

Abstract
Amyloid deposition and reduced β-cell mass are pathological hallmarks of the pancreatic islet in type 2 diabetes; however, whether the extent of amyloid deposition is associated with decreased β-cell mass is debated. We investigated the possible relationship and, for the first time, determined whether increased islet amyloid and/or decreased β-cell area quantified on histological sections is correlated with increased β-cell apoptosis. Formalin-fixed, paraffin-embedded human pancreas sections from subjects with (n = 29) and without (n = 39) diabetes were obtained at autopsy (64 ± 2 and 70 ± 4 islets/subject, respectively). Amyloid and β cells were visualized by thioflavin S and insulin immunolabeling. Apoptotic β cells were detected by colabeling for insulin and by TUNEL. Diabetes was associated with increased amyloid deposition, decreased β-cell area, and increased β-cell apoptosis, as expected. There was a strong inverse correlation between β-cell area and amyloid deposition (r = −0.42, P < 0.001). β-Cell area was selectively reduced in individual amyloid-containing islets from diabetic subjects, compared with control subjects, but amyloid-free islets had β-cell area equivalent to islets from control subjects. Increased amyloid deposition was associated with β-cell apoptosis (r = 0.56, P < 0.01). Thus, islet amyloid is associated with decreased β-cell area and increased β-cell apoptosis, suggesting that islet amyloid deposition contributes to the decreased β-cell mass that characterizes type 2 diabetes.
Type 2 diabetes is characterized by insulin resistance and β-cell failure,1 the latter resulting from reductions in β-cell function2,3 and/or β-cell mass.4–6 Together, these contribute to impaired insulin release and the inability to maintain euglycemia without glucose-lowering therapy.
A pathological hallmark of the pancreatic islet in type 2 diabetes is islet amyloid deposition. These deposits occur in the majority of patients with diabetes,5,7–10 but have also been reported in a small proportion of subjects who are apparently nondiabetic (but may have undiagnosed abnormalities in glucose metabolism), and especially in those who are older.7,11 The formation of islet amyloid occurs by aggregation of islet amyloid polypeptide (IAPP, or amylin),12,13 which is normally cosecreted with insulin by the β cell.14
In vitro studies have demonstrated that the process of IAPP aggregation is cytotoxic, resulting in β-cell apoptosis.15,16 In vivo studies of spontaneous islet amyloid deposition in nonhuman primates and in domestic cats,17–20 as well as in transgenic rodent models of islet amyloid formation,21–23 have shown that the accumulation of islet amyloid formation precedes fasting hyperglycemia and is associated with decreased β-cell function and β-cell loss.
Human studies investigating the relationship between β-cell mass and islet amyloid are more limited. Several studies have assessed β-cell area and islet amyloid deposition in histological sections from the same human pancreas samples.5,8,9,24,25 Only two studies have made assessments of correlations between these measures, however, and the findings are contradictory.8,24 One study identified a significant correlation between increased amyloid deposition and β-cell loss,24 but the other found that no such relationship exists.8 In addition, none of these studies examined whether the loss of β cells occurs selectively in amyloid-laden islets and whether islet amyloid deposition, or changes in β-cell area, are associated with increased β-cell apoptosis and/or decreased β-cell replication. With the present study, we sought to provide further insight into the relationship between islet amyloid deposition and decreased β-cell area in humans and to explore, for the first time, whether islet amyloid deposition is associated with increased β-cell apoptosis and/or reduced β-cell replication.
Materials and Methods
Subjects
We studied 29 patients with diabetes, identified by type 2 diabetes diagnosis in their medical records, with or without the use of antidiabetic medications. We also studied 39 nondiabetic control subjects who did not meet these criteria and who additionally had a random glucose of <7 mmol/L. Individuals with a history of pancreatic cancer, pancreatitis, end-stage liver disease, hepatitis, organ transplantation, or chronic glucocorticoid treatment were excluded. The study was approved by institutional review boards at the University of Washington and the VA Puget Sound Health Care System.
Pancreatic tissue was obtained during autopsies performed at the University of Washington and the VA Puget Sound Health Care System. Specimens were routinely sampled from the body of the pancreas; however, autopsy records did not always indicate from what specific region of the organ the pancreas samples had been obtained. Specimens were included in the study only if they showed no or minimal autolysis (as assessed by C.L.F.and R.L.H.). Pancreatic weight was not available; data are therefore presented as β-cell area, rather than β-cell mass.
Histological Assessments
Formalin-fixed, paraffin-embedded pancreas specimens were cut into sections 10-μm thick, and three consecutive sections were labeled. All sections were labeled with insulin antibody [I2018, clone K36AC10 at 1:2000 (Sigma-Aldrich, St. Louis, MO) or A0564 at 1:50 (Dako, Carpinteria, CA)] to visualize β cells and were colabeled with either thioflavin S to detect amyloid deposits, with deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) (In Situ Cell Death Detection Kit; Roche Applied Sciences, Indianapolis, IN) to detect apoptotic cells, or with Ki-67 (MIB-1 at 1:50; Dako) to detect replicating cells. Secondary antibodies for insulin immunolabeling were Cy3-conjugated goat-anti-mouse (Jackson ImmunoResearch, West Grove, PA) or Alexa Fluor 488-conjugated goat-anti guinea pig immunoglobulins (Invitrogen, Carlsbad, CA). For detection of Ki-67 labeling, biotinylated goat anti-mouse immunoglobulins followed by streptavidin-Cy3 were used (both from Jackson ImmunoResearch). All sections were counterstained with Hoechst 33258 (2 μg/mL; Sigma-Aldrich) to identify nuclei.
Microscopy
Histological assessments were made on all islets within a region of pancreas representative of the whole section. The data collector was blinded to the diabetes status of each specimen. The average number of islets scored per subject was 64 ± 2 in diabetic subjects and 70 ± 4 in the control group (P = 0.20). Neither the area of the sections assessed (17.8 ± 1.7 versus 20.6 ± 1.7 mm2, diabetes versus control; P = 0.27) nor islet density (4.4 ± 0.4 versus 4.2 ± 0.4 islets/mm2, diabetes versus control; P = 0.7) differed between groups. Islet area was defined by manually circumscribing each islet edge, identified visually by morphological characteristics of the tissue viewed on the thioflavin S channel and calculating the area of that region of interest. The lower cutoff for islet size was 1000 μm2 (∼15 cells). Insulin-positive area (β-cell area) and thioflavin S-positive area (amyloid area) were determined using a computer-based quantitative system as described previously.21,26
Amyloid prevalence was calculated as the number of islets containing amyloid divided by the total number of islets, amyloid severity as total amyloid area (μm2) divided by the total islet area (μm2), and β-cell area as insulin-positive area (μm2) or as total insulin-positive area (μm2) divided by the total islet area (μm2). All measures, except absolute β-cell area, were expressed as percentages.
Assessment of β-cell apoptosis and replication was performed in a randomly selected subset of samples. Power calculations showed that, for a twofold difference in apoptosis with variability similar to that in published studies,5 a sample size of approximately 15 subjects would be sufficient. β-Cell apoptosis was analyzed on sections from 16 control subjects and 13 diabetic subjects (comprising an average of 6322 ± 530 and 5667 ± 412 islet cells, respectively); for β-cell replication, sections from 15 control subjects and 17 diabetic subjects were examined (comprising an average of 5324 ± 561 and 5356 ± 587 islet cells, respectively). The demographic characteristics of these subjects and histological measures of islet area, islet amyloid deposition, and β-cell area in each subgroup did not differ from their respective whole cohorts. β-Cell apoptosis and replication were calculated as the average number of TUNEL-positive or Ki-67-positive β cells per islet, normalized to β-cell/islet area, according to approaches described previously.5 The range for TUNEL-positive β cells was 0% to 5.2%; the range for Ki-67-positive β cells was 0% to 0.75%. Note that for β-cell replication data, Ki-67 and insulin costained cells were absent in 20% of cases. In all these cases, however, Ki-67 positive cells were visible either in non-insulin-positive islet cells or in exocrine pancreas, indicating that this was a genuine lack of proliferating β cells and not a limitation of the Ki-67 labeling technique.
To determine islet size distribution, measures from islets of all diabetic subjects (n = 1844 islets) were compared with those of the control subjects (n = 2726 islets), using both raw and log-transformed data. To examine whether there was a difference in islet area and β-cell area between amyloid-positive and amyloid-negative islets, data from each individual islet from all diabetic subjects were categorized as amyloid positive (n = 859 islets) or negative (n = 985 islets). Islet area and β-cell area were then determined and compared with all islets in control subjects (n = 2726 islets) and with only those islets from control subjects that were amyloid negative (n = 2574 islets).
Statistical Analysis
Data for variables that were normally distributed are reported as means ± SD for subject characteristics and means ± SEM for all other data. Non-normally distributed data are presented as median and interquartile range (IQR). For data analysis purposes, non-normally distributed data were transformed (ln, square, or square root). Groups of data with normal distribution before or after transformation were compared using a Student's t-test. For data that were not normally distributed after transformation, a Mann-Whitney U-test was used. Regression analyses were performed using Spearman's correlation with or without adjustments for diabetes treatment and cause of death. A χ2 test was used to determine the difference in distributions for categorical variables between the diabetes and control groups. A P value of <0.05 was considered significant.
Results
Subject Characteristics
Group characteristics are presented in Table 1 and characteristics for individual subjects are presented in Table 2. Subjects were well matched for age and sex, although those with diabetes were heavier. As expected, glucose levels of diabetic subjects were significantly higher than control subjects. The mean time since diagnosis of diabetes was 10 years. The number of diabetic subjects treated with diet, oral medications, or insulin are reported in Tables 1 and 2; where subjects were treated with a combination of insulin and other antidiabetic medications, they were classified for the group data as insulin treated. The causes of death did not differ significantly between the two groups.
Table 1.
Subject Characteristics, Causes of Death, and Diabetes Medications
| Variable | Type 2 Diabetes | Controls | P value |
|---|---|---|---|
| Clinicodemographic characteristics | |||
| Sample size | n = 29 | n = 39 | |
| Age (years) | 63.9 ± 13.1 | 61.7 ± 19.3 | 0.58 |
| Sex (% female) | 34.5 | 46.2 | 0.34 |
| BMI (kg/m2) | 30.6 ± 6.8 | 26.6 ± 7.7 | 0.03 |
| Plasma glucose (mmol/L) | 8.9 ± 2.4 | 5.5 ± 0.6 | <0.001 |
| Diabetes duration (years)⁎ | 10 ± 2 | n/a | |
| Cause of death, n (%) | |||
| CVD/stroke | 10 (26) | 9 (23) | |
| Cancer | 6 (21) | 13 (33) | |
| Pulmonary | 3 (10) | 8 (21) | |
| Surgical | 2 (7) | 2 (5) | |
| Infection | 3 (10) | 6 (15) | |
| Gastrointestinal | 5 (17) | 1 (3) | |
| Diabetes medication, n (%) | |||
| None | 4 (14) | ||
| Oral | 11 (38) | ||
| Insulin | 11 (38) | ||
| Unknown | 3 (10) |
Data are reported as means ± SD.
BMI, body mass index; CVD, cardiovascular disease; n/a, not applicable.
Diabetes duration data were available only for 13/29 subjects.
Table 2.
Clinicodemographic Characteristics of Individual Subjects
| Age (years)/Sex | BMI (kg/m2) | Blood glucose (mmol/L) | Diabetes duration (years) | Diabetes medication | Cause of death |
|---|---|---|---|---|---|
| Diabetes group | |||||
| 62/F | 30.9 | 8.53 | ND | glyburide, insulin | pulmonary embolism |
| 78/F | 28.3 | 7.67 | 2 | diet | GI hemorrhage |
| 59/M | 32.6 | 7.90 | 8 | insulin | cardiac arrest |
| 59/M | 38.4 | 7.39 | ND | insulin | leukemia |
| 69/F | 21.2 | 9.50 | ND | metformin | malignancy |
| 81/M | 31.4 | 5.63 | ND | glyburide, metformin | aortic dissection |
| 68/M | 27.2 | 11.42 | ND | metformin | GI hemorrhage |
| 52/M | 26.3 | 9.07 | ND | none | cardiopulmonary failure |
| 61/M | 29.7 | 6.85 | 14 | insulin | respiratory failure |
| 71/F | 21.3 | 9.39 | 10 | glyburide | coronary artery disease |
| 51/F | 41.2 | 11.11 | ND | glyburide, insulin | malignancy |
| 62/M | 35.9 | 7.46 | ND | insulin in TPN | abdominal hemorrhage |
| 80/F | 29.1 | 8.89 | 20 | acetohexamide,insulin | coronary artery disease |
| 61/M | 36.3 | 8.92 | 12 | diet, glipizide | sepsis |
| 58/M | 34.4 | 8.29 | ND | insulin | coronary artery disease |
| 71/M | 37.5 | 6.50 | 27 | insulin | postoperative complications |
| 74/F | 36.0 | 13.17 | ND | unknown | aspiration pneumonia |
| 37/M | 39.7 | 7.69 | 3 | unknown | cardiomyopathy |
| 71/F | 21.6 | ND | 12 | glyburide | coronary artery disease |
| 70/F | 32.8 | 10.33 | ND | oral hypoglycemic, unspecified | abdominal hemorrhage |
| 69/M | 21.3 | 7.06 | 10 | glyburide | coronary artery disease |
| 64/M | 22.5 | 6.47 | ND | insulin | sepsis |
| 40/M | 41.2 | 10.17 | ND | diet, metformin | postoperative complications |
| 28/M | 35.8 | 7.67 | 1 | metformin | myelodysplastic syndrome |
| 82/M | 25.6 | 17.06 | ND | glipizide | sepsis |
| 63/F | 38.8 | 10.50 | 12 | insulin | malignancy |
| 63/M | 26.5 | 7.11 | ND | diet | malignancy |
| 68/M | 25.0 | ND | ND | diet | cardiac arrest |
| 82/M | 19.0 | 7.50 | 2 | diet | sepsis |
| Control group | |||||
| 87/F | 33.0 | 5.14 | NA | NA | valvular heart disease |
| 62/M | 23.4 | 5.58 | NA | NA | malignancy |
| 34/M | 17.5 | 5.14 | NA | NA | malignancy |
| 63/F | 18.7 | 5.23 | NA | NA | malignancy |
| 54/F | 49.3 | 5.94 | NA | NA | postoperative complications |
| 82/F | 40.9 | 4.72 | NA | NA | perforated duodenal ulcer |
| 61/F | 24.3 | 5.58 | NA | NA | abdominal hemorrhage |
| 65/M | 20.5 | 6.31 | NA | NA | stroke |
| 21/F | 32.2 | 4.63 | NA | NA | pulmonary embolism |
| 64/M | 18.7 | 6.47 | NA | NA | malignancy |
| 65/F | 40.1 | 4.79 | NA | NA | pulmonary veno-occlusive disease |
| 67/M | 24.1 | 5.12 | NA | NA | malignancy |
| 52/M | 24.1 | 5.26 | NA | NA | malignancy |
| 77/F | 26.3 | 6.08 | NA | NA | sepsis |
| 34/M | 28.8 | 5.44 | NA | NA | malignancy |
| 68/F | 18.6 | 4.86 | NA | NA | malignancy |
| 89/M | 17.8 | 6.07 | NA | NA | malignancy |
| 57/F | 19.2 | 5.87 | NA | NA | malignancy |
| 75/M | 22.6 | 4.94 | NA | NA | pneumonia |
| 66/F | 25.7 | 5.43 | NA | NA | respiratory failure |
| 50/M | 28.0 | 5.50 | NA | NA | cardiac failure |
| 45/M | 29.8 | 4.53 | NA | NA | cardiac arrest |
| 83/F | 19.8 | 5.15 | NA | NA | cardiac arrest |
| 44/F | 18.0 | 5.18 | NA | NA | meningitis |
| 20/F | 22.8 | 4.60 | NA | NA | pulmonary hypertension |
| 42/F | 34.0 | 5.39 | NA | NA | sepsis |
| 88/F | 29.2 | 6.16 | NA | NA | respiratory failure |
| 81/M | 27.1 | 5.56 | NA | NA | respiratory failure |
| 58/M | 24.9 | 5.44 | NA | NA | stroke |
| 19/M | 32.7 | 6.22 | NA | NA | malignancy |
| 46/F | 28.1 | 6.47 | NA | NA | sepsis |
| 78/F | 42.4 | 3.78 | NA | NA | abdominal hemorrhage |
| 75/M | 20.9 | 5.83 | NA | NA | malignancy |
| 72/M | 34.9 | 5.56 | NA | NA | multiple myeloma |
| 82/M | 32.5 | 6.06 | NA | NA | sepsis |
| 94/M | 21.9 | 5.22 | NA | NA | sepsis |
| 68/M | 24.0 | 5.17 | NA | NA | respiratory failure |
| 60/M | 15.9 | 6.94 | NA | NA | respiratory failure |
| 57/M | 22.8 | 5.56 | NA | NA | cardiac arrest |
F, female; GI, gastrointestinal; M, male; NA, not applicable; ND, no data; TPN, total parenteral nutrition.
To control for the difference in body mass index (BMI) between groups, a subset of subjects (n = 19 per group) were matched case by case for sex (47% females in each group), age (67 ± 12 versus 68 ± 9 years for diabetic versus control subjects), and BMI (29.5 ± 6.1 versus 29.1 ± 8.6 kg/m2). Blood glucose levels were significantly elevated in diabetic subjects in this smaller cohort (9.1 ± 2.7 versus 5.3 ± 0.62 mmol/L, P < 0.001). The remaining characteristics in these two subgroups did not differ between the groups and were not different from those of the remainder of their respective cohorts.
Islet Amyloid Deposition
Almost all individuals with diabetes (97%) demonstrated some islet amyloid deposition (Figure 1A); amyloid was also present in 25% of the nondiabetic control subjects, (Figure 1B). Both amyloid prevalence and severity were much higher in diabetic subjects (Figure 1, C and D). In the subset of subjects matched for BMI, islet amyloid prevalence and severity were still significantly higher in diabetic subjects (54 ± 8% versus 9 ± 5% and 12.5 ± 2.4% versus 1.2 ± 0.8%, respectively; P < 0.001 for both).
Figure 1.
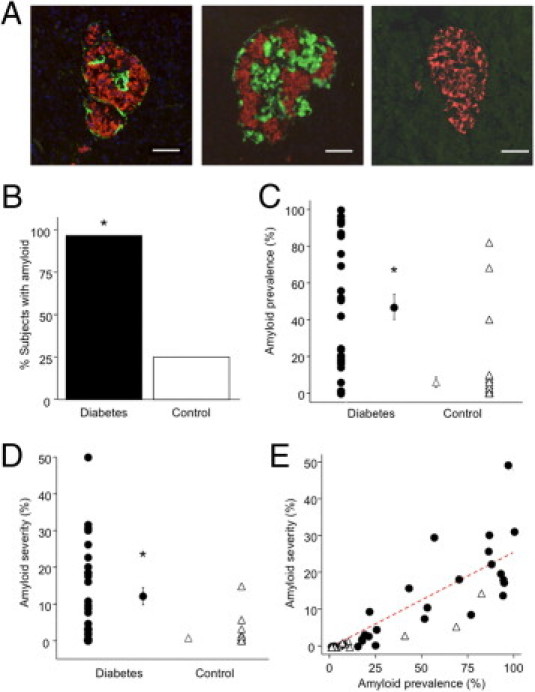
Islet amyloid deposition in individuals with diabetes (n = 29) and control subjects (n = 39). A: Representative images of islets stained for insulin (red) and thioflavin S (green). The left and middle images show islets from individuals with diabetes, with amyloid severities of 6% and 32%, respectively. The image at the right shows an islet from a control subject, amyloid negative. Scale bar = 50 μm. B–E: Quantitative analysis. The proportion of subjects with any detectable islet amyloid (B), the mean islet amyloid prevalence expressed as % amyloid-positive islets (C), and mean islet amyloid severity expressed as amyloid positive area/islet area × 100% (D) were all significantly greater in diabetic subjects than control subjects. Individual points represent data for each subject, with mean ± SEM for the two groups also depicted (C and D). Islet amyloid prevalence and severity were correlated in a linear manner (E); solid circles indicate individuals with diabetes, and open triangles indicate control subjects (r = 0.87). *P < 0.001 versus control subjects.
In the whole cohort, islet amyloid prevalence and severity were strongly correlated, both in all subjects (Figure 1E) and in diabetic subjects (r = 0.83, P < 0.001). Islet amyloid prevalence was positively correlated with age in all subjects (r = 0.31, P = 0.01) and in diabetic subjects (r = 0.43, P = 0.02). However, neither amyloid prevalence nor amyloid severity was associated with sex, BMI, blood glucose, diabetes duration, or diabetes medication. Given that islet amyloid was detected in a proportion of control subjects, we examined whether any characteristics were associated with the presence of amyloid deposition in this group. Those control subjects with islet amyloid did not differ from those without islet amyloid in terms of age, sex, BMI, or blood glucose.
Islet Area and β-Cell Area
The distribution of islet areas within each group was not normal; however, the distribution was very similar between groups (Figure 2, A and B). As a result, median islet area did not differ (P = 0.87, Figure 2C). Similarly, islet area did not differ in the subset of subjects matched for BMI [9345 (IQR 1678) versus 8399 (IQR 1843) μm2,P = 0.2], and islet area did not correlate with BMI in the whole cohort (r = 0.69, P = 0.58). Individuals with type 2 diabetes had a significantly lower absolute β-cell area, compared with control subjects (P < 0.05, Figure 2D). β-Cell area remained significantly lower in the diabetic group when normalized to islet area (P < 0.05, Figure 2E). In the subgroup of subjects matched for BMI, absolute β-cell area and β-cell/islet area were both significantly lower in individuals with diabetes [2770 (IQR 319) versus 3900 (IQR 926) μm2 and 34.7 ± 3.2 versus 47.5 ± 2.1%, respectively; P < 0.05 for both).
Figure 2.
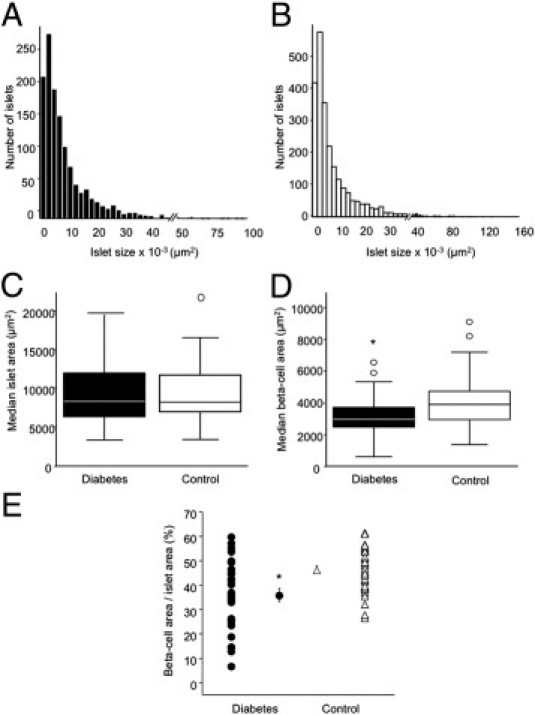
Measurements of cross-sectional islet and β-cell areas in individuals with diabetes (n = 29) and control subjects (n = 39). Islet size distribution was determined in individuals with diabetes (A) and in control subjects (B). Median islet area (C) was identical between groups. Median β-cell area (D) and mean β-cell area normalized to islet area, expressed as insulin positive area/islet area × 100% (E), were significantly lower in individuals with diabetes; individual points represent data for each subject, with mean ± SEM for the two groups also depicted. For box plots, boxes denote the 25th to 75th percentiles and whiskers denote 5th and 95th percentiles; data that fall outside the range of the 5th to 95th percentiles are outliers and are shown as circles (C and D). *P < 0.05 versus control subjects.
Relationship between Islet Amyloid Deposition and β-Cell Area
In the whole cohort, there was an inverse relationship between islet amyloid severity and both absolute β-cell area (Figure 3A) (r = −0.42, P < 0.001) and β-cell area/islet area (Figure 3B) (r = −0.76, P < 0.05). In individuals with diabetes, these correlations were stronger than for the whole cohort for both measures of β-cell area (r = −0.60, P < 0.001 for absolute β-cell area; r = −0.81, P < 0.001 for β-cell area/islet area). After adjustment for diabetes treatment and cause of death, the relationships between islet amyloid severity and both absolute β-cell area and β-cell area/islet area remained significant (P < 0.005 for both). In the control group, there was no significant association between islet amyloid severity and β-cell area (r = 0.07, P = 0.66 for absolute β-cell area and r = −0.12, P = 0.47 for β-cell area/islet area). Islet amyloid prevalence was also significantly negatively correlated with absolute β-cell area (r = −0.33, P < 0.01 for the whole cohort; r = −0.35, P = 0.07 for diabetic subjects) and was more strongly correlated with decreased β-cell area/islet area (r = −0.75, P < 0.001 for the whole cohort; r = −0.81, P < 0.001 for diabetic subjects). In the control group alone, there was no significant association between islet amyloid prevalence and β-cell area (r = 0.09, P = 0.61 for absolute β-cell area and r = −0.12, P = 0.45 for β-cell area/islet area).
Figure 3.
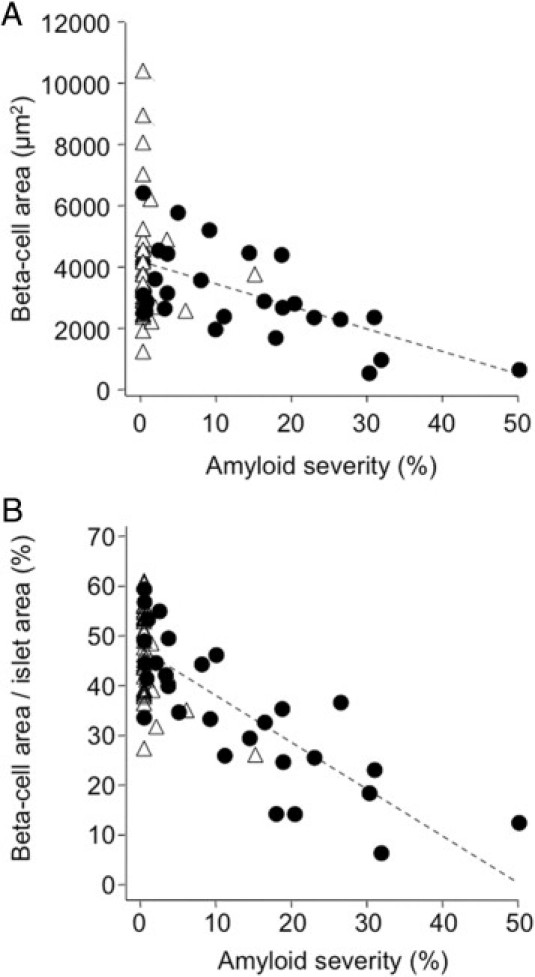
Increased islet amyloid deposition was associated with reduced β-cell area. Correlation is shown among all subjects between amyloid severity and absolute β-cell area (r = −0.42 and P < 0.001) (A), or β-cell area/islet area (r = −0.76 and P < 0.001) (B), in individuals with diabetes (solid circles) and in control subjects (open triangles).
To further examine the relationship between amyloid deposition and reduced β-cell area, we determined whether loss of β-cell area occurred selectively in amyloid-laden islets. Data from all islets from diabetic individuals, grouped as amyloid positive (n = 859 islets) or amyloid negative (n = 985 islets), were compared with all control islets (n = 2726, Figure 4). Amyloid-laden islets from diabetic subjects were bigger than islets without amyloid (P < 0.001, Figure 4A), and β-cell/islet area was significantly reduced in amyloid-laden islets (P < 0.001, Figure 4B). There was no difference in β-cell/islet area between amyloid-negative islets from diabetic subjects and all islets from control subjects (P = 0.99). When data from the islets obtained from the individuals with diabetes were compared with only amyloid-negative control islets (n = 2574), the findings were the same (data not shown).
Figure 4.
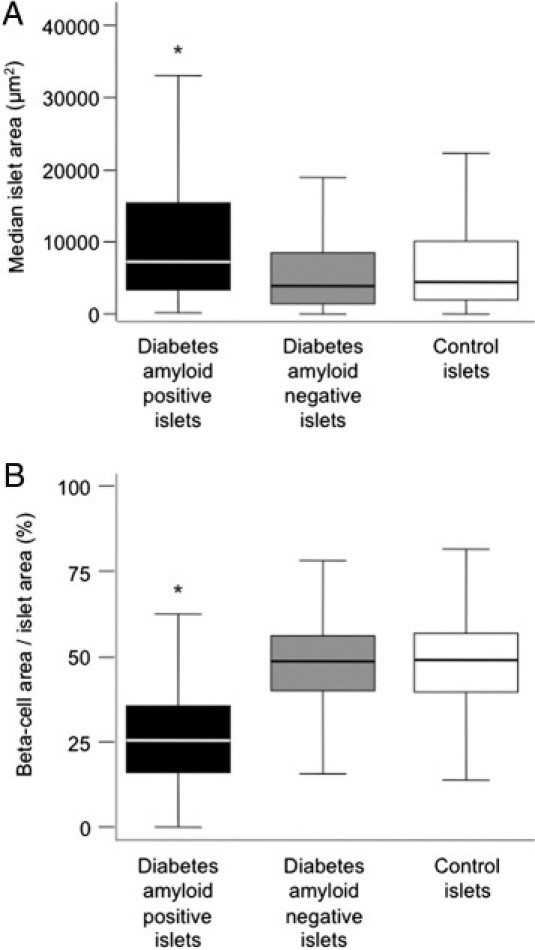
Median islet (A) and β-cell area/islet area (B) in amyloid-positive islets from individuals with diabetes (n = 859 islets), amyloid-negative islets from individuals with diabetes (n = 985 islets), and islets from control subjects (n = 2726 islets). Amyloid-laden islets were significantly larger (A), but had significantly lower β-cell area/islet area (B). Islet area and β-cell area/islet area were equivalent between amyloid-negative islets from individuals with diabetes and control subjects. For box plots, boxes denote the 25th to 75th percentiles and whiskers denote 5th and 95th percentiles. *P < 0.001 versus control subjects.
β-Cell Apoptosis and Replication
Finally, we sought to determine whether the increased islet amyloid severity and decreased β-cell area observed in individuals with diabetes was associated with changes in β-cell apoptosis or replication (Figure 5, A and B). The frequency of β-cell apoptosis in diabetic subjects was 4.5-fold higher than that for control subjects (P < 0.01, Figure 5C), whereas the average frequency of β-cell replication was similar between groups (P = 0.63, Figure 5D). For the subset of individuals matched for BMI, β-cell apoptosis was significantly higher in diabetic subjects (0.091 ± 0.022 versus 0.013 ± 0.010%, n = 8 and 6, respectively; P < 0.01), with no difference in β-cell replication (0.008 ± 0.004 versus 0.003 ± 0.001%, n = 10 and 5, respectively; P = 0.3).
Figure 5.
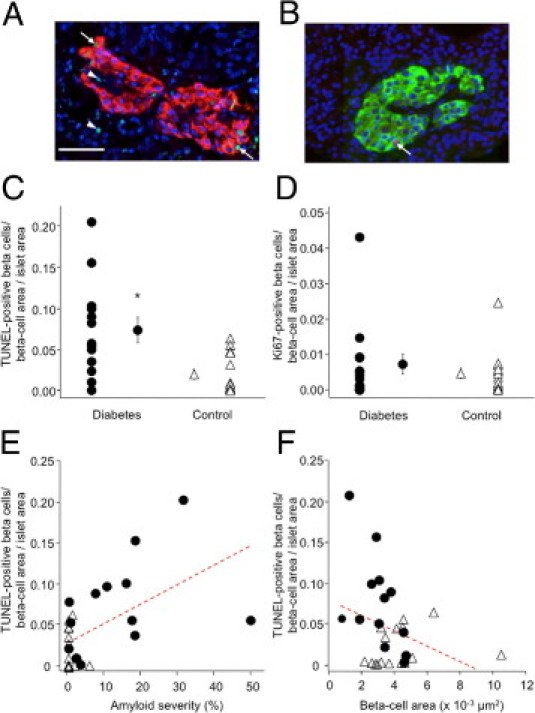
Representative examples of staining for β-cell apoptosis (A) with TUNEL (green), insulin (red), and Hoechst 33258 (blue) and for β-cell replication (B) with Ki-67 (red), insulin (green), and Hoechst 33258 (blue). Arrows indicate TUNEL- or KI-67-positive β cells; arrowheads indicate TUNEL-positive non-β cells. Mean β-cell apoptosis (C) was significantly higher in islets from individuals with diabetes (n = 13), compared with control subjects (n = 16). Mean β-cell replication (D) did not differ between islets from individuals with diabetes (n = 15) and control subjects (n = 17); P = 0.63. β-cell apoptosis was significantly associated with increased islet amyloid severity (E), r = 0.56 and P < 0.01, and tended to be associated with decreased absolute β-cell area (F), r = −0.33 and P = 0.07. Solid circles: diabetes; open triangles, controls. *P < 0.05 versus control subjects; scale bar = 50 μm.
In the whole cohort, increased amyloid severity was significantly associated with β-cell apoptosis (r = 0.56, P < 0.01; Figure 5E) and tended to be associated with increased β-cell apoptosis in diabetic subjects (r = 0.42, P = 0.16). Furthermore, β-cell apoptosis was negatively associated with β-cell area, both in all subjects (r = −0.33, P = 0.07; Figure 5F) and in those with diabetes (r = −0.59, P < 0.05). For these correlation analyses, absolute β-cell area was used, because the β-cell apoptosis measure incorporates the normalized β-cell area/islet area measure.
Discussion
We have shown that in type 2 diabetes the quantity of amyloid deposited in islets is strongly associated with decreased β-cell area. Previously published human studies investigating this relationship provided contradictory information.8,24 We think that our data help to resolve this discrepancy. Moreover, we made the novel observation that β-cell area is reduced only in amyloid-positive islets from diabetic subjects, highlighting the potential importance of amyloid in the loss of β cells in this disease. In addition, our study is the first to examine the relationship between amyloid deposition and β-cell apoptosis. We found, in accord with previous findings,5 that β-cell apoptosis was significantly increased in subjects with type 2 diabetes. In addition, and for the first time, we report that β-cell apoptosis is significantly associated with both increased islet amyloid severity and decreased β-cell area.
Islet amyloid was present to some degree in 97% of individuals with diabetes, in accord with previous reports.5,7,8 Increased islet amyloid prevalence was associated with age in both the whole cohort and the diabetes group alone, consistent with the progressive accumulation of amyloid deposits. Neither islet amyloid deposition nor β-cell/islet area was related to diabetes duration; however, data on diabetes duration were available in only 13 subjects, and even under optimal circumstances the measure is subjective, given that diabetes often goes undiagnosed for long periods of time. Thus, it is not surprising that we did not find a relationship between these two measures. Some studies have found a complete lack of islet amyloid in their control subjects.8,25 In the present study, however, we detected amyloid in at least one islet in 25% of nondiabetic subjects, in accord with other reports.7,11 Although islet amyloid deposition has previously been described in older subjects without known diabetes,7,11 we failed to observe a relationship between amyloid and age in our control subjects. This lack of correlation may be due to the small number of subjects with amyloid in this group. We also found a 22% decrease in β-cell area in individuals with diabetes, compared with control subjects, which is within the range reported previously.4–6,8,9 The large variation of β-cell loss reported in diabetes between and even within studies may be due to the large variability in β-cell area in both healthy control subjects and subjects with type 2 diabetes,6,9 which we observed also in our cohort.
We found a highly significant inverse relationship between the magnitude of islet amyloid deposition and each measure of β-cell area in diabetes, with 60% to 80% of the variance in β-cell area being explained by amyloid deposition. Furthermore, we made the novel observation that β-cell area in amyloid-negative islets from diabetes and control subjects was identical, but β-cell area was significantly reduced in amyloid-positive islets from diabetes subjects. Collectively, our findings strongly support a role for islet amyloid formation in the loss of β cells that is involved in the pathogenesis of type 2 diabetes.
IAPP aggregation/amyloid formation has been shown to result in β-cell apoptosis in cultured cells and islets, as well as in pancreas from animal models.15,16,23,27–30 Inhibition of islet amyloid formation results in decreased β-cell apoptosis in cultured transgenic mouse islets29 and human islets,28,30 further supporting a causal role between amyloid formation and β-cell apoptosis. In keeping with a previous study,5 we found β-cell apoptosis to be markedly increased in type 2 diabetes. Furthermore, we found that increased islet amyloid severity in all subjects was associated with increased β-cell apoptosis, suggesting that the process of amyloid deposition may also be an important cause of β-cell loss in humans. However, this correlation was not significant when the diabetes cohort was analyzed separately; this may be due to the smaller number of subjects for which β-cell apoptosis was determined.
In addition to the mature amyloid deposits detected in the present study by thioflavin S staining, other IAPP-derived aggregates may also contribute to β-cell toxicity in diabetes. Recent studies suggest that early aggregates of IAPP formed during the process of amyloid deposition (rather than the amyloid deposits themselves) are cytotoxic and thereby induce β-cell death.16 Small aggregates or oligomers of IAPP have been reported to be present in β cells from subjects with type 2 diabetes31,32 and are therefore postulated to represent the toxic form of IAPP. Of note, in the present study we showed that amyloid-negative islets from diabetic subjects did not have reduced β-cell area relative to those from nondiabetic control subjects. Thus, if small aggregates of IAPP were present in amyloid-negative islets from diabetic subjects, they do not seem to have resulted in measurable β-cell loss. However, and importantly, as we have discussed in a recent review,33 the reagents available to detect these small aggregates require further validation for use in human pancreas sections, and therefore any conclusions drawn regarding their location and role have to be somewhat circumspect.
A limitation of the present study is that diabetes status was determined from a retrospective medical record review. It is possible that we misclassified subjects, or that subjects treated with insulin may have had type 1 diabetes. If so, however, this should have made it more difficult to demonstrate the relationship between islet amyloid deposition and β-cell area that we observed in our study cohort. Another limitation of our work is that the region of the pancreas sampled was not recorded in all cases. Not having sections representing different portions of the pancreas may have resulted in an under- or overestimation of the amount of islet amyloid deposition. However, again we were still able to detect significant relationships between the variables of interest despite this potential source of variability. Furthermore, studies performed on both human IAPP transgenic mice and human pancreas have shown that amyloid develops throughout the pancreas,8,21 suggesting that not knowing the origin for all pancreas samples is unlikely to have substantially influenced our data.
Finally, the present study is cross-sectional and can provide only correlative data with respect to islet amyloid deposition and its role in β-cell loss in type 2 diabetes. Two important points are worth raising in this regard. The first is that, although our data support the concept that amyloid deposition contributes to β-cell toxicity in type 2 diabetes, in accord with data from in vitro studies and from in vivo animal data,15,16,23,27–30 the reverse may also be true; that is, amyloid deposition may occur as a result of the increased β-cell death that occurs in diabetes, or amyloid deposition may accompany but not contribute to β-cell loss. The second important point is that it is not possible from a study of this kind to determine whether islet amyloid deposition and/or the associated reduction in the β-cell mass are related (and if related, to what extent) to impaired insulin secretion and/or glycemic control.
In summary, we have demonstrated that islet amyloid deposition in human pancreatic islets is associated with decreased β-cell area and increased β-cell apoptosis. In addition, β-cell apoptosis was associated with decreased β-cell area. Our data therefore support a role for the accumulation of amyloid in the loss of β cells in type 2 diabetes and suggest that interventions aimed at limiting or preventing islet amyloid deposition may have beneficial effects in preserving β-cell mass in type 2 diabetes.
Acknowledgments
We thank Rahat Bhatti, Michael Peters, Christina Braddock, Maria Cone, Jaap Biesheuvel, Pier Woudstra, and Nicholas Wysham for excellent technical support. We thank Jane Shofer for providing statistical advice.
Footnotes
Supported by the U.S. Department of Veterans Affairs, VA Puget Sound Health Care System, Seattle, Washington; NIH grants P30-DK17047, K01-DK74404 (R.L.H.); R01-DK75998 (S.E.K.); T32-DK07247, and K99-DK80945 (S.Z.); the American Diabetes Association (S.E.K.); the Swedish Research Council (P.W. and G.T.W.); the Swedish Diabetes Association (G.T.W.); European Union FW6 program EURAMY (P.W. and G.T.W.); the Family Ernfors Fund; a Juvenile Diabetes Research Foundation Postdoctoral Fellowship Award (J.U.); a McAbee Fellowship (K.A.-M.); the University of Washington Medical Student Research Training Program (M.N.T); and by the NIH-National Institute of Diabetes and Digestive and Kidney Diseases Medical Research Program in Diabetes.
References
- 1.Kahn S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 2.Brunzell J.D., Robertson R.P., Lerner R.L., Hazzard W.R., Ensinck J.W., Bierman E.L., Porte D., Jr Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol Metab. 1976;42:222–229. doi: 10.1210/jcem-42-2-222. [DOI] [PubMed] [Google Scholar]
- 3.Jensen C.C., Cnop M., Hull R.L., Fujimoto W.Y., Kahn SE; American Diabetes Association GENNID Study Group. Beta-cell function is a major contributor to oral glucose tolerance in high-risk relatives of four ethnic groups in the U.S. Diabetes. 2002;51:2170–2178. doi: 10.2337/diabetes.51.7.2170. [DOI] [PubMed] [Google Scholar]
- 4.Klöppel G., Löhr M., Habich K., Oberholzer M., Heitz P.U. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res. 1985;4:110–125. doi: 10.1159/000156969. [DOI] [PubMed] [Google Scholar]
- 5.Butler A.E., Janson J., Bonner-Weir S., Ritzel R., Rizza R.A., Butler P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 6.Rahier J., Guiot Y., Goebbels R.M., Sempoux C., Henquin J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):32–42. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 7.Westermark P. Quantitative studies on amyloid in the islets of Langerhans. Ups J Med Sci. 1972;77:91–94. doi: 10.1517/03009734000000014. [DOI] [PubMed] [Google Scholar]
- 8.Clark A., Wells C.A., Buley I.D., Cruickshank J.K., Vanhegan R.I., Matthews D.R., Cooper G.J., Holman R.R., Turner R.C. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9:151–159. [PubMed] [Google Scholar]
- 9.Sakuraba H., Mizukami H., Yagihashi N., Wada R., Hanyu C., Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45:85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- 10.Zhao H.L., Lai F.M., Tong P.C., Zhong D.R., Yang D., Tomlinson B., Chan J.C. Prevalence and clinicopathological characteristics of islet amyloid in Chinese patients with type 2 diabetes. Diabetes. 2003;52:2759–2766. doi: 10.2337/diabetes.52.11.2759. [DOI] [PubMed] [Google Scholar]
- 11.Bell E.T. Hyalinization of the islets of Langerhans in nondiabetic individuals. Am J Pathol. 1959;35:801–805. [PMC free article] [PubMed] [Google Scholar]
- 12.Westermark P., Wernstedt C., Wilander E., Hayden D.W., O'Brien T.D., Johnson K.H. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci USA. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper G.J.S., Willis A.C., Clark A., Turner R.C., Sim R.B., Reid K.B.M. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci USA. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kahn S.E., D'Alessio D.A., Schwartz M.W., Fujimoto W.Y., Ensinck J.W., Taborsky G.J., Jr, Porte D., Jr Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes. 1990;39:634–638. doi: 10.2337/diab.39.5.634. [DOI] [PubMed] [Google Scholar]
- 15.Lorenzo A., Razzaboni B., Weir G.C., Yankner B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368:756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 16.Janson J., Ashley R.H., Harrison D., McIntyre S., Butler P.C. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48:491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- 17.Howard C.F., Jr Longitudinal studies on the development of diabetes in individual Macaca nigra. Diabetologia. 1986;29:301–306. doi: 10.1007/BF00452067. [DOI] [PubMed] [Google Scholar]
- 18.de Koning E.J., Bodkin N.L., Hansen B.C., Clark A. Diabetes mellitus in Macaca mulatta monkeys is characterised by islet amyloidosis and reduction in beta-cell population. Diabetologia. 1993;36:378–384. doi: 10.1007/BF00402271. [DOI] [PubMed] [Google Scholar]
- 19.Guardado-Mendoza R., Davalli A.M., Chavez A.O., Hubbard G.B., Dick E.J., Majluf-Cruz A., Tene-Perez C.E., Goldschmidt L., Hart J., Perego C., Comuzzie A.G., Tejero M.E., Finzi G., Placidi C., La Rosa S., Capella C., Halff G., Gastaldelli A., DeFronzo R.A., Folli F. Pancreatic islet amyloidosis, beta-cell apoptosis, and alpha-cell proliferation are determinants of islet remodeling in type-2 diabetic baboons. Proc Natl Acad Sci USA. 2009;106:13992–13997. doi: 10.1073/pnas.0906471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma Z., Westermark G.T., Johnson K.H., O'Brien T.D., Westermark P. Quantitative immunohistochemical analysis of islet amyloid polypeptide (IAPP) in normal, impaired glucose tolerant, and diabetic cats. Amyloid. 1998;5:255–261. doi: 10.3109/13506129809007298. [DOI] [PubMed] [Google Scholar]
- 21.Wang F., Hull R.L., Vidal J., Cnop M., Kahn S.E. Islet amyloid develops diffusely throughout the pancreas before becoming severe and replacing endocrine cells. Diabetes. 2001;50:2514–2520. doi: 10.2337/diabetes.50.11.2514. [DOI] [PubMed] [Google Scholar]
- 22.Hull R.L., Andrikopoulos S., Verchere C.B., Vidal J., Wang F., Cnop M., Prigeon R.L., Kahn S.E. Increased dietary fat promotes islet amyloid formation and beta-cell secretory dysfunction in a transgenic mouse model of islet amyloid. Diabetes. 2003;52:372–379. doi: 10.2337/diabetes.52.2.372. [DOI] [PubMed] [Google Scholar]
- 23.Udayasankar J., Kodama K., Hull R.L., Zraika S., Aston-Mourney K., Subramanian S.L., Tong J., Faulenbach M.V., Vidal J., Kahn S.E. Amyloid formation results in recurrence of hyperglycaemia following transplantation of human IAPP transgenic mouse islets. Diabetologia. 2009;52:145–153. doi: 10.1007/s00125-008-1185-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Westermark P., Grimelius L. The pancreatic islet cells in insular amyloidosis in human diabetic and non-diabetic adults. Acta Pathol Microbiol Scand A. 1973;81:291–300. doi: 10.1111/j.1699-0463.1973.tb03538.x. [DOI] [PubMed] [Google Scholar]
- 25.Deng S., Vatamaniuk M., Huang X., Doliba N., Lian M.M., Frank A., Velidedeoglu E., Desai N.M., Koeberlein B., Wolf B., Barker C.F., Naji A., Matschinsky F.M., Markmann J.F. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes. 2004;53:624–632. doi: 10.2337/diabetes.53.3.624. [DOI] [PubMed] [Google Scholar]
- 26.Hull R.L., Kodama K., Utzschneider K.M., Carr D.B., Prigeon R.L., Kahn S.E. Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia. 2005;48:1350–1358. doi: 10.1007/s00125-005-1772-9. [DOI] [PubMed] [Google Scholar]
- 27.Butler A.E., Janson J., Soeller W.C., Butler P.C. Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes. 2003;52:2304–2314. doi: 10.2337/diabetes.52.9.2304. [DOI] [PubMed] [Google Scholar]
- 28.Marzban L., Tomas A., Becker T.C., Rosenberg L., Oberholzer J., Fraser P.E., Halban P.A., Verchere C.B. Small interfering RNA-mediated suppression of proislet amyloid polypeptide expression inhibits islet amyloid formation and enhances survival of human islets in culture. Diabetes. 2008;57:3045–3055. doi: 10.2337/db08-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zraika S., Hull R.L., Udayasankar J., Aston-Mourney K., Subramanian S.L., Kisilevsky R., Szarek W.A., Kahn S.E. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia. 2009;52:626–635. doi: 10.1007/s00125-008-1255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Potter K.J., Scrocchi L.A., Warnock G.L., Ao Z., Younker M.A., Rosenberg L., Lipsett M., Verchere C.B., Fraser P.E. Amyloid inhibitors enhance survival of cultured human islets. Biochim Biophys Acta. 2009;1790:566–574. doi: 10.1016/j.bbagen.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Zhao H.L., Sui Y., Guan J., He L., Gu X.M., Wong H.K., Baum L., Lai F.M., Tong P.C., Chan J.C. Amyloid oligomers in diabetic and nondiabetic human pancreas. Transl Res. 2009;153:24–32. doi: 10.1016/j.trsl.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 32.Gurlo T., Ryazantsev S., Huang C.J., Yeh M.W., Reber H.A., Hines O.J., O'Brien T.D., Glabe C.G., Butler P.C. Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol. 2010;176:861–869. doi: 10.2353/ajpath.2010.090532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zraika S., Hull R.L., Verchere C.B., Clark A., Potter K.J., Fraser P.E., Raleigh D.P., Kahn S.E. Toxic oligomers and islet beta cell death: guilty by association or convicted by circumstantial evidence? Diabetologia. 2010;53:1046–1056. doi: 10.1007/s00125-010-1671-6. [DOI] [PMC free article] [PubMed] [Google Scholar]