

Abstract
Incisional hernia often occurs following laparotomy and can be a source of serious problems. Although there is evidence that a biological cause may underlie its development, the mechanistic link between the local tissue microenvironment and tissue rupture is lacking. In this study, we used matched tissue-based and in vitro primary cell culture systems to examine the possible involvement of fascia fibroblasts in incisional hernia pathogenesis. Fascia biopsies were collected at surgery from incisional hernia patients and non-incisional hernia controls. Tissue samples were analyzed by histology and immunoblotting methods. Fascia primary fibroblast cultures were assessed at morphological, ultrastructural, and functional levels. We document tissue and fibroblast loss coupled to caspase-3 activation and induction of apoptosis-like cell-death mechanisms in incisional hernia fascia. Alterations in cytoskeleton organization and solubility were also observed. Incisional hernia fibroblasts showed a consistent phenotype throughout early passages in vitro, which was characterized by significantly enhanced cell proliferation and migration, reduced adhesion, and altered cytoskeleton properties, as compared to non-incisional hernia fibroblasts. Moreover, incisional hernia fibroblasts displayed morphological and ultrastructural alterations compatible with autophagic processes or lysosomal dysfunction, together with enhanced sensitivity to proapoptotic challenges. Overall, these data suggest an ongoing complex interplay of cell death induction, aberrant fibroblast function, and tissue loss in incisional hernia fascia, which may significantly contribute to altered matrix maintenance and tissue rupture in vivo.
Incisional hernia (IH) is the most common complication of laparotomy, with a cumulative incidence of about 20%.1,2 It occurs when the myofascial layer deteriorates under intact skin and compromises tissue strength and compliance. No technique can completely prevent its formation, and, even with the use of mesh, repair is a challenge.3,4
The pathogenesis of this condition is difficult to study because biopsies are rarely obtained before the end stage, and available animal models do not fully represent the gradual course of the disease. Thus, the cellular and molecular mechanisms that control IH development remain elusive. At present, technical and patient-related risk factors have been reported.5 In addition, recent molecular-biochemical findings highlight biological alterations in patients with (recurrent) IH, which can help to explain its unchanging rates and recurrences. Disturbances in collagen metabolism have been described, and a relationship between mechanical failure and altered fibroblast function is theorized (reviewed in Refs 6–8). In this line, we recently demonstrated increased ratios of matrix metalloproteinases (MMPs) to their tissue inhibitors (TIMPs) concomitant with deregulated inflammatory signaling in the abdominal wall tissue (fascia and the skeletal muscle) of IH patients. Changes were tissue specific and were detectable at the mRNA and protein level (the substantial down-regulation of TIMP-3 detected in IH fascia could be of causal significance).9 We suggested that these dynamic alterations may significantly contribute to alter the local extracellular matrix (ECM) by posttranslational mechanisms, and trigger tissue loss and eventual rupture.
The fascia, a fiber-reinforced layer whose major function is to provide physical support to the skeletal muscles, is one of the most highly altered tissues in hernia.10 Traditionally, the fascia has received little scientific attention, but there is an increasing interest in the role it plays in several musculoskeletal disorders.11 Fascia structural integrity and function depend on the quality of its ECM, which is synthesized and maintained primarily by the resident fibroblasts. Any impediments to the phenotypic stability and survival of the fibroblasts because of internal or external factors can result in a defective ECM and prevent tissue repair.12–15 To our knowledge, very little is known regarding whether IH fibroblasts display a distinctive pathological phenotype.7
The etiology of multifactorial diseases is difficult to sort out because they are due to a combination of factors that individually only contribute in small part to the development of the illness. In this case, descriptive observational studies may be helpful to give rise to novel causal hypotheses, which subsequently can be tested to provide a progressively detailed mechanistic understanding of the disease.
With the aim to progress toward an evidence-based prophylactic approach to IH, in this study, we compared perioperative fascia samples obtained from IH patients and non-IH controls. We hypothesized that a malicious alliance of proinflammatory and proteolytic macromolecules9 within the IH fascia microenvironment may modulate IH fibroblast phenotype and result in stable pathological changes in the basic cellular machinery, which ultimately impede normal ECM functioning. Tissue-based and primary culture studies were combined to examine tissue at the morphological and subcellular level and to elucidate the biological characteristics of IH primary fibroblasts.
Materials and Methods
Tissue Acquisition
Fascia samples included in this study were retrieved from our incisional hernia tissue bank, which stores biopsies and primary fibroblasts from abdominal wall tissues of IH and non-IH subjects, and the associated clinical data.9 Specimens are taken intraoperatively by a specialized surgeon. Areas of visible necrosis or granulation tissue were intentionally avoided. IH samples were taken at least 8 cm away from the edge of the defect, when patients underwent the first repair and no mesh is present. The biopsies were divided into multiple pieces and fixed in 10% formalin, snap frozen in liquid nitrogen, or immersed in Dulbecco's modified Eagle's medium (DMEM) containing 20% heat-inactivated fetal bovine serum (FBS), 20 mmol/L HEPES, and 100 U/mL penicillin/streptomycin (Invitrogen/Gibco, Carlsbad, CA) for cell culture establishment.
The patients' characteristics are described in Table 1. IH was diagnosed in the outpatient clinic by physical examination. All hernias were in the midline. Previous operations leading to primary IH formation included resection of colon or rectal tumor (n = 17), cholecystectomy (n = 1), appendectomy (n = 1), caesarean section (n = 1), and abdominal trauma (n = 1). Hernia repair was performed with an open onlay technique.4 Reference tissue samples were obtained from voluntary donors undergoing elective abdominal surgery, who had not previously received a laparotomy.9 Patients with previous wound infection, diabetes, or connective or systemic inflammatory disease were not included in the study. The research protocol was reviewed and approved by the institutional ethics committee, and informed written consent was obtained from all participants.
Table 1.
Patient Characteristics
| Non-IH (n = 20) | IH (n = 21) | P value⁎ | |
|---|---|---|---|
| Age ± SEM (range) | 65 ± 3 (55–86) | 63 ± 3 (50–89) | 0.61 |
| Men | 13 | 16 | 0.51 |
| Smokers | 5 | 3 | 0.45 |
| Alcoholics | 2 | 3 | 1.00 |
| Obesity (body mass index >29) | 5 | 11 | 0.11 |
| Diabetes | 0 | 0 | 1.00 |
| Hyperlipidemia | 1 | 2 | 0.49 |
| Hyperuricemia | 1 | 0 | 1.00 |
Student's t-test or Fisher's exact test.
Histology and IHC
Paraffin-embedded sections (4 μm) underwent hematoxylin-eosin (H&E), PAS, Masson's trichrome, and Alcian Blue (pH 2.5) staining, as per routine clinical practice. Immunohistochemistry (IHC) was performed as described.9 The following primary antibodies were used: anti-vimentin (1:100, ab-2 clone V9; Dako, Glostrup, Denmark), anti-α-smooth muscle actin (anti−α-SMA) (1:100, clone 1A4; Dako), anti-Ki-67 (1:100, clone MIB-1; Dako), anti-proliferating cell nuclear antigen (anti-PCNA) (1:100, clone PC-10; Dako), anti-CD3 (1:25; Novocastra Laboratories, Newcastle upon Tyne, UK), anti-CD20 (1:100; Dako), anti-CD45 (1:100; Dako), anti-CD68 (1:80; Dako), and anti-CD138 (1:50; Dako). Cell death was quantified by terminal deoxynucleotidyl transferase-mediated dNTP-biotin nick end labeling (TUNEL), using a modification of the Roche in situ apoptosis detection kit (Roche, Lewes, UK).16 Cells were defined as apoptotic if the whole nuclear area of the cell labeled positively. An experienced soft-tissue pathologist (P.H.) inspected the biopsy specimens. A minimum of three sections (10 randomly chosen, noncontiguous, no-overlapping high-power fields per section; n = 30) were examined per patient. The TUNEL and PCNA indices,16,17 were based on the percentage of stained cells.
Fibroblast Isolation and Cell Culture
All reagents were obtained from Invitrogen/Gibco. Primary cultures were established according to an explant outgrowth protocol.18 Biopsies were minced and digested (0.625% trypsin, 0.02% EDTA; Sigma/Fluka, St. Louis, MO) in phosphate-buffered saline (PBS) without Ca2+/Mg2+ (20 hours, 4°C, mild agitation). Supernatants were centrifuged (150 × g, 10 minutes), resuspended in DMEM plus 20% FBS, 20 mmol/L HEPES, and 100 U/mL penicillin/streptomycin, plated into 25-cm2 flasks, and cultured (37°C, 5% CO2). Precipitates were redigested in trypsin solution (1 hour, 37°C), to increase fibroblast recruitment. At 50% confluence, 20% FBS was reduced to 10% FBS. Cells were passaged before reaching confluence. The medium was changed every other day. Frozen stocks were prepared and cryopreserved in 10% DMSO, in a Nalgene-Cryo-1°C Freezing Container (NALGENE Labware, Rochester, NY). Cells from passages 3 to 6 were used. Fibroblast identity was confirmed by vimentin and CD90 immunocytochemistry, as described below.
Cell Senescence Assay
β-Galactosidase (β-gal) staining19 was performed with a senescence staining kit (Sigma-Aldrich, St. Louis, MO), as per the manufacturer's protocol.
Electron Microscopy
Cells (1 × 106) were fixed in 0.5% glutaraldehyde, 0.1 mol/L sodium cacodylate, 8 mmol/L CaCl2, and 0.2% tannic acid for 1 hour. The solution was changed to 0.1 mol/L sodium-cacodylate buffer, 8 mmol/L CaCl2, 2.5% glutaraldehyde, and 4% paraformaldehyde (2 hours, 4°C). Cells were postfixed with osmium tetroxide, dehydrated through a graded series of ethanol, and then embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate, and examined with a JEM1010 transmission electron microscope (TEM) (JEOL, Ltd., Tokyo, Japan) at the Scientific and Technical Services of the University of Barcelona (Campus Casanova).
Fibroblast Immunocytochemistry
Cells seeded on glass coverslips (1 × 104/well) were incubated in DMEM and 10% FBS (36 hours), and then rinsed, fixed (4% paraformaldehyde, 30 minutes), quenched (20 nmol/L glycine, 15 minutes), permeabilized (0.05% Triton X-100, 5 minutes), and incubated with mouse monoclonal anti-vimentin (1:40, ab-2 clone V9; Dako), mouse monoclonal anti-CD90 (1:25; Serotec, Oxford, UK), or mouse monoclonal anti−α-SMA (1:40, CBL171; Chemicon/Millipore, Billerica, MA), in 5% BSA-PBS (1 hour, room temperature). After washing, (5 minutes, ×3), cells were incubated with Alexa-Fluor 488 goat anti-mouse IgG (1:100 in 0.05% Triton X-100; Invitrogen, Barcelona, Spain; 1 hour, room temperature, in dark). Coverslips were washed (10 minutes, ×3), treated with RNAase A (0.5 mg/mL, 15 minutes, 37°C; Applied Biosystems/Ambion, Austin, Texas), stained with propidium iodide (10 μg/mL; Molecular Probes, Eugene, OR), and mounted in Mowiol 4–88 (Calbiochem, VWR International Ltd., Lutterworth, UK). Actin filaments and focal contacts were visualized with Chemicon's Actin Cytoskeleton and Focal Adhesion Staining Kit (FAK100; Chemicon/Millipore). Cells were analyzed by laser confocal microscopy (Leica TCS-NT-UV; Leica Microsystems, Heidelberg, Germany) at the Scientific and Technical Services of the Institut de Recerca Hospital Universitari Vall d'Hebron (UCTS-IRHUVH). Control staining in the absence of primary antibody confirmed the specificity of immune labeling.
Cell Proliferation Assays
Population doubling (PD) was calculated as follows: PD = [ln(no. of harvested cells) − ln(no. of subcultivated cells)]/ln2. [3H]-thymidine and 5-bromo-2′-deoxyuridine (BrdU) (Calbiochem) incorporation into cell DNA was used to determine fibroblast proliferation rates. Cells were previously cultured in complete medium to allow cellular attachment (1 × 104 cells/well, 36 hours). They were then incubated in DMEM and 2% FBS (24 hours), and treated with 0.5 μCi/mL [3H]-thymidine (Amersham Pharmacia Biotech, Piscataway, NJ) for 18 hours, washed, and then removed by trypsinization. DNA was precipitated with cold trichloroacetic acid. The precipitate was air-dried overnight, dissolved in NCS tissue solubilizer (Amersham Pharmacia Biotech), and quantified in a scintillation β-counter. The BrdU cell proliferation ELISA kit (Calbiochem) was used as per the manufacturer's recommendations (5 × 103 cells/well). In a subset of cultures, cells were labeled with BrdU. Briefly, cells (1 × 104/well) were seeded on sterile glass coverslips and treated with 10-μm BrdU in DMEM and 10% FBS (12 hours). Cells were rinsed, fixed, quenched with NH4Cl 50 mmol/L (20 minutes), permeabilized (1% Triton X-100, 15 minutes), treated with 50 μL of DNase (1U/μL, 90 minutes, 37°C), and then incubated in 1% BSA (1 hour, room temperature). Mouse monoclonal anti-BrdU (1:100 in 1% BSA-PBS; Calbiochem) was added, and cells were treated as described in the immunocytochemistry section.
Migration Assays
For the in vitro scratch assay, cells (1 × 105/well) were seeded on fibronectin-coated wells (10 μg/mL; Boehringer Ingelheim GmbH, Ingelheim am Rhein, Germany) with markings on the outer bottom to be used as reference points during image acquisition.20 Cells were incubated with DMEM and 10% FBS (36 hours). To minimize proliferation, the medium was then changed to 2% FBS (12 hours). Cell layers were scraped in a straight line (p10 tip), rinsed (×3), and incubated (DMEM, 2% FBS, 24 hours). Plates were examined periodically and photographed (Nikon Eclipse TS100, Tokyo, Japan). The acquired images were analyzed with freeware (available at http://rsb.info.nih.gov/ij, last accessed October 29, 2009). For transwell migration assays, cells (5 × 103) were plated in DMEM and 2% FBS into the upper chamber of a Costar Transwell (8-μm-pore insert; Corning, Corning, NY) and incubated. The insert was coated with either type-I collagen (30 μg/mL; Calbiochem) or fibronectin (30 μg/mL; Boehringer Ingelheim). The lower chamber contained 10% FBS. Twelve hours later, nonmigrated cells were scraped. Cells remaining on the bottom surface were fixed (methanol 80% v/v, 30 minutes), stained (0.5% crystal violet w/v, 15 minutes), and analyzed by spectrophotometry (OD, 570 to 590 nm).
Adhesion Assay
Cells (1.5 × 103/well) were seeded on 24-well plates, and then incubated in DMEM and 2% FBS (12 hours). The plate was inversely centrifuged (900 × g, 10 minutes), and the amount of remaining attached cells was assessed by crystal violet staining.21,22
Immunoblotting
Cells or cryopreserved tissues were homogenized with RIPA buffer [50 mmol/L Tris-HCl (pH 7.6), 150 mmol/L NaCl, 1% Triton X-100, 0.25% sodium deoxycholate, 0.1% SDS, 1 mmol/L EDTA, plus protease inhibitors; Sigma] and centrifuged (12,000 × g, 4 minutes, 4°C). Protein concentration was determined by the bicinchoninic acid assay (BCA Protein Assay; Pierce, Madrid, Spain). Equal amounts of total protein were size-fractionated by SDS-PAGE under reducing conditions (120 V), transferred to nitrocellulose or polyvinylidene fluoride (PVDF) membranes (60 V, 4°C, overnight), and stained with red Ponceau. Membranes were blocked in TBS-T buffer [0.9% NaCl, 0.02 mol/L Tris (pH 7.5), 0.05% Tween 20, 5% to 10% skimmed milk, 1 hour, room temperature], before incubation with the following antibodies: mouse monoclonal anti-vimentin (1:700, ab-2 clone V9; NeoMarkers/Lab Vision, Thermo Fisher Scientific Inc., Fremont, CA), mouse monoclonal anti-poly (ADP-ribose) polymerase (PARP) (1:125; BD Biosciences, Franklin Lakes, NJ), mouse monoclonal anti-pan-actin (1:500; NeoMarkers/Lab Vision Corp), rabbit polyclonal anti−α-fodrin (1:700, 2122 Asp1185; Cell Signaling, Beverly, MA), rabbit polyclonal anti-caspase-3 (1:700; Cell Signaling), mouse monoclonal anti−β-tubulin (1:3000, 5H1; BD Biosciences), rabbit polyclonal anti-microtubule-associated protein 1 light chain 3 (LC3) (1:1000; PM036; MBL Medical & Biological Laboratories Co., Ltd., Nagoya, Japan); mouse monoclonal anti-flotillin-2 (1:5000; BD Biosciences). Detection was performed with appropriate peroxidase-labeled secondary antibodies in TBS-T buffer, using an enhanced chemiluminescence kit (Amersham Pharmacia Biotech). Bands were quantified with a digital image analyzer (Quantity One Quantitation Software; Bio-Rad, Barcelona, Spain), on unsaturated X-ray film. When appropriate, the ProteoExtract subcellular proteome extraction kit (Calbiochem) was used to obtain cytosolic, membrane/organelle, nuclear, and cytoskeletal protein fractions.23
Immunoprecipitation
Tissue samples (∼500 mg) or cells (∼1.5 × 106) were homogenized [20 mmol/L TrisHCl (pH 7.5), 150 mmol/L NaCl, 1% Triton X-100, 1 mmol/L EDTA, 1 mmol/L EGTA, 2.5 mmol/L Na4P2O7, 1 mmol/L β-glycerolphosphate, 1 mmol/L Na3VO4, 1 mmol/L PMSF, plus protease inhibitors; Sigma] and centrifuged (12,000 × g, 15 minutes, 4°C). Protein concentration was determined by the BCA assay (Pierce). Precipitating phospho-(Ser/Thr) Phe polyclonal antibody (1:100; Cell Signaling) was added to tissue (750 μg) or cell (500 μg) protein supernatants, and incubated with rocking (overnight, 4°C). Protein A-Agarose (Roche Applied Science, Mannheim, Germany) diluted in PBS (1:1 v/v) was added (1:20 v/v) and incubated with rocking (5 hours, 4°C). Beads were collected by centrifugation (12,000 × g, 1 minute) and washed (12,000 × g, 30 seconds, ×3). Twenty-five microliters of 3× sodium dodecyl sulfate (SDS) sample buffer [187.5 mmol/L Tris-HCl (pH 6.8 at 25°C), 6% w/v SDS, 30% glycerol, 150 mmol/L DTT, 0.03% w/v bromophenol blue] were added, and beads were denatured (10 minutes, 100°C). Aliquots of the supernatant were run on SDS-PAGE gels and immunoblotted, as described.
Apoptosis Induction
Cells (2 × 104/well) were seeded on glass coverslips and allowed to adhere (DMEM, 10% FBS, 12 hours). Medium was switched to DMEM, 2% FBS (12 hours, basal level of apoptosis ∼2% to 3%), and cells were treated with 0.1 μmol/L staurosporine (16 hours),24 or with tumor necrosis factor α and cycloheximide25 (TNFα/CHX, 0.25 to 50 ng/mL; Sigma; 72 hours). Cells were morphologically inspected, and necrosis/apoptosis was measured with the Vybrant Apoptosis Assay kit, n°2 (Molecular Probes) and laser confocal microscopy. Cells cultured in DMEM and 10% FBS were used as negative controls. To examine cytoskeletal cleavage, 5 × 104 cells were plated on 60-mm culture dishes and treated as described. At the end, adherent and floating cells were combined, lysed with the ProteoExtract subcellular proteome extraction kit (Calbiochem), and analyzed as described in the immunoblotting section.
Statistical Analysis
Data were quantified as the mean ± SE. Differences between group means were analyzed using the unpaired Student's t-test or Mann-Whitney U-test. Categorical data were analyzed by Fischer's exact test. A simple regression procedure was applied to assess the relationship between TUNEL and PCNA staining. P values for significance were designated at <0.05.
Results
The case and control groups were comparable for age, sex, and comorbidities (Table 1).
Degenerative Changes in IH Fascia Tissue Specimens
Relative to non-IH, IH fascia sections displayed disruption and thinning of the ECM together with a significant reduction in fibroblast density (IH: 207 ± 7 cells/field; non-IH: 372 ± 12 cells/field; P < 0.001) (Figure 1). Moreover, IH fibroblasts often displayed a round or oblong appearance and lacked cell-matrix contact (Figure 1D, inset). Alcian Blue staining was stronger in IH (semiquantitative analysis, as percentage of samples: non-IH: +60%, ++40%; IH: +10%, ++30%, +++60%; Figure 1, E and F). α-SMA labeling was limited to the vascular walls, and no myofibroblasts were seen (Figure 1, K and L). Inflammatory cell infiltration was negligible, as screened by PAS (Figure 1, G and H) and by CD3, CD20, CD45, CD68, and CD138 immunolabeling (data not shown).
Figure 1.
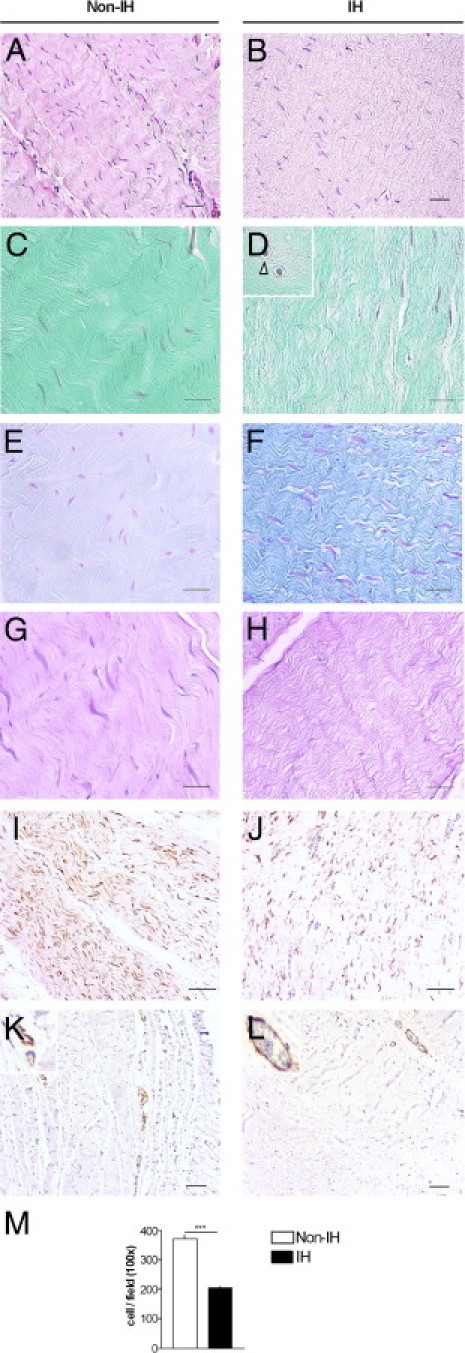
Fascia histological features. Sections of fascia from non-incisional hernia (non-IH) and IH patients were stained with H&E (A and B), Masson's trichrome (C and D), Alcian Blue (E and F), PAS (G and H), and vimentin (I and J). In IH fascia, fibroblasts tend to be round or oblong (arrowheads), and some are surrounded by an open space (D, inset); K and L: α-SMA immunostaining. Magnification: H&E, vimentin, and α-SMA, ×100, scale bar = 100 μm; Masson's trichrome and Alcian Blue, ×400, scale bar = 50 μm. M: Mean fibroblast cells per field from non-IH (n = 20) and IH (n = 21), as measured in H&E-stained fascia sections. ***P < 0.001, Student's t-test.
Cell death and proliferation rates were assessed by IHC (Figure 2). TUNEL and PCNA indices were higher in IH (Figure 2G). Ki-67 rates were low in both IH and non-IH (<1%; Figure 2, E and F). TUNEL-positive fibroblasts were densely labeled in their nuclei, accompanied by small particles resembling apoptotic bodies (Figure 2B), which is compatible with an apoptotic cell death. Only occasionally, a few cells showed a diffuse, nuclear and cytoplasmic, brown color, which is compatible with necrotic cells. PCNA and TUNEL indices correlated positively (r = 0.818; P = 0.007). Since fibroblasts were the predominant cellular components identified (Figure 1, I and J), we assumed that most of the PCNA+ and TUNEL+ cells were fibroblasts.
Figure 2.
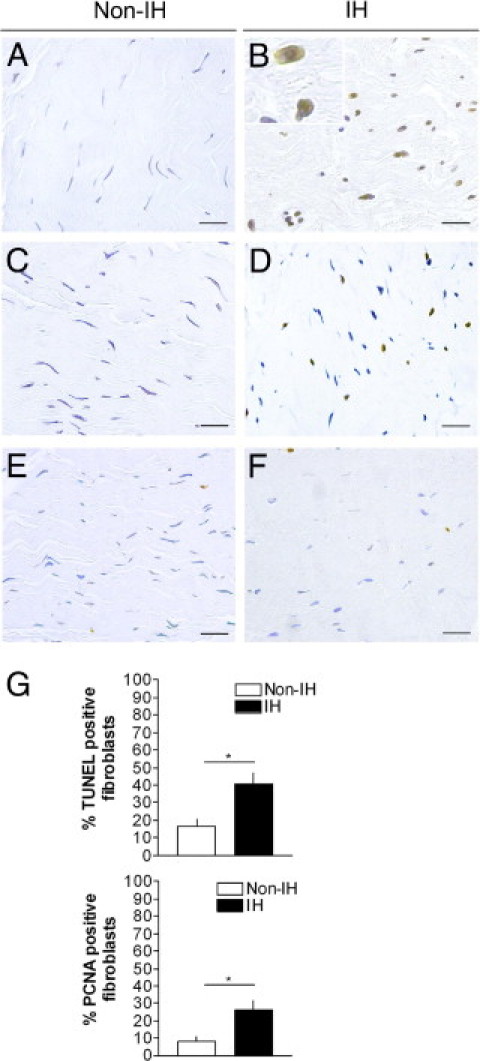
Apoptosis and cell proliferation markers in fascia tissue. Terminal deoxynucleotidyl transferase-mediated dNTP-biotin nick end labeling (TUNEL) (A and B), proliferating cell nuclear antigen (PCNA) (C and D), and Ki-67 (E and F) immunostaining of non-IH and IH fascia tissue (magnification, ×200; scale bars: 50 μm). G: Quantitative analyses of TUNEL-positive and PCNA-positive fibroblast nuclei, in non-IH (n = 20) and IH (n = 21) fascia; *P < 0.05 (Student's t-test).
Caspase-3 Activation and Cytoskeletal Cleavage in IH Fascia Tissue
Our results suggested that apoptotic cell death mechanisms may be involved in IH. Next, we examined caspase-3- and cytoskeleton-related components by immunoblotting. Of interest, IH homogenates displayed increased expression of procaspase-3 (32 kDa) and its major activated fragments (p20, p17), coincident with increased vimentin, α-fodrin, and PARP cleavage (Figure 3, A and B). Native (57 kDa) and fragmented vimentin (mainly the 53-kDa and <48- to 50-kDa bands) were higher in IH, as were intact (240 kDa) and cleaved (150 kDa, 120 kDa) α-fodrin and PARP (116 kDa) (Figure 3B). Neither β-tubulin nor actin cleavage was observed (Figure 3A). The increase of soluble-to-cytoskeletal vimentin ratios (Figure 3C) and phosphorylated vimentin levels in IH (especially an ∼48-kDa band, not detectably expressed in non-IH; Figure 3D) may reflect a comprehensive vimentin cytoskeleton disassembly.26
Figure 3.
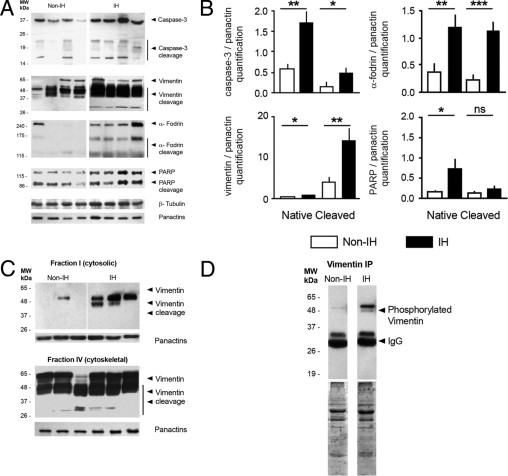
Proteolytic cleavage of caspase-3 and cytoskeletal substrates in fascia tissue. A: Representative immunoblot analyses from non-IH and IH fascia. Pan-actin was used as a loading control for normalization purposes. MW, molecular weight. B: Densitometric analysis of native and cleaved protein expression, in non-IH (n = 8) and IH (n = 8) samples (ns, nonsignificant; *P < 0.05, **P < 0.01, ***P < 0.001; Student's t-test). C: Representative immunoblots for the cytosol- (soluble) and cytoskeletal-enriched fractions from non-IH and IH fascia homogenates. D: Representative immunoblots of cytosol-enriched fractions, after immunoprecipitation with a specific polyclonal anti-phospho-Ser/Thr antibody (details in the Materials and Methods section).
IH Fibroblasts Display a Distinct Pathological Phenotype
Cells were positive for vimentin (Figure 4) and CD90 (data not shown). β-gal staining was very low (<1%) in both IH fibroblasts (IHF) and non-IH fibroblasts (non-IHF) (data not shown). A phenotypic transformation was observed in IHFs, which persisted throughout all passages studied (3 to 6; Table 2, Figure 4).
Figure 4.
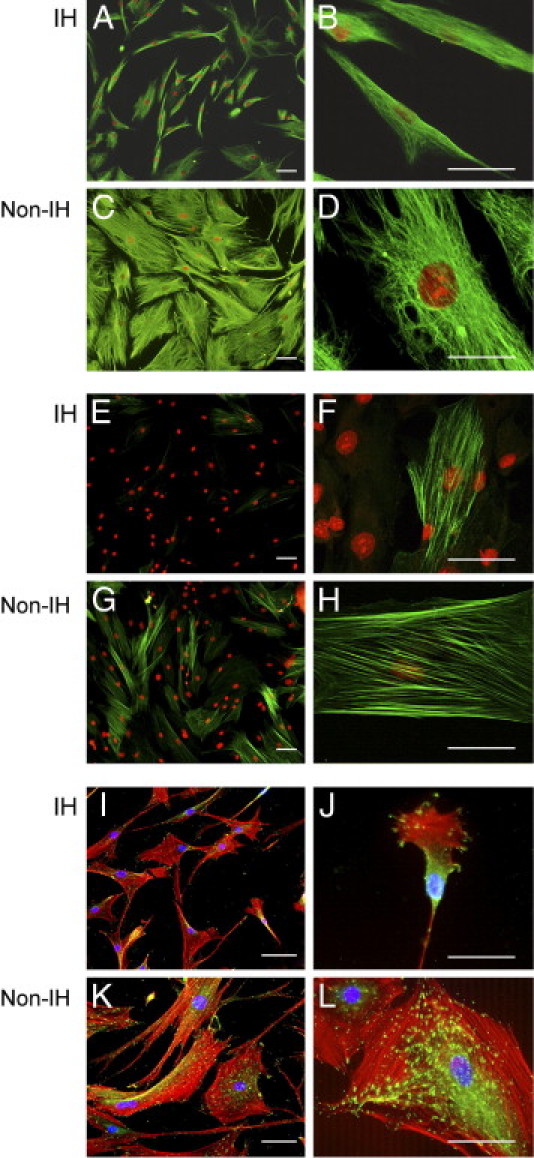
Fascia fibroblast characterization. Representative immunofluorescent staining of primary fibroblasts from non-IH and IH fascia, for vimentin (green, A–D), α-SMA (green, E–H), and vinculin/phalloidin (green/red, I–L). Magnification: ×100 (A, C, E, and G), ×200 (I and K), ×400 (B, D, F, H, J, and L); Scale bar = 50 μm.
Table 2.
Fascia Fibroblast Characterization
| Non-IH | IH | P value | |
|---|---|---|---|
| Cell culture efficiency | |||
| n | 20 | 21 | |
| Culture success, % | 40% | 70% | ⁎ |
| Time to emerge from the explant, days | 34.3 ± 27.0 | 18.2 ± 2.0 | ⁎⁎ |
| Cell yield, 106 cells/biopsy | 1.2 ± 0.3 | 2.6 ± 2.0 | ⁎ |
| Morphology analysis | |||
| n | 5 | 5 | |
| Spreading area, μm2 | 8463 ± 211 | 1008 ± 211 | ⁎ |
| Polarized cells, % | 7.5 ± 0.9 | 75.2 ± 2.1 | ⁎ |
| Actin microfilament network | |||
| n | 5 | 5 | |
| Cells with geodomes and stress fibers, % | 87 ± 1 | 54 ± 1 | ⁎ |
| Cells with stress fibers, % | 24.4 ± 1.1 | 4.6 ± 0.7 | ⁎ |
| α-SMA-positive cells, % | 17.1 ± 2.5 | 3.6 ± 1.0 | ⁎ |
| Focal adhesions | |||
| n | 5 | 5 | |
| Area, μm2 | 3.1 ± 0.1 | 2.2 ± 0.1 | ⁎ |
| Focal adhesion area/cell area, % | 6.3 ± 0.6 | 2.6 ± 0.2 | ⁎ |
P <0.05;
P <0.01, Mann-Whitney U-test.
Cell culture efficiency was higher in IH samples. Morphologically, non-IHFs showed a stellate morphology with long processes and contained large and well-organized actin fibers and a dense vimentin network throughout the cytoplasm. In contrast, IHFs displayed a bipolar spindle shape and significantly reduced surface area, as well as fewer actin microfilaments and a more shrunken vimentin network. Streaks of vinculin-positive focal adhesions were prominent and widely distributed in non-IHFs (Figure 4, K and L), whereas in IHFs, they were more diffused and confined to the leading edges (Figure 4, I and J). α-SMA expression, a feature of myofibroblasts,27 was higher in non-IHFs (Figure 4, G and H; P < 0.05).
Electron microscope observations showed an accumulation of vacuole/autophagosome-like structures, containing cytoplasm and encircled by multilayered lamellar structures, as well as fingerprint profiles and apparent mitochondrial swelling in IHFs (Figure 5).28 In non-IHFs, multilayered structures appeared only occasionally, and fingerprint profiles were absent. Outstandingly, IHFs rendered fibroblasts fragile and more prone to rupture. Simple preparation for TEM observation caused sudden cell membrane rupture (Figure 5B).
Figure 5.
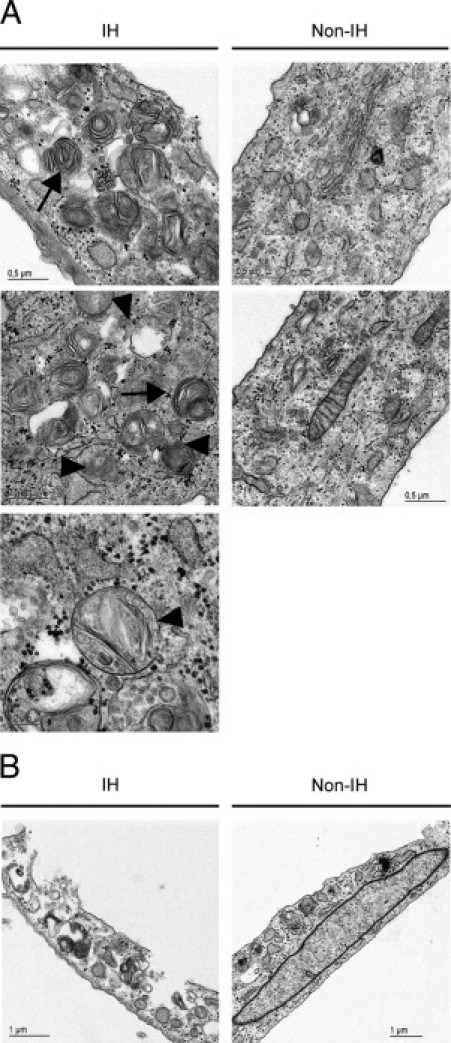
Ultrastructural appearance. A: Representative ultrastructural pictures of primary fascia fibroblasts. Outstandingly, IH fibroblasts showed autophagic vacuoles, autophagolysome-like structures, multilayered lamellar and fingerprint profiles (arrows), and mitochondrial swelling (arrowheads). B: Evidence of IH fibroblast fragility at preparation for transmission electron microscopy analysis.
Concerning the functional properties, the replicative rate was shorter in IHFs than in non-IHFs (PD, 11.9 ± 3.3 versus 32.8 ± 8.2 per day; P < 0.05), suggestive of increased IHF proliferative capacity, which was fully confirmed by [3H]-thymidine and BrdU incorporation assays and by direct BrdU labeling (Figure 6, A, B, and D). In addition, IHFs showed increased migration rates with respect to non-IHFs. Thus, in scratch wound assays, IHFs repopulated the wound faster than non-IHFs (Figure 6, C and E). Moreover, in transwell assays, IHFs migrated faster than non-IHFs in response to fibronectin and type I collagen (Figure 6G). Lastly, IHFs exhibited lower adhesiveness to plasticware substrate as compared to non-IHFs, as measured by reverse centrifugation (Figure 6F).
Figure 6.
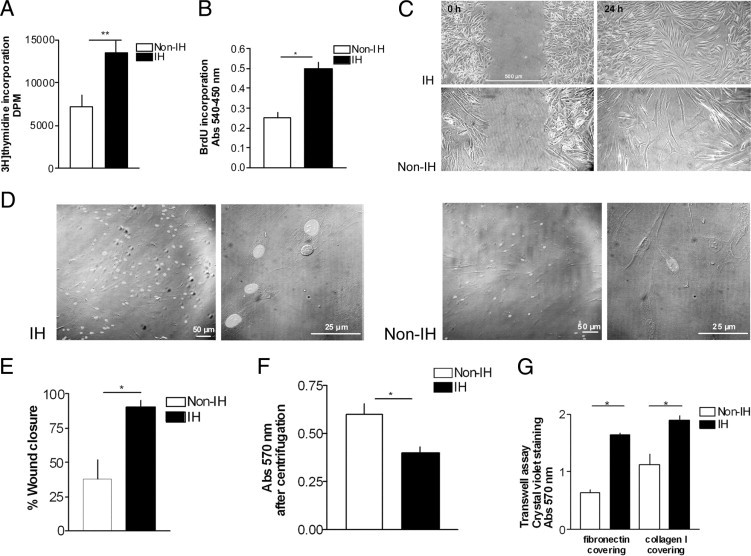
Analysis of primary fibroblast behavior. Proliferation assays: (A) [3H]-thymidine and (B) 5-bromo-2′-deoxy-uridine (BrdU) incorporation into DNA, in non-IHFs (n = 5) and IHFs (n = 5). Abs, absorption. Migration and adhesion assay: (C and E) representative images of the in vitro scratch assay captured at 0 hours to 24 hours postwounding, in non-IHFs and IHFs, and comparison of wound closure percentage between non-IHFs (n = 5) and IHFs (n = 5) (details in the Materials and Methods section). D: BrdU in situ immunostaining. Magnification: ×100, ×600. F: Cell adhesion of non-IHFs (n = 5) and IHFs (n = 5), quantified by inverted centrifugation. G: Migration of non-IHFs (n = 5) and IHFs (n = 5) across fibronectin- and type I collagen-coated inserts in transwell assays. *P < 0.05, **P < 0.01
Autophagy Marker LC3 Increases Its Expression in IH
LC3 has been proposed to be a biomarker of autophagy. To understand further the results obtained by electron microscopy, we examined the steady-state levels and intracellular localization of LC3 proteins, both in fascia fibroblasts and in fascia tissue biopsies by immunoblotting. An antibody raised against the recombinant human full-length LC3 (MAP1LC3B), was used.
First, we examined whole fibroblast homogenates (Figure 7) and whole tissue homogenates (Figure 8). As shown in Figure 7, IHF lysates displayed enhanced LC3-I expression relative to non-IHFs, but no changes were observed in LC3-II. Thus, the LC3-II to LC3-I ratio was significantly higher in non-IHFs as compared to IHFs (P = 0.037). Conversely, when whole tissue biopsies were examined, a significant increase in LC3-II to LC3-I ratio was observed in IH fascia (P = 0.045; Figure 8), which can be indicative of in vivo autophagosome formation.
Figure 7.
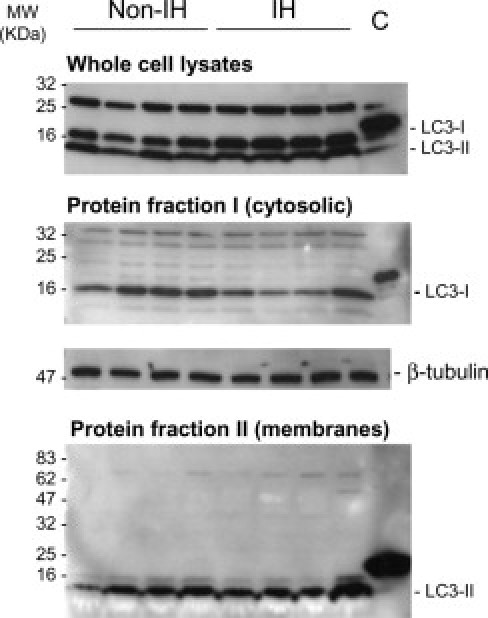
LC3 immunoblots in fascia fibroblasts. Representative immunoblots of LC3-I/LC3-II in whole-cell lysates (1 × 106 cells; RIPA lysis buffer), and cytosolic- and membrane-enriched fractions (ProteoExtract Subcellular Proteome Extraction Kit) from cultured non-IH fibroblasts (non-IHFs) and IHFs. Proteins were resolved on 4% to 20% polyacrylamide gradient SDS gels. Cytosolic and membrane protein fractions were concentrated to about 10-fold on Amicon Ultra 0.5-mL centrifugal filters (Millipore, Billerica, MA). Positive controls for anti-LC3 antibody were used (MBL International Corp.). β-Tubulin detection in cytosolic-enriched protein extracts was used as a loading control (C) for normalization purposes.
Figure 8.
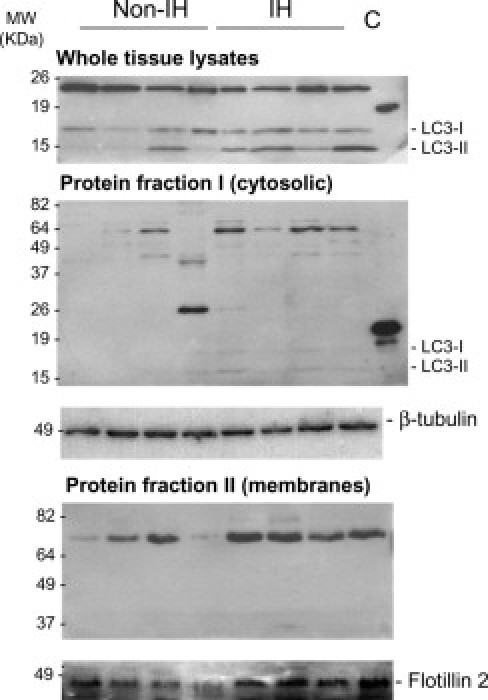
LC3 immunoblots in fascia tissue. Representative immunoblots of LC3-I/LC3-II in whole lysates (RIPA lysis buffer), cytosolic- and membrane-enriched fractions (ProteoExtract Subcellular Proteome Extraction Kit) from non-IH and IH fascia tissues. SDS-PAGE was performed using 12.5% polyacrylamide gels and positive controls for anti-LC3 antibody (MBL). The cytosolic and membrane protein fractions were further concentrated to about sevenfold on Amicon Ultra 0.5-mL centrifugal filters (Millipore) and resolved in 11% and 8% SDS polyacrylamide gels, respectively. β-Tubulin detection in cytosolic protein extracts was used as a loading control for normalization purposes. Flotillin-2 was used as a membrane marker.
LC3 is found in the cytoplasm (LC3-I) and membrane (LC3-II). Next, we aimed to test which subcellular fraction of LC3 was increased in IH samples. Both cytosolic and membrane/organelle fractions were evaluated. In fascia fibroblasts, LC3-I was detected in the cytosolic fraction and LC3-II in the membrane/organelle fraction (Figure 7). Intriguingly, LC3-I amounts were apparently lower in IHFs, as compared to non-IHFs (P = 0.006), but again, no changes were detected in LC3-II levels. Cytosolic fractions derived from fascia biopsies generally yielded no, or only faint, bands (Figure 8), excepting a specific ∼70-kDa band, which was observed in IH samples. Notably, in the membrane/organelle fraction, a strong signal corresponding to a specific ∼70-kDa band was observed, probably corresponding to the membrane-associated LC3-II form. This band was significantly higher in IH, as compared to non-IH samples (P = 0.007).
IH Fibroblasts Show Increased Sensitivity to Proapoptotic Challenges
Finally, we examined whether IHFs engaged cell death processes more readily than non-IHFs, after treatment with the proapoptotic agents staurosporine and TNFα/CHX. Staurosporine is a potent, cell-permeable protein kinase C inhibitor, which at high concentrations induces cell apoptosis. TNFα is a multifunctional proinflammatory cytokine that belongs to the tumor necrosis factor (TNF) superfamily.
Preliminary experiments in our laboratory revealed that both staurosporine and TNFα/CHX were competent, being that TNFα/CHX is a more potent inducer of programmed cell death in fascia fibroblasts. Since staurosporine induced a rapid transition from non-apoptotic to necrotic cell death, whereas TNFα/CHX acted more gradually, and due to sample limitations, further experiments to better characterize the response (ie, caspase activation, substrate cleavage) were exclusively performed with TNFα/CHX. As is shown in Figure 9, IHFs contained many loosely bound, detaching round vesicles after TNFα/CHX treatment, as compared with non-IHFs. The number of early and late apoptotic fibroblasts was also higher in IH. Immunoblotting experiments revealed more cytoskeletal cleaved products in IH following TNFα/CHX treatment (Figure 10). Of interest, the pattern of cleaved products observed during in vitro apoptosis induction resembled that seen in fascia tissues, suggesting that this process may occur in vivo.
Figure 9.
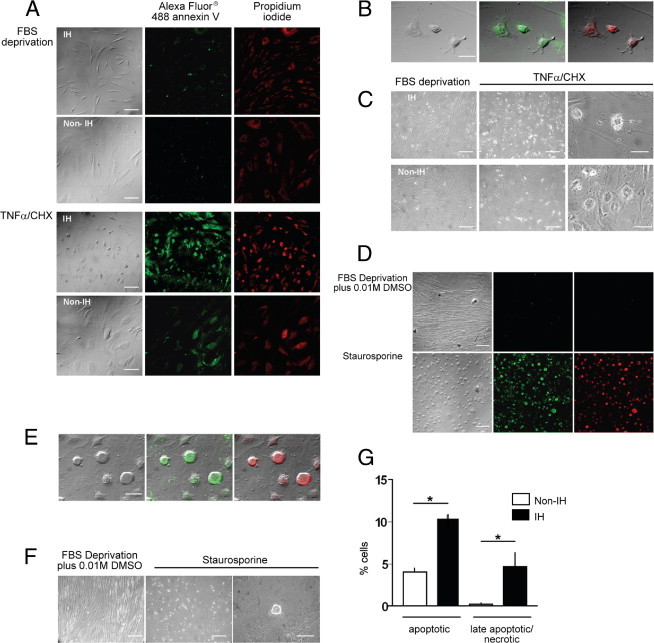
Fibroblast susceptibility to proapoptotic stimuli. Apoptotic response of non-IHFs (n = 5) and IHFs (n = 5), as measured by Vybrant Apoptosis Assay kit II (details in the Materials and Methods section). A–C, and G: TNF-α/CHX-treatment (72 hours): (A) Bright-field and immunofluorescence images, ×100; (B) merged images, ×600; (C) phase contrast images: left and center, ×100; right, ×600; (G) percentage of apoptotic cells (*P < 0.05, Mann-Whitney U-test). D–F: Staurosporine treatment (16 hours): (D) bright-field and immunofluorescence images, ×100; (E) merged images, ×600; (F) phase contrast images: left and center, ×100; right, ×600. Scale bars: 100 μm (×100); 50 μm (×600).
Figure 10.
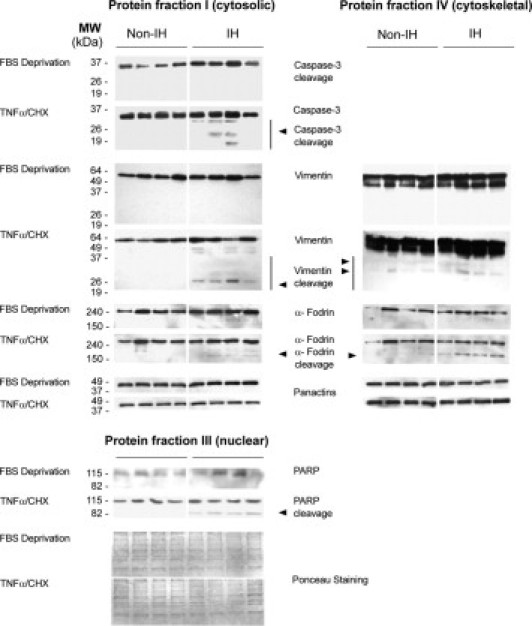
Immunoblots of proteins involved in apoptosis. Representative immunoblot analyses, obtained from cytosolic-, nuclear-, and cytoskeletal-enriched fractions from non-IHFs and IHFs, after TNFα/CHX treatment. Proteins were extracted by the ProteoExtractTM Subcellular Proteome Extraction Kit (details in Materials and Methods).
Discussion
Incisional hernia is a serious complication of laparotomies, which is characterized by the ongoing degeneration of the myofascial layer. Even though complex, multifactorial events have been associated with the disease, the pathomechanisms of its formation remain elusive. This relative lack of knowledge is surprising, but might be partly due to the availability of prosthetic biomaterials, and thus, the feeling in the surgical community that there is little need to understand the fundamental mechanisms of the disease (biopolymers revolutionized hernial repair, drastically reducing recurrences as compared to tissue mending and making it possible to reconstruct large ventral defects that were previously irreparable4). Within the scope of hernia biology,7 here, we tried to identify (new) cellular and molecular clues with a potential role in IH pathogenesis, which could be eventually useful for new preventive and therapeutic approaches. Our results demonstrate significant cell loss and matrix degradation in IH fascia. Also, new features such as aberrant fibroblast cell-death induction, and substantial in vitro morphological and functional changes have been detected in fascia-derived IHFs, which had not been previously described.
The upstream signals by which both tissue and cells are removed are unknown. Previous studies have reported the presence of unwarranted degradation and defective formation of newly synthesized proteins in IH samples, which may influence quantitative changes in the fascia microenvironment.7,9 Moreover, the intense Alcian Blue staining reported here entails additional qualitative alterations, such as enhanced glucososaminoglycans deposition in the ECM, which could putatively instigate prolonged proinflammatory signaling,29,30 or altered degradation activities31 in IH tissues.
An interesting and novel aspect of this study is the identification of apoptotic as well as autophagic signals in IH fascia, which can lead to fibroblast self-destruction without an efficient compensatory increase in cell proliferation levels.32,33 The observation of augmented TUNEL indices, together with activated caspase-3 and its associated cleavage signature, is compatible with apoptotic cell-death induction. Moreover, the present data also provide evidence that autophagy may be enhanced in vivo in IH fascia, as judged from the LC3-II membrane-associated content and the LC3-II to LC3-I ratio.34,35 Concerning cell proliferation, the DNA polymerase processivity factor proliferating cell nuclear antigen indices were increased in IH fascia samples. However, Ki-67 levels, a marker of actively cycling cells,36 were low and equivalent between patients and controls. PCNA is essential, not only for DNA replication, but also for several forms of DNA repair. Of interest, different binding affinities for PCNA in DNA replication and repair have been described (a surprisingly longer residence time of PCNA at damaged areas than at replication foci has been observed).37 Overall, these results are compatible with processes related to cellular and DNA damage. Further studies in molecular and genetic terms are required to dissect these aspects.
The observed fragmentation of cytoskeleton components informs on a dynamic process of cell disruption, which ultimately may weaken IH fibroblast function and lead to fascia atrophy.38 In particular, we report vimentin proteolysis together with increased soluble and phosphorylated vimentin (indices of vimentin disassembly) in IH fascia homogenates.39 Vimentin is the major intermediate filament of fibroblasts. As such, it acts as a scaffold for organelles and a massive number of signaling molecules, and may modulate the response to specific proapoptotic signals.40–44 Whether the changes observed here imply a cytoprotective mechanism to damp down proapoptotic signaling,41,42 or a way to perpetuate IH progression requires further investigation. α-Fodrin proteolysis is considered a molecular feature of apoptosis, leading to membrane malfunction and cell shrinkage.45 PARP-1 is a nuclear enzyme that participates in both DNA repair and cell death, depending on the amount of cell damage, and promotes the expression of proinflammatory agents.46
At present, the initiation factors or the molecular pathways involved in these cell-death processes can only be speculated on. The deregulated proinflammatory and proteolytic signaling reported in the tissues microenvironment of IH patients9 may favor the release of products that trigger caspase activation.13,47 Another mechanism might arise from detachment of internal cytoskeleton structures from the external ECM.7,48–50 Also, evidence from our laboratory indicates transcriptional changes in IHFs, which may tip the balance into a proapoptotic direction (unpublished observations). Whether these and/or other inductive factors, such as loss of growth factors or excessive levels of reactive oxygen species,51,52 regulate fibroblasts death in vivo remains to be established.
With respect to the in vitro experiments, our findings indicate that fascia-derived IHFs acquire an imprinted phenotype in vivo that is consistent in culture throughout several passages, and impairs fundamental cellular processes.15,53,54 Previous work has established altered type I/III collagen ratios in IHFs.55 Here, we report that fascia IHFs display reduced cell size, increased proliferation and migration rates, and decreased strength of adhesion, in parallel with an altered cytoskeleton network, fewer α-SMA-positive stress fibers, and smaller focal contacts.56,57 Moreover, they show enhanced sensitivity to proapoptotic challenges, as well as features compatible with compromised autophagic/lysosomal processes.28
Fibroblast hyperproliferation may be due to prolonged exposure to a low fibril density ECM in vivo,49 to altered transcription of genes involved in the cell cycle, or DNA repair regulation, as was recently seen in gene expression profiling experiments in our laboratory (unpublished observations), or to a combination of all these factors.58,59 Increased migratory and invasive capacity may be a consequence of defective cytoskeleton organization,60–62 or to other factors such as extreme MMP activation,9 or ECM alterations. Moreover, cytoskeletal network defects may cause fibroblasts to become fragile, and this would account for their trauma-induced rupture.62–64 Although we could not measure the autophagic flux, the results obtained by combined steady-state measurements (electron microscopy and LC3 immunoblotting) suggest that autophagy induction, and/or inhibition of autophagosome clearance, may have a role in IH. Autophagy may contribute to cell death or be advantageous for cell survival during ECM detachment; also, autophagy is critical for the presentation of engulfment signals that mediate the phagocytic clearance of apoptotic cells.65–69 Further studies are needed to further illuminate this previously unrecognized aspect of IH pathology.
Finally, we show that fascia primary IHFs were more sensitive to proapoptotic agents (either staurosporine or TNFα/CHX) than non-IHFs, as an indirect sign of DNA damage. The observed differences in the responses suggest that the level of initial DNA damage in the two cell types may have differed, or perhaps, the repair of the damaged DNA in the two cell types varied kinetically. On the other hand, the differences in the responses by the two cell types may be due to relative differences in the abilities of IH and non-IH cells to mount a response to a common level of DNA damage. In summary, the apoptosis-like abnormalities seen in IH homogenates are recapitulated in vitro in IHFs under proapoptotic stimuli, supporting the idea that these events are relevant to in vivo findings.70 Interestingly, our previous results demonstrated deregulated TNFA expression and signaling in abdominal wall tissue of IH patients,9 which would be consistent with the increased susceptibility of IHFs to TNFα-induced apoptosis reported here.
Conclusions are limited by the chronic nature of the disease process (tissue was sampled at a point in time when IH had already developed). Nonetheless, we believe that our findings are representative, supported by the fact that no differences were present between IH and non-IH control patients (eg, age, sex, and comorbidities), and because in vivo and ex vivo analyses were performed simultaneously. It will be a challenge to determine the specific role these events play in the very early phase of the disease, even before the onset of symptoms.
To date, most studies dealing with IH biology have evaluated parameters related to ECM metabolism (reviewed in reference 7). Our data provide indirect, yet compelling evidence on how the local ECM microenvironment may influence IH development. Our findings suggest that fascia atrophy and dysmorphology may be an active driver of IH, by means of transducing signals that affect cell survival, functional phenotype, and (probably) gene expression. Also, they identify fibroblasts and cell fragility as major pathogenic contributors to IH.63 Damage to fascia fibroblasts may trigger excess cell death in the form of classical apoptosis or autophagy, in which caspases and lysosomal enzymes, respectively, play the main role in the cells' self-digestion. The accumulation of apoptotic-prone defective fascia fibroblasts may not support normal fascial structure and function, and thus enhance proteolytic fascial destruction in a context of low cell proliferation.71,72 These findings are important because they identify new players in the IH process as it occurs in vivo, and provide information to define new targets to eventually solve treatment failure and persistent recurrence.
Acknowledgments
We thank Celine L. Cavallo for English language review, Marta Rebull for technical assistance, and Isabel Quiles for help with artwork.
Footnotes
Supported in part by grants from the Carlos III Spanish Institute of Health (projects PI030290 and PI070507).
R.D. and M.T.Q. contributed equally to this work.
References
- 1.Muysoms F.E., Miserez M., Berrevoet F., Campanelli G., Champault G.G., Chelala E., Dietz U.A., Eker H.H., El Nakadi I., Hauters P., Hidalgo Pascual M., Hoeferlin A., Klinge U., Montgomery A., Simmermacher R.K., Simons M.P., Smietański M., Sommeling C., Tollens T., Vierendeels T., Kingsnorth A., Muysoms F.E., Miserez M., Berrevoet F. Classification of primary and incisional abdominal wall hernias. Hernia. 2009;13:407–414. doi: 10.1007/s10029-009-0518-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flum D.R., Horvath K., Koepsell T. Have outcomes of incisional hernia repair improved with time?: A population-based analysis. Ann Surg. 2003;237:129–135. doi: 10.1097/00000658-200301000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arbos M.A., Ferrando J.M., Quiles M.T., Vidal J., López-Cano M., Gil J., Manero J.M., Peña J., Huguet P., Schwartz-Riera S., Reventós J., Armengol M. Improved surgical mesh integration into the rat abdominal wall with arginine administration. Biomaterials. 2006;27:758–768. doi: 10.1016/j.biomaterials.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 4.den Hartog D., Dur A.H., Tuinebreijer W.E., Kreis R.W. Open surgical procedures for incisional hernias. Cochrane Database Syst Rev. 2008;(3) doi: 10.1002/14651858.CD006438.pub2. CD006438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yahchouchy-Chouillard E., Aura T., Picone O., Etienne J.C., Fingerhut A. Incisional hernias: I. Related risk factors. Dig Surg. 2003;20:3–9. doi: 10.1159/000068850. [DOI] [PubMed] [Google Scholar]
- 6.Jansen P.L., Mertens P., Klinge U., Schumpelick V. The biology of hernia formation. Surgery. 2004;136:1–4. doi: 10.1016/j.surg.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Franz M.G. The biology of hernia formation. Surg Clin North Am. 2008;88:1–15. doi: 10.1016/j.suc.2007.10.007. vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fricke M., Langer C., Brunner E., Sakai L.Y., Füzesi L., Reinhardt D.P., Quondamatteo F. Fibrillin-1 in incisional hernias: an immunohistochemical study in scar and non-scar regions of human skin and muscle fasciae. J Anat. 2008;212:674–685. doi: 10.1111/j.1469-7580.2008.00885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guillen-Marti J., Diaz R., Quiles M.T., Lopez-Cano M., Vilallonga R., Huguet P., Ramon-y-Cajal S., Sanchez-Niubo A., Reventós J., Armengol M., Arbos M.A. MMPs/TIMPs and inflammatory signalling deregulation in human incisional hernia tissues. J Cell Mol Med. 2009;13:4432–4443. doi: 10.1111/j.1582-4934.2008.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rath A.M., Chevrel J.P. The healing of laparotomies: review of the literature. Hernia. 1998;2:145–149. [Google Scholar]
- 11.Grimm D. Biomedical research: Cell biology meets rolfing. Science. 2007;318:1234–1235. doi: 10.1126/science.318.5854.1234. [DOI] [PubMed] [Google Scholar]
- 12.Fisher G.J., Quan T., Purohit T., Shao Y., Cho M.K., He T., Varani J., Kang S., Voorhees J.J. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am J Pathol. 2009;174:101–114. doi: 10.2353/ajpath.2009.080599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobaczewski M., Gonzalez-Quesada C., Frangogiannis N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010;48:504–511. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lerman O.Z., Galiano R.D., Armour M., Levine J.P., Gurtner G.C. Cellular dysfunction in the diabetic fibroblast: impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am J Pathol. 2003;162:303–312. doi: 10.1016/S0002-9440(10)63821-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flack E.C., Lindsey M.L., Squires C.E., Kaplan B.S., Stroud R.E., Clark L.L., Escobar P.G., Yarbrough W.M., Spinale F.G. Alterations in cultured myocardial fibroblast function following the development of left ventricular failure. J Mol Cell Cardiol. 2006;40:474–483. doi: 10.1016/j.yjmcc.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 16.Bateman A.C., Turner S.M., Thomas K.S., McCrudden P.R., Fine D.R., Johnson P.A., Johnson C.D., Iredale J.P. Apoptosis and proliferation of acinar and islet cells in chronic pancreatitis: evidence for differential cell loss mediating preservation of islet function. Gut. 2002;50:542–548. doi: 10.1136/gut.50.4.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bravo R. Synthesis of the nuclear protein cyclin (PCNA) and its relationship with DNA replication. Exp Cell Res. 1986;163:287–293. doi: 10.1016/0014-4827(86)90059-5. [DOI] [PubMed] [Google Scholar]
- 18.Rolf C.G., Fu B.S., Pau A., Wang W., Chan B. Increased cell proliferation and associated expression of PDGFR beta causing hypercellularity in patellar tendinosis. Rheumatology (Oxford) 2001;40:256–261. doi: 10.1093/rheumatology/40.3.256. [DOI] [PubMed] [Google Scholar]
- 19.Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubelj I., Pereira-Smith O., Peacocke M., Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang C.C., Park A.Y., Guan J.L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 21.Hinz B., Dugina V., Ballestrem C., Wehrle-Haller B., Chaponnier C. Alpha-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol Biol Cell. 2003;14:2508–2519. doi: 10.1091/mbc.E02-11-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simpson K.J., Dugan A.S., Mercurio A.M. Functional analysis of the contribution of RhoA and RhoC GTPases to invasive breast carcinoma. Cancer Res. 2004;64:8694–8701. doi: 10.1158/0008-5472.CAN-04-2247. [DOI] [PubMed] [Google Scholar]
- 23.Abdolzade-Bavil A., Hayes S., Goretzki L., Kröger M., Anders J., Hendriks R. Convenient and versatile subcellular extraction procedure, that facilitates classical protein expression profiling and functional protein analysis. Proteomics. 2004;4:1397–1405. doi: 10.1002/pmic.200300710. [DOI] [PubMed] [Google Scholar]
- 24.Belmokhtar C.A., Hillion J., Segal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene. 2001;20:3354–3362. doi: 10.1038/sj.onc.1204436. [DOI] [PubMed] [Google Scholar]
- 25.Holtmann H., Hahn T., Wallach D. Interrelated effects of tumor necrosis factor and interleukin 1 on cell viability. Immunobiology. 1988;177:7–22. doi: 10.1016/S0171-2985(88)80087-1. [DOI] [PubMed] [Google Scholar]
- 26.Li Q.F., Spinelli A.M., Wang R., Anfinogenova Y., Singer H.A., Tang D.D. Critical role of vimentin phosphorylation at Ser-56 by p21-activated kinase in vimentin cytoskeleton signaling. J Biol Chem. 2006;281:34716–34724. doi: 10.1074/jbc.M607715200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomasek J.J., Gabbiani G., Hinz B., Chaponnier C., Brown R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 28.Todde V., Veenhuis M., van der Klei I.J. Autophagy: principles and significance in health and disease. Biochim Biophys Acta. 2009;1792:3–13. doi: 10.1016/j.bbadis.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 29.Tomasek J.J., Gabbiani G., Hinz B., Chaponnier C., Brown R.A. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015–H1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 30.Wang J.Y., Roehrl M.H. Glycosaminoglycans are a potential cause of rheumatoid arthritis. Proc Natl Acad Sci U S A. 2002;99:14362–14367. doi: 10.1073/pnas.222536599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z., Yasuda Y., Li W., Bogyo M., Katz N., Gordon R.E., Fields G.B., Brömme D. Regulation of collagenase activities of human cathepsins by glycosaminoglycans. J Biol Chem. 2004;279:5470–5479. doi: 10.1074/jbc.M310349200. [DOI] [PubMed] [Google Scholar]
- 32.Galluzzi L., Maiuri M.C., Vitale I., Zischka H., Castedo M., Zitvogel L., Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–1243. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 33.Taylor R.C., Cullen S.P., Martin S.J. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 34.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barth S., Glick D., Macleod K.F. Autophagy: assays and artifacts. J Pathol. 2010;221:117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cattoretti G., Becker M.H., Key G., Duchrow M., Schlüter C., Galle J., Gerdes J. Monoclonal antibodies against recombinant parts of the Ki-67 antigen (MIB 1 and MIB 3) detect proliferating cells in microwave-processed formalin-fixed paraffin sections. J Pathol. 1992;168:357–363. doi: 10.1002/path.1711680404. [DOI] [PubMed] [Google Scholar]
- 37.Essers J., Theil A.F., Baldeyron C., van Cappellen W.A., Houtsmuller A.B., Kanaar R., Vermeulen W. Nuclear dynamics of PCNA in DNA replication and repair. Mol Cell Biol. 2005;25:9350–9359. doi: 10.1128/MCB.25.21.9350-9359.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ndozangue-Touriguine O., Hamelin J., Bréard J. Cytoskeleton and apoptosis. Biochem Pharmacol. 2008;76:11–18. doi: 10.1016/j.bcp.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 39.Omary M.B., Ku N.O., Tao G.Z., Toivola D.M., Liao J. “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem Sci. 2006;31:383–394. doi: 10.1016/j.tibs.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 40.Blain E.J., Gilbert S.J., Hayes A.J., Duance V.C. Disassembly of the vimentin cytoskeleton disrupts articular cartilage chondrocyte homeostasis. Matrix Biol. 2006;25:398–408. doi: 10.1016/j.matbio.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 41.Herrmann H., Bär H., Kreplak L., Strelkov S.V., Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol. 2007;8:562–573. doi: 10.1038/nrm2197. [DOI] [PubMed] [Google Scholar]
- 42.Eriksson J.E., Dechat T., Grin B., Helfand B., Mendez M., Pallari H.M., Goldman R.D. Introducing intermediate filaments: from discovery to disease. J Clin Invest. 2009;119:1763–1771. doi: 10.1172/JCI38339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Omary M.B. “IF-pathies”: a broad spectrum of intermediate filament-associated diseases. J Clin Invest. 2009;119:1756–1762. doi: 10.1172/JCI39894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byun Y., Chen F., Chang R., Trivedi M., Green K.J., Cryns V.L. Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death Differ. 2001;8:443–450. doi: 10.1038/sj.cdd.4400840. [DOI] [PubMed] [Google Scholar]
- 45.Jänicke R.U., Ng P., Sprengart M.L., Porter A.G. Caspase-3 is required for alpha-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J Biol Chem. 1998;273:15540–15545. doi: 10.1074/jbc.273.25.15540. [DOI] [PubMed] [Google Scholar]
- 46.Scovassi I.A., Diederich M. Modulation of poly(ADP-ribosylation) in apoptotic cells. Biochem Pharmacol. 2004;68:1041–1047. doi: 10.1016/j.bcp.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 47.Day C.P. From fat to inflammation. Gastroenterology. 2006;130:207–210. doi: 10.1053/j.gastro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 48.Chiarugi P., Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. 2008;76:1352–1364. doi: 10.1016/j.bcp.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 49.Pizzo A.M., Kokini K., Vaughn L.C., Waisner B.Z., Voytik-Harbin S.L. Extracellular matrix (ECM) microstructural composition regulates local cell-ECM biomechanics and fundamental fibroblast behavior: a multidimensional perspective. J Appl Physiol. 2005;98:1909–1921. doi: 10.1152/japplphysiol.01137.2004. [DOI] [PubMed] [Google Scholar]
- 50.Fisher G.J., Varani J., Voorhees J.J. Looking older: fibroblast collapse and therapeutic implications. Arch Dermatol. 2008;144:666–672. doi: 10.1001/archderm.144.5.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–1388. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolf B.B., Schuler M., Echeverri F., Green D.R. Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation factor-45/inhibitor of caspase-activated DNase inactivation. J Biol Chem. 1999;274:30651–30656. doi: 10.1074/jbc.274.43.30651. [DOI] [PubMed] [Google Scholar]
- 53.Brown R.D., Ambler S.K., Mitchell M.D., Long C.S. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 54.Vasilopoulos Y., Gkretsi V., Armaka M., Aidinis V., Kollias G. Actin cytoskeleton dynamics linked to synovial fibroblast activation as a novel pathogenic principle in TNF-driven arthritis. Ann Rheum Dis. 2007;66S3:iii23–iii28. doi: 10.1136/ard.2007.079822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Si Z., Bhardwaj R., Rosch R., Mertens P.R., Klosterhalfen B., Klinge U. Impaired balance of type I and type III procollagen mRNA in cultured fibroblasts of patients with incisional hernia. Surgery. 2002;131:324–331. doi: 10.1067/msy.2002.121376. [DOI] [PubMed] [Google Scholar]
- 56.Zimerman B., Volberg T., Geiger B. Early molecular events in the assembly of the focal adhesion-stress fiber complex during fibroblast spreading. Cell Motil Cytoskeleton. 2004;58:143–159. doi: 10.1002/cm.20005. [DOI] [PubMed] [Google Scholar]
- 57.Tsuruta D., Jones J.C. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J Cell Sci. 2003;116:4977–4984. doi: 10.1242/jcs.00823. [DOI] [PubMed] [Google Scholar]
- 58.Parsonage G., Falciani F., Burman A., Filer A., Ross E., Bofill M., Martin S., Salmon M., Buckley C.D. Global gene expression profiles in fibroblasts from synovial, skin and lymphoid tissue reveals distinct cytokine and chemokine expression patterns. Thromb Haemost. 2003;90:688–697. doi: 10.1160/TH03-04-0208. [DOI] [PubMed] [Google Scholar]
- 59.Flavell S.J., Hou T.Z., Lax S., Filer A.D., Salmon M., Buckley C.D. Fibroblasts as novel therapeutic targets in chronic inflammation. Br J Pharmacol. 2008;153:S241–S246. doi: 10.1038/sj.bjp.0707487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ivaska J., Pallari H.M., Nevo J., Eriksson J.E. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp Cell Res. 2007;313:2050–2062. doi: 10.1016/j.yexcr.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 61.McInroy L., MÄättÄ A. Down-regulation of vimentin expression inhibits carcinoma cell migration and adhesion. Biochem Biophys Res Commun. 2007;360:109–114. doi: 10.1016/j.bbrc.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 62.Goffin J.M., Pittet P., Csucs G., Lussi J.W., Meister J.J., Hinz B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J Cell Biol. 2006;172:259–268. doi: 10.1083/jcb.200506179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coulombe P.A., Kerns M.L., Fuchs E. Epidermolysis bullosa simplex: a paradigm for disorders of tissue fragility. J Clin Invest. 2009;119:1784–1793. doi: 10.1172/JCI38177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Geiger B., Spatz J.P., Bershadsky A.D. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 65.Fung C., Lock R., Gao S., Salas E., Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19:797–806. doi: 10.1091/mbc.E07-10-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He B., Lu N., Zhou Z. Cellular and nuclear degradation during apoptosis. Curr Opin Cell Biol. 2009;21:900–912. doi: 10.1016/j.ceb.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eskelinen E.L. To be or not to be?: Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy. 2008;16:257–260. doi: 10.4161/auto.5179. [DOI] [PubMed] [Google Scholar]
- 69.Mizushima N., Yoshimori T., Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huber L.C., Distler O., Tarner I., Gay R.E., Gay S., Pap T. Synovial fibroblasts: key players in rheumatoid arthritis. Rheumatology (Oxford) 2006;45:669–675. doi: 10.1093/rheumatology/kel065. [DOI] [PubMed] [Google Scholar]
- 71.Lockshin R.A., Zakeri Z. Cell death in health and disease. J Cell Mol Med. 2007;11:1214–1224. doi: 10.1111/j.1582-4934.2007.00150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Terman A., Brunk U.T. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res. 2005;68:355–365. doi: 10.1016/j.cardiores.2005.08.014. [DOI] [PubMed] [Google Scholar]