

Abstract
Pulmonary arterial hypertension (PAH) is a disorder of the pulmonary vasculature associated with elevated pulmonary vascular resistance. Despite recent advances in the treatment of PAH, with eight approved clinical therapies and additional therapies undergoing clinical trials, PAH remains a serious life-threatening condition. The lack of pulmonary vascular selectivity and associated systemic adverse effects of these therapies remain the main obstacles to successful treatment. Peptide-mediated drug delivery that specifically targets the vasculature of PAH lungs may offer a solution to the lack of drug selectivity. Herein, we show highly selective targeting of rat PAH lesions by a novel cyclic peptide, CARSKNKDC (CAR). Intravenous administration of CAR peptide resulted in intense accumulation of the peptide in monocrotaline-induced and SU5416/hypoxia-induced hypertensive lungs but not in healthy lungs or other organs of PAH rats. CAR homed to all layers of remodeled pulmonary arteries, ie, endothelium, neointima, medial smooth muscle, and adventitia, in the hypertensive lungs. CAR also homed to capillary vessels and accumulated in the interstitial space of the PAH lungs, manifesting its extravasation activity. These results demonstrated the remarkable ability of CAR to selectively target PAH lung vasculature and effectively penetrate and spread throughout the diseased lung tissue. These results suggest the clinical utility of CAR in the targeted delivery of therapeutic compounds and imaging probes to PAH lungs.
Pulmonary arterial hypertension (PAH) is a disease of the pulmonary vasculature defined by elevated pulmonary vascular resistance, which eventually leads to right heart failure and premature death. The cause of this disease remains unknown. PAH can be idiopathic or associated with other conditions or exposures (secondary pulmonary hypertension), including connective tissue diseases, HIV infection, portal hypertension, and anorexigenic drug ingestion. Most cases of severe PAH, especially idiopathic PAH, are associated with aberrant proliferation of pulmonary arterial endothelial cells and smooth muscle cells, leading to narrowing or even obliteration of the precapillary pulmonary vessel lumen.1 Thus, intimal and medial thickening of resistant arteries is typically found in the lesions. The adventitia is often markedly remodeled in patients with certain forms of collagen vascular diseases associated with severe PAH, most notably scleroderma.2 Inflammatory mechanisms seem to play a significant role in the pathogenesis and progression of PAH. The involvement of leukocytes, such as macrophages and lymphocytes, in complex lesions of PAH has been described.3,4
The current therapeutic approaches to PAH consist of the administration of a variety of systemic vasodilators, which reduce pulmonary vascular resistance in some patients. However, the systemic use of vasodilators can produce adverse effects, such as hypotension, impaired intrapulmonary gas exchange, depressed cardiac function, and even death.5 Drugs such as endothelin receptor antagonist may cause serious liver damage.5 These limitations of the current PAH regimens necessitate the development of target-specific interventions.6
Delivering drugs to a diseased tissue by coupling the drug to a compound that specifically binds at the target tissue (synaphic targeting) can overcome the limitations of nonselective drug activity.7,8 Such a technology would provide significant therapeutic advantages, such as concentrating the drug at the targeted site, which increases efficacy while decreasing adverse effects in other tissues. However, to our knowledge, no compounds that specifically bind to the blood vessels in PAH lungs are known. We set out to develop such a compound.
We have been using in vivo screening of phage peptide libraries to identify specific molecular markers in the vasculature in different organs and diseased tissues. The “molecular zip codes” revealed by these screens can be used in organ-specific or lesion-specific delivery of systemically administered diagnostics and therapeutics.7 For example, αv integrins are highly expressed in tumor vasculature, where they have been accessed with peptides that contain the RGD integrin recognition motif to deliver drugs, biologicals, viruses, and nanoparticles to tumor vasculature.8
Previously, we identified a cyclic peptide, CARSKNKDC (CAR), via in vivo screening of a phage peptide library for homing to angiogenic blood vessels during wound repair.9 CAR accumulates in tendon and skin wounds in rats and mice with excellent selectivity, suggesting that the peptide targets the vasculature of injured tissues/lesions.9,10 CAR specifically binds to heparin, and its binding to angiogenic endothelial cells and tumor cells requires the glycosaminoglycan heparan sulfate,9 suggesting that this peptide recognizes a specific form of heparan sulfate in the target tissues. Because endothelial activation is associated with PAH,4,11 we hypothesized that the CAR peptide may be useful for targeting vascular lesions in the diseased lung. Herein, we show that CAR enables highly effective and selective targeting of PAH lesions.
Materials and Methods
Animals
Monocrotaline-Induced PAH Model
Monocrotaline (MCT; Sigma-Aldrich Corp., St. Louis, MO) was dissolved in 0.3 mol/L hydrochloride solution and neutralized with 0.3 mol/L sodium hydroxide solution (approximate adjusted pH 7.0). Adult male Sprague-Dawley rats (150 to 200 g; Harlan Laboratories, Indianapolis, IN) were administered a single s.c. injection of MCT solution, 60 mg/kg body weight, and control rats were administered 0.9% saline.12 Rats were randomly selected and examined for peptide targeting at 1, 3, 7, 14, or 21 days after treatment with MCT.
SU5416/Hypoxia-Induced PAH Model
Adult male Sprague-Dawley rats (approximately 200 g) were injected s.c. with SU5416 (20 mg/kg body weight; SUGEN Inc., South San Francisco, CA) suspended in carboxymethylcellulose (0.5% carboxymethylcellulose sodium, 0.9% sodium chloride, 0.4% polysorbate, and 0.9% benzyl alcohol in deionized water). The rats were then exposed to chronic hypoxia in a hypobaric chamber (barometric pressure, 410 mmHg: inspired O2 tension, 76 mmHg) for 3 weeks followed by an additional 6 weeks of reexposure to normoxia. Rats were examined 9 weeks after the injection of SU5416, when they develop severe occlusive pulmonary arterial lesions.13
Peptide Targeting Study
The following peptides were labeled with 5-carboxyfluorescein (FAM) and were used for the lung targeting studies: CAR, CARSKNKDC; vascular cell adhesion molecule 1 (VCAM-1), CVHSPNKKCGGSK; CG7C control, CGGGGGGGC; and CAR mutant, CAQSNNKDC. Peptides were dissolved in PBS at concentrations of 0.5 mg/mL. Hypertensive rats were injected with peptide solution through the tail vein (3.3 mg/kg). Two hours after injection, the rats were perfused with PBS containing 1% bovine serum albumin while under deep anesthesia, and tissues were fixed by systemic perfusion with 10% buffered formalin. The organs were excised and fixed for an additional 24 hours and were processed for anti-fluorescein immunohistochemical (IHC) analysis.
IHC Analysis
To determine the localization of the peptides, paraffin-embedded tissue sections were immunostained with rabbit anti-fluorescein isothiocyanate (FITC) antibody (Invitrogen, Carlsbad, CA) followed by horseradish peroxidase–labeled anti-rabbit IgG secondary antibody. The peptide localization was then visualized by diaminobenzidine. Sections were counterstained with hematoxylin to visualize the region of cells. A Discovery XT automated staining system (Ventana Medical Systems Inc., Tucson, AZ) was used for staining. To examine the co-localization of the peptides with macrophages or alveolar type II cells, tissue sections were double stained with rabbit anti-FITC antibody and mouse anti-rat CD68 antibody (ED1; AbD Serotec, Raleigh, NC) or rabbit anti-prosurfactant protein C (Millipore, Temecula, CA), respectively. For FITC/anti-prosurfactant protein C double staining, FITC staining was completed first. The tissue sections were then treated with citrate buffer (pH 6.0) at 98°C for 10 minutes before proceeding to anti-prosurfactant protein C staining. Biotinylated species-specific secondary antibodies and alkaline phosphatase-labeled streptavidin (Vector Laboratories, Burlingame, CA) were used to visualize macrophages and alveolar type II cells with Permanent Red (Dako, Carpinteria, CA).
Data Analysis
Images of stained slides were observed and captured using an ECLIPSE 90i or 80i microscope with a CCD camera DS-5M (Nikon Instruments, Melville, NY). To quantify the targeting efficiency of the peptides to the lung, the immunostained sections were scanned using an Aperio ScanScope XT system and were analyzed using ImageScope software (Aperio Technologies Inc., Vista, CA). For the quantification of area and density of peptide staining, a color deconvolution tool (Aperio Technologies Inc.) was used to compare total peptide-positive area and intensity of peptide staining in each animal. The thresholds were set empirically for strong, medium, and weak staining. The area of each level of staining was presented as a percentage of such area in the total cell area. The total stained area was normalized by hematoxylin-stained areas of the same regions.
Results
Specific Accumulation of CAR in MCT-Induced PAH Lesions
PAH was induced in the rat by s.c. injection of MCT, which resulted in considerable remodeling of the pulmonary vasculature approximately 3 to 4 weeks later (see Supplemental Figure S1 at http://ajp.amjpathol.org). FAM-labeled CAR peptide or the control FAM-CG7C peptide was i.v. injected into these animals to determine peptide homing to the lung lesions. The strong green autofluorescence of the lungs made it difficult to accurately interpret the data of direct FAM imaging. For this reason, and to preserve the histologic features, we used IHC staining with anti-fluorescein antibodies to detect the FAM-labeled peptides.
In this analysis, we found extensive accumulation of CAR in PAH lungs (Figures 1 and 2). CAR accumulated in the endothelium and medial smooth muscle of pulmonary arteries (Figure 2A). The accumulation of CAR was also prominent at the adventitia of pulmonary arteries (Figures 1A and 2C). In addition, capillary vessel walls and infiltrating macrophages were strongly positive for CAR staining (Figure 2, B and E). Furthermore, CAR accumulated significantly in the extravascular space (interstitial space and alveolar lumen) of the PAH lungs (Figure 1G), indicating that CAR penetrates through the vessel wall and binds to macrophages and extracellular matrix components deposited in the injured lung; the putative receptors for CAR are heparan sulfate proteoglycans upregulated at the site of tissue injury.9 Staining with a type II alveolar cell marker indicates that these cells are also positive for CAR in MCT-treated hypertensive rats (Figure 2G). In comparison, little accumulation of CAR was detected in the normal lung of non–MCT-treated rats (Figure 1, D and I). Moreover, only very weak staining was detected for the control CG7C peptide in the MCT-treated lungs (Figures 1, C and I, and 2D). These results demonstrate the specificity of CAR targeting to PAH lung lesions.
Figure 1.
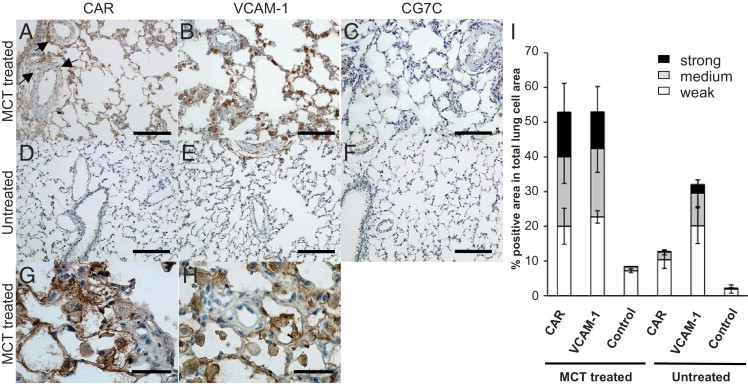
Accumulation of targeting peptides in rat lung tissue. A–H: Peptide distribution in hypertensive and healthy lungs. Twenty-one days after MCT induction of PAH, MCT-treated rats (A–C) and untreated control rats (D–F) were injected with FAM-conjugated targeting peptides via the tail vein (3.3 mg/kg). Two hours after peptide administration, the lungs were perfused, excised, and processed for IHC analysis. The accumulation of FAM-conjugated CAR peptide (A and D), VCAM-1 homing peptide (B and E), and CG7C control peptide (C and F) was detected by anti-fluorescein immunostaining. G and H: MCT-treated rats with CAR or VCAM-1 homing peptide in higher magnification. Arrows indicate homing of CAR to adventitia of pulmonary arteries. Scale bars: 100 μm (A–F); 50 μm (G and H). I: Lung homing efficiency and specificity of targeting peptides. Immunostained lung sections were analyzed for MCT-treated and untreated rats using the Aperio color deconvolution tool to quantify staining intensity. The peptide-positive area relative to the total lung cell area is shown. Three detection thresholds were set to determine weak to strong accumulations of the peptides. Multiple sections were analyzed for each group. Data are given as mean ± SD (error bars).
Figure 2.
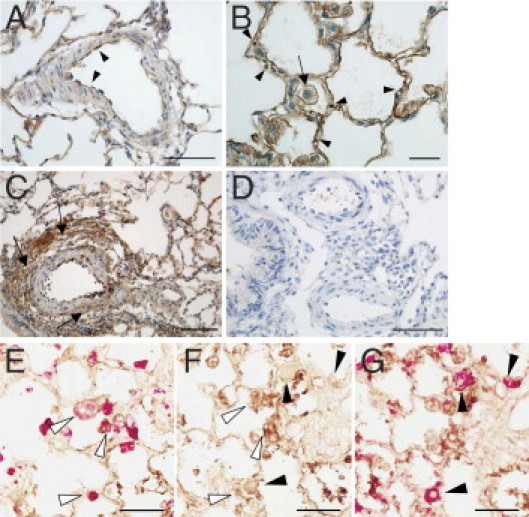
CAR accumulation in the lung vasculature. A: Strong accumulation of CAR at the pulmonary arterial endothelium (arrowheads). Accumulation of CAR was also found in the medial smooth muscle layers of pulmonary arteries and arterioles. B: High-magnification image of CAR accumulation at capillary vessel walls. Circulating red blood cells in the capillaries (arrowheads) and interstitial macrophage (arrow) are indicated. C: The site of extensive adventitial remodeling shows intense accumulation of CAR (arrows). D: The absence of CG7C control peptide accumulation in MCT-treated rat pulmonary arteries. E–G: Accumulation of CAR in macrophages and alveolar type II cells. Sequential sections were stained with anti-FITC antibody to visualize CAR peptide (brown). Infiltrated macrophages or alveolar type II cells were probed with anti-ED1 (E) or anti-prosurfactant protein C (G) antibodies, respectively, and were visualized by Permanent Red (red). Staining with anti-FITC antibody only (F). Co-localization of CAR with infiltrated macrophages (open arrowheads in E and F) and alveolar type II cells (closed arrowheads in F and G) is observed in the hypertensive lungs of MCT-treated animals. Scale bars: 50 μm (A, D, and E–G); 20 μm (B); 100 μm (C).
We next examined the PAH targeting activity of VCAM-1–binding peptide (VHSPNKK)14,15 to compare with that of CAR. This peptide has been shown to target atherosclerotic plaques in ApoE−/− mice. VCAM-1 is highly expressed in inflammatory endothelium.16,17 Clinical data indicate that this is also true for the activated endothelium in PAH,11 suggesting that VCAM-1 may be useful in PAH targeting. VCAM-1–binding peptide accumulated in PAH lungs to a similar extent as did CAR (Figure 1, B, H, and I). However, most of the peptide accumulation was in infiltrating macrophages and/or type II alveolar cells, and the vasculature was less positive (Figure 1, B and H). A fair amount of the VCAM-1–binding peptide also accumulated in normal lungs, indicating the lack of specificity to the diseased lung, which was in contrast to the results of CAR (Figure 1, E and I). These observations demonstrate the excellent PAH selectivity and targeting ability of CAR.
Spatiotemporal Pattern of PAH Targeting by CAR
During PAH lesion development, weak to moderate accumulation of CAR was detected in a few small discrete areas in the injured lung 3 days after injury (data not shown). This accumulation was absent 7 days after injury, suggesting that CAR homes to acute lung lesions. More significant and broad accumulation of CAR to the pulmonary vasculature was detected 2 weeks after injury, and the intensity of the accumulation significantly increased in the chronic lesions in the third week (Figure 3A). The temporal pattern of CAR homing coincided with the progressive remodeling of the pulmonary vasculature and with an increase in the number of infiltrating macrophages.12 These observations demonstrate that lung targeting by CAR is dependent on formation of the lung lesions. Despite the extensive homing of CAR to the injured lung, accumulation in other organs was absent or very weak, except in the kidney (Figure 3B). Peptides administered i.v. get excreted through the kidney. Thus, CAR accumulation in the kidney was observed in MCT-treated PAH rats and in untreated healthy animals. The control peptide also accumulated in the kidney (see Supplemental Figure S2 at http://ajp.amjpathol.org). Little accumulation in healthy lungs and other organs further supports that CAR is useful for selectively targeting PAH lesions.
Figure 3.
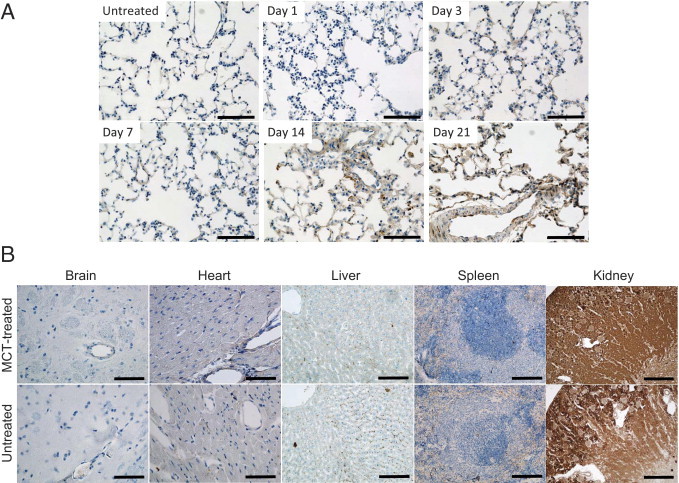
Lung homing of CAR during hypertensive lesion formation and distribution of the peptide in other organs. A: Targeting of CAR peptide to a developing pulmonary lesion. FAM-labeled CAR was injected into the tail vein at the indicated time points after MCT induction of PAH. Two hours after the injection of peptide, rats were sacrificed and lung tissues were processed for IHC analysis as described in Materials and Methods. No significant accumulation of CAR peptide was detected in the control lung (untreated). Extensive vascular remodeling and many interstitial macrophages were observed between days 14 and 21. Coinciding with pulmonary lesion formation, CAR accumulation increased significantly and progressively from days 14 to 21. Scale bars = 100 μm. B: Tissue distribution of CAR. CAR was i.v. administered to rats 21 days after MCT induction of PAH. Various organs were collected 2 hours later and were processed for IHC analysis. Tissue sections of brain (striatum), heart, liver, spleen, and kidney from MCT-treated rats and untreated control rats were stained with anti-fluorescein antibody. Scale bars: 50 μm (brain and heart); 100 μm (liver and spleen); 500 μm (kidney).
CAR Targets Vascular Endothelial Growth Factor Receptor Inhibition/Hypoxia-Induced PAH
To examine whether CAR also exhibits selective targeting to PAH lesions induced by a different mechanism, we used another rat PAH model: severe occlusive disease induced by the vascular endothelial growth factor receptor blocker SU5416 and hypoxia.13 After a single subcutaneous injection of SU5416, rats were exposed to hypoxia for 3 weeks followed by an additional 6 weeks of reexposure to normoxia. Rats were examined 9 weeks after the injection of SU5416, when they develop severe occlusive pulmonary arterial lesions. In this model, sustained pulmonary hypertension is accompanied by the formation of complex plexiform lesions in addition to the medial wall thickening and neointima formation, thus well recapitulating pulmonary lesion development in the pulmonary arteriopathy of human PAH.13 The i.v. CAR administration resulted in prominent accumulation of the peptide throughout PAH lungs (Figure 4). CAR accumulation was detected at high intensity in all layers of remodeled pulmonary arteries, ie, endothelium, neointima, media, and adventitia. Furthermore, CAR-positive lesions included not only the endothelium and thickening medial wall but plexiform-like lesions and occluded arterioles. In contrast, only negligible signal was detected in the PAH lesion when CAR mutant, an inactive CAR derivative with site-directed mutations (CAQSNNKDC), was tested, demonstrating the specificity of CAR homing. Thus, CAR exhibited specific targeting to various forms of pulmonary arterial lesions in two distinct PAH models.
Figure 4.
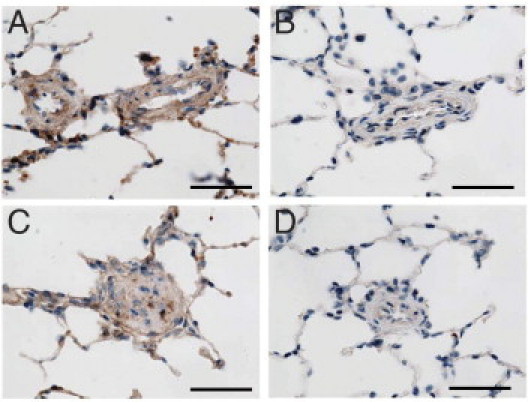
CAR peptide accumulates in SU5416/hypoxia-induced PAH lesions. PAH was induced in rats by a single s.c. injection of SU5416 and 3-week exposure to hypoxia (10% O2) followed by 6 weeks of normoxia.13 CAR was accumulated in remodeled pulmonary arteries (A: medial wall thickening and C: occlusive neointimal formation). Little signal was detected in lung lesions of PAH rats administered CAR mutant peptide (B, medial wall thickening; D, occlusive neointimal formation). Scale bars = 50 μm.
Binding and Penetration of CAR into Human Cells
To assess the potential utility of CAR in targeting human PAH, we tested binding of CAR to human endothelial cells in culture. When grown in culture, these endothelial cells are highly activated and mitotic, with elevated expression of angiogenic genes, recapitulating the characteristics of the endothelial cells of pathologically regenerating blood vessels.18,19 CAR specifically bound to the growing endothelial cells in culture and was internalized into the cells (see Supplemental Figure S3 at http://ajp.amjpathol.org). Reinforcing the ability of CAR to target human cells in vivo, we showed that i.v. injected CAR homes to human tumor xenografts in mice with high selectivity and efficiency. Significantly, the peptide was found to be associated with human tumor cells (see Supplemental Figure S4 at http://ajp.amjpathol.org). These results indicate the expression of CAR receptor in human cells, suggesting that the application of CAR targeting to human PAH is feasible.
Discussion
In this study, we demonstrate the highly selective PAH-targeting and tissue-penetration abilities of a recently described cyclic peptide, CAR. To our knowledge, this is the first report of a technology that enables selective targeting of the life-threatening lung disorder PAH. We found that CAR homes to the pulmonary arterial endothelium and smooth muscle of the diseased lung, which are the primary tissue targets of current PAH interventions.20 CAR also accumulates in the adventitia of pulmonary arteries and in interstitial macrophages. The remodeling of the adventitia is important in the development of PAH, and macrophages recruited to the PAH lesion play a crucial role in the pathogenesis of the disease through mediating inflammatory responses. The CAR targeting of the lung lesions may, therefore, offer opportunities to target multiple cell types important in the pathogenesis of PAH.
Because inflammation is an element in the PAH models we used, there could be a correlation between inflammation in the lungs and CAR accumulation. However, the inflammatory response in the lungs is significantly stronger in MCT-induced PAH than in the SU5416/hypoxia model or in human idiopathic PAH.13 Nonetheless, CAR effectively targets the PAH lungs in both rat models. Moreover, CAR showed excellent specificity for the lungs in the MCT model, although this treatment is known to cause inflammation in the heart and liver as well.21,22 Therefore, inflammation alone does not seem to account for the selective CAR homing to hypertensive lungs.
The molecular target of CAR homing in the PAH lesion is unknown. A BLAST analysis revealed a high homology of CAR sequence with the heparin-binding motif of bone morphogenetic protein-4.9 Whether CAR has any effect on bone morphogenetic protein-4 signaling is unknown. In agreement with its putative heparin-binding sequence, cell binding of CAR is dependent on heparan sulfate expression of the cells,9 suggesting that the mechanism for the vascular homing to the site of hypertensive lesions may be due to the expression of a unique glycosaminoglycan in the diseased lung. Thus, PAH vasculature seems to express a molecular zip code not present in normal lung vasculature. Identification of the receptor for CAR is an important goal of future work that could further advance PAH targeting technology.
Currently, there are several pharmacologic agents for the treatment of PAH, some of which hold promise for improved efficacy.5,20 However, the lack of pulmonary vascular selectivity and associated systemic adverse effects imposes significant obstacles to successful outcomes of these therapies. When conjugated with CAR, a systemically administered fluorescence probe (FAM) was successfully delivered to and concentrated in the lung lesions. This suggests that CAR will also be useful for delivering other imaging probes and therapeutic compounds to PAH lesions. CAR accumulated significantly in the interstitial space of PAH lungs, indicating that CAR, having specifically bound to the vasculature of the diseased lungs, extravasates and penetrates into lung parenchyma. This deep tissue penetration may make it possible to overcome the additional problem of inadequate drug penetration into the target tissues. The targeting technology described in this article may be useful for developing drugs specifically engineered for targeted treatment of PAH.
Acknowledgments
We thank Dr. Venkata Ramana Kotamraju for peptide synthesis and Ms. Akane Urakami for technical assistance. Histologic preparation and immunostaining were performed at the Histology Core Facility of Sanford-Burnham Medical Research Institute at Lake Nona.
Footnotes
Supported by grants from the National Heart, Lung, and Blood Institute (R41 HL088771 to M.K. and D.M.), the National Cancer Institute (R01 CA125255 to M.K.), the Department of Defense (USAMRAA/AFIRM W81XWH-08-2-0032 to E.R.), and the Sigrid Juselius Foundation, Competitive Research Funding of the Pirkanmaa Hospital District, Tampere University Hospital Finnish Medical Foundation Academy of Finland, and Instrumentarium Research Foundation (T.A.H.J.).
D.M., E.R., and M.K. are shareholders in VBS Pharmaceuticals.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.02.032.
Supplementary data
Pulmonary vascular remodeling induced by monocrotaline. H&E staining of normal healthy control lung (untreated) and monocrotaline-treated rat lung (MCT) sections is shown. The thickening of arterial smooth muscle was observed in the MCT-treated rats indicating the development of hypertensive vascular lesions. Numerous macrophage infiltrations were found in the interstitial space and alveolar lumen, indicating significant inflammatory response in the lung. Scale bar = 50 μm. PA, pulmonary arteries; Br, bronchus; Arrows, arterioles and macrophages.
Specificity of anti-FITC antibody staining in the kidney. Rats were administered with FAM-conjugated CAR (A and D) or CG7C control peptide (B and E), and the kidneys were harvested two hours after the peptide administration. A control animal group received no peptide (C and F). The sections were stained with anti-FITC antibody. No significant signal was detected in the kidney of control rats (C and F) confirming the specificity of the antibody staining. Scale bars: 500 μm (A-C), 200 μm (D-F).
Internalization of CAR peptide into human endothelial cells. CAR peptide (A) but not the mutant CAR control (B) was internalized by cultured human umbilical cord vein endothelial cells (HUVEC) 30 min after peptide addition to the culture media (10 μM). To use activated endothelial cells for this study, HUVECs were maintained in complete endothelial cell growth media EGM-2, which is rich in angiogenic growth factors. Green, FAM-labeled CAR; blue, dapi (nuclei).
In vivo targeting and penetration of CAR into human tumor xenograft. CAR peptide (green) specifically homed and penetrated into tumor tissue when i.v. injected into nude mice bearing human breast cancer MDA-MB-235 xenografts. FAM-labeled CAR (A) or control peptide (B). Blood vessels, CD-31 (magenta); DAPI (blue).
References
- 1.Humbert M., Morrell N.W., Archer S.L., Stenmark K.R., MacLean M.R., Lang I.M., Christman B.W., Weir E.K., Eickelberg O., Voelkel N.F., Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 2.Cool C.D., Groshong S.D., Oakey J., Voelkel N.F. Pulmonary hypertension: cellular and molecular mechanisms. Chest. 2005;128:565S–571S. doi: 10.1378/chest.128.6_suppl.565S. [DOI] [PubMed] [Google Scholar]
- 3.Tuder R.M., Groves B., Badesch D.B., Voelkel N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 4.Dorfmuller P., Perros F., Balabanian K., Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–363. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 5.McLaughlin V.V., Archer S.L., Badesch D.B., Barst R.J., Farber H.W., Lindner J.R., Mathier M.A., McGoon M.D., Park M.H., Rosenson R.S., Rubin L.J., Tapson V.F., Varga J., Harrington R.A., Anderson J.L., Bates E.R., Bridges C.R., Eisenberg M.J., Ferrari V.A., Grines C.L., Hlatky M.A., Jacobs A.K., Kaul S., Lichtenberg R.C., Lindner J.R., Moliterno D.J., Mukherjee D., Pohost G.M., Rosenson R.S., Schofield R.S., Shubrooks S.J., Stein J.H., Tracy C.M., Weitz H.H., Wesley D.J. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119:2250–2294. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- 6.Rubin L.J. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–117. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- 7.Ruoslahti E. Vascular zip codes in angiogenesis and metastasis. Biochem Soc Trans. 2004;32:397–402. doi: 10.1042/BST0320397. [DOI] [PubMed] [Google Scholar]
- 8.Ruoslahti E., Bhatia S.N., Sailor M.J. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Järvinen T.A., Ruoslahti E. Molecular changes in the vasculature of injured tissues. Am J Pathol. 2007;171:702–711. doi: 10.2353/ajpath.2007.061251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Järvinen T.A., Ruoslahti E. Target-seeking antifibrotic compound enhances wound healing and suppresses scar formation in mice. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1016233107. [DOI] [PMC free article] [PubMed] [Google Scholar]; [Epub ahead of press]
- 11.Kato G.J., Martyr S., Blackwelder W.C., Nichols J.S., Coles W.A., Hunter L.A., Brennan M.L., Hazen S.L., Gladwin M.T. Levels of soluble endothelium-derived adhesion molecules in patients with sickle cell disease are associated with pulmonary hypertension, organ dysfunction, and mortality. Br J Haematol. 2005;130:943–953. doi: 10.1111/j.1365-2141.2005.05701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bader M. Rat models of cardiovascular diseases. Methods Mol Biol. 2010;597:403–414. doi: 10.1007/978-1-60327-389-3_27. [DOI] [PubMed] [Google Scholar]
- 13.Abe K., Toba M., Alzoubi A., Ito M., Fagan K.A., Cool C.D., Voelkel N.F., McMurtry I.F., Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–2754. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 14.Kelly K.A., Nahrendorf M., Yu A.M., Reynolds F., Weissleder R. In vivo phage display selection yields atherosclerotic plaque targeted peptides for imaging. Mol Imaging Biol. 2006;8:201–207. doi: 10.1007/s11307-006-0043-6. [DOI] [PubMed] [Google Scholar]
- 15.Kelly K.A., Allport J.R., Tsourkas A., Shinde-Patil V.R., Josephson L., Weissleder R. Detection of vascular adhesion molecule-1 expression using a novel multimodal nanoparticle. Circ Res. 2005;96:327–336. doi: 10.1161/01.RES.0000155722.17881.dd. [DOI] [PubMed] [Google Scholar]
- 16.Cybulsky M.I., Gimbrone M.A., Jr Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- 17.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 18.Chappey O., Wautier M.P., Boval B., Wautier J.L. Endothelial cells in culture: an experimental model for the study of vascular dysfunctions. Cell Biol Toxicol. 1996;12:199–205. doi: 10.1007/BF00438146. [DOI] [PubMed] [Google Scholar]
- 19.Schoenfeld J., Lessan K., Johnson N.A., Charnock-Jones D.S., Evans A., Vourvouhaki E., Scott L., Stephens R., Freeman T.C., Saidi S.A., Tom B., Weston G.C., Rogers P., Smith S.K., Print C.G. Bioinformatic analysis of primary endothelial cell gene array data illustrated by the analysis of transcriptome changes in endothelial cells exposed to VEGF-A and PlGF. Angiogenesis. 2004;7:143–156. doi: 10.1007/s10456-004-1677-0. [DOI] [PubMed] [Google Scholar]
- 20.Girgis R.E. Emerging drugs for pulmonary hypertension. Expert Opin Emerg Drugs. 2010;15:71–85. doi: 10.1517/14728210903551271. [DOI] [PubMed] [Google Scholar]
- 21.Campian M.E., Hardziyenka M., de Bruin K., van Eck-Smit B.L., de Bakker J.M., Verberne H.J., Tan H.L. Early inflammatory response during the development of right ventricular heart failure in a rat model. Eur J Heart Fail. 2010;12:653–658. doi: 10.1093/eurjhf/hfq066. [DOI] [PubMed] [Google Scholar]
- 22.Copple B.L., Ganey P.E., Roth R.A. Liver inflammation during monocrotaline hepatotoxicity. Toxicology. 2003;190:155–169. doi: 10.1016/s0300-483x(03)00164-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pulmonary vascular remodeling induced by monocrotaline. H&E staining of normal healthy control lung (untreated) and monocrotaline-treated rat lung (MCT) sections is shown. The thickening of arterial smooth muscle was observed in the MCT-treated rats indicating the development of hypertensive vascular lesions. Numerous macrophage infiltrations were found in the interstitial space and alveolar lumen, indicating significant inflammatory response in the lung. Scale bar = 50 μm. PA, pulmonary arteries; Br, bronchus; Arrows, arterioles and macrophages.
Specificity of anti-FITC antibody staining in the kidney. Rats were administered with FAM-conjugated CAR (A and D) or CG7C control peptide (B and E), and the kidneys were harvested two hours after the peptide administration. A control animal group received no peptide (C and F). The sections were stained with anti-FITC antibody. No significant signal was detected in the kidney of control rats (C and F) confirming the specificity of the antibody staining. Scale bars: 500 μm (A-C), 200 μm (D-F).
Internalization of CAR peptide into human endothelial cells. CAR peptide (A) but not the mutant CAR control (B) was internalized by cultured human umbilical cord vein endothelial cells (HUVEC) 30 min after peptide addition to the culture media (10 μM). To use activated endothelial cells for this study, HUVECs were maintained in complete endothelial cell growth media EGM-2, which is rich in angiogenic growth factors. Green, FAM-labeled CAR; blue, dapi (nuclei).
In vivo targeting and penetration of CAR into human tumor xenograft. CAR peptide (green) specifically homed and penetrated into tumor tissue when i.v. injected into nude mice bearing human breast cancer MDA-MB-235 xenografts. FAM-labeled CAR (A) or control peptide (B). Blood vessels, CD-31 (magenta); DAPI (blue).