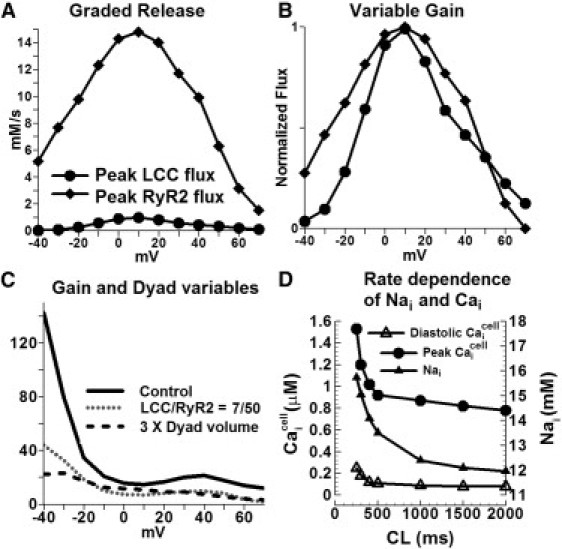
Graded release, variable gain, effect of dyad properties on ECC, and rate dependence of myoplasmic Nai and Cai. (A) Peak LCC Ca flux and peak Ca release flux as a function of transmembrane voltage. (B) Normalized peak LCC Ca flux and peak Ca release flux. (C) ECC gain for three conditions: 1), control (100/15 RyR2/LCC ratio, 2 × 10−19 L dyadic volume); 2), 50/7 RyR2/LCC ratio, 2 × 10−19 L dyadic volume; and 3), 100/15 RyR2/LCC ratio, 6 × 10−19 L dyadic volume. Initial conditions were those of a paced myocyte at 1 Hz. After 5 ms the cell was clamped at the voltage indicated on the x axis. (D) Myoplasmic Nai and Cai rate dependence. Steady-state (after 1000 beats of pacing) Nai, peak Cai and diastolic Cai as a function of cycle length (CL).
Simulated Nai, peak , and diastolic as a function of pacing rate are shown in Fig. 5 D. Both Na and Ca ions accumulate increasingly in the cell as pacing frequency is increased, consistent with previous results (13).
Macroscopic consequences of changes in microscopic dyadic function
Effects of alterations of dyadic structure on ECC
Because ECC occurs at local domains of the dyad, changes in structural dyadic properties that alter the functional interaction between LCCs and RyR2s during CICR can have significant effects on the Ca release process. Such structural alterations are observed in failing or hypertrophic hearts (21,22). Fig. 5 C shows the ECC gain curve (defined as the ratio of peak RyR2 Ca release flux to peak LCC Ca influx) when the dyadic space volume is increased by a factor of 3 (solid line: control) simulating increased separation between LCCs and RyR2s during tubular disarray. The dotted line shows the gain curve when the numbers of RyR2s/LCCs are reduced to 50/7 (from 100/15 in control), simulating structural derangement of channels in the dyad. Gain values are lower than control at all voltages. Taken together, the results support the hypothesis that reduced ECC gain of CICR in certain pathologies that involve structural changes (e.g., hypertrophy) may be due to microscopic structural defects in the dyads (21).
Effects of structural derangement of channels in the dyad on the whole-cell Ca release process are investigated further in Fig. 6. In the first set of simulations (dashed line; control is indicated by solid black), the number of functional channels in the dyad is reduced to 4/25 LCC/RyR2, whereas the Ca entry via individual LCCs stays the same. During pacing, CaTs are severely blunted, indicating a near absence of SR Ca release, and only a few dyads are activated. The likely explanation for such impaired function is that the reduced number of LCCs in the dyad reduces the functional coupling between LCCs and RyR2s during CICR. We then investigated whether increasing Ca entry per LCC could rescue the impaired CICR, as suggested by the positive inotropic effect of Ca channel agonist BayK8644 (23). The second set of simulations (gray line) shows a scenario where Ca entry through LCC is increased such that is similar to control conditions, whereas the number of channels in the dyad is still reduced to 4/25 LCC/RyR2. With this manipulation, CaTs are rescued only partially, indicating that Ca release is still impaired due to asynchronous activation of dyads (Fig. 6 D). This implies that the number of functional channels, via its effect on synchrony of release, has a greater influence on global CaT than the magnitude of Ca entry. These results support the hypothesis that in a failing heart, despite similar whole-cell ICaL-values, smaller amplitude and slowed kinetics of CaT are due to asynchronous activation of Ca sparks (24).
Figure 6.
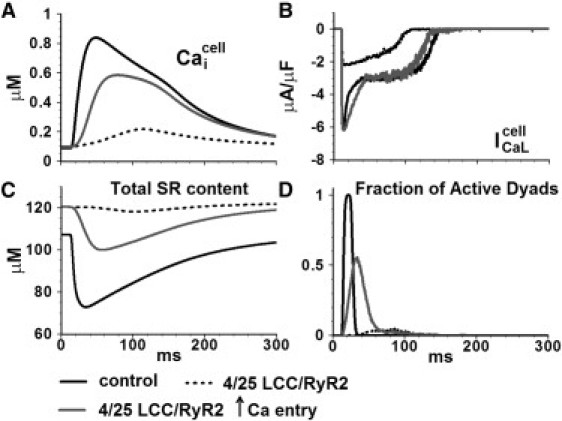
Effect of changing the number of functional RyR2s and LCCs in the dyad on whole-cell behavior. (A) Myoplasmic CaT (B) L-type Ca current (C) Total SR Ca content. (D) Fraction of active dyads.
Role of interdyad coupling in ECC
When the number of LCCs in the dyad was reduced to seven (from 15 in control) and the interdyad coupling was impaired, SR Ca release became asynchronous (Fig. 7, black line) resulting in a smaller peak and slower time to peak of CaT. With intact interdyad coupling, SR Ca release became more synchronous (gray line), indicating that interdyad coupling had a synchronizing effect on SR Ca release in this simulated pathology. Similar results were obtained when the dyadic volume was increased threefold (Fig. S5). Pathologic structural defects associated with heart failure, such as detubulation, may result in a reduced number of functional LCCs or an increased dyadic space volume. The simulation results suggest that under such circumstances, SR Ca release becomes asynchronous and interdyad coupling serves as a compensatory mechanism that improves synchrony.
Figure 7.
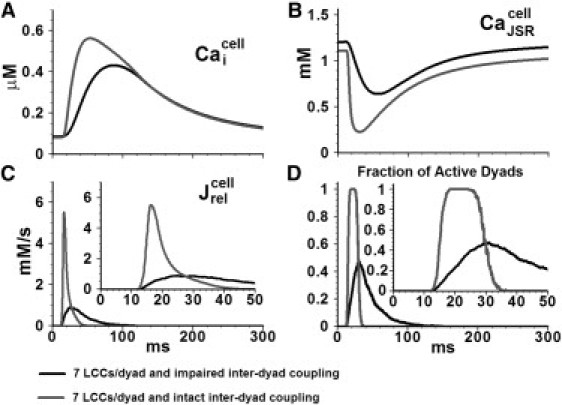
Effect of impaired interdyad coupling on whole-cell behavior. (A) Myoplasmic CaT (B) JSR Ca concentration (C) Ca release flux (D) Fraction of active dyads. The myocyte model is paced at 1 Hz. Insets show and the fraction of active dyads on an expanded timescale.
Effects of impaired CSQN function
In SR, CSQN can function as both a Ca buffer and a regulator of RyR2 openings (9,10). Experimentally, it is very difficult, if not impossible, to isolate and study separately the effects of CSQN as a buffer or a regulator on the SR Ca release process. The model allowed us to differentiate these effects in a controlled manner.
Simulated impaired CSQN buffering ability resulted in a smaller Ca release flux, smaller CaT, and higher diastolic JSR load (Fig. 8). Faster recovery of the JSR load after release was observed. The reduced CICR was not associated with asynchronous activation of dyads (Fig. 8 D); rather, it was reduced due to a reduced amount of releasable Ca from the SR. Diastolic Ca release events were not observed, despite the faster recovery of the JSR Ca load.
Figure 8.
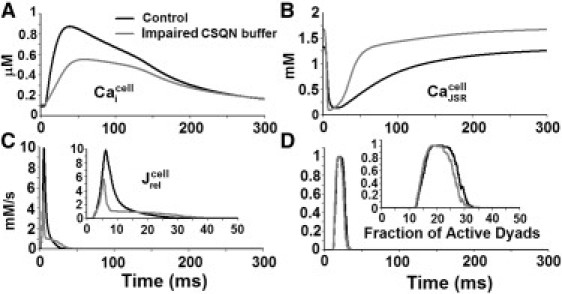
Effect of impaired CSQN buffering capacity on whole-cell behavior. (A) Myoplasmic CaT (B) JSR Ca concentration (C) Ca release flux (D) Fraction of active dyads. The myocyte model is paced at 1 Hz. Insets show and the fraction of active dyads on an expanded timescale.
Fig. 9 shows the effect of an impaired luminal Ca sensor function on whole-cell behavior. In the simulation, the effect of CSQN on RyR2 openings was examined in the presence of an intact (Fig. 9, A and B) or impaired (Fig. 9, C and D) CSQN buffering function, in an attempt to separate the two processes. Multiple diastolic Ca release events in the form of Ca sparks (indicated by yellow arrows) and Ca waves (indicated by white arrows) occur when the CSQN buffer is intact (Fig. 9 A). Panel B shows that the dyad reactivates during the AP (indicated by #) and activates spontaneously during diastole (indicated by ∗), indicating abnormal restitution of SR Ca release. In a paced control myocyte model, Ca sparks rarely occur in diastole, due to the refractoriness of the RyR2s. In a quiescent myocyte, when RyR2s are in the active state, the spontaneous Ca spark frequency is calculated to be 40/cell/s, which is similar to the experimentally recorded frequency in guinea-pigs (25).
Figure 9.
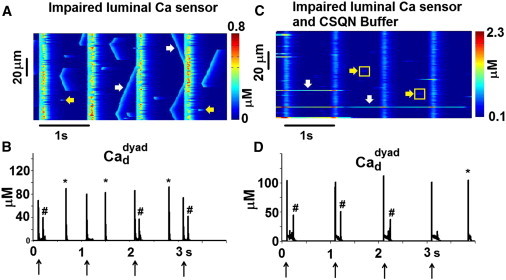
Effect of impaired luminal Ca sensor function on whole-cell behavior. (A and B) CSQN buffer is intact. (C and D) CSQN buffer is impaired. (A and C) Line-scan image of . (B and D) Dyadic space Ca concentration in one of the dyads. Arrows at the bottom of panels B and D indicate pacing stimuli. Spontaneous diastolic Ca release events in the form of Ca sparks (yellow arrows) and waves (white arrows) are seen in panel A. Long-lasting Ca release events in a few dyads (white arrows) and dim Ca sparks (yellow arrows) are seen in panel C. The dyad reactivates during the paced AP (#) and spontaneously activates during diastole (∗) in panels B and D.
When the CSQN buffer is also impaired, long-lasting Ca release events occur in few dyads, indicating a defective termination process of Ca release (white arrows in Fig. 9 C). Ca sparks occur during diastole (yellow arrows). However, in contrast to the case of impaired Ca sensor function alone (but with intact buffering), Ca waves do not develop, because in the absence of CSQN buffer, the JSR Ca content and the amount of released Ca is small and insufficient to trigger release from adjacent dyads to generate propagating waves. in one of the dyads is plotted in panel D. The dyad reactivates during the AP (indicated by #) and can also activate spontaneously (indicated by ∗) during diastole, indicating that in addition to the abnormal termination, there is an abnormal restitution of Ca release at the dyadic level. In a dyad undergoing a long-lasting Ca release event (Fig. S3), the open fraction of RyR2s oscillates between 0.8 and 0.1, and the release lasts for >3.8 s.
Role of interdyad coupling when CSQN function is impaired
When impaired interdyad coupling is present in addition to impaired luminal Ca sensor function (but CSQN buffering is intact), Ca waves no longer occur. However, dyads can still reactivate during the AP or spontaneously activate during diastole (results not shown). Thus, interdyad coupling promotes the formation of Ca waves when CSQN sensor function is impaired, but its buffering ability is intact. When CSQN is dysfunctional as both a luminal Ca sensor and buffer, impaired interdyad coupling prevents the formation of long-lasting release events. We conclude that abnormal patterns of Ca release due to impaired CSQN function are modulated by the interaction between CSQN as a buffer and interdyad coupling.
Discussion
In cardiac ventricular myocytes, Ca release occurs at local domains along t-tubules where LCCs and RyR2s interact via Ca in dyadic spaces. Pathologies such as heart failure may involve detubulation of the myocyte t-tubule system (6), changes in dyadic structure, and impaired functional coupling between LCCs and RyR2s (22). These subcellular local changes can alter CICR and ECC, and disrupt normal whole-cell macroscopic behavior. Ca release can also occur spontaneously during diastole, and impaired CSQN function, both as a regulator of RyR2 gating and as a buffer of SR Ca (11,12), has been implicated in such aberrant Ca release events. At present, it is extremely difficult, if not impossible, to study microscopic processes in subcellular compartments experimentally. With experimental approaches, the ability to integrate across scales from microscopic processes to whole-cell behavior is very limited. To study these phenomena through computer modeling, we constructed a multiscale model of Ca cycling that takes into account local dyadic Ca release activities and interactions between dyads via Ca diffusion. The model is stochastic in nature, and its relation to other existing models is discussed in the Supporting Material.
Effect of structural changes in dyads on whole-cell behavior
In normal cardiac myocytes, depolarization produces a rapid rise in myoplasmic Ca as a consequence of coordination and summation of localized Ca release events (18). Disruption of coordinated Ca spark initiation may underlie abnormalities of CaTs, as observed in myocytes from hearts with various pathologies. Several membrane factors can reduce the synchrony of activation and summation of Ca sparks, including a depressed phase-1 AP notch due to a reduced transient outward current, Ito1 (26), which decreases the driving force for ICaL, thereby affecting the trigger for SR Ca release. Changes in ICaL due to mutations, remodeling, or drugs can also have a similar effect on Ca release synchrony. Intracellularly, altered Ca sensitivity and gating kinetics of RyR2s can affect the synchrony of release (27). Besides such molecular changes, impaired coupling between LCCs and RyR2s (22), or structural remodeling of t-tubules in failing hearts (6) can result in Ca release asynchrony. In this work, we simulated several structural defects in the dyad and found support for the hypothesis that ECC gain is lower with such defects. Simulations also showed that even with a normal level of whole-cell Ca entry via ICaL, as might occur in failing or hypertrophic hearts (22), a reduced dyadic density of LCCs can alter CICR, resulting in smaller CaTs due to asynchronous activation of dyads. In normal ventricular myocytes, Ca influx through LCCs is thought to be the primary synchronizing mechanism of Ca release (28). Our simulation results suggest that propagated CICR via interdyad diffusive coupling increases synchronization of Ca release when Ca influx sensed by RyR2s is reduced due to a decreased number of dyadic LCCs or increased dyadic volume. Such t-tubular or dyadic structural changes occur in failing hearts, and interdyad coupling may serve as a compensatory mechanism for rescuing ECC gain to improve contractility in these hearts.
Effects of impaired CSQN function
CSQN is a high-capacity Ca-binding protein in the SR of cardiac myocytes. When CSQN buffering ability was impaired, SR Ca release flux was smaller in magnitude and duration, consistent with CSQN's role as a Ca buffer. This resulted in depressed CaT, which can suppress cardiac contractility. Impaired CSQN buffering ability caused little change in the synchronization of Ca release. CaJSR recovery was faster, which could have led to premature recovery of RyR2s from luminal Ca-dependent refractoriness (29) and diastolic release events. This possibility was not supported by our simulation results, because restitution of Ca release was preserved by intact CSQN regulator function.
Multiple processes have been implicated in restitution of Ca release, including cytosolic Ca-dependent or use-dependent inactivation of RyR2s, luminal Ca-dependent refractoriness of RyR2s, inactivation of LCCs, and slower refilling of SR Ca after a previous SR Ca release event (30). We found that in the presence of intact CSQN buffering ability, an impaired luminal Ca sensor function of CSQN resulted in increased diastolic Ca release activity in the form of Ca waves. This suggests that luminal Ca-dependent refractoriness may be a potent mechanism for restitution of Ca release.
When both CSQN buffering ability and luminal Ca sensor function were impaired, increased diastolic Ca sparks, but not Ca waves, were present. Both the frequency of RyR2 openings and the SR Ca load are important factors in propagation of Ca waves, which are usually observed only in Ca-overloaded cells (3). In this case, despite the increased frequency of diastolic Ca sparks, the reduced SR load due to impaired CSQN buffer may have been insufficient to sustain Ca waves.
Our results indicate that impaired CSQN regulation function, but with preserved buffering ability, is necessary for formation of arrhythmogenic Ca waves. Ca-wave-induced delayed after-depolarizations and triggered activity have been implicated as the arrhythmogenic mechanism in catecholaminergic polymorphic ventricular tachycardia (CPVT). The D307H missense mutation in CSQN, presumably leading to impaired CSQN buffering capacity (12), was also implicated in CPVT. It is possible that the mutation also affects CSQN regulation of RyR2, and that the degree of buffering impairment is insufficient to prevent the formation of Ca waves. It is also possible that compensatory mechanisms such as increased SR volume (31) and/or overexpression of the SR-binding protein calreticulin (32) may also preserve SR load to sustain propagated CICR in such mutants.
Ca sparks occur as brief, spatially restricted elevations of Ca, suggesting that a potent mechanism terminates Ca release. Depletion of SR Ca has been proposed as a key mechanism of release termination (16). Long-lasting Ca release events occurred in the simulations only when luminal Ca sensor function was impaired, indicating its role in signaling Ca release termination.
Interdyad coupling
In the model, interdyad coupling in the SR was implemented as rapid diffusion of Ca between NSRs of adjacent dyads (τdiff_NSR_NSR = 1 ms), and slower diffusion of Ca between NSR and JSR in a given dyad (τrefill = 10 ms). Recent reports have indicated rapid diffusion of Ca in the SR (33), suggesting a functionally continuous structure. Our choice to distinguish between NSR and JSR is based on two considerations: 1), CSQN localizes only at the terminal cisternae of the SR (we define JSR as the volume (compartment) of CSQN distribution (Fig. S1)); and 2), structural considerations restrict connectivity between JSR and NSR (34). Experiments have also indicated that some junctions are better connected to the rest of the SR than others (35). During global CICR, the simulated local SR Ca depletion in individual dyads (including contributions from JSR and NSR) is 47% (computed as the ratio of Ca released to free SR Ca before release), 33% (computed as the ratio of Ca released to total SR Ca before release), or 23% (computed as the ratio of free SR Ca depletion to free SR Ca before release; Fig. 3 F). The whole-cell SR Ca depletion (contributed from JSR and NSR) during a global CaT constitutes a similar percentage of diastolic CaSR content. These numbers are consistent with measurements indicating that diastolic CaSR is only partially depleted (24–63%) during contraction (19).
Ca waves and long-lasting Ca release events are not present when interdyad coupling is impaired. Local Ca release disturbances still remain in the form of dyad reactivation during the AP and spontaneous Ca release during diastole. These results demonstrate that interdyad coupling can amplify local stochastic Ca release disturbances. This deleterious amplification can occur at the level of the dyad, causing long-lasting Ca release events, or across the dyads to form Ca waves in the whole cell. On the other hand, under pathological conditions when the dyadic structure is impaired, interdyad coupling may serve as a beneficial mechanism to maintain normal synchronization of Ca release. Thus, in a functioning cardiac ventricular myocyte, optimal tuning of interdyad coupling may be a factor in maintaining proper systolic function without amplification of local Ca release events during diastole.
Limitations
The stochastic multiscale model presented here goes beyond previous efforts to construct a myocyte Ca cycling model that captures the spatial dynamics of Ca release and CSQN-mediated regulation of RyR2 gating. However, it has important limitations that should be considered. The model represents the complex three-dimensional spatial organization of dyads and their distribution in the myocyte by simplified linear geometry. Interdyad coupling in the cytoplasm is implemented by diffusion of Ca between adjacent dyadic spaces. Although this implementation is phenomenological, it is consistent with the experimental observation that Ca waves occur in Ca-overloaded cells (3) (Fig. S4). Finally, luminal Ca regulation of RyR2 gating is assumed to occur exclusively via CSQN. Alternative regulation mechanisms of RyR2 gating, such as luminal Ca feed-through or Ca inactivation sites (36), were not considered.
Acknowledgments
We thank our laboratory members Leonid Livshitz, Ashwin Mohan, Ali Nekouzadeh, Pan Li, Jiajing Xu, Thomas O'Hara, and Smiruthi Ramasubramanian for helpful discussions.
This research was supported by the National Heart, Lung, and Blood Institute, National Institutes of Health (grants R01-HL-33343-26 and R01-HL-49054-18), a Fondation Leducq Award to the Alliance for Calmodulin Kinase Signaling in Heart Disease (grant 08CVD01 to Y.R.), and an American Heart Association predoctoral fellowship (103600009 to N.G.). This material is also based in part on work supported by the National Science Foundation under grant No. CBET-0929633. Any opinions, findings, and conclusions or recommendations expressed here are those of the authors and do not necessarily reflect the views of the National Science Foundation. Yoram Rudy is the Fred Saigh Distinguished Professor at Washington University in St. Louis.
Supporting Material
References
- 1.Franzini-Armstrong C., Protasi F., Tijskens P. The assembly of calcium release units in cardiac muscle. Ann. N. Y. Acad. Sci. 2005;1047:76–85. doi: 10.1196/annals.1341.007. [DOI] [PubMed] [Google Scholar]
- 2.Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng H., Lederer W.J., Cannell M.B. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 4.Soeller C., Cannell M.B. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ. Res. 1999;84:266–275. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- 5.Cheng H., Cannell M.B., Lederer W.J. Propagation of excitation-contraction coupling into ventricular myocytes. Pflugers Arch. 1994;428:415–417. doi: 10.1007/BF00724526. [DOI] [PubMed] [Google Scholar]
- 6.Song L.S., Sobie E.A., Cheng H. Orphaned ryanodine receptors in the failing heart. Proc. Natl. Acad. Sci. USA. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeshima H., Komazaki S., Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 8.van Oort R.J., Garbino A., Wehrens X.H. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacLennan D.H., Wong P.T. Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 1971;68:1231–1235. doi: 10.1073/pnas.68.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Györke I., Hester N., Györke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rizzi N., Liu N., Priori S.G. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ. Res. 2008;103:298–306. doi: 10.1161/CIRCRESAHA.108.171660. [DOI] [PubMed] [Google Scholar]
- 12.Dirksen W.P., Lacombe V.A., Periasamy M. A mutation in calsequestrin, CASQ2D307H, impairs sarcoplasmic reticulum Ca2+ handling and causes complex ventricular arrhythmias in mice. Cardiovasc. Res. 2007;75:69–78. doi: 10.1016/j.cardiores.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faber G.M., Silva J., Rudy Y. Kinetic properties of the cardiac L-type Ca2+ channel and its role in myocyte electrophysiology: a theoretical investigation. Biophys. J. 2007;92:1522–1543. doi: 10.1529/biophysj.106.088807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang S.Q., Song L.S., Cheng H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 15.Smith G.D., Keizer J.E., Cheng H. A simple numerical model of calcium spark formation and detection in cardiac myocytes. Biophys. J. 1998;75:15–32. doi: 10.1016/S0006-3495(98)77491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobie E.A., Dilly K.W., Jafri M.S. Termination of cardiac Ca(2+) sparks: an investigative mathematical model of calcium-induced calcium release. Biophys. J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Györke I., Györke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys. J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wier W.G., Balke C.W. Ca(2+) release mechanisms, Ca(2+) sparks, and local control of excitation-contraction coupling in normal heart muscle. Circ. Res. 1999;85:770–776. doi: 10.1161/01.res.85.9.770. [DOI] [PubMed] [Google Scholar]
- 19.Shannon T.R., Guo T., Bers D.M. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ. Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- 20.Wier W.G., Egan T.M., Balke C.W. Local control of excitation-contraction coupling in rat heart cells. J. Physiol. 1994;474:463–471. doi: 10.1113/jphysiol.1994.sp020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gómez A.M., Guatimosim S., Lederer W.J. Heart failure after myocardial infarction: altered excitation-contraction coupling. Circulation. 2001;104:688–693. doi: 10.1161/hc3201.092285. [DOI] [PubMed] [Google Scholar]
- 22.Gómez A.M., Valdivia H.H., Lederer W.J. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 23.Thomas G., Chung M., Cohen C.J. A dihydropyridine (Bay k 8644) that enhances calcium currents in guinea pig and calf myocardial cells. A new type of positive inotropic agent. Circ. Res. 1985;56:87–96. doi: 10.1161/01.res.56.1.87. [DOI] [PubMed] [Google Scholar]
- 24.Litwin S.E., Zhang D., Bridge J.H. Dyssynchronous Ca(2+) sparks in myocytes from infarcted hearts. Circ. Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. [DOI] [PubMed] [Google Scholar]
- 25.Ogrodnik J., Niggli E. Increased Ca(2+) leak and spatiotemporal coherence of Ca(2+) release in cardiomyocytes during β-adrenergic stimulation. J. Physiol. 2010;588:225–242. doi: 10.1113/jphysiol.2009.181800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sah R., Ramirez R.J., Backx P.H. Modulation of Ca(2+) release in cardiac myocytes by changes in repolarization rate: role of phase-1 action potential repolarization in excitation-contraction coupling. Circ. Res. 2002;90:165–173. doi: 10.1161/hh0202.103315. [DOI] [PubMed] [Google Scholar]
- 27.Kubalova Z., Terentyev D., Györke S. Abnormal intrastore calcium signaling in chronic heart failure. Proc. Natl. Acad. Sci. USA. 2005;102:14104–14109. doi: 10.1073/pnas.0504298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bers D.M. Kluwer Academic Publishers; Dordrecht/Boston: 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. [Google Scholar]
- 29.Györke S., Györke I., Wiesner T.F. Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle. Front. Biosci. 2002;7:d1454–d1463. doi: 10.2741/A852. [DOI] [PubMed] [Google Scholar]
- 30.Sobie E.A., Song L.S., Lederer W.J. Restitution of Ca(2+) release and vulnerability to arrhythmias. J. Cardiovasc. Electrophysiol. 2006;17(Suppl 1):S64–S70. doi: 10.1111/j.1540-8167.2006.00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knollmann B.C., Chopra N., Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song L., Alcalai R., Seidman J.G. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J. Clin. Invest. 2007;117:1814–1823. doi: 10.1172/JCI31080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu X., Bers D.M. Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ. Res. 2006;99:283–291. doi: 10.1161/01.RES.0000233386.02708.72. [DOI] [PubMed] [Google Scholar]
- 34.Brochet D.X., Yang D., Cheng H. Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc. Natl. Acad. Sci. USA. 2005;102:3099–3104. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zima A.V., Picht E., Blatter L.A. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ. Res. 2008;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laver D.R. Luminal Ca(2+) activation of cardiac ryanodine receptors by luminal and cytoplasmic domains. Eur. Biophys. J. 2009;39:19–26. doi: 10.1007/s00249-009-0417-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.