

Abstract
We consider the problem of aligning two metabolic pathways. Unlike traditional approaches, we do not restrict the alignment to one-to-one mappings between the molecules (nodes) of the input pathways (graphs). We follow the observation that, in nature, different organisms can perform the same or similar functions through different sets of reactions and molecules. The number and the topology of the molecules in these alternative sets often vary from one organism to another. With the motivation that an accurate biological alignment should be able to reveal these functionally similar molecule sets across different species, we develop an algorithm that first measures the similarities between different nodes using a mixture of homology and topological similarity. We combine the two metrics by employing an eigenvalue formulation. We then search for an alignment between the two input pathways that maximizes a similarity score, evaluated as the sum of the similarities of the mapped subnetworks of size at most a given integer k, and also does not contain any conflicting mappings. Here we prove that this maximization is NP-hard by a reduction from the maximum weight independent set (MWIS) problem. We then convert our problem to an instance of MWIS and use an efficient vertex-selection strategy to extract the mappings that constitute our alignment. We name our algorithm SubMAP (Subnetwork Mappings in Alignment of Pathways). We evaluate its accuracy and performance on real datasets. Our empirical results demonstrate that SubMAP can identify biologically relevant mappings that are missed by traditional alignment methods. Furthermore, we observe that SubMAP is scalable for metabolic pathways of arbitrary topology, including searching for a query pathway of size 70 against the complete KEGG database of 1,842 pathways. Implementation in C++ is available at http://bioinformatics.cise.ufl.edu/SubMAP.html.
Key words: alternative reaction sets, maximum weight independent set, metabolic pathway alignment, one-to-many mappings, subnetwork mappings
1. Introduction
Biological networks show how different molecules interact with each other to perform vital functions. In the literature, the terms network and pathway are used interchangeably. Metabolic pathways, an important class of biological networks, represent how different compounds are transformed through various reactions. Analyzing these pathways is essential in understanding the machinery of living organisms.
The efforts on analyzing pathways can be classified into two types. The first type considers one pathway at a time and explores its important properties such as its robustness (Edwards and Palsson, 2000), steady states (Devloo et al., 2003; Garg et al., 2007; Ay et al., 2009b), modular structure (Lu et al., 2006; Schuster et al., 2002; Ay et al., 2010), network motifs (Milo et al., 298; Wernicke and Rasche, 2006; Grochow and Kellis, 2007), as well as its representation (Michal, 1998; Babur et al., 2010). The second type is the comparative approach which considers multiple pathways to identify their frequent subgraphs (Koyuturk et al., 2004; Qian and Yoon, 2009) and their alignments (Singh et al., 2008, 2007; Liao et al., 2009; Kalaev et al., 2008; Sharan et al., 2005; Kalaev et al., 2009; Dost et al., 2007; Flannick et al., 2006; Koyuturk et al., 2005; Pinter et al., 2005; Berg and Lassig, 2004; Dandekar et al., 1999; Tohsato and Nishimura, 2008; Tohsato et al., 2000; Cheng et al., 2009; Ay et al., 2008, 2009a). Alignment is a fundamental type of comparative analysis that aims to identify similar parts between pathways. For metabolic pathways, these similarities provide insights for drug target identification (Sridhar et al., 2007; Watanabe et al., 2007), metabolic reconstruction of newly sequenced genome (Francke et al., 2005), phylogenic reconstruction (Clemente et al., 2005; Heymans and Singh, 2003), and identification of enzyme clusters and missing enzymes (Ogata et al., 2000; Green and Karp, 2004).
In the literature, performing an alignment is often considered as finding one-to-one mappings between the molecules of two pathways. In this case, the global/local pathway alignment problems are GI (Graph isomorphism)/NP complete as the graph/subgraph isomorphism problems can be reduced to them in polynomial time (Damaschke, 1991). A number of studies have been done to systematically align different types of biological networks. For alignment of protein-protein interaction (PPI) networks, a number of methods such as IsoRank (Singh et al., 2008, 2007), QNet (Dost et al., 2007), Greamlin (Flannick et al., 2006), and that of Koyuturk et al. (2005) have been successfully applied to identify conserved parts among PPI networks of different organisms. For metabolic pathways, Pinter et al. (2005) devised an algorithm that aligns query pathways with specific topologies by using a graph theoretic approach. Tohsato and colleagues proposed two algorithms for metabolic pathway alignment, one relying solely on Enzyme Commission (EC) (Webb, 1992) numbers of enzymes and the other considering only the chemical structures of compounds of the query pathways (Tohsato and Nishimura, 2008; Tohsato et al., 2000). Cheng et al. (2009) developed a tool, MetNetAligner, for metabolic pathway alignment that allows a certain number of insertions and deletions of enzymes. However, most of these methods limit the query pathways to certain topologies, such as trees, non-branching paths or limited cycles. In order to alleviate this limitation, they either change the topology of the networks (e.g., breaking the cycles) or only consider queries up to a certain size which degrades their accuracy and applicability to complex networks. A number of these methods avoid the restrictions on queries and use heuristic algorithms to scale for real size problems. For instance, IsoRank (Singh et al., 2008, 2007) uses an iterative framework that combines topological features of the networks and the sequence similarity of proteins to do the global alignment of two PPI networks. They map the alignment problem to graph isomorphism and align genome-wide PPI networks of well-studied organisms. We extended this framework in our earlier work to align metabolic pathways while considering the interactions between non-homogenous entities such as reactions, enzymes, and compounds (Ay et al., 2008, 2009a). These two methods showed that formulating the similarity score in this manner and not restricting the query topologies improve accuracy and applicability of the alignment algorithm.
However, all the methods discussed above limit the possible molecule mappings to only one-to-one mappings. As also pointed out by Deutscher et al. (2008) considering each molecule one by one fails to reveal its function(s) in complex pathways. This restriction prevents all the above methods from identifying biologically relevant mappings when different organisms perform the same function through a varying number of steps. As an example, there are alternative paths for LL-2,6-diaminopimelate production in different organisms (Watanabe et al., 2007; McCoy et al., 2006). LL-2,6-diaminopimelate is a key intermediate compound since it lies at the intersection of different paths on the synthesis of L-lysine. Figure 1 illustrates two paths both producing LL-2,6-diaminopimelate starting from 2,3,4,5-tetrahydrodipicolinate. The upper path represents the shortcut used by plants and Chlamydia to synthesize L-lysine. This shortcut is not an option, for example, for E. coli or H. sapiens, due to the lack of the gene encoding LL-DAP aminotransferase (2.6.1.83). E. coli and H. sapiens have to use a three-step process shown with the gray path in Figure 1 to do this transformation. Thus, a meaningful alignment should map the two paths when the lysine biosynthesis pathways of human and a plant are aligned. However, since these two paths have a different number of reactions, traditional alignment methods that are limited to one-to-one mappings fail to identify this mapping.
FIG. 1.

A portion of lysine biosynthesis pathway. Each reaction is represented by the Enzyme Commission (EC) number of the enzyme that catalyzes it. Circles represent compounds (intermediate compounds are not shown). E. coli and H. sapiens (human) use the path colored with gray with three reactions, whereas plants and Chlamydia achieve this transformation directly through the path with a single reaction shown in white.
For PPI networks, the idea of extending alignments beyond one-to-one mappings and allowing multiple alignments has been proposed by Liao et al. (2009) (IsoRankN), by Sharan et al. (2005), and by Kalaev et al. (2009) (NetworkBLAST-M). IsoRankN creates a functional similarity graph from input networks and searches for near cliques of highly similar proteins using spectral clustering on the induced graph of pairwise alignment scores. NetworkBLAST-M constructs a network alignment graph from queries and uses a heuristic seed-extension method to search for conserved paths or cliques. To the best of our knowledge, no current method explicitly allows mappings beyond one-to-one for metabolic pathways.
Our aim in this article is to design an algorithm that can accurately identify such biologically relevant mappings by allowing one-to-many mappings of molecules in metabolic pathways. Note that, in Figure 1, both reaction sets form linear paths. It is possible to have reaction sets with different topologies performing a certain function (see Fig. 5 below). Therefore, we use the term subnetwork to denote all types of topologies. Also, since we only consider the sets of reactions that are connected, we will simply use the term subnetwork instead of connected subnetwork.
FIG. 5.

Visual representations of subnetwork mappings reported in Table 2. (a–j) Correspond to rows 1–10 of Table 2. Enzymes are represented by their Enzyme Commission (EC) numbers (Webb, 1992).
1.1. Problem definition
We consider the problem of aligning two metabolic pathways. Unlike traditional alignment approaches, we allow aligning a molecule of one pathway to a connected subnetwork of the other. More formally, let 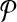 and
and 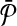 be two query pathways, and k be a positive integer. We want to find the mapping between molecules of
be two query pathways, and k be a positive integer. We want to find the mapping between molecules of 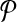 and
and 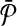 with the largest alignment score such that:
with the largest alignment score such that:
Each molecule in
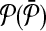 can map to a subnetwork of
can map to a subnetwork of 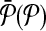 with at most k molecules.
with at most k molecules.Each molecule can appear in at most one mapping.
The first condition above allows one-to-many mappings. The reason for having one-to-many mappings in our alignment is not only that they capture functionally similar parts, but also they enable us to construct many-to-many mappings of arbitrary sizes. Identifying many-to-many mappings of molecule sets of different sizes is essential and is not possible with only one-to-one mappings as their combinations enforce both sides of a many-to-many mapping to be of the same size. The second condition enforces consistency. That is, if a molecule is already mapped alone or as a part of a subnetwork, it cannot map to another molecule. We elaborate on consistency and the problem definition later in Section 2. Note that, allowing one-to-many mappings in an alignment introduces new computational challenges (e.g., exponential increase in the problem size, conflicting mappings) that cannot be addressed using existing methods and hence novel methods are needed to tackle this problem.
1.2. Contributions
In this article, we propose a novel algorithm that finds subnetwork mappings in alignment of pathways. SubMAP accounts for both the effect of pairwise similarities (homology) and the organization of pathways (topology). This combination is motivated by its successful applications on network alignment by Singh et al. (2007, 2008) and Ay et al. (2008, 2009a). However, allowing one-to-many mappings makes it impossible to trivially extend these methods to our problem. Here, we describe our method that addresses this challenge. Similar to IsoRank and our earlier work, we first formulate the alignment as an eigenvalue problem and solve it using an iterative technique called power method. The result of the power method provides us an eigenvector that defines a weighted bipartite graph where each node corresponds to a molecule or a subnetwork. The edges are only between two nodes from different pathways and their weights define the similarity of these nodes. Unlike the case of only one-to-one molecule mappings, the nodes on the same side of the bipartite graph can be intersecting as the same molecule can appear in more than one subnetwork. If two such intersecting nodes on one side are mapped to two different nodes of the other side, they create inconsistent mappings for the elements of the intersection. We term such pairs of mappings as conflicting mappings. Our aim is to extract a set of mappings that has no conflicts and maximizes the sum of the similarity scores of the mappings. We prove that this maximization is NP-hard (see Theorem 1). We then construct a vertex weighted conflict graph with nodes representing a mapping of two subnetworks, one from each pathway, and edges representing a conflict between two mappings. The weights of the nodes in this graph are the edge weights of the bipartite graph from the earlier step. At this point, our alignment problem is equivalent to finding a maximum weight independent set (MWIS) of the conflict graph. To extract an independent set from the conflict graph, we use a vertex-selection strategy proposed for MWIS problem. We report the mappings that correspond to the selected set of nodes from the conflict graph as the alignment of the query pathways. Our experiments on the metabolic pathways from KEGG (Ogata et al., 1999) database suggest that SubMAP finds biologically meaningful alignments efficiently. Also, SubMAP is scalable for subnetwork sizes up to three or four as it can align two real size metabolic pathways in about a minute.
The rest of the article is organized as follows. Section 2 describes our algorithm. Section 3 presents experimental results. Section 4 concludes the paper.
Implementation in C++ is available at http://bioinformatics.cise.ufl.edu/SubMAP.html.
2. Our Algorithm: SubMAP
In this section, we present our algorithm for pairwise metabolic pathway alignment that allows one-to-many molecule mappings. We begin by introducing some notation that we use throughout this section. Then, we formally state the problem and describe the SubMAP algorithm in detail (Table 1).
Table 1.
Commonly Used Symbols in this Article
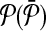 |
Query metabolic pathways |
| k | Parameter for the largest subnetwork size |
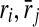 |
Reactions of query pathways |
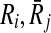 |
Subnetworks of query pathways |
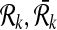 |
Sets of all subnetworks with size at most k |
| n, m | Numbers of the reactions in query pathways |
| Nk, Mk | Numbers of all subnetworks of size at most k |
| φk | Set of all possible one-to-many mappings for a given k |
| ∣φk∣ | Number of all possible one-to-many mappings for a given k |
| S | Support matrix |
| H | Homological similarity vector |
| Gc | Conflict graph |
| α | Parameter adjusting relative weights of homology and topology |
Let, 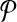 be a pathway which is represented by a directed unweighted graph G = (V, E). Here, we only use the reactions of the pathway in graph representation. Hence, the vertex set
be a pathway which is represented by a directed unweighted graph G = (V, E). Here, we only use the reactions of the pathway in graph representation. Hence, the vertex set 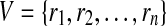 is the set of all reactions of
is the set of all reactions of 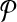 . We include a directed edge eij from ri to rj in E if and only if at least one output compound of ri is an input compound of rj. We call ri a backward neighbor of rj and rj a forward neighbor of ri if
. We include a directed edge eij from ri to rj in E if and only if at least one output compound of ri is an input compound of rj. We call ri a backward neighbor of rj and rj a forward neighbor of ri if 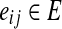 . Note that reactions can be reversible (bi-directional) and hence both eij and eji can exist.
. Note that reactions can be reversible (bi-directional) and hence both eij and eji can exist.
A subnetwork of a pathway is a subset of its reaction set such that the induced undirected graph of the elements of this subset forms a connected graph. Let Ri ⊆ V be such a subnetwork of 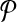 . We define
. We define 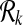 , the set of all subnetworks of
, the set of all subnetworks of 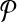 that have at most k reactions, as
that have at most k reactions, as  where ∣Ri∣ ≤ k for all
where ∣Ri∣ ≤ k for all  . Here, ∣Ri∣ denotes the cardinality of the reaction set Ri. Using this notation, we define a binary relation that maps a reaction of a query pathway to a subnetwork of the other as follows:
. Here, ∣Ri∣ denotes the cardinality of the reaction set Ri. Using this notation, we define a binary relation that maps a reaction of a query pathway to a subnetwork of the other as follows:
Definition 1
Let  and
and 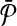 be two pathways and k be a positive integer. Also, let
be two pathways and k be a positive integer. Also, let 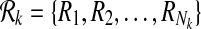 and
and 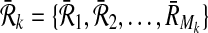 be the sets of subnetworks with size at most k of
be the sets of subnetworks with size at most k of  and
and 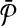 respectively. We define a binary relation φ between
respectively. We define a binary relation φ between 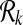 and
and 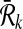 that allows one-to-many reaction mappings as
that allows one-to-many reaction mappings as 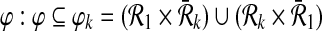 .
.
Let us denote the number of reactions of 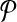 and
and 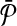 with n and m respectively. The number of all possible one-to-many mappings between
with n and m respectively. The number of all possible one-to-many mappings between 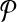 and
and 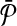 is:
is:
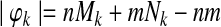 |
(1) |
The alignment of  and
and  is a binary relation φ that is a subset of all these possible mappings and satisfies certain criteria that we describe next.
is a binary relation φ that is a subset of all these possible mappings and satisfies certain criteria that we describe next.
Recall that for a mapping 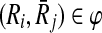 one of the Ri or
one of the Ri or 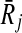 can contain more than one reaction. Reporting this mapping as a part of our alignment implies that all the reactions of the subnetwork with multiple reactions are aligned to a single reaction of the other. To have a consistent alignment none of the reactions of these subnetworks can be included in any other mapping. Next, we formally define the term conflict to characterize this property.
can contain more than one reaction. Reporting this mapping as a part of our alignment implies that all the reactions of the subnetwork with multiple reactions are aligned to a single reaction of the other. To have a consistent alignment none of the reactions of these subnetworks can be included in any other mapping. Next, we formally define the term conflict to characterize this property.
Definition 2
Let φ be a binary relation and 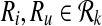 and
and 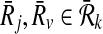 . The distinct pairs
. The distinct pairs 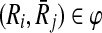 and
and 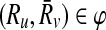 conflict if and only if
conflict if and only if 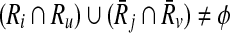 .
.
Conflicts can cause inconsistencies about which reaction subset of one pathway should be aligned to the one of the other pathway. If φ has a conflicting pair of elements, we say φ is inconsistent. Since this is not a desirable property, we limit our alignment to the consistent relations only.
In addition to discarding the conflicting mappings, we also need to use a meaningful scoring score in order to gather biologically relevant alignments. One standard scoring scheme for this purpose incorporates the homology of the aligned molecules with their topologies (Singh et al., 2007, 2008; Ay et al., 2008, 2009a). Here, we generalize this scheme to one-to-many mappings. We will elaborate on this similarity score later in Section 2.4. Next, we state our problem formally.
Problem formulation
Given k and two pathways 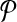 and
and 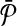 , let
, let 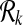 and
and 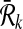 be the sets of subnetworks with size at most k of
be the sets of subnetworks with size at most k of 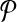 and
and 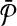 respectively. We want to find the consistent binary relation
respectively. We want to find the consistent binary relation 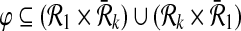 that maximizes the summation of the similarity scores of the aligned subnetworks.
that maximizes the summation of the similarity scores of the aligned subnetworks.
In the following, we present our algorithm SubMAP. Section 2.1 explains the enumeration of the subnetworks of query pathways. Section 2.2 and 2.3 discuss homological and topological similarities, respectively. Section 2.4 describes the eigenvalue formulation that combines these similarities and explains the extraction of subnetwork mappings.
2.1. Enumeration of connected subnetworks
The first step of SubMAP is to create the sets of all connected subnetworks of size at most k for each query pathway. Here, we describe the enumeration process for a single query pathway. Let G = (V, E) represent a pathway and k be a positive integer. We construct the set of subnetworks 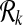 as follows. For k = 0,
as follows. For k = 0, 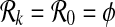 and for k = 1,
and for k = 1, 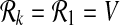 . For k > 1 we define
. For k > 1 we define 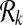 recursively by using
recursively by using 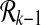 . At each recursive step we check for each reaction in V if it can be added to the already enumerated subnetworks of size k − 1 to create a new connected subnetwork of size k. This way the kth recursive step takes
. At each recursive step we check for each reaction in V if it can be added to the already enumerated subnetworks of size k − 1 to create a new connected subnetwork of size k. This way the kth recursive step takes 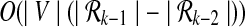 time.
time.
The size of the set 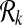 can be exponential in k when G is dense. However, metabolic pathways are usually sparse (on the average there are 2.5 forward neighbors per reaction). We observe that the number of subnetworks of the metabolic pathways in our dataset for k = 3 is around 5∣V∣ and for k = 4 is 10∣V∣ on the average. In Section 3.2, we provide a detailed discussion of how
can be exponential in k when G is dense. However, metabolic pathways are usually sparse (on the average there are 2.5 forward neighbors per reaction). We observe that the number of subnetworks of the metabolic pathways in our dataset for k = 3 is around 5∣V∣ and for k = 4 is 10∣V∣ on the average. In Section 3.2, we provide a detailed discussion of how 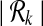 changes with different pathway sizes and different k values.
changes with different pathway sizes and different k values.
2.2. Homological similarity of subnetworks
Recall that the relation φ maps a reaction to a subnetwork that can contain multiple reactions. This necessitates computing the similarity between reaction sets. Since reactions are defined by their input and output compounds (i.e., substrates and products) and the enzymes that catalyze them, we measure the homological similarity between reactions using the similarities of these components.
In the literature, there are alternative pairwise similarity scores for compounds, enzymes and reactions. Particularly, two well known measures are information content similarity for enzyme pairs (Pinter et al., 2005) and SIMCOMP (Hattori et al., 2003) for compound pairs. We denote these measures by SimE and SimC, respectively. We defer the readers to Ay et al. (Ay et al., 2009a) for details on computing these similarities. Here, we utilize these similarity measures to compute the homological similarity between two reaction sets. To calculate this, we first construct three sets for both reaction sets. These are the union of (1) the input compounds (Ii), (2) the output compounds (Oi), and (3) the enzymes (Ei) of the reactions in each subnetwork Ri. For instance, in Figure 1 if we take the upper path as the subnetwork Ri, then Ei = {2.3.1.117, 2.6.1.17, 3.5.1.8}.
Next, we compute the similarity of each of these three set pairs and combine them using weights (i.e., non-negative real numbers) to calculate the homological similarity of the two reaction sets. Let W(A, B, SimX) denote the similarity between two sets A and B with respect to the similarity score SimX, where W is calculated as the sum of the similarities of the pairs returned by their maximum weight bipartite matching (MWBM). Also, let γe, γi, γo denote the relative weights of the similarities of enzymes, input and output compounds respectively with the constraint γe + γi + γo = 1. We define the similarity of the reaction sets Ri and 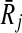 as
as
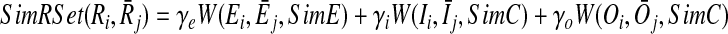 |
(2) |
In this article, we use γi = γo = 0.3 and γe = 0.4 as they provide a good balance between enzymes and compounds. However, in general we prefer to leave the choice of these parameters to the user as it provides flexibility to our method and allows it to be used in different scenarios. For instance, setting γE = 1 and 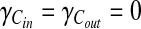 means that the reaction similarities are determined by enzymes and the alignment will be enzyme driven.
means that the reaction similarities are determined by enzymes and the alignment will be enzyme driven.
We calculate SimRSet for all possible one-to-many mappings between the subnetworks of two pathways. Therefore, we do this calculation ∣φk∣ times in total. This way, we assess the homological similarities between all possible subnetwork mappings. Even though this scoring can be considered a good measure of similarity, relying solely on this score ignores the topology similarity which we explain next.
2.3. Topological similarity of subnetworks
We follow the intuition of IsoRank that if the subnetwork Ri is mapped to 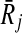 , then their neighbors in the corresponding pathways should also be similar. With this motivation, we utilize the topological similarity to favor mappings of subnetworks that induce similar topologies. We do this by extending the formulation of IsoRank and our earlier work to account for subnetwork mappings. We first expand the neighborhood definition of reactions to reaction subnetworks. Then, we generalize the notion of support to include subnetwork mappings. Using these definitions, we formally describe how we calculate the support matrix and show it on an illustrative example.
, then their neighbors in the corresponding pathways should also be similar. With this motivation, we utilize the topological similarity to favor mappings of subnetworks that induce similar topologies. We do this by extending the formulation of IsoRank and our earlier work to account for subnetwork mappings. We first expand the neighborhood definition of reactions to reaction subnetworks. Then, we generalize the notion of support to include subnetwork mappings. Using these definitions, we formally describe how we calculate the support matrix and show it on an illustrative example.
Definition 3
Let 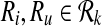 . Then, Ru is a forward neighbor of Ri
. Then, Ru is a forward neighbor of Ri 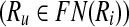 if and only if there exists
if and only if there exists 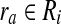 and
and 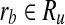 such that rb is a forward neighbor of ra or
such that rb is a forward neighbor of ra or 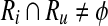 . Ri is a backward neighbor of
. Ri is a backward neighbor of 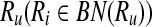 if and only if Ru is a forward neighbor of Ri.
if and only if Ru is a forward neighbor of Ri.
Definition 4
Let Ri, 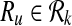 and
and 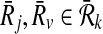 . The mapping (
. The mapping (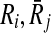 ) supports the mapping (
) supports the mapping (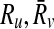 ) if and only if both
) if and only if both 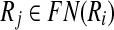 and
and 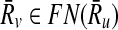 or both
or both 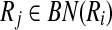 and
and 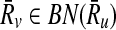 .
.
Definition 5
Let  , be two pathways and
, be two pathways and 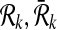 be the sets of their subnetworks that have at most k reactions. The support matrix S is a ∣φk∣ × ∣φk∣ matrix with each entry S[i,j][u,v] identifying the fraction of the total support provided by (
be the sets of their subnetworks that have at most k reactions. The support matrix S is a ∣φk∣ × ∣φk∣ matrix with each entry S[i,j][u,v] identifying the fraction of the total support provided by (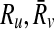 ) mapping to (
) mapping to (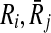 ) mapping. Let
) mapping. Let 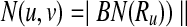
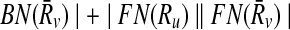 denote the number of all possible mappings between backward neighbors of Ru and
denote the number of all possible mappings between backward neighbors of Ru and 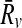 plus the ones between their forward neighbors. Then, each entry of S is computed as:
plus the ones between their forward neighbors. Then, each entry of S is computed as:
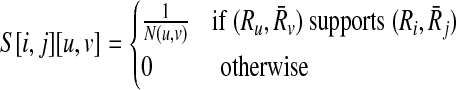 |
(3) |
Definition 4 states that the mapping of Ri to  favors all possible mappings of forward (backward) neighbors of Ri to those of
favors all possible mappings of forward (backward) neighbors of Ri to those of 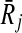 . To see it on an example, let us consider the case when k = 1 and focus on the mapping (
. To see it on an example, let us consider the case when k = 1 and focus on the mapping (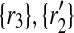 ) in Figure 2. We see that
) in Figure 2. We see that 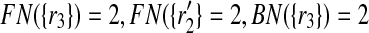 and
and 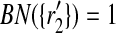 . Then, by Definition 5 we distribute the support of the mapping (
. Then, by Definition 5 we distribute the support of the mapping (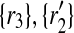 ) to 2 × 2 + 2 × 1 = 6 other mappings by placing
) to 2 × 2 + 2 × 1 = 6 other mappings by placing 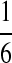 in the corresponding entries of S. Namely, these mappings are (
in the corresponding entries of S. Namely, these mappings are (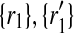 ), (
), (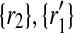 ), (
), (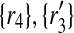 ), (
), (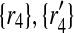 ), (
), (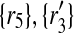 ) and (
) and (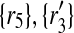 ).
).
FIG. 2.

Reaction-based representation of two hypothetical metabolic pathways (a,b). An arrow from ri to rj represents that an output of ri is used as an input in rj.
There can be cases when one mapping does not provide support to any others. In such cases, we simply distribute its support equally to all possible mappings (∣φk∣). Notice that, by construction, the entries in each column of S sums up to 1. This is important as it ensures the stability and convergence of our algorithm as we explain in Section 2.4. Interested reader can find a detailed description of the properties of support matrix in a previous work of ours (Ay et al., 2009a).
Before moving into describing how we use the support matrix, it is important to explain how we create it in our implementation. A trivial but costly way of doing this is to check each mapping against all the others to calculate the support values. However, such an exhaustive strategy will require computing a huge matrix S of size ∣φk∣ × ∣φk∣. Since the creation of S will incur prohibitive computational costs, we do not construct this matrix literally. Instead, for each mapping Ri, 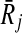 , we take the sets FN(Ri),
, we take the sets FN(Ri), 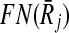 and BN(Ri),
and BN(Ri), 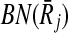 to generate only the pairs supported by Ri,
to generate only the pairs supported by Ri, 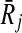 . In other words, we use the sparse matrix form of the support matrix S.
. In other words, we use the sparse matrix form of the support matrix S.
2.4. Aligning two pathways
We discussed what homology and topology similarities are and how we calculate them. In the following, we first describe how we combine these two similarities to get a better similarity measure for subnetworks. Then, we explain the heuristic method we use to extract the set of subnetwork mappings (i.e., the alignment) that maximizes the sum of the similarities of the mapped pairs.
2.4.1. Combining homology and topology
Both the homological similarities of subnetworks and their topological organization provide us significant information for the alignment of metabolic pathways. A good alignment algorithm needs to combine these two factors in an efficient and accurate way. Recently, the authors of IsoRank (Singh et al., 2007, 2008) have described an iterative technique named power method that achieves this combination. Here, we describe how we use power method to obtain this combination for alignment of metabolic pathways with subnetwork mappings.
Let k be a given parameter and 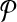 ,
, 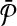 be two pathways with connected subnetwork sets
be two pathways with connected subnetwork sets 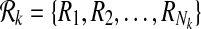 and
and 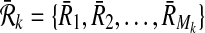 respectively. We represent the homological similarity of all subnetwork pairs by the column vector H of size ∣φk∣. Each entry of H denotes the homological similarity between two subnetworks one from each pathway.
respectively. We represent the homological similarity of all subnetwork pairs by the column vector H of size ∣φk∣. Each entry of H denotes the homological similarity between two subnetworks one from each pathway.
Let S be the ∣φk∣ × ∣φk∣ support matrix as described in Section 2.3. Given a parameter 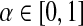 to adjust the relative weights of homology and topology, we combine them through power method iterations as follows:
to adjust the relative weights of homology and topology, we combine them through power method iterations as follows:
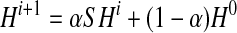 |
(4) |
In this equation, 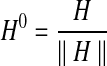 . We iterate this equation till Hi+1 = Hi (i.e., it converges). When S and H0e are both column stochastic matrices (i.e., sum of the entries of each column is one and all entries are non-negative) this system converges to the principal eigenvector of the matrix αS +(1 − α)H0e where e is a row vector of size ∣φk∣ with all entries equal to 1 (for proof of existence and uniqueness, see Ay et al., 2009a). S is column stochastic by our construction since it has non-negative entries with each column adding up to one. We ensure the column stochasticity of H0e by normalizing H0. The resulting vector Hi gives us a combination of homological and topological similarities for each possible mapping (i.e., each entry corresponds to a mapping score). For the choice of weight parameter α we follow the literature and use α = 0.6. This value has been shown to provide a good combination of the two similarities both for PPI networks (Singh et al., 2007, 2008) and metabolic pathways (Ay et al., 2008, 2009a).
. We iterate this equation till Hi+1 = Hi (i.e., it converges). When S and H0e are both column stochastic matrices (i.e., sum of the entries of each column is one and all entries are non-negative) this system converges to the principal eigenvector of the matrix αS +(1 − α)H0e where e is a row vector of size ∣φk∣ with all entries equal to 1 (for proof of existence and uniqueness, see Ay et al., 2009a). S is column stochastic by our construction since it has non-negative entries with each column adding up to one. We ensure the column stochasticity of H0e by normalizing H0. The resulting vector Hi gives us a combination of homological and topological similarities for each possible mapping (i.e., each entry corresponds to a mapping score). For the choice of weight parameter α we follow the literature and use α = 0.6. This value has been shown to provide a good combination of the two similarities both for PPI networks (Singh et al., 2007, 2008) and metabolic pathways (Ay et al., 2008, 2009a).
2.4.2. Extracting subnetwork mappings
Recall that our aim is to find an alignment that maximizes the summation of the similarity scores defined by Hi while preserving the consistency between different mappings. In case of pairwise alignments with only one-to-one mappings, maximum weight bipartite matching provides an optimal and an efficient way to extract the alignment (Singh et al., 2007; Ay et al., 2009a). However, efficient heuristics are needed for this extraction phase when multiple networks are aligned or one-to-many mappings are allowed. For aligning multiple networks, IsoRank (Singh et al., 2008) used a greedy strategy to extract a maximum weight k-partite matching and IsoRankN (Liao et al., 2009) employed spectral clustering to find the alignment clusters.
Here, we first show that finding an optimal alignment while allowing one-to-many mappings is NP-hard by a reduction from the MWIS problem in bounded degree graphs. MWIS problem, even for graphs with largest degree 3, is NP-hard (Lovasz, 1994) and there is no constant factor approximation to the optimal solution in polynomial time (Austrin et al., 2009; Berman and Karpinski, 1999). We then describe how we construct a conflict graph from our alignment problem and apply a greedy vertex-selection strategy to extract a MWIS of the conflict graph which gives us the set of mappings that generates the alignment.
Theorem 1
(Finding the best alignment is NP-hard). Let 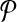 and
and 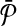 be two pathways with reaction sets
be two pathways with reaction sets 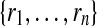 and
and 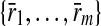 respectively. Let
respectively. Let 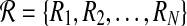 and
and 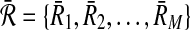 be all possible reaction subsets of
be all possible reaction subsets of  and
and  with size at most a given positive integer k. Also, let
with size at most a given positive integer k. Also, let 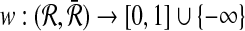 be a similarity function defining the score for each mapping and the conflicts between mappings be defined according to Definition 2. Then, finding a set of mappings (i.e., alignment) that maximizes the sum of mapping scores (i.e., alignment score) and has no conflicting pairs is NP-hard.
be a similarity function defining the score for each mapping and the conflicts between mappings be defined according to Definition 2. Then, finding a set of mappings (i.e., alignment) that maximizes the sum of mapping scores (i.e., alignment score) and has no conflicting pairs is NP-hard.
Proof
We prove the NP-hardness of our problem by a reduction from the MWIS problem in bounded degree graphs. Let G = (V, E, w) be a vertex weighted undirected graph with largest degree k − 1 (i.e., 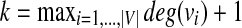 ). Let us set n = ∣V∣ + ∣E∣, m = ∣V∣. We will construct two hypothetical pathways
). Let us set n = ∣V∣ + ∣E∣, m = ∣V∣. We will construct two hypothetical pathways 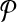 and
and 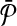 through a polynomial time reduction such that their best alignment is equivalent to the MWIS of G.
through a polynomial time reduction such that their best alignment is equivalent to the MWIS of G.
Reduction. Following from the notation we used elsewhere in this paper, let us denote the pathways 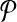 and
and 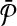 as
as 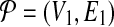 and
and 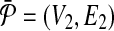 . Here, Vi and
. Here, Vi and 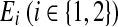 denote the set of reactions (vertices) and interactions (edges), respectively.
denote the set of reactions (vertices) and interactions (edges), respectively.
We initialize V1, V2, E1 and E2 as the empty set. We then insert a vertex ri in V1 for each 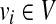 . Similarly, we insert a vertex
. Similarly, we insert a vertex 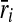 in V2 for each
in V2 for each 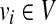 . At this point we have completed the construction of
. At this point we have completed the construction of 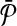 but not
but not 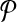 . We continue by inserting a new vertex in V1 for each edge
. We continue by inserting a new vertex in V1 for each edge 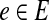 . Thus, each vertex in V1 corresponds to either a vertex or an edge in G while each vertex in V2 corresponds to a vertex in G.
. Thus, each vertex in V1 corresponds to either a vertex or an edge in G while each vertex in V2 corresponds to a vertex in G.
Next, we populate E1. Let ri and rj be two vertices in V1 which correspond to a vertex v in G and to an edge e in G respectively. We include an edge between ri and rj in E1 if e has v at one of its ends in G. This completes the construction of 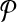 .
.
Figure 3 illustrates the construction of the two hypothetical pathways 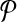 and
and 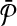 from a simple instance of G. Figure 3a shows a sample graph G with four vertices. Figure 3b presents the resulting pathways. We do not show the weights of the vertices of G to simplify Figure 3a.
from a simple instance of G. Figure 3a shows a sample graph G with four vertices. Figure 3b presents the resulting pathways. We do not show the weights of the vertices of G to simplify Figure 3a.
FIG. 3.

An illustrative example for the reduction from the MWIS problem to the metabolic pathway alignment problem. (a) A vertex-weighted graph G that is an input to the MWIS problem. Four vertices labeled from “1” to “4” have weights from w(1) to w(4) respectively. The edges are labeled from “a” to “e” and are unweighted and undirected. (b) Two pathways 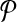 and
and 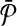 constructed from G. We create one vertex for each vertex of G in both pathway
constructed from G. We create one vertex for each vertex of G in both pathway 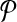 and
and 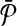 . Then, for
. Then, for 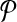 we add a vertex labeled with a letter for each edge of G and add edges from it to the vertices on its both ends in G. In order to simplify the figure, we match the label of each vertex in
we add a vertex labeled with a letter for each edge of G and add edges from it to the vertices on its both ends in G. In order to simplify the figure, we match the label of each vertex in 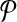 with that of its corresponding vertex or edge in G. Similarly, we match the label of each vertex in
with that of its corresponding vertex or edge in G. Similarly, we match the label of each vertex in 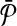 with that of its corresponding vertex in G. (c) The assignment of similarity scores for subnetwork pairs one from
with that of its corresponding vertex in G. (c) The assignment of similarity scores for subnetwork pairs one from 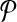 and the other from
and the other from 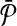 . Each vertex here shows a subnetwork from
. Each vertex here shows a subnetwork from 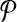 or
or 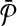 . The label of each vertex lists the vertices contained in that subnetwork. For instance, label “1ab” indicates the subnetwork of
. The label of each vertex lists the vertices contained in that subnetwork. For instance, label “1ab” indicates the subnetwork of 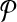 that consists of the three vertices labeled as “1,” “a,” and “b.” The edge weights show the similarity of the two subnetworks corresponding to the two vertices at its end points.
that consists of the three vertices labeled as “1,” “a,” and “b.” The edge weights show the similarity of the two subnetworks corresponding to the two vertices at its end points.
Following from the construction, we ensure that there is a connected subnetwork in 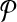 that contains the reactions corresponding to each vertex and its edges in G. For instance, in Figure 3a, the vertex labeled as “1” has two edges labeled as “a” and “b.” In Figure 3b, there are three reactions that have the labels “1,” “a,” and “b,” and they make up a (connected) subnetwork. Thus, the set of subnetworks of
that contains the reactions corresponding to each vertex and its edges in G. For instance, in Figure 3a, the vertex labeled as “1” has two edges labeled as “a” and “b.” In Figure 3b, there are three reactions that have the labels “1,” “a,” and “b,” and they make up a (connected) subnetwork. Thus, the set of subnetworks of 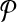 with size up to k is guaranteed to contain each vertex of G jointly with all of its edges. Figure 3c demonstrates this. The vertices on the left side are the subnetworks of
with size up to k is guaranteed to contain each vertex of G jointly with all of its edges. Figure 3c demonstrates this. The vertices on the left side are the subnetworks of 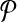 and those on the right side are the subnetworks of
and those on the right side are the subnetworks of 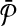 .
.
We complete our reduction by assigning similarities to subnetwork pairs one from 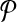 and the other from
and the other from 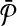 . The similarities we assign correspond to the entries of the column vector H described in Section 2.4.1.
. The similarities we assign correspond to the entries of the column vector H described in Section 2.4.1.
Let Ri be a subnetwork of 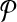 that corresponds to a vertex v in G and all edges of G which have v on one end. In Figure 3b, the subnetwork that contains the reactions labeled “1,” “a,” and “b” is an example to such Ri. Also, let
that corresponds to a vertex v in G and all edges of G which have v on one end. In Figure 3b, the subnetwork that contains the reactions labeled “1,” “a,” and “b” is an example to such Ri. Also, let 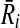 be the subnetwork in
be the subnetwork in 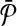 that corresponds to vertex v as well. We assign the similarity of Ri and
that corresponds to vertex v as well. We assign the similarity of Ri and 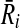 as the weight of vertex v (i.e., w(v)). We repeat this process for all
as the weight of vertex v (i.e., w(v)). We repeat this process for all 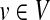 . We assign the similarity between all remaining pairs of subnetworks (Ri,
. We assign the similarity between all remaining pairs of subnetworks (Ri, 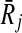 ) as −∞. Figure 3c depicts the assignment of similarities for the pathways in Figure 3b.
) as −∞. Figure 3c depicts the assignment of similarities for the pathways in Figure 3b.
Correctness of reduction. We need to address two issues to prove the correctness of our reduction.
Cost of reduction. For a given vertex weighted graph G, we can reduce the MWIS problem on G to our problem in polynomial time. This is because we create one reaction for each edge and two reactions for each vertex in G. We also create two interactions for each edge in G. Thus, we conclude that the reduction is polynomial in the size of G.
Equivalence of the result. Next, we prove that the optimal alignment of
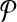 and
and 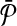 produces the optimal solution to the MWIS problem on G. An alignment is a subset of subnetwork pairs from one from
produces the optimal solution to the MWIS problem on G. An alignment is a subset of subnetwork pairs from one from 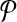 and the other from
and the other from 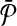 . By construction,
. By construction, 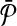 contains only subnetworks of size one. Each subnetwork of
contains only subnetworks of size one. Each subnetwork of 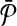 corresponds to a vertex in G. We claim that the vertices of G corresponding to the subnetworks of
corresponds to a vertex in G. We claim that the vertices of G corresponding to the subnetworks of 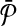 in the optimal alignment constitute the MWIS of G.
in the optimal alignment constitute the MWIS of G.
Clearly, the optimal alignment cannot contain a subnetwork pair whose similarity is −∞. This is because it is possible to choose an arbitrary subnetwork pair that has a positive score. Also, the optimal alignment cannot contain two overlapping subnetworks from the same pathway as they will conflict with each other. By construction, two subnetworks from 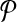 conflict only if they share a common reaction for the same vertex or edge in G. For instance the subnetworks labeled “1ab” and “2acd” in Figure 3c conflict since they both contain “a” in Figure 3b. The subnetworks in the optimal alignment cannot contain such reactions. Hence, the optimal alignment constitute an independent set in G. The score of the alignment is the sum of the weights of the corresponding vertices in G. Therefore, we conclude that by maximizing the alignment score
conflict only if they share a common reaction for the same vertex or edge in G. For instance the subnetworks labeled “1ab” and “2acd” in Figure 3c conflict since they both contain “a” in Figure 3b. The subnetworks in the optimal alignment cannot contain such reactions. Hence, the optimal alignment constitute an independent set in G. The score of the alignment is the sum of the weights of the corresponding vertices in G. Therefore, we conclude that by maximizing the alignment score 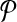 and
and 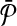 , we optimally solve the MWIS problem for G. ▪
, we optimally solve the MWIS problem for G. ▪
We demonstrate our result on the hypothetical example in Figure 3. Assume that all the vertices in Figure 3a have the same weight w. The optimal alignment of 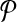 and
and 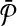 contains the following set of subnetwork pairs
contains the following set of subnetwork pairs 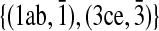 and has an alignment score of 2w. This is because the remaining subnetwork pairs either conflict with each other or have a −∞ score. The optimal alignment suggests that the MWIS of G is {1, 3}.
and has an alignment score of 2w. This is because the remaining subnetwork pairs either conflict with each other or have a −∞ score. The optimal alignment suggests that the MWIS of G is {1, 3}.
Construction of conflict graph
Now that we proved extracting the best alignment is NP-hard, the next part is to describe how we tackle this problem. In the first step, we use the scores of mappings represented by Hi and the definition of conflict (Definition 2) to create a vertex weighted undirected graph Gc = (Vc, Ec, w), which we name as the conflict graph as follows. We define a one-to-one correspondence from the mappings (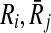 ) to the vertices in Vc. We also set the weight of each vertex
) to the vertices in Vc. We also set the weight of each vertex 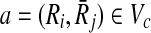 (i.e., w(a)) to the similarity between Ri and
(i.e., w(a)) to the similarity between Ri and 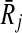 as computed in Hi. We draw an undirected edge between two vertices
as computed in Hi. We draw an undirected edge between two vertices 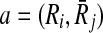 and
and 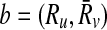 if
if 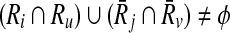 (i.e., a and b conflict). For instance, in Figure 4 there is an edge between a and b representing the fact that they conflict since reaction r1 is common to both a and b.
(i.e., a and b conflict). For instance, in Figure 4 there is an edge between a and b representing the fact that they conflict since reaction r1 is common to both a and b.
FIG. 4.

(a) Each row corresponds to a possible mapping between subnetworks from two hypothetical metabolic pathways. The first column is the unique label for each mapping. Second and third columns are the reactions in the two subnetworks that can be mapped. The last column is the similarity between two subnetworks. (b) The conflict graph Gc for the mappings in (a).
Handling conflicting mappings
In the second step, we explain the greedy vertex-selection strategy we adopt from Sakai et al. (2003) in order to extract the MWIS of Gc as our alignment. Let N(v) denote the set of vertices that are connected to 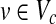 . At each iteration of this algorithm, we pick a vertex v that maximizes
. At each iteration of this algorithm, we pick a vertex v that maximizes  . This strategy implies that a vertex is more likely to be picked if the mapping it represents has large similarity score and conflicts with a small number of other mappings with small similarity scores. After picking a vertex v, we put v into the resulting set and remove v and all the vertices connected to it (i.e., v ∪ N(v)). We also remove all the edges incident to at least one of the removed vertices. When there are no more vertices to remove from Gc, the result set contains a maximal weight independent set. For our alignment problem, this vertex set corresponds to a list of non-conflicting subnetwork mappings. As an example, in Figure 4d is the first vertex to be picked. Then, we remove d and
. This strategy implies that a vertex is more likely to be picked if the mapping it represents has large similarity score and conflicts with a small number of other mappings with small similarity scores. After picking a vertex v, we put v into the resulting set and remove v and all the vertices connected to it (i.e., v ∪ N(v)). We also remove all the edges incident to at least one of the removed vertices. When there are no more vertices to remove from Gc, the result set contains a maximal weight independent set. For our alignment problem, this vertex set corresponds to a list of non-conflicting subnetwork mappings. As an example, in Figure 4d is the first vertex to be picked. Then, we remove d and 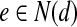 from the graph and insert d to the result set. Next, we pick the vertex b as
from the graph and insert d to the result set. Next, we pick the vertex b as 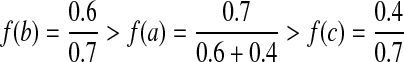 . We remove b and
. We remove b and 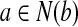 and include b in the result set. Finally, only c is left and taking it into our result set, we have our alignment as the mappings
and include b in the result set. Finally, only c is left and taking it into our result set, we have our alignment as the mappings 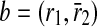 ,
, 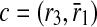 and
and 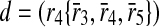 . Note that b and c are one-to-one mappings, and d is a one-to-many mapping where one reaction (r4) of first pathway is aligned to three reactions (
. Note that b and c are one-to-one mappings, and d is a one-to-many mapping where one reaction (r4) of first pathway is aligned to three reactions (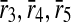 ) of the second pathway.
) of the second pathway.
3. Experimental Results
In this section, we experimentally evaluate the performance of SubMAP. We use the metabolic pathways of 20 organisms taken from the KEGG database. Our dataset contains 1,842 pathways in total. The average number of reactions per pathway is 21, and the largest pathway has 72 reactions. We also combined 12 different pathways under the metabolism of cofactors and vitamins for 10 different organisms. The average number of reactions of the largest connected components of these 10 pathways is 98, and the biggest one has 130 reactions.
3.1. Alternative subnetworks
Different organisms can perform the same function through different subnetworks. We name such altered parts that have similar functions as alternative subnetworks. An accurate alignment should reveal alternative subnetworks in different pathways. In our first experiment, we evaluate whether SubMAP can find them in real metabolic pathways. We align the pathway pairs which are known to contain functionally similar parts with different reaction sets and topologies. Table 2 presents a subset of reaction subnetwork mappings that are found by our algorithm. Figure 5 visualizes the topologies of these mappings by using an enzyme based representation.
Table 2.
Alternative Subnetworks that Produce Same or Similar Output Compounds from the Same or Similar Input Compounds in Different Organisms
| Pathway | Organisms | Input comp.a | Output comp.b | Reaction mappingsc |
|---|---|---|---|---|
| Lysine biosynthesis |
A. thaliana H. Sapiens |
2,3,4,5-Tetra-hydrodipico. | LL-2,6-Di aminopimelate | R07613 ⇔ R02734 + R04365 + R04475 |
| Lysine biosynthesis |
A.thaliana H.sapiens |
L-Saccharo.meso-2,6-Di. | L-Lysine | R00451 + R00715 + R00716 ⇔ R00451 |
| Pyruvate metabolism |
E.coli H.sapiens |
Pyruvate | Oxaloacetate | R00199 + R00345 ⇔ R00344 |
| Pyruvate metabolism |
E.coli H.sapiens |
Oxaloacetate | Phosphoenol-pyruvate | R00341 ⇔ R00431 + R00726 |
| Pyruvate metabolism |
T.acidophilum A.tumefaciens |
Pyruvate | Acetyl-CoA | R01196 ⇔ R00472 + R00216 + R01257 |
| Glycine, serine, threonine met. |
H.sapiens R.norvegicus |
Glycine | Serine L-Threonine |
R00945 ⇔ R00751 + R00945 + R06171 |
| Fructose and mannose met. |
E.coli H.sapiens |
L-Fucose | L-Fucose 1-p L-Fuculose 1-p |
R03163 + R03241 ⇔ R03161 |
| Citrate cycle |
S.aureus N315 S.aureus COL |
Isocitrate | 2-Oxoglutarate | R00268 + R01899 ⇔ R00709 |
| Citrate cycle |
H.sapiens A.tumefaciens |
Succinate | Succinyl-CoA | R00432 + R00727 ⇔ R00405 |
| Citrate cycle | H.sapiens | Isocitrate | 2-Oxoglutarate | R00709 ⇔ R00362 |
| A. tumefaciens | Citrate | Oxaloacetate |
Main input compound utilized by the given set of reactions.
Main output compound produced by the given set of reactions.
Reactions mappings that corresponds to alternative paths. Reactions are represented by their KEGG identifiers.
The first row of Table 2 corresponds to alternative subnetworks in Figure 5a (also in Fig. 1). The reaction R07613 represents the top path in Figure 5a that plants and Chlamydia use to produce LL-2,6-diaminopimelate from 2,3,4,5-tetrahydrodipicolinate. This path is discovered and reported as a shortcut on the L-lysine synthesis path for plants and Chlamydia, which is not present in humans or E. coli (Watanabe et al., 2007; McCoy et al., 2006). Also, Watanabe et al. (2007) suggested that, since humans lack the catalyzer of the reaction R07613, namely LL-DAP aminotransferase (EC:2.6.1.83), this is an attractive target for the development of new drugs (antibiotics and herbicides). When we aligned the lysine biosynthesis pathways of H. sapiens and A. thaliana, our algorithm mapped the reaction R07613 of A. thaliana to the three reactions that H. sapiens has to use to transform 2,3,4,5-tetrahydrodipicolinate to LL-2,6-diaminopimelate (R02734, R04365, R04475). In other words, SubMAP successfully identified the alternative subnetworks of different size (one for A. thaliana and three for H. sapiens) that perform the same function.
Another interesting example is the second row that is extracted from the same alignment described above. In this case, the three reactions that can independently produce L-lysine for A. thaliana are aligned to the only reaction that produces L-lysine for H. sapiens (Fig. 5b). R00451 is common to both organisms and it utilizes meso-2,6-diaminopimelate to produce L-lysine. The reactions R00715 and R00716 take place and produce L-lysine in A. thaliana in the presence of L-saccharopine (Saunders and Broquist, 1966).
For the alignment of pyruvate metabolisms of E. coli and H. sapiens, the third and fourth rows show two mappings that are found by SubMAP. The first one maps the two-step process in E. coli that first converts pyruvate to orthophosphate (R00199) and then orthophosphate to oxaloacetate (R00345) to the single reaction that directly produces oxaloacetate from pyruvate (R00344) in H. sapiens (Fig. 5c). The second one shows another mapping in which a single reaction of E. coli is replaced by two reactions of H. sapiens (Fig. 5d). The first two rows for citrate cycle also report similar mappings for other organism pairs (Fig. 5e).
Note that all the above examples (rows 6–9 of Table 2 depicted in Fig. 5f–i) are one-to-many reaction mappings and hence a merit of the new algorithm we propose here. Our algorithm SubMAP also reports one-to-one mappings. The last row of Table 2 is an example in which one reaction of an organism is replaced by exactly one reaction of another organism. Aligning citrate cycles of H. sapiens and A. tumefaciens reveals that even though both the input and output compounds of the two reactions R00709 and R00362 are different, SubMAP maps these reactions (Fig. 5j). Also, if we look at the EC numbers of the enzymes catalyzing these reactions (1.1.1.41 and 4.1.3.6) their similarity is zero (see Information content enzyme similarity) (Ay et al., 2009a). If we were to consider only the homological similarities, these two reactions could not have been mapped to each other. However, both of these reactions are the neighbors of the two other reactions R01325 and R01900 that are present in both organisms. The mappings of R01325 to R01325 and R01900 to R01900 support the mapping of their neighbors R00709 to R00362. Therefore, by incorporating the topological similarity, our algorithm is able to find meaningful mappings with similar topologies and distinct homologies. An algorithm not considering pathway topologies would fail to identify such mappings.
These results suggest the following: (i) By allowing one-to-many mappings, our method identifies functionally similar subnetworks even if they have a different number of reactions. (ii) Incorporation of topological similarity makes it possible to find mappings that can be missed by only considering pairwise similarities of different entities.
3.2. Number of connected subnetworks
Given the parameter k, our algorithm enumerates all connected reaction subnetworks of size at most k for each query pathway. One question that we need to answer is: How many such subnetworks exist? Figure 6 plots this average for different pathway sizes in our dataset. When k = 1, the figure shows the number of reactions. For k > 1 the results demonstrate that the number of subnetworks increase exponentially with k. However, the increase is significantly lower than the theoretical worst case 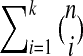 (i.e., n choose i). For instance, the number of subnetworks we obtained for n = 72 and k = 5 is around 750 times less than the theoretical worst case. For n = 130 and k = 5 the number of subnetworks is 0.027% of the worst case.
(i.e., n choose i). For instance, the number of subnetworks we obtained for n = 72 and k = 5 is around 750 times less than the theoretical worst case. For n = 130 and k = 5 the number of subnetworks is 0.027% of the worst case.
FIG. 6.

The number of subnetworks with at most k reactions for pathways of different sizes. Fitting curves are drawn using second degree polynomials.
The figure also suggests that the number of subnetworks increases as a low-degree polynomial of the size of the pathway. This is mainly because the average number of edges (i.e., neighbors) of a node (i.e., subnetwork) remains roughly the same as the size of the network increases. As a result, we conclude that for k ≤ 4, we can enumerate and store all the subnetworks of the pathways in KEGG dataset. In practice it is unlikely for a single reaction to replace a subnetwork with more than three or four reactions. Therefore, we expect that using k ≤ 4 would be sufficient to find most of the biologically relevant alternative subnetworks.
3.3. One-to-many mappings within and across major clades
In Section 3.1, we demonstrated that our algorithm can find alternative subnetworks on a number of examples. An obvious question that follows is: How frequent are such alternative subnetworks and what are their characteristics? In other words, is there really a need to allow one-to-many mappings in alignment. In this experiment, we aim to answer these questions.
We conduct an experiment as follows. We first pick nine different organisms, three from each major phylogenic clade. These organisms are T. acidophilum, Halobacterium sp., and M. thermoautotrophicum from Archaea; H. sapiens, R. norvegicus, and M. musculus from Eukaryota; and E. coli, P. aeruginosa, and A. tumefaciens from Bacteria.
We then extract 10 common pathways for each of these nine organisms from KEGG. For each of these common pathways, we choose all possible pairs of organisms 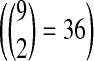 and align that specific pathway for all organism pairs. In these alignments we exclude the self alignments and the alignment with parameter k = 1 since those will definitely incur a bias favoring the number of one-to-one alignments. We computed all possible alignments (10 × 36 = 360) for k = 2, 3 and 4 (360 × 3 = 1,080 alignments in total). Finally, we calculated the number of four possible types of subnetwork mappings which are 1-to-1, 1-to-2, 1-to-3, and 1-to-4. We hypothesize that the metabolisms of the organisms within a clade will tend to perform the same function through the same (or similar) sized sets of reactions while those across different clades will perform through alternative subnetworks of varying sizes.
and align that specific pathway for all organism pairs. In these alignments we exclude the self alignments and the alignment with parameter k = 1 since those will definitely incur a bias favoring the number of one-to-one alignments. We computed all possible alignments (10 × 36 = 360) for k = 2, 3 and 4 (360 × 3 = 1,080 alignments in total). Finally, we calculated the number of four possible types of subnetwork mappings which are 1-to-1, 1-to-2, 1-to-3, and 1-to-4. We hypothesize that the metabolisms of the organisms within a clade will tend to perform the same function through the same (or similar) sized sets of reactions while those across different clades will perform through alternative subnetworks of varying sizes.
Table 3 summarizes the results of this experiment. The percentages of each mapping type between two clades is shown as a row in this table. The first three rows correspond to alignments within a clade and the last three represents alignments across two different clades. An important outcome of these results is that there are considerably large number of one-to-many mappings between organisms of different clades. In the extreme case (last row), nearly half of the mappings are one-to-many. The results also support our hypothesis that one-to-one mappings are more frequent for alignments within the clades compared to across clades due to high similarity between the organisms of the same clade. For instance, for both the first and last row, one side of the query set is the Eukaryota. However, going from first row to last, we see around 40% decrease in the number of one-to-one mappings and 250%, 850%, and 450% increase in the number of 1-to-2, 1-to-3, and 1-to-4 mappings, respectively. Considering Archaea are single-cell microorganisms (e.g., Halobacteria) and Eukaryota are complex organisms with cell membranes (e.g., animals and plants), these jumps in the number of one-to-many mappings suggest that the individual reactions in Archaea are replaced by a number of reactions in Eukaryota. These results have two major implications. (i) One-to-many mappings are frequent in nature. To obtain biologically meaningful alignments we need to allow such mappings. (ii) The characteristics of the alterative subnetworks can help in inferring the phylogenic relationship among different organisms.
Table 3.
Percentages of 1-to-1, 1-to-2, 1-to-3, and 1-to-4 Mappings in Between and Across Three Major Clades
| 1-to-1 | 1-to-2 | 1-to-3 | 1-to-4 | |
|---|---|---|---|---|
| E-E | 89.6 | 8.8 | 1.1 | 0.5 |
| B-B | 80.1 | 16.0 | 3.1 | 0.8 |
| A-A | 78.3 | 15.7 | 4.7 | 1.3 |
| B-E | 69.1 | 23.1 | 6.3 | 1.5 |
| A-B | 60.5 | 28.3 | 8.5 | 2.7 |
| A-E | 55.8 | 31.0 | 10.4 | 2.8 |
A, Archaea; E, Eukaryota; B, Bacteria.
3.4. Running time and memory utilization
SubMAP allows one-to-many mappings to find biologically relevant alignments. This however comes at the expense of increased computational cost. Theoretically, this increase can be exponential in k. The worst case happens when the pathway is highly connected. Metabolic pathways however are sparse and their connectivity follows power law distribution (Jeong et al., 2000). In order to understand the capabilities and limitations of our method, we examine its performance on real datasets in terms of its running time and memory usage.
We evaluate the performance of our method for querying a database of pathways as follows. We create a query set by selecting 50 pathways of varying sizes from KEGG and adding it to the combined pathways for cofactors and vitamins metabolism of 10 different organisms as described at the beginning of this section. We then select another 50 pathways of different sizes to use as our database set for this experiment. We pick the latter 50 pathways such that the average reactions per pathway is 21.4, which is very close to that of the entire database. We then align each of the 60 query pathways with all the database pathways one by one for different values of k. We measure the average running time and the average memory usage for each query pathway and k value combination. Note that we do not present any performance comparison with an existing method as the existing methods do not allow one-to-many mappings. However, our results for k = 1 show the performance of our algorithm when we restrict it to one-to-one mappings similar to the traditional alignment methods.
Figure 7a shows the average running time of SubMAP for query pathways with increasing number of reactions. When k = 1 (i.e., only one-to-one mappings as in existing methods), it runs in less than a few seconds even for the largest query pathway in our query set. As k increases, the running time increases significantly. This is because the number of subnetworks and the average number of forward and backward neighbors of subnetworks increase with k. We observe that our method can perform alignments in practical time even when k = 4 for pathways with around 70 reactions. Hence, it is scalable for all the individual pathways in KEGG. However, for the 10 combined pathways of cofactors and vitamins metabolism, the running time and memory use becomes a bottleneck when we consider k > 3.
FIG. 7.

(a) Average running time and (b) memory use of SubMAP when a query pathway is aligned with a database containing 50 pathways. Pathway size is measured by the number of reactions. k is the size of the largest subnetwork allowed.
We also measure the actual memory usage of our algorithm for real pathways of varying sizes and k values in Figure 7b. For k = 1 or 2, the memory usage is negligible (100 MB or less) for all pathways. Although the memory usage increases quickly with k, it remains feasible for query pathways with around 70 reactions for k = 4. For k = 3, the biggest pathway of 130 reactions requires 1GB of memory per query on the average. These results show that, SubMAP can run on a standard computer for aligning real-sized metabolic pathways for k ≤ 4.
4. Conclusion
In this article, we considered the problem of aligning two metabolic pathways. The distinguishing feature of our work from the literature is that we allow mapping one molecule of one pathway to a set of molecules of the other. To address this problem, given two metabolic pathways  and
and  and an upper bound k on the size of the connected subnetworks, we developed the SubMAP algorithm that can find the consistent mapping of the subnetworks of
and an upper bound k on the size of the connected subnetworks, we developed the SubMAP algorithm that can find the consistent mapping of the subnetworks of 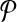 and
and 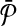 with the maximum similarity. We transformed the alignment problem to an eigenvalue problem. The solution to this eigenvalue problem produced a good mixture of homological and topological similarities of the subnetworks. Using these similarity values, we constructed a vertex weighted graph that connects conflicting mappings with an edge. Then, our alignment problem is transformed into finding the maximum weight independent set of this graph. We employed a heuristic method that is used to solve MWIS problem. The result of this method provided us an alignment that has no conflicting pair of mappings (i.e., consistent). Our experiments on real datasets suggested that our method can identify biologically relevant mappings of alternative subnetworks that are missed by traditional alignment methods. Furthermore, even though SubMAP does not restrict the topologies of query pathways, it is still scalable for real size metabolic pathways when the reaction subsets of size at most four are considered.
with the maximum similarity. We transformed the alignment problem to an eigenvalue problem. The solution to this eigenvalue problem produced a good mixture of homological and topological similarities of the subnetworks. Using these similarity values, we constructed a vertex weighted graph that connects conflicting mappings with an edge. Then, our alignment problem is transformed into finding the maximum weight independent set of this graph. We employed a heuristic method that is used to solve MWIS problem. The result of this method provided us an alignment that has no conflicting pair of mappings (i.e., consistent). Our experiments on real datasets suggested that our method can identify biologically relevant mappings of alternative subnetworks that are missed by traditional alignment methods. Furthermore, even though SubMAP does not restrict the topologies of query pathways, it is still scalable for real size metabolic pathways when the reaction subsets of size at most four are considered.
Acknowledgments
This work was supported partially by the National Science Foundation (grants CCF-0829867 and IIS-0845439).
Disclosure Statement
No conflicting financial interests exist.
References
- Austrin P. Khot S. Safra M. Inapproximability of vertex cover and independent set in bounded degree graphs. Proc. IEEE Conf. Comput. Compl. 2009:74–80. [Google Scholar]
- Ay F. Dinh T. Thai T., et al. Finding dynamic modules of biological regulatory networks. Proc. IEEE Int. Symp. Bioinformatics Bioeng. 2010:136–143. [Google Scholar]
- Ay F. Kahveci T. de Crecy-Lagard V. Consistent alignment of metabolic pathways without abstraction. Proc. 7th Annu. Int. Conf. Comput. Syst. Bioinformatics. 2008;7:237–248. [PubMed] [Google Scholar]
- Ay F. Kahveci T. de Crecy-Lagard V. A fast and accurate algorithm for comparative analysis of metabolic pathways. J. Bioinformatics Comput. Biol. 2009a;7:389–428. doi: 10.1142/s0219720009004163. [DOI] [PubMed] [Google Scholar]
- Ay F. Xu F. Kahveci T. Scalable steady state analysis of Boolean biological regulatory networks. PLoS ONE. 2009b;4:e7992. doi: 10.1371/journal.pone.0007992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babur O. Dogrusoz U. Demir E., et al. ChiBE: interactive visualization and manipulation of BioPAX pathway models. Bioinformatics. 2010;26:429–431. doi: 10.1093/bioinformatics/btp665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J. Lassig M. Local graph alignment and motif search in biological networks. Proc. Natl. Acad. Sci. U.S.A. 2004;101:14689–14694. doi: 10.1073/pnas.0305199101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman P. Karpinski M. On some tighter inapproximability results [Extended Abstract] Lect. Notes Comput. Sci. 1999;1644:705. [Google Scholar]
- Cheng Q. Harrison R. Zelikovsky A. MetNetAligner: a web service tool for metabolic network alignments. Bioinformatics. 2009;25:1989–1990. doi: 10.1093/bioinformatics/btp287. [DOI] [PubMed] [Google Scholar]
- Clemente J. Satou K. Valiente G. Reconstruction of phylogenetic relationships from metabolic pathways based on the enzyme hierarchy and the Gene Ontology. Genome Inform. 2005;16:45–55. [PubMed] [Google Scholar]
- Damaschke P. Graph-theoretic concepts in computer science. Lect. Notes Comput. Sci. 1991;484:72–78. [Google Scholar]
- Dandekar T. Schuster S. Snel B., et al. Pathway alignment: application to the comparative analysis of glycolytic enzymes. Biochem. J. 1999;343:115–124. [PMC free article] [PubMed] [Google Scholar]
- Deutscher D. Meilijson I. Schuster S., et al. Can single knockouts accurately single out gene functions? BMC Syst. Biol. 2008;2:50. doi: 10.1186/1752-0509-2-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devloo V. Hansen P. Labbe M. Identification of all steady states in large networks by logical analysis. Bull. Math. Biol. 2003;65:1025–1051. doi: 10.1016/S0092-8240(03)00061-2. [DOI] [PubMed] [Google Scholar]
- Dost B. Shlomi T. Gupta N. Qnet: a tool for querying protein interaction networks. Proc. RECOMB 07. 2007:1–15. doi: 10.1089/cmb.2007.0172. [DOI] [PubMed] [Google Scholar]
- Edwards J. Palsson B. Robustness analysis of the Escherichia coli metabolic network. Biotechnol. Prog. 2000;16:927–939. doi: 10.1021/bp0000712. [DOI] [PubMed] [Google Scholar]
- Flannick J. Novak A. Srinivasan B. Greamlin: general and robust alignment of multiple large interaction networks. Genome Res. 2006;16:1169–1181. doi: 10.1101/gr.5235706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francke C. Siezen R. Teusink B. Reconstructing the metabolic network of a bacterium from its genome. Trends Microbiol. 2005;13:550–558. doi: 10.1016/j.tim.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Garg A. Xenarios I. Mendoza L., et al. An efficient method for dynamic analysis of gene regulatory networks and in silico gene perturbation experiments. Proc. RECOMB 07. 2007:62–76. [Google Scholar]
- Green M. Karp P. A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases. BMC Bioinformatics. 2004;5:76. doi: 10.1186/1471-2105-5-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grochow J. Kellis M. Network motif discovery using subgraph enumeration and symmetry-breaking. Proc. RECOMB 07. 2007:92–106. [Google Scholar]
- Hattori M. Okuno Y. Goto S., et al. Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. J. Am. Chem. Soc. 2003;125:11853–11865. doi: 10.1021/ja036030u. [DOI] [PubMed] [Google Scholar]
- Heymans M. Singh A. Deriving phylogenetic trees from the similarity analysis of metabolic pathways. Bioinformatics. 2003;19:138–146. doi: 10.1093/bioinformatics/btg1018. [DOI] [PubMed] [Google Scholar]
- Jeong H. Tombor B. Albert R., et al. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- Kalaev M. Bafna V. Sharan R. Fast and accurate alignment of multiple protein networks. J. Comput. Biol. 2009;8:989–999. doi: 10.1089/cmb.2009.0136. [DOI] [PubMed] [Google Scholar]
- Kalaev M. Smoot M. Ideker T., et al. NetworkBLAST: comparative analysis of protein networks. Bioinformatics. 2008;24:594–596. doi: 10.1093/bioinformatics/btm630. [DOI] [PubMed] [Google Scholar]
- Koyuturk M. Grama A. Szpankowski W. An efficient algorithm for detecting frequent subgraphs in biological networks. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2004:200–207. doi: 10.1093/bioinformatics/bth919. [DOI] [PubMed] [Google Scholar]
- Koyuturk M. Grama A. Szpankowski W. Pairwise local alignment of protein interaction networks guided by models of evolution. Proc. RECOMB 05. 2005:48–65. [Google Scholar]
- Liao C. Lu K. Baym M., et al. IsoRankN: spectral methods for global alignment of multiple protein networks. Bioinformatics. 2009;25:253–258. doi: 10.1093/bioinformatics/btp203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovasz L. Stable set and polynomials. Discr. Math. 1994;124:137–153. [Google Scholar]
- Lu H. Shi B. Wu G., et al. Integrated analysis of multiple data sources reveals modular structure of biological networks. Biochem. Biophys. Res. Commun. 2006;345:302–309. doi: 10.1016/j.bbrc.2006.04.088. [DOI] [PubMed] [Google Scholar]
- McCoy A. Adams N. Hudson A., et al. L,L-diaminopimelate aminotransferase, a trans-kingdom enzyme shared by Chlamydia and plants for synthesis of diaminopimelate/lysine. Proc. Natl. Acad. Sci. U.S.A. 2006;103:17909–17914. doi: 10.1073/pnas.0608643103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michal G. On representation of metabolic pathways. Biosystems. 1998;47:1–7. doi: 10.1016/s0303-2647(98)00019-7. [DOI] [PubMed] [Google Scholar]
- Milo R. Shen-Orr S. Itzkovitz S., et al. Network motifs: simple building blocks of complex networks. Science. 2002;47:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- Ogata H. Fujibuchi W. Goto S., et al. A heuristic graph comparison algorithm and its application to detect functionally related enzyme clusters. Nucleic Acids Res. 2000;28:4021–4028. doi: 10.1093/nar/28.20.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H. Goto S. Sato K., et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter R. Rokhlenko O. Yeger-Lotem E., et al. Alignment of metabolic pathways. Bioinformatics. 2005;21:3401–3408. doi: 10.1093/bioinformatics/bti554. [DOI] [PubMed] [Google Scholar]
- Qian X. Yoon B. Effective identification of conserved pathways in biological networks using hidden Markov models. PLoS ONE. 2009;4:e8070. doi: 10.1371/journal.pone.0008070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai S. Togasaki M. Yamazaki K. A note on greedy algorithms for the maximum weighted independent set problem. Discr. Appl. Math. 2003;126:313–322. [Google Scholar]
- Saunders P. Broquist H. Saccharopine, an intermediate of aminoadipic acid pathway of lysine biosynthesis. J. Biol. Chem. 1966;241:3435–3440. [PubMed] [Google Scholar]
- Schuster S. Pfeiffer T. Koch A., et al. Exploring the pathway structure of metabolism: decomposition into subnetworks and application to Mycoplasma pneumoniae. Bioinformatics. 2002;18:351–361. doi: 10.1093/bioinformatics/18.2.351. [DOI] [PubMed] [Google Scholar]
- Sharan R. Suthram S. Kelley R., et al. Conserved patterns of protein interaction in multiple species. Proc. Natl. Acad. Sci. U.S.A. 2005;102:1974–1979. doi: 10.1073/pnas.0409522102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R. Xu J. Berger B. Pairwise global alignment of protein interaction networks by matching neighborhood topology. Proc. RECOMB 07. 2007:16–31. [Google Scholar]
- Singh R. Xu J. Berger B. Global alignment of multiple protein interaction networks with application to functional orthology detection. Proc. Natl. Acad. Sci. U.S.A. 2008;105:12763–12768. doi: 10.1073/pnas.0806627105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar P. Kahveci T. Ranka S. An iterative algorithm for metabolic network-based drug target identification. Pac. Symp. Biocomput. 2007;12:88–99. [PubMed] [Google Scholar]
- Tohsato Y. Matsuda H. Hashimoto A. A multiple alignment algorithm for metabolic pathway analysis using enzyme hierarchy. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2000:376–383. [PubMed] [Google Scholar]
- Tohsato Y. Nishimura Y. Metabolic pathway alignment based on similarity of chemical structures. Info. Media Tech. 2008;3:191–200. [Google Scholar]
- Watanabe N. Cherney M. van Belkum M., et al. Crystal structure of LL-diaminopimelate aminotransferase from Arabidopsis thaliana: a recently discovered enzyme in the biosynthesis of L-lysine by plants and Chlamydia. J. Mol. Biol. 2007;371:685–702. doi: 10.1016/j.jmb.2007.05.061. [DOI] [PubMed] [Google Scholar]
- Webb E. Enzyme Nomenclature 1992. Academic Press; New York: 1992. [Google Scholar]
- Wernicke S. Rasche F. FANMOD: a tool for fast network motif detection. Bioinformatics. 2006;22:1152–1153. doi: 10.1093/bioinformatics/btl038. [DOI] [PubMed] [Google Scholar]