

Abstract
We report superresolution fluorescence microscopy in an intact living organism, namely Caenorhabditis elegans nematodes expressing green fluorescent protein (GFP)-fusion proteins. We also superresolve, by stimulated emission depletion (STED) microscopy, living cultured cells, demonstrating that STED microscopy with GFP can be widely applied. STED with GFP can be performed with both pulsed and continuous-wave lasers spanning a wide wavelength range from at least 556–592 nm. Acquiring subdiffraction resolution images within seconds enables the recording of movies revealing structural dynamics. These results demonstrate that numerous microscopy studies of live samples employing GFP as the marker can be performed at subdiffraction resolution.
Although it is the most widely applied marker in the biological sciences, few reports exist in which the green fluorescent protein (GFP) is used in superresolution microscopy (1–3). This stems from the fact that the popular methods (F)PALM (4,5) and STORM (6) are not designed for nonphotoactivatable proteins, and stimulated emission depletion (STED) microscopy (7) has not yet been demonstrated with GFP-labeled living samples. Taking advantage of developments in fiber laser technology, we now demonstrate the viability of GFP for performing nanoscopy with living samples using STED and leverage this finding to extend superresolution fluorescence microscopy to the nervous system of an intact, living, multicellular organism. We determine that the spectral range suitable for STED with GFP is broad, and employ light sources which are easy to implement both in pulsed- and continuous-wave (CW) STED imaging modalities.
STED microscopy uses a beam of light for disallowing the occupation of the marker's fluorescent state by stimulated emission. The role of this so-called STED beam is to transiently turn off the fluorescence capability of markers (7). This transient fluorescence silencing is induced throughout the region of excitation except at a subdiffraction-sized region of minimum intensity. The remaining region in which markers can be fluorescent, defining the resolution, is a function of the wavelength λ of the STED light applied; ISTED, the intensity of the STED light IS at which the fluorescence is reduced by half; and the numerical aperture (NA) of the lens, and is given by Δr ≈ λ/2NA√(1+ISTED/IS). The diffraction-limited excitation and STED beams are scanned through regions of interest and the markers in region Δr are registered to form the image. With fast beam scanning, STED is well suited for observing the processes of life on the nanoscale in real-time, in three dimensions, and in vivo.
The wavelength of the STED beam is usually tuned to the tail of the emission spectrum where the cross section for stimulated emission is appreciable, but red-shifted enough to avoid excitation of the fluorophore from the ground state. Also important, it has been shown that appreciable excited state absorption (ESA) at the STED wavelength has a negative impact on the stability of fluorophores for use with STED (8). To explore the optimal spectral region for STED with GFP with minimum ESA we performed pump-probe spectroscopy of enhanced GFP (eGFP). The ESA spectrum is shown in Fig. 1, and it can be seen that ESA is negligible from ∼500 nm to 580 nm and small in the spectral region between 580 nm and 650 nm, strongly suggesting that this wavelength range is optimal for STED.
Figure 1.
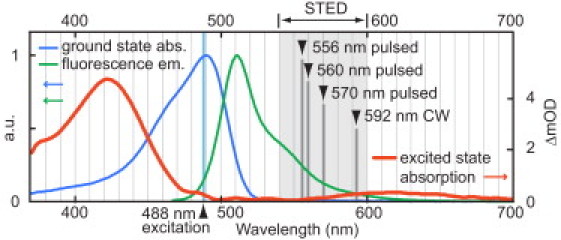
Excited state absorption (ESA) of eGFP and spectral rationale for employed stimulated emission depletion (STED) wavelengths. Normalized ground state absorption and fluorescence emission spectra are juxtaposed with the ESA spectrum measured 100 ps after excitation. STED wavelengths reported in this Letter are indicated. The unit on the ordinate at right is change in optical density ×10−3.
In contrast to the earlier work which imaged fixed GFP-labeled samples with STED using a complex light source consisting of an optical parametric oscillator tuned to 572 nm pumped by a titanium-sapphire laser operating at 80 MHz (1), we applied substantially simpler light sources. Three types of laser systems were demonstrated to be suitable using wavelengths ranging from 556 nm to 592 nm, either pulsed at 20 MHz with pulse durations of ∼1 ns or CW. We note the wide parameter range suitable for live-cell imaging of GFP-labeled structures that can be applied using STED, and add that the conditions demonstrated here are by no means exhaustive.
We screened the STED light sources by imaging live baker's yeast, Saccharomyces cerevisiae, expressing GFP on eisosomes, protein assemblies in the plasma membrane which determine the site of endocytosis. Typical STED and corresponding confocal images are shown in Fig. 2 A and show eisosome structures with full width at half-maximum (FWHM) values below 60 nm. Electron microscopy studies of eisosomes demonstrate a diameter of ∼50 nm (9), meaning that, conservatively, the obtained resolution is <60 nm. Images of comparable quality were obtained using STED light at 570 nm from a pulsed source based on stimulated Raman scattering, producing a comb spectrum conveniently located in the emission spectrum of GFP, similar to a previously reported source (10) but operating at 20 MHz, and a CW laser source operating at 592 nm (MPB Photonics, Pointe-Claire, Canada).
Figure 2.
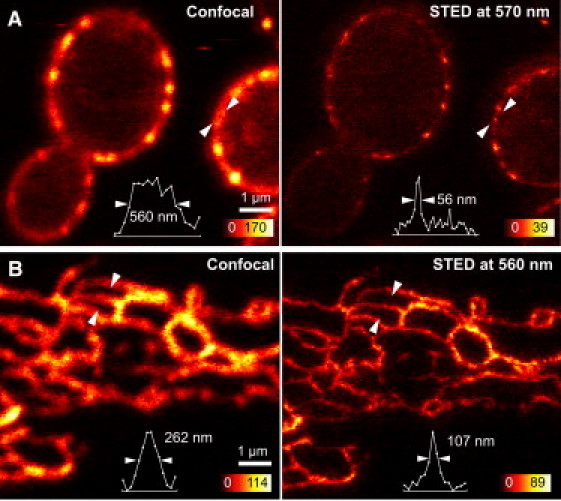
STED images of single cells. (A) STED image (right) of living yeast cells expressing GFP (S65T) on eisosomes, with confocal image for comparison (left). Line profiles (below) were taken of the image features (open arrows); values indicated are FWHM. Pulsed STED light at 570 nm was used, with applied excitation and STED powers of 6 μW and 8 mW, respectively. (B) Living Vero cells expressing eGFP in the endoplasmic reticulum measured using a STED wavelength of 560 nm. The original 20 × 20 μm STED recording took ∼8 s to acquire. All data shown is raw.
Next we imaged living mammalian cells. STED images were obtained using a dedicated pulsed source operating at 560 nm and producing 900-ps pulses at 20 MHz (MPB Photonics), 570-nm light from the stimulated Raman scattering source, and 592 nm from the CW laser. Representative data are shown in Fig. 2 B, showing the endoplasmic reticulum of a living Vero cell taken using 560-nm STED light; the original 20 × 20 μm STED recording took ∼8 s to acquire. We were able to record >20 STED images in succession of endoplasmic reticulum movement, imaging continuously for 13 min to record morphological dynamics. A video is shown in Movie S1 in the Supporting Material.
After establishing the robustness of GFP for STED imaging of live samples, we performed imaging on the roundworm Caenorhabditis elegans to demonstrate nanoscale imaging within a living multicellular organism. C. elegans lends itself as an in vivo model system because its small size and transparency render all points within the organism accessible under an optical microscope. Its nervous system is well characterized, consisting of 302 neurons that form ∼7000 chemical synapses and 600 gap junctions, and is the only animal with an available comprehensive neural connectivity map (11).
We used a C. elegans strain expressing cytoplasmic GFP cell- specifically in a bilateral pair of serotonergic neurons called NSMs. The NSM neurons extend axon arbors off the main axon shaft. Similar to serotonergic arbors in vertebrates, the NSM axon arbors display plastic autoregulation at an ultrastructural level, which is why they have been used as a model to understand the biomedically important question of serotonergic feedback autoregulation of neuronal morphology (12). These NSM axon arbors are complex, small structures, and aspects of their morphology and development cannot be discerned using conventional microscopy. Fig. 3 A shows the location of the NSM neurons within the animal with a superposition of a transmission-illumination image and an epifluorescent image. A STED recording of the arbor structures is shown in Fig. 3 B with confocal microscopy comparisons in areas of interest (insets). STED reveals the lack of connectivity between adjacent arbors and morphological features not distinguishable by confocal microscopy. Additionally, we took image stacks of the NSM neurons, recording their complex three-dimensional morphology, as shown in Fig. S1 in the Supporting Material.
Figure 3.
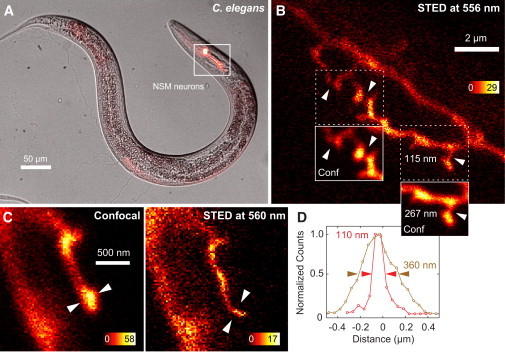
STED imaging of neurons in live C. elegans. (A) Combined transmission and fluorescence images showing the location of NSM neurons. (B) A STED image of the branched areas of the ventral nerve process of the NSM neurons. (Insets) Confocal comparisons of areas of interest; STED reveals a lack of connectivity not seen in the confocal image (above) and a subdiffraction spine-neck diameter (below) with FWHM values measured at the location indicated by arrows. (C) Confocal and STED images taken using a dedicated 560-nm source, with FWHM values given for the location indicated by arrows. Employed STED powers for panels B and C were ∼4 mW. (D) Line profiles and FWHM values for data shown in panel C.
Adding GFP as a fluorescent label for nanoscale imaging in living cells and organisms is a valuable extension of the subdiffraction resolution tool kit, opening up the possibility to leverage the numerous readily available samples expressing GFP to pursue biological questions which can be answered by observation at the nanoscale. Exploiting recent advancements in laser technology, employing GFP with STED is no more challenging than using other fluorophores. The imaging parameters demonstrated here are not exhaustive and further technological developments will provide additional options and improvements to the STED imaging presented here. Further possibilities for exploiting GFP for STED imaging are numerous. Facilitated by the broad spectral range suitable for STED with GFP, employing additional fluorescent proteins for colocalization imaging is within reach. Thus our results have prepared the ground for an important step in quantitative systems biology: nanoscale colocalization of proteins of small animals in vivo.
Acknowledgments
We thank A. Schönle for development of the Imspector data acquisition software, S. Berning and Peng Xi for help implementing beam scanning, and S. Jakobs for eGFP samples. We thank R. Lindquist, J. Moresco, and M. Koelle (Yale) for C. elegans strain LX837.
D.A.C.-R. and J.C.N. were funded by grant Nos. R00 NS057931 and T32-NS41228. S.W.H. acknowledges a Deutsche Gemeinschaft Leibniz prize.
Supporting Material
References and Footnotes
- 1.Willig K.I., Kellner R.R., Hell S.W. Nanoscale resolution in GFP-based microscopy. Nat. Methods. 2006;3:721–723. doi: 10.1038/nmeth922. [DOI] [PubMed] [Google Scholar]
- 2.Kner P., Chhun B.B., Gustafsson M.G. Super-resolution video microscopy of live cells by structured illumination. Nat. Methods. 2009;6:339–342. doi: 10.1038/nmeth.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Q., Wu S.S.H., Chou K.C. Subdiffraction-limit two-photon fluorescence microscopy for GFP-tagged cell imaging. Biophys. J. 2009;97:3224–3228. doi: 10.1016/j.bpj.2009.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Betzig E., Patterson G.H., Hess H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 5.Hess S.T., Girirajan T.P.K., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rust M.J., Bates M., Zhuang X.W. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hell S.W., Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- 8.Hotta J., Fron E., Hofkens J. Spectroscopic rationale for efficient stimulated-emission depletion microscopy fluorophores. J. Am. Chem. Soc. 2010;132:5021–5023. doi: 10.1021/ja100079w. [DOI] [PubMed] [Google Scholar]
- 9.Strádalová V., Stahlschmidt W., Malinsky J. Furrow-like invaginations of the yeast plasma membrane correspond to membrane compartment of Can1. J. Cell Sci. 2009;122:2887–2894. doi: 10.1242/jcs.051227. [DOI] [PubMed] [Google Scholar]
- 10.Rankin B.R., Hell S.W. STED microscopy with a MHz pulsed stimulated-Raman-scattering source. Opt. Express. 2009;17:15679–15684. doi: 10.1364/OE.17.015679. [DOI] [PubMed] [Google Scholar]
- 11.White J.G., Southgate E., Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. Roy. Soc. (Lond.). B Biol. Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 12.Axäng C., Rauthan M., Pilon M. Developmental genetics of the C. elegans pharyngeal neurons NSML and NSMR. BMC Dev. Biol. 2008;8:38. doi: 10.1186/1471-213X-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.