

Abstract
ATP-binding cassette transporters use the energy of ATP hydrolysis to transport substrates across cellular membranes. They have two transmembrane domains and two cytosolic nucleotide-binding domains. Biochemical studies have characterized an occluded state of the transporter in which nucleotide is tenaciously bound in one active site, whereas the opposite active site is empty or binds nucleotide loosely. Here, we report molecular-dynamics simulations of the bacterial multidrug ATP-binding cassette transporter Sav1866. In two simulations of the ATP/apo state, the empty site opened substantially by way of rotation of the nucleotide-binding domain (NBD) core subdomain, whereas the ATP-bound site remained occluded and intact. We correlate our findings with elastic network and molecular-dynamics simulation analyses of the Sav1866 NBD monomer, and with existing experimental data, to argue that the observed transition is physiological, and that the final structure observed in the ATP/apo simulations corresponds to the tight/loose state of the NBD dimer characterized experimentally.
Introduction
ATP-binding cassette (ABC) transporters use the energy of ATP hydrolysis to translocate substrates across cellular membranes, although in some atypical cases they are gated channels or transmembrane signal receptors. ABC transporters comprise one of the largest protein superfamilies, are found in all living organisms, and transport a wide range of substrates (1–4). Members of this superfamily are involved in multidrug resistance (MDR) in both human cancers and pathogenic microbes, and significant human genetic disorders are caused by mutations in many ABC genes (5).
The general architecture of ABC transporters is dimeric, with each half comprising one domain that spans the membrane and one cytosolic nucleotide-binding domain (NBD) or ABC. These domains are configured in various combinations of two transmembrane domains (TMDs) and two NBDs as four separate subunits, two half transporters, or a single polypeptide, with the canonical configuration TMD-NBD-TMD-NBD, although exceptions with the opposite order or additional N-terminal or regulatory domains also exist (6,7). The TMDs form the membrane channel and contain substrate-binding sites. In contrast to the TMD, the NBD is highly conserved in sequence and structure. Its bilobal architecture consists of a catalytic core subdomain containing the Walker A (P-loop) and B consensus nucleotide-binding motifs, and a flexibly attached α-helical subdomain containing the LSGGQ ABC signature sequence (6).
In the assembled transporter, two composite ATP-binding active sites are formed by the interaction of each LSGGQ motif with an ATP molecule bound to the Walker A and B motifs of the opposite NBD (8,9). Structural studies indicate that although this head-to-tail NBD dimer is a common feature, at least three distinct TMDs architectures exist within the ABC superfamily. Nevertheless, in all whole transporter structures, the TMDs and NBDs interact through intracytoplasmic loops (ICLs) that are associated with the TMDs (10–16), and this interaction is expected to mediate the coupling of ATP hydrolysis to substrate translocation. In ABC exporters, two α-helices, which are known as coupling helices (CHs) and originate from the ICLs, form a substantial part of the interface between the TMDs and the NBDs (Fig. 1 A).
Figure 1.
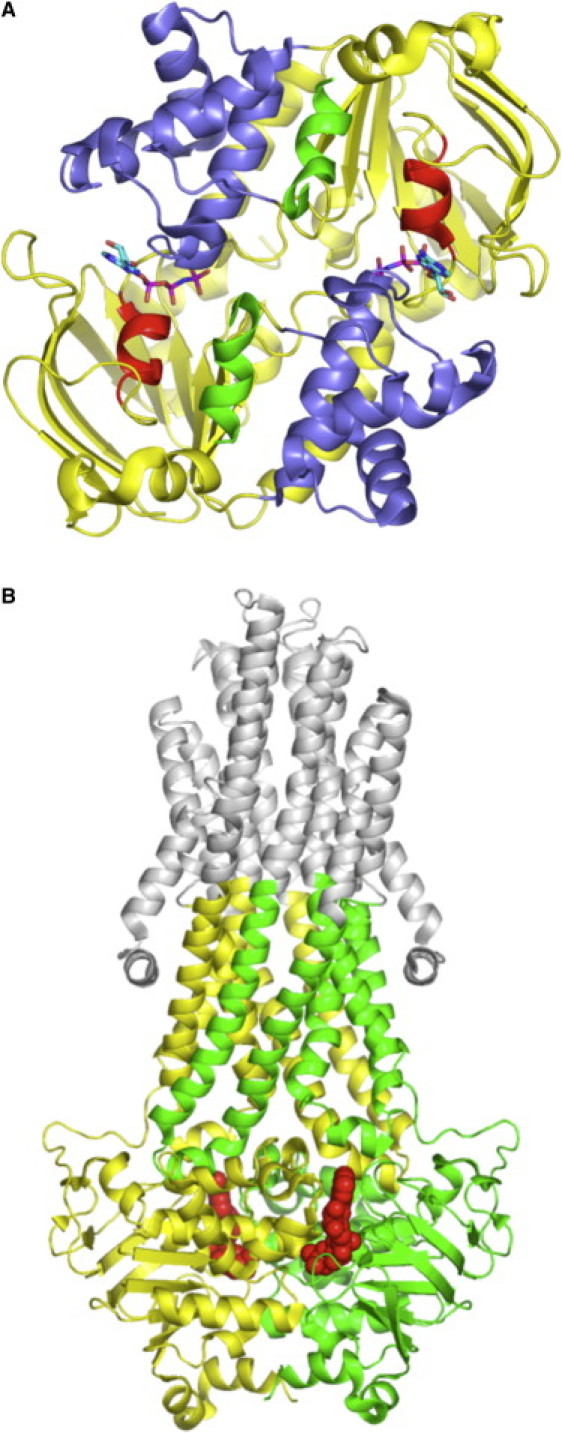
Sav1866. (A) View from the membrane of the Sav1866 NBD dimer and both CH2s from the TMDs. The interface between the monomers runs approximately horizontally through the middle of the dimer. The core subdomains are colored yellow, the HDs blue, the α-helix following the P-loop red, and the CH2s green. The CH2 binds in the cleft between the core and HDs of the NBD covalently attached to the opposite monomer. Nucleotides are shown in stick form and are bound between the P-loop of the core subdomain and the LSGGQ of the HD of the opposite monomer. (B) ICL-NBD structure. View of Sav1866 parallel to the plane of the membrane, with regions used in the simulations colored green (monomer A) and yellow (monomer B). The nucleotides are red space-filled spheres. Regions not included in the simulations are in gray (Color online.)
A number of experimental studies of MDR-related ABC exporters have indicated that a state exists in which ATP is tightly bound in an occluded active site, whereas the opposite active site is unoccupied or binds nucleotide loosely in a nonoccluded state. This tight/loose state of the active sites is likely to be important for understanding the enzyme's catalytic cycle, but it has not been observed in crystal structures. To try to gain insights into its structural basis, we performed molecular-dynamics (MD) simulations using the crystal structure of the Staphylococcus aureus Sav1866, a homodimer of two half-transporters whose primary sequence and biochemical characteristics identify it as an MDR ABC transporter (17).
In the Sav1866 crystal structure, two nucleotides are sequestered in active sites formed by both NBDs and ICLs in an essentially symmetrical manner with respect to the two half-transporters. In two simulations beginning with the predicted cytosolic regions of this structure, with ATP bound in one site and the opposite site empty, the empty site opened partially while the ATP-bound site remained occluded and intact. The transition from the crystal structure to this asymmetric state occurred substantially by way of movement of the core subdomain of the NBD, which did not contain ATP bound to its P-loop, away from the LSGGQ of the opposite, ATP-bound NBD and also away from the ICLs. We correlate these findings with an elastic network (EN) analysis of the NBD monomer, simulations of the isolated Sav1866 NBD monomer, and existing experimental data to argue that the observed transition is likely to have physiological relevance, and that the final structure observed in the ATP/apo simulations corresponds to the tight/loose state of the NBD dimer characterized experimentally.
Methods
System setup
The starting coordinates for MD simulations were taken from the x-ray structure of the AMP-PNP-bound ABC exporter Sav1866 (11) (PDB 2ONJ). We prepared a truncated structure comprising the predicted cytosolic regions using coordinates for residues 82–138 (ICL1), 183–246 (ICL2), and 300–578 (ICL3-NBD) from each monomer (Fig. 1 B). This structure includes a short buffer zone at the truncated ends of the ICLs, which may embody part of the TMD region. The N-terminal residues of the ICLs were acetylated, and the C-terminal residues of ICLs 1 and 2 were N-methylamidated. To place ATP in the consensus orientation in the active site, we root mean-square (RMS)-fitted the P-loop and ATP molecule of ATP-bound MJ0796 (PDB 1L2T; residues 32–49) (9) to Sav1866 using the Cα coordinates of the P-loop residues (368–385 in Sav1866). The resultant coordinates of the pyrophosphate moiety were used in the starting structure. Systems of the NBD monomer used coordinates for residues 338–578 from monomer A, with the N-terminal residue acetylated. Each complex was solvated in a truncated octahedral periodic cell with a minimum of 20 Å between periodic images of the protein, and neutralized with a 0.2 M NaCl solution. All histidine residues were neutral and protonated at the ɛ nitrogen, with the exception of active-site histidine 534, which was ionized; all other ionizable residues were in the default ionization state.
Simulation parameters
We performed MD simulations using NAMD versions 2.6 and 2.7b3 (18) with the CHARMM27 force field (19), including ϕ/ψ cross-term map corrections (20), and the TIP3P model for water (21). The SHAKE and SETTLE algorithms were used to constrain the bonds containing hydrogens to equilibrium length (22). A cutoff of 11 Å (switching function starting at 9.5Å) for van der Waals and real-space electrostatic interactions was used. The particle-mesh Ewald method (23) was used to compute long-range electrostatic forces with a grid density of ∼1/Å3. An integration time step of 2 fs was used with a multiple time-stepping algorithm. Interactions involving covalent bonds and short-range nonbonded interactions were computed every time step, and long-range electrostatic forces were computed every two time steps. Langevin dynamics was utilized to maintain a constant temperature of 310 K with a friction coefficient of 5 ps−1 on all nonhydrogen atoms. A Langevin piston was used to control pressure with a target of 1 atm, a decay period of 100 fs, and a damping timescale of 50 fs.
Equilibration
To ensure that the canonical coordination sphere of the catalytic divalent cation would be in ATP-bound active sites, during the equilibration we applied harmonic forces with a force constant of 10 kcal/mol·Å2 to the coordinating atoms of the Mg2+ ion (i.e., two water molecules, a β- and γ-phosphate oxygen, the amido side-chain oxygen of conserved glutamine 422, and the side-chain hydroxyl oxygen of Walker A serine 381), constraining them to an equilibrium distance of 2.2 Å from the Mg2+. The solvated starting structure was minimized by conjugate gradient minimization to a 0.5 kcal/mol·Å RMS gradient with all protein and Mg2+ATP heavy atoms fixed. Water molecules, NaCl ions, and hydrogens were then further minimized during a 50 ps MD run at 310 K, in which all protein and Mg2+ATP heavy atoms were again fixed. This starting model was then minimized with harmonic positional constraints on the NCαCO backbone. A 100 kcal/mol·Å2 force constant was used to minimize the system to a 0.5 kcal/mol·Å RMS gradient. The constraints were gradually removed by subsequent minimizations to a 0.1 kcal/mol·Å RMS gradient, with the initial force constants scaled by factors of 0.5, 0.15, 0.05, and 0. The minimized structure was then heated from 50 K to 310 K in steps of 25 K using velocity reassignment during a 30 ps MD run. This system was then simulated for 500 ps using Langevin temperature and pressure controls, with the harmonic restraints on the coordinating atoms of the catalytic Mg2+ remaining.
Production runs
The equilibrated system was used for the production runs without restraints. Five systems (three using the truncated structure and two using the monomer structure) were simulated for 100 ns. The truncated-structure systems comprised one ATP/ATP system (simulation 1) and two ATP/apo systems in which the nucleotide was bound to the P-loop of either monomer A or monomer B (simulations 2A and 2B, respectively). For the NBD monomer, the ATP-bound and apo states were simulated (simulations 3A and 3B). Simulation 2B was run for a further 20 ns, as discussed in Results. All simulations remained stable to completion. For analysis, the coordinates were saved every 10 ps.
Analysis
We performed a principal component analysis (PCA) of the simulation Cα atom trajectories using the GROMACS package (20). The program Hingefinder was used to analyze domain rotations using Cα coordinates, the slow partitioning algorithm and a tolerance of 70% (24). VMD (25), Xplor-NIH (26), and Simulaid (27) were used to prepare the system and analyze MD trajectories. All structural figures and Movie S2 in the Supporting Material were prepared using PyMol (http://www.pymol.org/pymol), and Movie S1 was made with VMD. We performed an EN analysis of the monomer structure (residues 338–578 from monomer A) using the online facility with default settings (28) available at http://ignm.ccbb.pitt.edu/.
Results
To investigate conformational states in an ABC NBD dimer in the presence of its interaction with the ICLs, we used the AMP-PNP-bound Sav1866 whole transporter structure (PDB 2ONJ) (11) as a starting point for MD simulations. A system using only the cytosolic regions of the transporter, which comprised the NBD dimer and the ICLs, was simulated (Fig. 1 B). This approach is similar to that used in a previous MD study of the Sav1866 homolog, the MsbA lipid A transporter (29), in which only the predicted membrane-spanning segments were simulated, as well as in many studies in which isolated TMD (30) or NBD (31–39) domains of ABC transporters were simulated.
Binding mode of ATP
In the AMP-PNP-bound Sav1866 whole transporter, the ATP-analog is not bound in the active site in the consensus configuration observed in other ABC NBDs (Table S1). In particular, the plane described by the two P-N bonds of the bridging PNP β-nitrogen, and the plane described by the two P-O bonds of the bridging phosphoester α-oxygen are both approximately orthogonal to that universally observed for the corresponding phosphoester bonds in P-loop protein-bound ATP (Fig. S1). Thus, the oxygen atoms of the pyrophosphate moiety do not interact with the P-loop in the characteristic manner. Also, the structure does not contain a water molecule that coordinates the catalytic metal ion and is also hydrogen-bonded to the conserved Walker B aspartate. To set up a canonical nucleotide-bound active site, we generated coordinates for Mg2+ATP and the missing water molecule using the active site of ATP-bound MJ0796 as a template. Throughout the equilibration for each MD run, harmonic restraints were applied between the catalytic magnesium ion and its six coordinating ligands. The resultant structure was used without restraints in the production run.
To provide a point of comparison with respect to the binding mode of ATP with two recent simulations of the ATP/ATP bound Sav1866 full transporter (40,41), we performed a 100 ns simulation of the truncated structure described above, with two ATP molecules bound (simulation 1). In pointed contrast to the other MD studies, we found that ATP remained bound in both active sites in the consensus configuration with respect to their interaction with the Walker A motif and Walker B aspartate (Table S2). Moreover, the coordinating interactions between the catalytic metal and conserved glutamine side-chain oxygen were stable and remained intact. In addition, the direct interactions between the γ-phosphate and atoms of the LSGGQ motif remained analogous to those observed in the MJ0796 NBD dimer (9). These findings were also true for all ATP sites simulated, as discussed below (Table S2).
Simulations of the ATP/apo state show that rotation of the core subdomain partially opens the empty active site
Initially, we performed two 100 ns simulations of the ATP/apo state: one with ATP bound to the P-loop of monomer A (simulation 2A), and one with ATP bound to the P-loop of monomer B (simulation 2B). Simulation 2B was extended to 120 ns (see below). Fig. 2, A and B, show, for simulations 2A and 2B, the time series of the RMS deviation (RMSD) of Cα atoms of the whole structure from the starting structure, as well as the breakdown for each NBD and the ICLs separately. The plots for overall RMSD show that both structures remain stable. The plots for the NBDs show that in both cases, the NBD without nucleotide bound to its P-loop (apo NBD) undergoes greater changes than the ATP-bound NBD. This is also illustrated by the time course for each NBD of the solvent-accessible surface area and the radius of gyration, the latter giving a measure of the compactness of the NBD (Fig. S2). The ICL plots include ICLs 1 and 2 from each half, but not the first and last four residues in the buffer zone at the truncated ends of each ICL, thus giving a measure of the stability of the predicted cytosolic TMD regions. These plots show that in both cases, the (unrestrained) ICLs remain very stable and close to the starting structure throughout the simulations. Thus, the truncation of the protein at the level of the membrane did not introduce instabilities into or significantly alter the regions of the ICLs that form the NBD/TMD interface.
Figure 2.
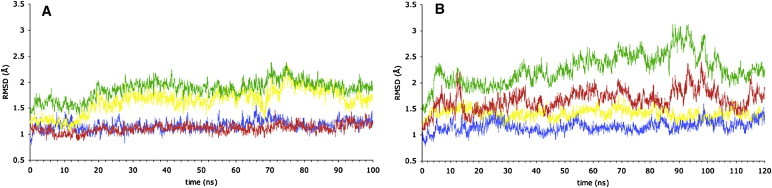
Time course of RMSD of Cα atoms in the ATP/apo simulations. Cα atoms are in green, NBD monomer A red, NBD monomer B yellow, ICLs blue, sampled every 50 ps (color online). (A) Simulation 2A, ATP bound in NBD monomer A. (B) Simulation 2B, ATP bound in NBD monomer B.
Throughout both ATP/apo simulations, ATP interactions with the catalytic metal and the protein, including the ATP interaction with the LSGGQ motif of the opposite NBD, remained essentially as they were at the beginning (Table S2). Thus, the relative disposition of the helical subdomain (HD) and the core subdomain that comprised the ATP-bound active sites did not alter significantly. In contrast, the core subdomain of the apo NBD moved significantly relative to the HD of the opposite NBD. This is illustrated by plotting movements relative to the HDs. The frames of the simulation trajectory were structurally aligned using the coordinates of the HD, and the movement of the helix immediately downstream from the P-loop within the core subdomain of the opposite NBD (Fig. 1 A), relative to its starting position, was plotted (Fig. 3, A and B). Similar plots of the movement of CH2 from the opposite monomer, relative to each HD are also shown (Fig. 3, A and B). CH2 from the opposite monomer sits alongside the HD in the cleft dividing it from the core subdomain within its NBD monomer (Fig. 1 A). These plots for both simulations show that both CH2s do not move significantly relative to the proximal HD. Because the ICLs also remain close to their starting conformation (Fig. 2, A and B), this shows that the core subdomain of the apo NBD moves relative to the rest of the complex, which remains close to the starting structure. This movement of the apo NBD core subdomain is illustrated in Fig. 4, A and B), which shows the transition of the core subdomain in simulation 2B and in Movie S1, A and B, which show the NBD dimer in simulations 2A and 2B, respectively. The plots of the time course of the solvent accessibility of each NBD also illustrate how the apo NBD becomes more accessible to the solvent as its core subdomain rotates away from the rest of the transporter (Fig. S2). It is notable that the transition does not progress to a stable state, and that opening and closing of the empty active site occurred.
Figure 3.
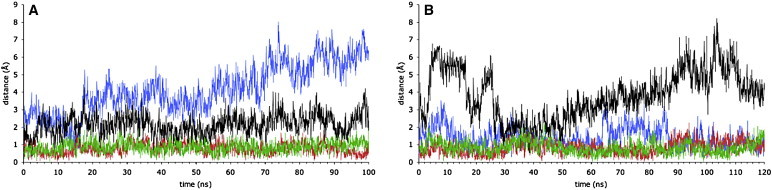
Conformational changes in the ATP/apo simulations. Time course of movement relative to the HDs of the opposite core subdomain and the proximal CH2. The core subdomain movement is monitored by the distance moved from the starting structure by the α-helix immediately C-terminal to the P-loop (Cα atoms of residues 380–387; see Fig. 1A). The CH2 movement is monitored by the distance moved from the starting structure by the Cα atoms of the CH2 α-helix (residues 210–217). Where trajectory frames were structurally aligned using coordinates of the HD (Cα atoms of residues 426–438 and residues 444–494) of monomer A, the distance moved by the core subdomain of monomer B is blue and by the CH2 of monomer B red. Where trajectory frames were aligned to the HD of monomer B, the distance moved by the core subdomain of monomer A is black and by the CH2 of monomer A, green, sampled every 50 ps (color online). (A) Simulation 2A, ATP bound in NBD monomer A. (B) Simulation 2B, ATP bound in NBD monomer B.
Figure 4.
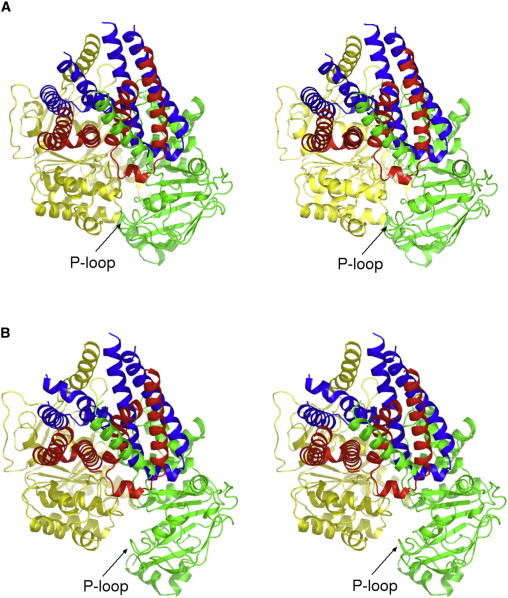
Opening of the apo active site via rotation of the core subdomain. Stereo pairs of the starting structure (A) and from the apo/ATP simulation 2B at t = 92 ns; (B) showing rotation of the core subdomain in the apo active site. ICLs 1 and 2 from monomer A are in red and ICLs 1 and 2 from monomer B are blue. ICL3 and the NBD from monomer A are in green and ICL3 and the NBD from monomer B are yellow. The core subdomain of the monomer A NBD (apo NBD) is in the bottom right quadrant with its P-loop indicated by an arrow. The view is from the membrane, angled to illustrate the movement of the P-loop away from the opposite NBD.
The apo NBD monomer moves along its fundamental dynamic mode to open the active site
Because the simulations were performed using a truncated structure, the observed conformational changes require further verification that they may be physiologically relevant. To that end, we performed an anisotropic network model (ANM (42)) analysis of the Sav1866 NBD monomer. ANM is a normal-mode analysis that is based on a coarse-grained model in which the protein is modeled as a set of points, corresponding to the Cα atom positions, connected to their nearest neighbors by simple harmonic springs (43). This model incorporates the geometry and distributions of interresidue contacts and determines the paths of least resistance to motion that are imprinted within the particular architecture. These structural transitions predominantly embody the protein's functional motions.
We plotted the projection of the motion described by the ANM mode 1 onto the trajectories of the Cα atoms for each NBD in both simulations (Fig. 5, A and B). The plots illustrate that in both simulations, the structure equilibrated to a conformation in which the apo NBD had moved significantly along the ANM global mode, whereas the ATP-bound NBD had not (simulation B was extended by 20 ns to ensure that this was indeed the case). This is also illustrated in Table S3, which shows for each simulation the percentage of all Cα atom fluctuations along the ANM mode 1. This indicates that for both simulations 2A and 2B, fluctuations along the ANM mode 1 are greater for the apo NBD compared with the ATP-bound NBD. Table S3 also shows the correlation between the global mode derived from PCA of the Cα atom trajectory from the simulations, which shows the largest concerted global motion occurring in the simulation, with the ANM mode 1. Again, in simulations 2A and 2B, the correlation of the PC mode 1 with the ANM mode 1 is greater for the apo NBD than for the ATP-bound NBD.
Figure 5.

Movement of the NBDs along the ANM global mode in the ATP/apo simulations. Projection of the ANM mode 1 on the Cα trajectory of the NBDs (residues 338–578) from the ATP/apo simulations. Monomer A, black, monomer B, red (color online). Projection is in nanometres. (A) Simulation 2A, ATP bound in NBD monomer A. (B) Simulation 2B, ATP bound in NBD monomer B.
To further illustrate the relationship between the ANM global mode and the conformational changes observed in the MD simulations, we produced a trajectory of one Sav1866 NBD monomer moving along its ANM global mode. We then structurally aligned the frames of this trajectory to one monomer in the Sav1866 NBD dimer structure using the coordinates of the HD. An animation was then made in which the opposite NBD monomer was static. This static monomer thus approximated the behavior of the ATP-bound monomer observed in the ATP/apo simulations in not moving significantly or sustainably along the ANM global mode. This animation shows clearly, when in situ with respect to the NBD dimer, how the motion of one NBD along its ANM global mode results in movement of its core subdomain away from the LSGGQ of the opposite NBD, whereas its HD remains engaged with the ATP in the opposite NBD and stationary, approximately as observed in our MD simulations of the ATP/apo state (Movie S2).
Both the ANM global mode and the conformational change that occurred in the apo monomers in the ATP/apo simulations appeared qualitatively similar to changes observed between ATP and apo structures of other ABC NBDs in both crystallographic structures (9,44–46) and MD simulations (31–33,37). This change is characterized by a movement of the HD relative to the core subdomain, such that the HD pivots about the loop at its C-terminus, moving the LSGGQ away from the P-loop (31). To examine this, we performed 100 ns unrestrained MD simulations of the apo and ATP-bound Sav1866 NBD monomer (residues 338–578). In these simulations, as in simulations of the ATP/apo complexes, the apo monomer underwent a greater change in conformation than the ATP-bound monomer (Fig. 6 A), becoming less compact (Fig. S3). The apo monomer also moved along ANM mode 1 significantly more than the ATP-bound monomer (Fig. 6 B and Table S3). To characterize the motion observed in the apo monomers in all simulations, we calculated the hinge axes of the rotation of the HD from the starting structure and compared them with the hinge axis of the rotation of the HD described by ANM mode 1. Fig. 6 C illustrates how the motions observed in the apo NBDs in the monomer and the ICL-NBD complexes are similar to the motion described by ANM mode 1, in that the hinge detection algorithm defines the core and HDs as moving relative to each other about an axis located approximately in the vicinity of the loop at the C-terminus of the HD.
Figure 6.
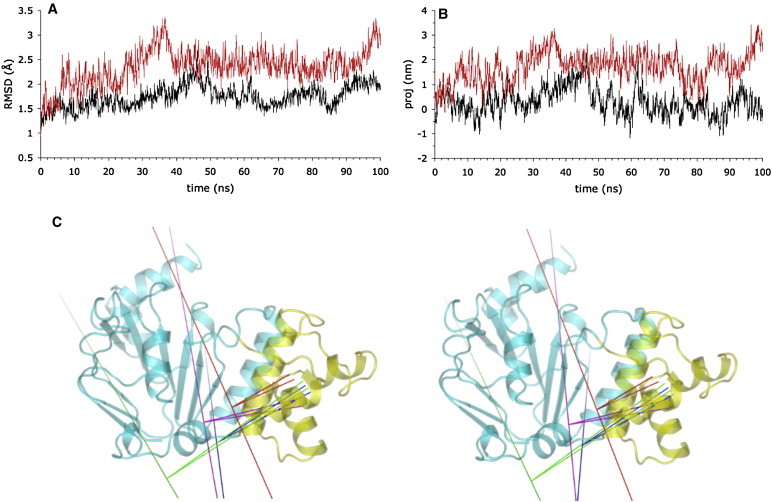
Conformational changes in the isolated ATP-bound and apo NBD. (A) RMSD of Cα atoms in the simulations of the isolated Sav1866 NBD monomer; ATP-bound (black), apo (red). (B) Projection of the ANM mode 1 on the Cα trajectory of the simulations of the isolated Sav1866 NBD monomer; ATP-bound (black), apo (red). Projection is in nanometres. (C) Comparison of rotation axes of the HD in the simulations and the ANM analysis. Stereo representation of the Sav1866 NBD with the HD depicted as the three differently shaded helices on the right side of each structure and the remainder of the NBD shaded differently, on the left and centre of each panel. The rotation axes of the HD are shown by long lines. The shorter lines connect the centre of mass of the rotating domain with the pivot point on the rotation axis. The pivot points are proximal to the loop joining the C-terminal of the HD to the core subdomain approximately at the bottom middle of the NBD. Principal component analysis of the Cα trajectories were performed and the hinge axes calculated between the maximum and minimum projection structures. ANM mode 1 (blue), apo NBD simulation 3B (purple), apo NBD simulation 2A (blue), apo NBD simulation 2A (green) (Color online.)
Discussion
The nature of the molecular processes whereby the cycle of ATP hydrolysis in the NBDs is coupled to the cycle of substrate binding and translocation in the TMDs is a central and unsolved conundrum in current ABC transporter research.
Recent findings concerning the occluded state of the nucleotide are likely to have relevance for understanding the molecular mechanism. Experiments with the Sav1866 homolog P-glycoprotein (ABCB1) showed that orthovanadate and beryllium fluoride trap nucleotide by forming stable complexes that mimic the catalytic transition state (47–49). In the trapped state, one nucleotide is tenaciously bound or occluded, whereas the opposite active site can bind nucleotide that is easily removed, and this site is not occluded (49). In addition, for ABCB1 in which hydrolysis is abrogated by mutation of the putative catalytic base in both active sites, ATP can be occluded in a conformation similar to that observed in trapping with transition-state analogs, with a stoichiometry of one occluded nucleotide per transporter (50,51). In recent studies, investigators found that they could also generate the occluded nucleotide conformation in wild-type ABCB1 using the slowly hydrolyzed ATP analog ATPγS, also with a stoichiometry of one occluded nucleotide per transporter (52,53). The ATPγS-bound ABCB1 was in an asymmetric conformation that could bind nucleotide at the unoccupied site with low affinity (53).
Although these enzymological studies established the existence of an asymmetric tight/loose state of the transporter in both ATP-bound and post-hydrolysis states, its structural basis is not known. The occluded site involves the engagement of the LSGGQ with the nucleotide (54) and additionally requires interaction with the TMDs (55,56). In Sav1866, the nucleotide is almost completely sequestered in a pocket formed by residues from both NBDs and the ICLs (Fig. S4), and appears likely to represent the occluded site in MDR exporters. In the trapped state, nucleotide can freely enter and exit the nonoccluded site; therefore, the LSGGQ and P-loop that comprise this site must be further apart than they are in the occluded site. In current models of NBD function, separation of the LSGGQ and P-loop is required to allow nucleotide exit and entry, and closure to form the tight hydrolysis-capable active site.
To investigate the structural and mechanistic basis of the tight/loose state of the transporter active sites, we performed unrestrained MD simulations of the Sav1866 structure. Although previous MD simulations of Sav1866 (40) and BtuCD (57) revealed asymmetries in the active sites of the ATP/ATP bound transporter, no significant opening of either site was reported. Access to the tight/loose state observed experimentally is probably not achievable with current MD simulations beginning with the ATP/ATP-bound state. In an attempt to access the tight/loose state, we performed simulations of the ATP/apo state. Our rationale for using this approach was that the loose site would be empty during the period between nucleotide exit and entry, which presumably would be a significant fraction of the time. In addition, because previous membrane-embedded MD simulations of Sav1866 (41) and BtuCD (58) did not reveal any significant active-site opening between the ATP/ATP and apo/apo states on the 100 ns timescale, only the predicted cytosolic regions of Sav1866 were simulated in this study. While enabling the ICLs to stabilize the NBD dimer, this strategy could also allow conformational changes in the NBDs to occur that might not otherwise occur on accessible timescales, due to their dependence on global changes involving the TMDs. In this regard, a recent experimental study indicated that rotation of the NBD HD is a part of a global conformational change in the maltose permease (59), and although HD rotation was reported in 10–40 ns simulations of isolated NBD monomers and dimers (31–33,37), it was not seen in 30–100 ns MD simulations of the membrane-embedded Sav1866 (40,41) and BtuCD transporters (57,58,60).
We ran a 100 ns simulation of the ATP/ATP-bound truncated structure to verify that the tight/loose state is not readily accessible from the ATP/ATP-bound state on this timescale or probably well beyond. In this simulation, both nucleotides remained tightly bound to the Walker A and B residues and the LSGGQ signature motif, whereas the core and HDs of the opposing NBDs maintained their spatial relationship. In contrast, in two 100 ns simulations of the ATP/apo structure, the core subdomain of the apo NBD moved significantly relative to the opposite HD, partially opening the active site, whereas the ATP remained occluded in the opposite active site. The opening of the apo site occurred by way of an intramonomer conformational change in the apo NBD in which the core subdomain rotated relative to the HD. The ATP-bound NBD remained relatively close to the starting structure, and less marked alterations between its core and HDs occurred. The HD of the apo NBD remained approximately in place with respect to the rest of the transporter, and remained engaged with the ATP in the opposite NBD. The internal rotation in the apo NBD thus resulted in its core subdomain moving away from the opposite LSGGQ and also away from the ICLs.
Support for the idea that the conformational change observed in the NBDs in the ATP/apo simulations is physiologically relevant, and not simply an artifact due to their interactions with truncated TMDs, comes from a number of sources. The NBD monomers in the ATP/apo simulations essentially behaved as the NBD in the isolated NBD simulations: the ATP-bound monomers did not undergo significant alterations with respect to the core and HDs, whereas all the apo monomers underwent similar significant changes in this respect. This suggests that the behavior of the NBDs observed in the ATP/apo simulations is intrinsic to the NBD structure and is largely influenced by its nucleotide-bound and unbound states. This idea is supported by the fact that the change in the apo monomers from the initial ATP-bound state in all simulations was qualitatively similar to that observed between ATP-bound and apo monomers of other ABC NBDs in both crystallographic analyses (9,44–46) and MD simulations (31–33,37). The EN analysis strengthens the idea that the motion observed in the apo monomer is fundamental and intrinsic to its architecture. Projection of the ANM global mode on all NBD monomer trajectories revealed that motion along this mode was consistently greater in the apo state than in the ATP-bound state, suggesting that ATP-binding stops motion along this mode (Figs. 4 and 5 B). In the ATP/apo NBD dimer, this leads to a similar movement of the core subdomain of the apo monomer away from the opposite monomer's LSGGQ, as observed in the simulations (Movie S1 and Movie S2).
The changes observed in the ATP/apo simulations are consistent with experimental data. Although the opening of the apo active site does not appear sufficient to allow nucleotide binding, the manner of its opening, in allowing ATP to remain sequestered in the opposite site, indicates that it is partway toward forming the occluded nucleotide state, as discussed above (51,53). The final conformation of the ATP/apo state is consistent with the finding for beryllium fluoride-trapped ABCB1, which is expected to be close to the prehydrolysis ATP-bound state, in that the unoccupied binding site is in a hydrophilic environment, whereas the occluded site is hydrophobic (49). In addition, changes in the relative orientation of the core and HDs between the apo and ATP-bound state in a functional ABC transporter were recently demonstrated in experiments with the maltose permease system (59).
Rotation of the core subdomain relative to the CHs in the ATP/apo simulations is consistent with a growing body of evidence indicating that the NBD/TMD interface alters during the catalytic cycle. Such dynamic changes have been observed experimentally between the NBDs and the CHs for MalK (61), TAP (62), P-glycoprotein (ABCB1) (63), CFTR (64), LmrA (65), and BmrA (66). The results of the ATP/apo simulations are supported by cysteine cross-linking experiments with ABCB1 (63) that suggested that, while CH2 remains in contact with the cleft between the helical and core subdomains, nucleotides affected the ability of residues located in the core subdomain and CH2 to cross-link. In addition, the MD simulations indicate that the ATP/apo state involves a dynamic opening and closing of the empty active site (Fig. 3, A and B). These dynamic changes are consistent with NMR data for the bacterial MDR LmrA, which showed that the NBDs are highly dynamic relative to the ICLs in the unliganded state (65). Moreover, the observation in the ATP/apo simulations that the P-loop and downstream α-helix were mobile in the apo NBD core subdomain while the opposite ATP-bound NBD remained relatively static is consistent with EPR experiments with MsbA, which consistently showed the existence of both immobile and mobile components in the spectra of spin-label pairs located in the equivalent regions in the ATP-bound state (67).
The structural basis of the tight/loose state of the occluded ABC transporter is pertinent for elucidating the molecular mechanism because the biochemical data suggest that it spans the crucial step between the reactant and product state of the hydrolysis of at least one ATP molecule. It might be suggested that, relative to the ATP/ATP sandwich dimer, this could occur by rigid-body movements of the NBDs, or simply by cracking of the NBD dimer. However, some flexibility in the NBD monomers would be required to maintain the engagement of the LSGGQ with the ATP in the occluded site, as observed in ATP/ATP sandwich dimers, and which is expected to be required for hydrolysis to occur. Alternatively, because the HD is known to be flexibly attached to the core subdomain, the HD of the ATP-bound monomer could rotate away from the opposite unoccupied core subdomain to allow nucleotide to enter and exit the nonoccluded site. However, crystallographic analyses and MD simulations of isolated NBD dimers and monomers, as well as whole ABC transporters, indicate that ATP holds the NBD monomer in a closed state with respect to rotation of the HD. In contrast, structural and theoretical analyses have shown that the apo NBD monomer is in a more open conformation in which the HD is rotated outward (9,31–33,37,44–46). Here the data indicate that the conformations of the closed ATP-bound and open apo NBD monomer are characteristically and inherently distinct, in particular with respect to the core and helical subdomains. We suggest that this accounts for the tight/loose or occluded state of the transporter by promoting rotation of the core subdomain of the apo NBD to open the unoccupied active site while the opposite ATP-bound site remains occluded. Finally, it should be noted that, while the protein will equilibrate to the lowest energy conformation for a particular state, the transition between any two states may not correspond to one occurring usually in the catalytic cycle.
Acknowledgments
This work was supported by a grant from the Australian Partnership for Advanced Computing, and performed at the NCI National Facility in Canberra, Australia.
Supporting Material
References
- 1.Higgins C.F. ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 2.Holland I.B., Blight M.A. ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol. 1999;293:381–399. doi: 10.1006/jmbi.1999.2993. [DOI] [PubMed] [Google Scholar]
- 3.Aravind L., Walker D.R., Koonin E.V. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999;27:1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones P.M., O'Mara M.L., George A.M. ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends Biochem. Sci. 2009;34:520–531. doi: 10.1016/j.tibs.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Dean M., Rzhetsky A., Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–1166. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 6.Jones P.M., George A.M. The ABC transporter structure and mechanism: perspectives on recent research. Cell. Mol. Life Sci. 2004;61:682–699. doi: 10.1007/s00018-003-3336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toyoda Y., Hagiya Y., Ishikawa T. MRP class of human ATP binding cassette (ABC) transporters: historical background and new research directions. Xenobiotica. 2008;38:833–862. doi: 10.1080/00498250701883514. [DOI] [PubMed] [Google Scholar]
- 8.Jones P.M., George A.M. Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol. Lett. 1999;179:187–202. doi: 10.1111/j.1574-6968.1999.tb08727.x. [DOI] [PubMed] [Google Scholar]
- 9.Smith P.C., Karpowich N., Hunt J.F. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Locher K.P., Lee A.T., Rees D.C. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 11.Dawson R.J., Locher K.P. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 12.Pinkett H.W., Lee A.T., Rees D.C. An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science. 2007;315:373–377. doi: 10.1126/science.1133488. [DOI] [PubMed] [Google Scholar]
- 13.Hollenstein K., Frei D.C., Locher K.P. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446:213–216. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- 14.Oldham M.L., Khare D., Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 15.Gerber S., Comellas-Bigler M., Locher K.P. Structural basis of trans-inhibition in a molybdate/tungstate ABC transporter. Science. 2008;321:246–250. doi: 10.1126/science.1156213. [DOI] [PubMed] [Google Scholar]
- 16.Kadaba N.S., Kaiser J.T., Rees D.C. The high-affinity E. coli methionine ABC transporter: structure and allosteric regulation. Science. 2008;321:250–253. doi: 10.1126/science.1157987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Velamakanni S., Yao Y., van Veen H.W. Multidrug transport by the ABC transporter Sav1866 from Staphylococcus aureus. Biochemistry. 2008;47:9300–9308. doi: 10.1021/bi8006737. [DOI] [PubMed] [Google Scholar]
- 18.Kale L., Skeel R., Shulten K. NMAD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999;151:283–312. [Google Scholar]
- 19.MacKerell A.D., Bashford D., Bellott R.L., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 20.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 21.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 22.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 23.Darden T., York D., Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 24.Wriggers W., Schulten K. Protein domain movements: detection of rigid domains and visualization of hinges in comparisons of atomic coordinates. Proteins. 1997;29:1–14. [PubMed] [Google Scholar]
- 25.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–38. [DOI] [PubMed] [Google Scholar]
- 26.Schwieters C.D., Kuszewski J.J., Clore G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 27.Mezei M. Simulaid: a simulation facilitator and analysis program. J. Comput. Chem. 2010;31:2658–2668. doi: 10.1002/jcc.21551. [DOI] [PubMed] [Google Scholar]
- 28.Haliloglu T., Bahar I., Erman B. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 1997;79:3090. [Google Scholar]
- 29.Haubertin D.Y., Madaoui H., Orlowski S. Molecular dynamics simulations of E. coli MsbA transmembrane domain: formation of a semipore structure. Biophys. J. 2006;91:2517–2531. doi: 10.1529/biophysj.106.084020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun T., Liu M., Wang C. Molecular dynamics simulation of the transmembrane subunit of BtuCD in the lipid bilayer. Sci. China. Life Sci. 2010;53:620–630. doi: 10.1007/s11427-010-0103-7. [DOI] [PubMed] [Google Scholar]
- 31.Jones P.M., George A.M. Nucleotide-dependent allostery within the ABC transporter ATP-binding cassette: a computational study of the MJ0796 dimer. J. Biol. Chem. 2007;282:22793–22803. doi: 10.1074/jbc.M700809200. [DOI] [PubMed] [Google Scholar]
- 32.Jones P.M., George A.M. Mechanism of ABC transporters: a molecular dynamics simulation of a well characterized nucleotide-binding subunit. Proc. Natl. Acad. Sci. USA. 2002;99:12639–12644. doi: 10.1073/pnas.152439599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campbell J.D., Deol S.S., Sansom M.S. Nucleotide-dependent conformational changes in HisP: molecular dynamics simulations of an ABC transporter nucleotide-binding domain. Biophys. J. 2004;87:3703–3715. doi: 10.1529/biophysj.104.046870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell J.D., Sansom M.S. Nucleotide binding to the homodimeric MJ0796 protein: a computational study of a prokaryotic ABC transporter NBD dimer. FEBS Lett. 2005;579:4193–4199. doi: 10.1016/j.febslet.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 35.Oloo E.O., Fung E.Y., Tieleman D.P. The dynamics of the MgATP-driven closure of MalK, the energy-transducing subunit of the maltose ABC transporter. J. Biol. Chem. 2006;281:28397–28407. doi: 10.1074/jbc.M513614200. [DOI] [PubMed] [Google Scholar]
- 36.Wen P.C., Tajkhorshid E. Dimer opening of the nucleotide binding domains of ABC transporters after ATP hydrolysis. Biophys. J. 2008;95:5100–5110. doi: 10.1529/biophysj.108.139444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones P.M., George A.M. Opening of the ADP-bound active site in the ABC transporter ATPase dimer: evidence for a constant contact, alternating sites model for the catalytic cycle. Proteins. 2009;75:387–396. doi: 10.1002/prot.22250. [DOI] [PubMed] [Google Scholar]
- 38.Newstead S., Fowler P.W., Iwata S. Insights into how nucleotide-binding domains power ABC transport. Structure. 2009;17:1213–1222. doi: 10.1016/j.str.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliveira A.S., Baptista A.M., Soares C.M. Insights into the molecular mechanism of an ABC transporter: conformational changes in the NBD dimer of MJ0796. J. Phys. Chem. B. 2010;114:5486–5496. doi: 10.1021/jp905735y. [DOI] [PubMed] [Google Scholar]
- 40.Aittoniemi J., de Wet H., Sansom M.S. Asymmetric switching in a homodimeric ABC transporter: a simulation study. PLOS Comput. Biol. 2010;6:e1000762. doi: 10.1371/journal.pcbi.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Becker J.-P., Van Bambeke F., Prévost M. Dynamics and structural changes induced by ATP binding in SAV1866, a bacterial ABC exporter. J. Phys. Chem. B. 2010;114:15948–15957. doi: 10.1021/jp1038392. [DOI] [PubMed] [Google Scholar]
- 42.Atilgan A.R., Durell S.R., Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bahar I., Atilgan A.R., Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 44.Chen J., Lu G., Quiocho F.A. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell. 2003;12:651–661. doi: 10.1016/j.molcel.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Karpowich N., Martsinkevich O., Hunt J.F. Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure. 2001;9:571–586. doi: 10.1016/s0969-2126(01)00617-7. [DOI] [PubMed] [Google Scholar]
- 46.Zaitseva J., Oswald C., Schmitt L. A structural analysis of asymmetry required for catalytic activity of an ABC-ATPase domain dimer. EMBO J. 2006;25:3432–3443. doi: 10.1038/sj.emboj.7601208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Urbatsch I.L., Sankaran B., Senior A.E. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 1995;270:19383–19390. doi: 10.1074/jbc.270.33.19383. [DOI] [PubMed] [Google Scholar]
- 48.Sankaran B., Bhagat S., Senior A.E. Inhibition of P-glycoprotein ATPase activity by beryllium fluoride. Biochemistry. 1997;36:6847–6853. doi: 10.1021/bi970034s. [DOI] [PubMed] [Google Scholar]
- 49.Russell P.L., Sharom F.J. Conformational and functional characterization of trapped complexes of the P-glycoprotein multidrug transporter. Biochem. J. 2006;399:315–323. doi: 10.1042/BJ20060015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sauna Z.E., Müller M., Ambudkar S.V. Importance of the conserved Walker B glutamate residues, 556 and 1201, for the completion of the catalytic cycle of ATP hydrolysis by human P-glycoprotein (ABCB1) Biochemistry. 2002;41:13989–14000. doi: 10.1021/bi026626e. [DOI] [PubMed] [Google Scholar]
- 51.Tombline G., Bartholomew L.A., Senior A.E. Properties of P-glycoprotein with mutations in the “catalytic carboxylate” glutamate residues. J. Biol. Chem. 2004;279:46518–46526. doi: 10.1074/jbc.M408052200. [DOI] [PubMed] [Google Scholar]
- 52.Sauna Z.E., Kim I.W., Ambudkar S.V. Catalytic cycle of ATP hydrolysis by P-glycoprotein: evidence for formation of the E.S reaction intermediate with ATP-γ-S, a nonhydrolyzable analogue of ATP. Biochemistry. 2007;46:13787–13799. doi: 10.1021/bi701385t. [DOI] [PubMed] [Google Scholar]
- 53.Siarheyeva A., Liu R., Sharom F.J. Characterization of an asymmetric occluded state of P-glycoprotein with two bound nucleotides: implications for catalysis. J. Biol. Chem. 2010;285:7575–7586. doi: 10.1074/jbc.M109.047290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fetsch E.E., Davidson A.L. Vanadate-catalyzed photocleavage of the signature motif of an ATP-binding cassette (ABC) transporter. Proc. Natl. Acad. Sci. USA. 2002;99:9685–9690. doi: 10.1073/pnas.152204499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hunke S., Döse S., Schneider E. Vanadate and bafilomycin A1 are potent inhibitors of the ATPase activity of the reconstituted bacterial ATP-binding cassette transporter for maltose (MalFGK2) Biochem. Biophys. Res. Commun. 1995;216:589–594. doi: 10.1006/bbrc.1995.2663. [DOI] [PubMed] [Google Scholar]
- 56.Davidson A.L., Laghaeian S.S., Mannering D.E. The maltose transport system of Escherichia coli displays positive cooperativity in ATP hydrolysis. J. Biol. Chem. 1996;271:4858–4863. [PubMed] [Google Scholar]
- 57.Oloo E.O., Tieleman D.P. Conformational transitions induced by the binding of MgATP to the vitamin B12 ATP-binding cassette (ABC) transporter BtuCD. J. Biol. Chem. 2004;279:45013–45019. doi: 10.1074/jbc.M405084200. [DOI] [PubMed] [Google Scholar]
- 58.Kandt C., Tieleman D.P. Holo-BtuF stabilizes the open conformation of the vitamin B12 ABC transporter BtuCD. Proteins. 2010;78:738–753. doi: 10.1002/prot.22606. [DOI] [PubMed] [Google Scholar]
- 59.Orelle C., Alvarez F.J., Davidson A.L. Dynamics of α-helical subdomain rotation in the intact maltose ATP-binding cassette transporter. Proc. Natl. Acad. Sci. USA. 2010;107:20293–20298. doi: 10.1073/pnas.1006544107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sonne J., Kandt C., Tieleman D.P. Simulation of the coupling between nucleotide binding and transmembrane domains in the ATP binding cassette transporter BtuCD. Biophys. J. 2007;92:2727–2734. doi: 10.1529/biophysj.106.097972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daus M.L., Grote M., Schneider E. ATP-driven MalK dimer closure and reopening and conformational changes of the “EAA” motifs are crucial for function of the maltose ATP-binding cassette transporter (MalFGK2) J. Biol. Chem. 2007;282:22387–22396. doi: 10.1074/jbc.M701979200. [DOI] [PubMed] [Google Scholar]
- 62.Oancea G., O'Mara M.L., Tampé R. Structural arrangement of the transmission interface in the antigen ABC transport complex TAP. Proc. Natl. Acad. Sci. USA. 2009;106:5551–5556. doi: 10.1073/pnas.0811260106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zolnerciks J.K., Wooding C., Linton K.J. Evidence for a Sav1866-like architecture for the human multidrug transporter P-glycoprotein. FASEB J. 2007;21:3937–3948. doi: 10.1096/fj.07-8610com. [DOI] [PubMed] [Google Scholar]
- 64.Serohijos A.W., Hegedus T., Dokholyan N.V. Diminished self-chaperoning activity of the DeltaF508 mutant of CFTR results in protein misfolding. PLOS Comput. Biol. 2008;4:e1000008. doi: 10.1371/journal.pcbi.1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Siarheyeva A., Lopez J.J., Glaubitz C. Probing the molecular dynamics of the ABC multidrug transporter LmrA by deuterium solid-state nuclear magnetic resonance. Biochemistry. 2007;46:3075–3083. doi: 10.1021/bi062109a. [DOI] [PubMed] [Google Scholar]
- 66.Dalmas O., Orelle C., Jault J.M. The Q-loop disengages from the first intracellular loop during the catalytic cycle of the multidrug ABC transporter BmrA. J. Biol. Chem. 2005;280:36857–36864. doi: 10.1074/jbc.M503266200. [DOI] [PubMed] [Google Scholar]
- 67.Buchaklian A.H., Klug C.S. Characterization of the Walker A motif of MsbA using site-directed spin labeling electron paramagnetic resonance spectroscopy. Biochemistry. 2005;44:5503–5509. doi: 10.1021/bi047568v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.