

Abstract
Powerful noninvasive imaging technologies enable real-time tracking of pathogen-host interactions in vivo, giving access to previously elusive events. We visualized the interactions between wild-type Bacillus anthracis and its host during a spore infection through bioluminescence imaging coupled with histology. We show that edema toxin plays a central role in virulence in guinea pigs and during inhalational infection in mice. Edema toxin (ET), but not lethal toxin (LT), markedly modified the patterns of bacterial dissemination leading, to apparent direct dissemination to the spleen and provoking apoptosis of lymphoid cells. Each toxin alone provoked particular histological lesions in the spleen. When ET and LT are produced together during infection, a specific temporal pattern of lesion developed, with early lesions typical of LT, followed at a later stage by lesions typical of ET. Our study provides new insights into the complex spatial and temporal effects of B. anthracis toxins in the infected host, suggesting a greater role than previously suspected for ET in anthrax and suggesting that therapeutic targeting of ET contributes to protection.
Noninvasive imaging technologies have recently enabled real-time tracking of the interactions between pathogens and their host in vivo.1 The power of these technologies has given access to previously elusive events, difficult to detect by the simple tests usually available, such as lethal dose determination, mean time to death, or CFU counts. They have helped unravel key steps in the infectious processes.
Bacillus anthracis, the spore-forming causative agent of anthrax, produces major virulence factors: a poly-γ-D-glutamic acid capsule and two toxins composed of three proteins: a common receptor-binding domain, protective antigen (PA), combines with the lethal factor (LF) to form the lethal toxin (LT) and combines with the edema factor (EF) to form the edema toxin (ET).2 The poorly immunogenic capsule shields the bacterium from the immune system and protects against phagocytosis.3 The toxins modulate host immune responses leading to immune paralysis.4,5
The mouse model has been, and still is, extensively used and has allowed significant advances in the understanding of key steps of anthrax. Using bioluminescent imaging, we recently followed in real time the kinetics and dissemination pattern of encapsulated nontoxinogenic or nonencapsulated toxinogenic B. anthracis during murine cutaneous, inhalational, and gastrointestinal infections.6–8 We showed that spores germinate and establish infections at the initial site of inoculation in inhalational, cutaneous, and gastrointestinal infections (nasal-associated lymphoid tissues, skin, and Peyer's patches, respectively) without needing to be transported to the draining lymph nodes. Spore entry also occurs through the alveolar space, with germination taking place either en route to or in the mediastinal/thoracic lymph nodes in the first steps of infection, as primitively considered. All routes of infection with encapsulated strains progress first to the draining lymph node; the spleen then acts as a reservoir, and ultimately the lung is colonized by a hematogenic route, leading to death. Absence of capsule markedly modifies the dissemination patterns,6–8 the bacteria being initially confined to the portal of entry for a long period, then reaching specific organs (kidneys, gastrointestinal tract) with minimal colonization of the spleen and, late in the infection, to the lungs.
Histopathological observations in human anthrax cases and in various animal models9–15 show the most prominent lesions at the late stage of infection to be edema and hemorrhage in a wide array of organs, whatever the route of infection: spleen, lungs, lymph nodes (especially in the mediastinum), meninges, or gastrointestinal tract. The inflammatory response is dominated by neutrophilic infiltrates, but is disproportionally mild, despite the large numbers of bacteria. Serosanguineous pleural effusions are prominent, with hemorrhagic mediastinitis, pulmonary infiltrates, and intra-alveolar edema. Edema and hemorrhage appear to originate from multifocal necrotizing vasculitis. Lymphoid depletion is often mentioned in spleen and mediastinal lymph nodes. All these lesions are considered to be caused primarily by the bacterial toxins, as observed after systemic administration of purified ET and LT.4,16,17 Infection with nonencapsulated B. anthracis suggests that LT is a key virulence factor in cutaneous and inhalational murine infection.18,19 Other studies, using an encapsulated laboratory strain, suggest that the toxins do not modify either dissemination or virulence.20–22
In the present study, we visualized in real time the effects of concomitant expression of both capsule and toxins, and their respective contribution, on the early and late interactions between wild-type B. anthracis and its host during cutaneous and inhalational infections. Our approach, which couples histology with bioluminescence imaging under biosafety level 3 (BSL3) confinement conditions, enabled us to focus the analysis on the spleen, which acts as a reservoir during infection, at particular time points of infection; this approach provided, for the first time, insights into the spatial and temporal effects of the toxins on the host immune system and tissues. We showed that ET plays a pivotal role in virulence in guinea pigs that could be partially reversed by an anti-ET therapy. Edema toxin, but not LT, markedly modified the patterns of dissemination in the mouse, compared with a nontoxinogenic strain, and each toxin provoked particular lesions in the spleen. Finally, when ET and LT were expressed together during inhalational infection, an ET-dominated pattern of dissemination was observed in the majority of the animals. In contrast, the toxins did not modify the dissemination pattern during cutaneous infection. Finally, the lesions produced by the toxins occurred with a specific temporal pattern, with lesions consistent with LT occurring first, then followed by lesions typical of ET.
Materials and Methods
Animal and Bacterial Strains
Female BALB/c mice (6 to 8 weeks old) and Hartley guinea pigs (200 to 250 g) were purchased from Charles River (L'Arbresle, France). The animals were housed in the BSL3 animal facilities of the Institut Pasteur licensed by the French Ministry of Agriculture and complying with European Union regulations. The protocols were approved by the Institut Pasteur Safety Committee, according to the standard procedures recommended by the Institut Pasteur Animal Care and Use Committee.
The B. anthracis strains used were the wild-type clinical isolate 9602 and the isogenic derivative mutants inactivated in the pagA gene 9602P(Tox−); their LD50 is <25 to 40 spores.23 Strains constructed in this study were the toxin-inactivation mutants in the cya gene [9602C(LF+PA+)] and in the lef gene [9602L(EF+PA+)] by insertion of a spectinomycin and erythromycin resistance cassette, respectively, by conjugation as described previously19,23 (Table 1). The gene inactivations were checked by PCR on bacterial lysates and by Western blot on secreted bacterial proteins in R medium as described previously.24,25 Luminescent B. anthracis strains used in this study were constructed by insertion of the luxABCDE operon of Photorhabdus luminescens as described previously.6,8 Expression of the lux operon did not affect the virulence of the strains, as evidenced by equivalent mean time to death compared with the parental strains (data not shown).
Table 1.
Bacillus anthracis Strains Used in This Study
| Strain | Capsule | Toxin components | Toxins | ||
|---|---|---|---|---|---|
| 9602WT | Cap+ | EF+ | LF+ | PA+ | ET+ LT+ |
| 9602L | Cap+ | EF+ | Inactivated | PA+ | ET+ |
| 9602C | Cap+ | Inactivated | LF+ | PA+ | LT+ |
| 9602P | Cap+ | EF+ | LF+ | Inactivated | None |
Cap+ denotes the presence of the cap operon coding for the enzymes necessary for synthesis of the polyglutamate capsule. EF+, LF+, and PA+ denote functional cya, lef, and pagA genes coding for the toxin components edema factor EF, lethal factor LF, and protective antigen PA, respectively.
Mouse Infection and Bioluminescence Imaging
Mice were challenged with each luciferase-expressing bacterial strain either into the ear pinna (3.40 ± 0.29 log10 CFU, mean ± SD, in 10 μL) or by intranasal route (6.52 ± 0.21 log10 CFU, mean ± SD, in 20 μL) as described previously7,8 and were analyzed at least twice a day. The challenge inocula were lethal, and all mice displayed bioluminescence on infection. For CFU determination, organs were immediately placed in ice-cold saline solution and homogenized in a chilled glass tube. Spore enumeration was performed after heat treatment of a fraction of each tissue homogenate (65°C, 30 minutes). Samples were then serially diluted and plated on brain heart infusion (BHI) agar plates. Immunization with PA was performed by intranasal and subcutaneous injection of rPA with cholera toxin and alum as the respective adjuvants, as described previously.26 Mortality after intranasal infection with the parental nonbioluminescent strains (5.31 ± 0.19 log10 CFU) was monitored for 15 days, three times per day.
Bioluminescent imaging was acquired using an IVIS 200 system (Xenogen, Hopkinton, MA) according to the manufacturer's instructions. Analysis and acquisition were performed using Living Image 3.0 software (Xenogen). Before image acquisition, mice were anesthetized and placed in a mobile BSL3 confinement ventilated box compatible with luminescence technology (Tem Sega, Lormont, France) approved by the Institut Pasteur Safety Committee. Ventilation was performed by aspiration with an optimal electronic pump with a 150 L/h air output (Schego, Offenbach am Main, Germany) and 0.22-μm filters for inflow and outflow. Images were acquired with a binning of 16. Luminescent signals from the exterior of mice were acquired for 1 minute; when luminescence signal intensity was too high, it was integrated for 10 seconds. All other photographic parameters were held constant. Quantifying photons per second emitted by each organ was performed by defining regions of interest corresponding to the organ of interest. Image analysis was performed only on animals for which the entire kinetics of bacterial dissemination could be followed from the portal of entry to the spleen and lungs. Animals that died between the experimental observation windows were excluded from analysis, because information on the kinetic patterns was missing. For example, the fulminant nature of inhalational infection progression with the 9602L-lux(EF+PA+) strain resulted in exclusion of a significant proportion of the infected BALB/c mice (as described in the Results section).
Guinea Pig Infection and Therapeutic Intervention
For determination of the median lethal dose LD50 of each toxin-deficient strain, guinea pigs were infected subcutaneously in the flank with an inoculum (200 μL) of increasing concentration. Survival was monitored twice daily for 15 days. LD50 was calculated according to the method of Reed and Muench.27 Immunization with PA was performed by subcutaneous injection of 30 μg PA (kindly provided by Dr. Les Baillie, Cardiff University, Wales) at 28 and 14 days before spore challenge. Therapeutic administration of adefovir dipivoxyl (Bis-POM-PMEA called PMEA throughout the text; 9-[2-[bis[(pivaloyloxy)methoxy]-phosphinyl]-methoxy]ethyl]adenine, kindly provided by Craig Gibbs, Gilead Sciences, Foster City, CA), 30 mg/kg per day, was performed by intraperitoneal injection twice daily in 500 μL PBS; controls were injected with PBS alone. Challenge was performed subcutaneously with the 9602L(EF+PA+) strain (2.25 ± 0.32 log10 CFU).
Flow Cytometry Analysis
The cervical draining lymph node was excised 20 hours after spore inoculation into the ear pinna and was dissociated to obtain a single cell suspension. Cell-surface labeling was performed with the following antibodies anti-F4/80 for macrophages and anti-CD4 and anti-CD8 for T lymphocytes (all from Pharmingen, BD Biosciences, Le Pont de Claix, France) and anti-Ly-6G for neutrophils (Biolegend, San Diego, CA). Analysis was performed on a FACSCalibur flow cytometry system (BD Biosciences, San Jose, CA).
Histological Analysis
Organs were taken at specific time points of infection as defined by bioluminescence (Table 2). After fixation in 10% neutral buffered formalin and embedding in paraffin, serial 5-μm sections were cut and stained with H&E and Gram stain. Encapsulated bacilli were detected by immunohistochemistry (IHC) using a rabbit polyclonal antiserum specific for capsule polyglutamate as described previously.8 For detection of apoptosis processes, an in situ cell death detection kit (Roche, Mannheim, Germany) was used, based on labeling of DNA strand breaks (TUNEL technology: terminal deoxynucleotidyl transferase dUTP nick end-labeling), according to the manufacturer's instructions.
Table 2.
Edema Toxin or Lethal Toxin Alone Restores Full Virulence in the Guinea Pig Cutaneous Infection
| Strain | Doses tested | LD50 (log10 CFU) (95% CI) | Mean time to death (days ± SEM) | Animals |
|---|---|---|---|---|
| 9602WT(EF+LF+PA+) | 4 | 1.8 (<1.0–2.2) | 2.8 ± 0.6 | 12 |
| 9602L(EF+PA+) | 28 | 2.3 (2.2–2.4) | 3.1 ± 0.8 | 53 |
| 9602C(LF+PA+) | 6 | 1.9 (<1.0–2.3) | 3.3 ± 1.0 | 15 |
| 9602P(Tox−) | 14 | 4.4 (4.3–4.5) | 6.5 ± 3.0 | 14 |
Guinea pigs were infected subcutaneously in the flank with increasing doses of spores (4 animals per dose) of the wild-type 9602 strain and the toxin inactivation mutants 9602L(EF+PA+), 9602C(LF+PA+) and 9602P(Tox−). Survival was followed over a 15-day period. LD50 values were calculated according to Reed and Muench.27 Linear regression with 95% confidence limits (95% CI) was calculated with Prism software. Results represent the synthesis of at least three experiments for each strain.
Splenic histological lesions were scored as described by Paola Roccabianca (Descriptive Technique in Pathology, http://www.cldavis.org/cgi-bin/download.cgi?pid=65; last accessed January 25, 2011), coded as follows: − for no lesion and +/− for minimal, + for mild, ++ for moderate, +++ for marked, and ++++ for severe lesions.
Statistical Analysis
Statistical analysis and graphing were performed with GraphPad Prism software (version 4.0; GraphPad Software, San Diego, CA) with Student's unpaired t-test, log-rank test for survival analysis, linear regression for mean lethal dose determination, and Mann-Whitney test for mean time to death determination.
Results
ET or LT Toxin Alone Restores Full Virulence in the Guinea Pig Cutaneous Infection
The guinea pig is sensitive to infection with wild-type strains and has been the animal of choice for the discovery and purification of the toxins.28 It is often used for Sterne vaccine efficacy testing (European Pharmacopeia). Guinea pigs efficiently control nontoxinogenic encapsulated B. anthracis.23 It is thus a highly sensitive model for characterizing the respective roles of ET and LT secreted during an infection with the wild-type encapsulated strain 9602WT(EF+LF+PA+) and its isogenic toxin-mutants lacking either LT [9602L(EF+PA+)], or ET [9602C(LF+PA+)].
The LD50 of the toxin-inactivated mutants 9602L(EF+PA+), 9602C(LF+PA+), and 9602P(Tox−) and the 9602WT(EF+LF+PA+) strains were determined for a cutaneous infection (Table 2). We confirmed the high virulence of the 9602WT(EF+LF+PA+) wild-type strain and the attenuation of the 9602P(Tox−) strain. Surprisingly, production of ET alone was able to restore nearly full virulence; LT alone was also able to restore virulence. The mean time to death was similar for a comparable infectious dose for the 9602WT(EF+LF+PA+), 9602L(EF+PA+), and 9602C(LF+PA+) encapsulated strains, but was extended for the nontoxinogenic encapsulated 9602P(Tox−) strain even with a 100-fold increased infectious dose (Table 2).
Restoration of virulence was due to production of EF in addition to PA, because neutralization of toxin activity by immunization with protective antigen (PA), the common cell-binding toxin component, led to protection against infection with the 9602L(EF+PA+) strain (Figure 1A; P = 0.0003). Inhibition of ET activity through therapeutic intervention could thus be of high beneficial value for the infected host. Adefovir dipivoxyl (PMEA), an inhibitor of the enzymatic activity of EF in vitro,29 has been used for hepatitis B treatment in humans.30 Treatment with PMEA led to significant partial protection against cutaneous 9602L(EF+PA+) infection (Figure 1B; P = 0.027).
Figure 1.
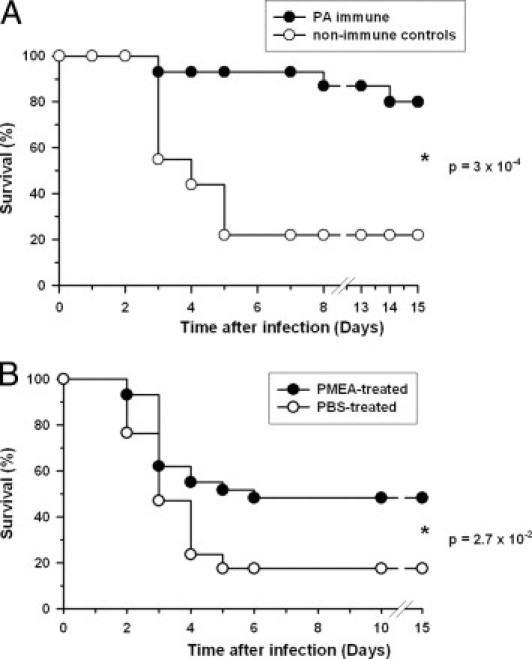
Control of edema toxin activity leads to protection. A: Guinea pigs were immunized with PA and infected subcutaneously in the flank with spores of the 9602L(EF+PA+) strain as described in Materials and Methods. Survival was followed over a 15-day period. Results are the synthesis of two independent experiments giving similar results (controls, n = 9; PA-immune, n = 15). B: Guinea pigs were infected and followed as in A; one group was then treated twice a day with the EF inhibitor bis-POM-PMEA (PMEA) as described in Materials and Methods, the other control group was treated with PBS under the same conditions. Results are the synthesis of four independent experiments giving similar results (controls, n = 17; PMEA-treated, n = 29). Statistical significance was calculated by the log-rank test with GraphPad Prism software.
Specific Effects of ET and LT in Cutaneous Anthrax
To address in greater depth the specific role of edema toxin in anthrax, we used the powerful imaging technology of bioluminescence for tracking in real time the toxin-mutant bacteria through its host. Bioluminescent imaging cannot be performed in the guinea pig, however, because the depth of internal organs and the thickness of the skin hinder light transmission. We therefore had to resort to the mouse model, which is more amenable to detailed analysis. We have previously characterized in real time the infection in the mouse model.6–8 The three bioluminescent strains used in the present study were the wild-type strain 9602WT-lux(EF+LF+PA+) and the encapsulated isogenic derivative mutants lacking either ET [9602C-lux(LF+PA+)] or LT [9602L-lux(EF+PA+)]. The luxABCDE operon was under control of the toxin promoter pagA, which is highly expressed in vivo. Bioluminescence signal is detected when the bacilli are metabolically active.
After inoculation into the subcutis of the ear pinna, bioluminescence was detected at the site of injection of the three toxin-secreting lux strains within 24 hours in 97% of the 88 infected mice, showing that spore germination was triggered and that the bacteria became metabolically active (Figure 2). Production of LT by the 9602C-lux(LF+PA+) strain led to a progression of infection similar to that previously reported for the nontoxinogenic encapsulated 9602P-lux(Tox−)8 (Figure 2, A and E). Bioluminescence was observed in the ear within 21 hours after inoculation (Figure 2A); dissemination to the draining cervical lymph node occurred within 26 hours in 89% of the mice and systemic dissemination to the spleen and lungs within 48 ± 4 hours (mean ± SEM, n = 6; range, 45 to 68 hours). In contrast, production of ET during infection with the encapsulated 9602L-lux(EF+PA+) strain clearly modified the dissemination pattern. Bioluminescence was observed in the ear within 20 hours; however, it was never detected in the cervical lymph node for any of the 32 mice tested, even when systemic dissemination was clearly detected in the spleen and the lungs (45 ± 4 hours; range, 24 to 68 hours; n = 13) (Figure 2B). Nevertheless, bacteria could be detected in the cervical lymph node (2.29 ± 0.3 log10 CFU per lymph node, n = 3) when the spleen displayed bioluminescence; this shows that the bacteria transited through the lymph node, although the bacterial load remained below the detection threshold of the bioluminescence signal.8 This suggests that ET did not allow extensive local multiplication in the draining lymph node.
Figure 2.

Specific dissemination patterns induced by edema and lethal toxins from cutaneous anthrax. A–C and E: Top row: ventral view of a single representative mouse. Bottom row: dorsal view of the same mouse. Mice were inoculated into the ear with spores of the 9602C-lux(LF+PA+) (A), 9602L-lux(EF+PA+) (B), 9602WT-lux(EF+LF+PA+) (C), and 9602P-lux(Tox−) (E) strains and were analyzed for bioluminescence at the indicated times. These image series are representative of at least 20 infected mice. Black and white photographs are overlaid with false-color representation of luminescence intensity expressed in photon/s/cm2/sr. D: Surface labeling of the cervical draining lymph node cells 20 hours after cutaneous inoculation of spores of each toxin-secreting strain. Results are expressed as means ± SEM, n = 5. *P < 0.05; **P < 0.005.
To address the immunological events occurring in the lymph node, we analyzed the host innate cell response in the lymph node 20 hours after inoculation of mice with the three toxin-secreting strains (Figure 2D). Infection with the 9602C-lux(LF+PA+) strain provoked an increase in total cell number (2.4-fold), because of recruitment of neutrophils (eightfold increase) and CD4 and CD8 T cells (2.6-fold increase). Notably, infection with the 9602L-lux(EF+PA+) strain did not induce significant increase in total cell number, neutrophils, and CD4 T cells; in contrast, CD8 T-cell number was increased (threefold), leading to an inversion in the CD4/CD8 ratio. No macrophage recruitment was observed for the three toxin-secreting strains (data not shown).
To address the potential synergistic role of both toxins during infection, mice were infected with the wild-type strain 9602WT-lux(EF+LF+PA+). Progression of infection was similar to that observed with the 9602C-lux(LF+PA+) strain. After detection of luminescence in the ear within 24 hours, bacteria spread to the cervical lymph nodes in 88% of the animals (Figure 2C). Systemic dissemination to the spleen and lungs was then observed (range, 24 to 68 hours; 42 ± 3 hours, mean ± SEM; n = 13).
Specific Temporal Relationship of the ET- and LT-Induced Lesions in Vivo during Infection
To gain insight into how the toxins produced during a spore-initiated infection alter the integrity of the host tissues and subvert the inflammatory response, we performed a histopathological analysis on the spleen. We have previously shown that, whatever the route of infection with an encapsulated strain, the spleen is the first organ displaying bioluminescence at the stage of systemic dissemination, acting as a reservoir for bacterial multiplication.8 Progression of infection was slightly different for each mouse, and bioluminescence quantification in spleen and lungs thus allowed us to focus on defined time windows of the systemic dissemination process (Table 3). Lung bioluminescence defines a late infection stage, death occurring in the following few hours. A synthesis of the observations is presented in Table 4. Although some traits of the lesions observed with the three toxin-secreting strains were indeed common with the no-toxin 9602P strain, additional traits due to the toxic effects of the toxins specifically modified the lesion pattern of the no-toxin strain.
Table 3.
Bioluminescence Parameters for the Mice Used for Histology
| Strain | Route of infection | Time (hours) | Signal (photon/s/cm2/sr) |
Morphotype | |
|---|---|---|---|---|---|
| Lungs | Spleen | ||||
| 9602WT-lux(EF+LF+PA+) | SC | 50 | 1.9 × 105 | 1.3 × 106 | 1 |
| SC | 48 | 1.7 × 106 | 8.6 × 106 | 1 and 2 | |
| SC | 50 | 2.2 × 106 | 6.0 × 106 | 2 | |
| SC | 72 | 3.0 × 106 | 2.1 × 107 | 2 | |
| IN | 48 | 3.7 × 104 | 4.8 × 104 | 1 | |
| IN | 48 | 1.2 × 107 | 8.3 × 106 | 2 | |
| 9602C-lux(LF+PA+) | SC | 44 | 4.0 × 105 | 2.6 × 106 | NA |
| SC | 44 | 4.0 × 105 | 1.6 × 106 | NA | |
| 9602L-lux(EF+PA+) | SC | 50 | 9.0 × 104 | 1.7 × 105 | NA |
| SC | 40 | 1.7 × 105 | 4.2 × 105 | NA | |
| SC | 40 | 5.5 × 105 | 1.8 × 106 | NA | |
| 9602P-lux(Tox−) | SC | 40 | 4.1 × 105 | 5.4 × 106 | NA |
| SC | 40 | 1.1 × 107 | 6.7 × 106 | NA | |
Values of the bioluminescence signal (photon/s/cm2/sr) at the time of organ removal during cutaneous (SC) and intranasal (IN) infection with the four bioluminescent strains. Quantification of the bioluminescence signal was performed by defining regions of interest spanning the lungs and the spleen. Definition of morphotypes for the 9602WT-lux(EF+LF+PA+) lesions is given in the Results section. Background bioluminescence level was 4.5 × 104 ± 1.3 photon/s/cm2/sr for the lungs and 8.3 × 104 ± 3.0 for the spleen (mean ± SD, n = 13). NA, not applicable.
Table 4.
Patterns of Splenic Lesions According to the Bacterial Strain
| Bacterial strains | Red pulp (RP) |
White pulp (WP) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Localization | Severity | Necrosis | Edema | Neutrophils |
Bacteria |
Capsule | Cell debris | TUNEL | |||
| Number | Fragmentation | Localization | Density | ||||||||
| 9602C-lux (LF+PA+) | Multifocal | +++ | ++ | − | ++++ | ++++ | Multifocal | +++ | ++ | − | − |
| 9602L-lux (EF+PA+) | Multifocal to coalescing | +++ | +/− | ++++ | + | +/− | Diffuse | ++++ | ++ | Periphery ++ | Periphery ++ |
| 9602P-lux (Tox−) | Multifocal to coalescing | +++ | + | ++ | ++ | ++ | Diffuse | ++++ | ++ | − | − |
| 9602WT-lux (EF+LF+PA+) morphotype 1 | Multifocal | +/− | +/− | − | + | +++ | Multifocal | ++ | ++ | − | − |
| 9602WT-lux (EF+LF+PA+) morphotype 2 | Multifocal to coalescing | +++ | ++ | ++ | ++ | + | Diffuse | +++ | ++ | Periphery +++ | Periphery +++ |
Scoring of lesions: −, none; +/−, minimal; +, mild; ++, moderate; +++, marked; ++++, severe.
The nontoxinogenic encapsulated strain provoked marked splenic inflammatory lesions in the red pulp (Figure 3, G–I). The bacteria were located at the immediate periphery of the white pulp and were surrounded by a peripheral rim of well-delineated cellular infiltrates of numerous neutrophils; few bacteria were found in these infiltrates, and few neutrophils were present in the bacterial aggregates (Figure 3, H, I, and inset in I). Infection with the LT-secreting strain provoked large cellular infiltrates (up to 500 μm in diameter) of fragmented neutrophils invading the red pulp (Figure 3, A–C); their center was necrotic, containing dying neutrophils, extensive cell debris, and many encapsulated bacteria. Some of these infiltrates formed microabscesses, leading to almost total destruction of the red pulp (Figure 3, B and C). IHC staining with an anti-capsular polyglutamate immune serum showed that the bacteria were encapsulated and surrounded by extracellular capsular material (ie, stained material not associated with the bacterial cell) (Figure 3C, inset). No significant histological lesion was observed in the white pulp for the nontoxinogenic and LT-secreting strains (Figure 4, E–H).
Figure 3.
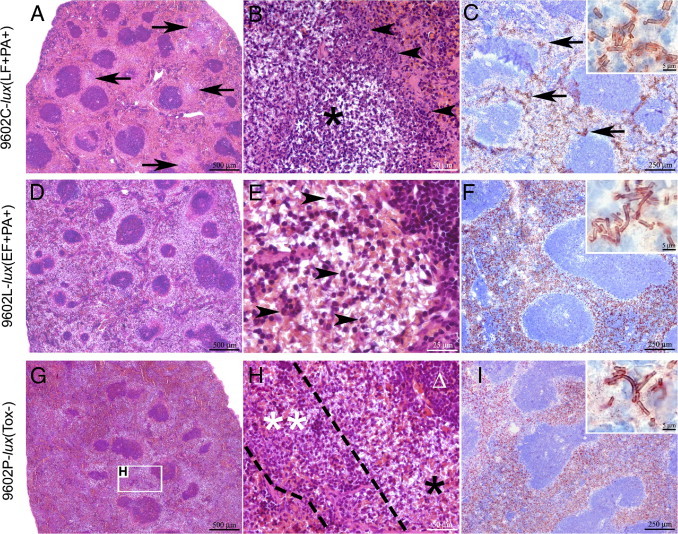
Edema and lethal toxins elicit particular patterns of lesions in the spleen during cutaneous spore infection. H&E staining (A, B, D, E, G, and H) or IHC using a polyclonal antibody directed against B. anthracis polyglutamate (C, F, and I). A–C: At 44 hours after inoculation with 9602C-lux(LF+PA+), multifocal lesions are distributed in the red pulp (A, arrows) displaying a concentric organization (B), characterized by a necrotic center (asterisk) surrounded by a very high number of neutrophils (arrowheads), often karyorrhectic, forming microabscesses; multifocal to coalescing bacteria colonies (C, arrows), included in extracellular capsular material (C, inset), were observed in the red pulp. D–F: At 40 hours after inoculation with 9602L-lux(EF+PA+), multifocal to coalescing lesion was observed in the red pulp (D), characterized by a replacement of red pulp cells by an abundant extracellular and poorly stained material (edema) (E) and containing rare normal neutrophils (E, arrowheads); bacteria were diffusely infiltrating the red pulp (F) and were included in capsular material (F, inset). G–H: At 40 hours after inoculation with 9602P-lux(Tox−), multifocal to coalescing lesion centered on the red pulp was observed (G), characterized by a high number of bacteria (H, singleasterisk) located at the immediate periphery of the lymphoid structures (Δ), and circled by a high number of neutrophils, very often karyorrhectic (double asterisks); high infiltration of the red pulp by the bacteria was observed (I). Scale bars: 500 μm (A, D, and G); 250 μm (C, F, and I); 50 μm (B and H); 25 μm (E); and 5 μm (insets).
Figure 4.
Edema toxin provokes apoptosis at the periphery of the white pulp during cutaneous infection. H&E staining (left) or TUNEL labeling (right). A and B: At 50 hours after inoculation with 9602WT-lux(EF+LF+PA+) morphotype 2, there was extensive cell debris (arrowheads) at the periphery of the lymphoid structures (A) and significant TUNEL labeling at the periphery of the white pulp (B). No labeling was observed for morphotype 1 lesions (data not shown). C and D: At 40 hours after inoculation with 9602L-lux(EF+PA+), cell debris (C) associated with a TUNEL-positive signal (D) was observed at the periphery of the white pulp structures. E–J: At 44 hours after inoculation with 9602C-lux(LF+PA+) (E and F), at 40 hours after inoculation with 9602P-lux(Tox−) (G and H), and in control spleen (I and J), neither cell debris nor TUNEL signal were detected. Scale bars = 25 μm.
Infection with the ET-secreting strain provoked distinctive lesions (Figure 3, D–F). They were characterized by an abundant multifocal edema in the splenic red pulp close to the white pulp (Figure 3D). It contained few scattered neutrophils, little cell debris, and some residual erythrocytes and was diffusely infiltrated by encapsulated bacteria and capsular material (Figure 3, E, F, and inset in F). Most interestingly, the peripheral zone of the white pulp contained extensive cell debris (Figure 4C) and numerous TUNEL-positive apoptotic lymphoid cells (Figure 4D). Production of ET or LT alone during infection thus resulted in marked differences in the nature of the lesions.
We then addressed the synergistic role of ET and LT produced during infection with the wild-type strain. Two markedly different inflammatory lesion patterns or morphotypes were observed in the spleen: either minimal lesions (defined as morphotype 1) or marked and necrotizing lesions (defined as morphotype 2). These two morphotypes were related to the intensity of the bioluminescence signal in the lung, and therefore to the stage of the infection8,31 (Table 3). Morphotype 1 represented lesions from an early stage of systemic dissemination (low or no bioluminescence signal in the lungs) and displayed similarities with those observed with the LT-alone secreting strain, albeit of lower severity (Figure 5, A–C). Small multifocal infiltrates of fragmented neutrophils (<100 μm in diameter) were located in the splenic red pulp close to the white pulp, without edema and with minimal necrosis (Figure 5, A and B); they contained a limited number of encapsulated bacteria, mostly forming small microcolonies, and extracellular capsular material (Figure 5, B, C, and inset in C). The white pulp displayed no histological lesion (data not shown).
Figure 5.
Secretion of edema and lethal toxins by wild-type B. anthracis elicits a temporal-dependent pattern of lesions in the spleen during cutaneous spore infection (50 hours after inoculation). H&E staining (A, B, D, E, G, and left part of H–J), IHC using a polyclonal antibody directed against B. anthracis polyglutamate (C and F), and Gram staining (right column of H-J). A–C: For morphotype 1, almost no lesions were observed at low magnification (A); at higher magnification, small infiltrates of karyorrhectic neutrophils (arrowheads) without edema were observed (B); bacteria (C) were observed as small microcolonies (arrows) surrounded by capsular material (inset) in the red pulp at the periphery of the white pulp. D–F: For morphotype 2, multifocal lesions (D, arrows) were located in the red pulp and sparing the white pulp; large infiltrates of neutrophils with abundant edema (E) were observed, and at higher magnification foci of red pulp necrosis (E, inset); a high number of bacteria (F) were observed within lightly labeled extracellular capsular material (inset) at the periphery of the white pulp (arrows). G: For mixed morphotypes 1 and 2, at low magnification, numerous inflammatory foci with various morphologies were observed H–J: Higher magnification views of these features (left column), with two different stainings, reveal small infiltrates of often karyorrhectic neutrophils, associated with rare bacteria (reminiscent of morphotype 1 lesions) (H), large infiltrates of neutrophils and bacteria within limited edema (intermediate lesion between morphotypes 1 and 2) (I), and a large lesion, with a low number of neutrophils, rarely karyorrhectic, but a very high number of bacteria, within an abundant edema (reminiscent of morphotype 2 lesions) (J). Scale bars: 500 μm (A and D); 250 μm (C, F, and G); 50 μm (E, E inset, and H–J, left); 25 μm (B and H–J, right); and 5 μm (C and F, insets).
Morphotype 2 represented lesions from a late stage of infection (high levels of bioluminescence in both spleen and lungs). They displayed similarities with those observed with the ET-secreting strain, associated with some traits observed with the LT-secreting strain (Figure 5, D–F). The lesions were characterized by large multifocal foci (50 to 300 μm in diameter) of abundant edema in the red pulp close to the white pulp, with moderate red pulp destruction (Figure 5, D, E, and inset in E). The foci contained more numerous neutrophils, rarely fragmented, than in ET-induced lesions, sometimes forming small infiltrates within the edema (Figure 5E). A high number of encapsulated bacteria diffusely infiltrated the edema, the neutrophilic infiltrates, and the remaining red pulp (Figure 5F). Capsular material was present in the edema and around the inflammatory cells and the bacteria (Figure 5F, inset). The white pulp was markedly atrophied, with extensive cell debris (Figure 4A), and numerous TUNEL-positive cells (Figure 4B) were observed at the periphery of most atrophic lymphoid structures, indicating apoptosis (and/or necrosis) of lymphoid cells.
The transitory time point between morphotypes 1 and 2 could be observed and was characterized by coexistence of morphotype 1 and -2 lesions in the red pulp (Figure 5, G–J): i) small (<50 μm) neutrophil infiltrates, often fragmented, and associated with a very low number of bacteria, resembling morphotype 1 lesion (Figure 5, G and H); ii) an intermediate lesion characterized by larger infiltrates (≤75 μm) of neutrophils, with a lower density of fragmented cells, a higher number of bacteria, and limited edema, resembling mixed morphotype 1 and 2 lesions (Figure 5, G and I); and iii) a large lesion (≤800 μm), characterized by an abundant edema containing very few rarely fragmented neutrophils, and a high number of bacteria, resembling morphotype 2 lesion (Figure 5, G and J).
Evidence for a Prominent Role of ET in Inhalational Anthrax
We have previously shown that, in inhalational infection, spore entry and bacterial multiplication occur in the nasopharynx both for the encapsulated nontoxinogenic 9602P-lux(Tox−) strain8 (Figure 6J) and the toxinogenic nonencapsulated Sterne-equivalent 9602R-lux(Tox+) strain.7,8 It strongly suggested that expression of either toxin or capsule does not interfere with spore capture and multiplication in the nasopharynx. Dissemination patterns and kinetics were therefore studied after intranasal inoculation of spores of the three bioluminescent wild type-derived strains.
Figure 6.

Dissemination pattern typical of ET during intranasal anthrax. Mice were inoculated intranasally with spores of the bioluminescent strains secreting LT (A), ET (B), both toxins (C–E), or 9602P(Tox−) (J) and were analyzed for bioluminescence as described for Figure 2. C and D: For the wild-type strain, two different patterns of dissemination were observed; a series of two representative mice are shown displaying initial bioluminescence either in the cervical lymph nodes (C) or in the spleen (D). E: Dissection of a mouse showing bioluminescence in the cervical lymph nodes at 44 hours of infection with the wild-type strain. F–H: Histological analysis of the bioluminescent lymph node presented in E, with H&E (F and G) or Gram staining (H). I: When luminescence began to be detected in the spleen (as discussed under Results), total CFUs of wild-type B. anthracis unheated (UnH) or spores only, heated (HT), were enumerated in the cervical lymph node (cLN), tracheobronchial lymph nodes (tLN), lungs, and spleen. Each symbol represents the CFU derived from one mouse; results are expressed as log10 CFU per organ; horizontal lines indicate the mean. *P < 0.05, UnH versus HT. Scale bars: 100 µm (F); 25 µm (G); and 10 µm (H).
Surprisingly, no detectable luminescence could be observed in the nasopharynx of mice infected with the encapsulated bacteria producing ET or LT alone, or both toxins (Figure 6, A–C; 41 mice). We hypothesized that the initial site of spore capture at the respiratory epithelium in the nasopharynx was not modified, but that coexpression of toxins and capsule after epithelium crossing enabled the bacteria to readily disseminate, thus maintaining the metabolically active bacterial load in the nasopharynx under the detection threshold of our bioluminescence system.8 Indeed, neutralization of toxin activity through PA immunization prior intranasal challenge with the wild-type strain led to detection of luminescence in the nasopharynx in 4/5 immunized mice, whereas none of the nonimmunized controls displayed luminescence in this location (Figure 7). These results thus confirmed the nasopharynx to be the main portal of spore entry. Furthermore, they strongly suggest that expression of the toxins was able to inhibit the local confinement of bacterial growth at the initial site of entry.
Figure 7.
Toxin neutralization enables detection of bioluminescent B. anthracis in the nasopharynx. Mice were immunized with PA and challenged intranasally with spores of the wild-type bioluminescent strain and analyzed for bioluminescence as described for Figure 2. A: Focused view of the nasopharynx of five individual PA-immunized mice (Top) or control mice (Bottom), with the same intensity scale for all mice. B: Quantification of the bioluminescence signal was performed at 36 hours by defining regions of interest of the same size spanning the nasopharynx. Luminescence intensity is expressed in photon/s/cm2/sr. The five individual mice are represented in the graph as differently colored bars (open: PA-immunized mice; hatched: nonimmunized mice). Error bar indicates the mean ± SD of the nasopharynx bioluminescence signal for the five nonimmunized mice is indicated. Asterisks mark values of the nasopharynx bioluminescence signal for the PA-immunized mice greater than the mean ± 2 SD of the nasopharynx bioluminescence signal for the nonimmunized mice.
Production of LT alone during inhalational infection led to a progression of infection similar to that observed with the nontoxinogenic encapsulated strain8 (Figure 6, A and J). Detectable bacterial growth within the cervical lymph nodes was observed within 45 hours, and systemic dissemination to the spleen within 50 hours (Figure 6A). In contrast, all mice infected by the strain secreting only ET showed an apparent direct spread of the bacteria to the spleen. No bioluminescence was detected in the cervical lymph nodes (Figure 6B). Further dissemination to the lungs and septicemia occurred within 5 hours. Death occurred so rapidly during infection with the ET-secreting strain that sufficient time-point data for kinetic analysis could be collected for only 33% of the mice studied (8/24). In contrast, in the same experimental time frame, bioluminescence could be detected before death in 64% of the mice infected with similar inocula of the LT-secreting or the nontoxinogenic strains. This suggested a shorter mean time to death for mice infected with the ET-secreting strain. Indeed, mean time to death after intranasal inoculation was significantly shorter for the ET-secreting strain (42 ± 1 hours, n = 7), compared with the LT-secreting strain (54 ± 5 hours, n = 6), the wild type (49 ± 1 hours, n = 6), and the nontoxinogenic strain (54 ± 4 hours, n = 6) (mean ± SEM; P = 0.01; representative from two independent experiments).
To address the synergistic role of both toxins during inhalational infection, mice were infected with the wild-type strain. Two different patterns of dissemination occurred. The majority of the mice displayed a pattern similar to the ET-alone secreting strain (61%, or 14/23 mice) (Figure 6D), a pattern characterized by absence of luminescence in the cervical lymph nodes and initial light detection in the spleen. The remaining mice displayed a pattern similar to the nontoxinogenic or the strain secreting only LT (39%, or 9/23 mice) (Figure 6C), a pattern characterized by initial detection of light in the cervical lymph nodes (Figure 6E) before systemic dissemination to the spleen. The bacteria were located in the subcapsular sinus (Figure 6F), which was focally dilated and harbored many neutrophils (Figure 6, G and H). Both patterns of dissemination ended with detection of bioluminescence in the lungs within 5 hours after initial detection in the spleen (Figure 6, C and D).
To further determine the route of spread of B. anthracis after intranasal inoculation, which would give indications on the route of entry, we quantified the bacterial load in the cervical and tracheobronchial lymph nodes, which drain the upper and lower parts of the respiratory tract, respectively. This was performed at the early stage of systemic dissemination (ie, when bioluminescence began to be detected in the spleen, but not yet in the lung).31 As expected, at this stage of infection, vegetative cells were found in the spleen, but not in the lung (Figure 6I). Both cervical and tracheobronchial lymph nodes harbored vegetative cells as well as spores (Figure 6I). These findings suggest that entry of the spores could occur throughout the entire respiratory tract.
Discussion
Previous reports on the role of ET during infection with either encapsulated20,21 or nonencapsulated strains18,19 have attributed largely an accessory role to ET, as working primarily in synergy with LT, which acts as the key toxin virulence factor. Evidence provided by the present study highlights a prominent role of ET in anthrax. Production of ET alone by the encapsulated B. anthracis restored almost full virulence in the guinea pig cutaneous infection. In the mouse, production of ET was sufficient to markedly modify the pattern of dissemination: local multiplication in the draining lymph nodes (cutaneous and intranasal infections) and/or the nasopharynx (intranasal infection) could not be detected.
Previous reports in cell culture models have indicated that ET increases maturation and chemotaxis of dendritic cells32 and motility of macrophages.33 Such increase in cell motility could thus favor bacterial dissemination, if these cells were carrying phagocytosed spores. On the other hand, ET may also hinder the local innate immune response in the lymph node, thus promoting further bacterial dissemination in the lymphatic system. Indeed, we found that neutrophil recruitment was weaker in the 9602L(EF+PA+) lesion than in the 9602P(Tox−) and 9602C(LF+PA+) lesions. ET impairs chemokinesis, chemotaxis, and ability to polarize of neutrophils, due to defects in actin assembly.34 The abundant edema produced during the infection with the ET-secreting strain may also decrease the ability for chemokines to diffuse in the extracellular environment and/or for recruited cells to migrate toward the infected foci. Furthermore, ET exerted a specific cytotoxic effect against lymphoid cells, leading to apoptosis in the peripheral zone of the white pulp. This effect was not observed with the LT-secreting strain, indicating that ET is the main cause of white pulp apoptosis during infection. Given that the bacteria initially multiply in the marginal zone, close to the white pulp but sparing it, this finding provides strong evidence that the toxic effect is mediated through local diffusion of ET.
In contrast, the LT-secreting strain presented a pattern and kinetics of dissemination similar to the nontoxinogenic strain. Of note, neutrophil recruitment was not inhibited in vivo, in contrast to the reported effects of LT on cell recruitment in vitro.4 Cytotoxicity was nevertheless observed, because the majority of these infiltrating neutrophils were dying in situ and forming abscesses. Possibly, cell death triggered by the toxic effects of LT induces a strong inflammatory reaction, further enhancing neutrophil recruitment, as recently suggested.35
When both ET and LT toxins were coproduced by the wild-type strain, markedly different patterns of dissemination were observed, depending on the route of infection. In cutaneous infection, the pattern was similar to that of the nontoxinogenic strain, confirming previous reports on the central pathogenic role of the capsule over the toxins in murine cutaneous infections.23,36 Nevertheless, our present results show that ET and LT were able to exert their toxic effects locally in the spleen. In contrast, in inhalational infection, the pattern of dissemination resembled the 9602L(EF+PA+) pattern in the majority of animals (no detection in the draining lymph node, apparent direct spread to the spleen), with the nontoxinogenic pattern (detection in the lymph node) observed in the remaining animals. Through the use of the highly powerful and sensitive bioluminescence system, we were thus able to show that ET exerted an overall predominant effect during the initial stages of inhalational infection in a significant proportion of animals. Furthermore, no bioluminescence signal could be detected in the nasopharynx, regardless of whether the infecting strains expressed ET, LT, or both.
We have previously shown that expression of toxins7 or capsule8 alone does not hinder capture and multiplication of B. anthracis in the nasopharynx. Production of ET and/or LT in conjunction with capsule inhibits local confinement of bacterial growth in the nasopharynx, thereby maintaining the bacterial load below the detection limits of our bioluminescent system.8 Indeed, toxin neutralization resulted in the detection of bioluminescence in the nasopharynx. Finally, mean time to death after inhalational infection was significantly shorter with the ET-secreting strain than with the wild-type, the LT-secreting, or the no-toxin strains (all of which are encapsulated). Taken together, our results strongly suggest that the tissue microenvironment that bacteria initially encounter during infection influences the outcome of the first steps of the infection and that ET contributes to this pathogenic effect.
Each toxin secreted alone provoked particular lesions, and, when produced together, induced lesions in a specific temporal pattern during the time course of infection. The pattern observed was dependent on the stage of infection being analyzed. Morphotype 1 lesions were observed when sampling was performed at an early stage of dissemination (ie, when bioluminescence was initially detected in the spleen, but no signal or a low signal was observed in the lung). They resembled the LT-induced lesions in terms of recruitment of neutrophils with a high level of cell death. Conversely, morphotype 2 lesions were observed at a later stage of infection (ie, when a high level of bioluminescence was detected in the lung), indicating terminal dissemination of the bacteria. They resembled the ET-induced lesions, with large foci of edema, weak inflammatory cell recruitment with low level of cell death, and apoptosis in the periphery of the white pulp. The biological effects of LT and ET thus appear to develop differently with time in each focus of bacterial multiplication in the spleen.
What mechanisms could be at the origin of these temporal differences? During the course of natural spore infection, bacilli exit from the draining lymph nodes by the efferent lymphatics in a continuous manner31; that is, bacterial arrival in the spleen is not synchronous, but continuous. Initially, the low number of bacteria in each focus of infection leads to secretion of only small amounts of toxins; then, because of bacterial growth, the levels of toxins gradually increase locally. The different cytotoxic effects could thus be dependent on the concentrations, and the gradients generated, of the toxins in the tissue. A low level of LT has been shown to induce toxin resistance in macrophages, suggesting that cells may adapt so as to tolerate toxic doses of LT.37 Whether this could apply in vivo, and whether other cell types could be induced to tolerate LT, remains to be explored. By contrast, a low dose of ET can sensitize mice to LT.38 The cumulative biological effects of ET leading to formation of extensive edema combined with the release of capsular material by the bacteria (observed with all mutants) may also decrease the ability of LT to diffuse extracellularly and interact with its cell receptors, thus further decreasing its toxic effects.
Our approach attempts to explore the short-range effects of the toxins in the tissues, when B. anthracis intimately interacts with eukaryotic cells through its capsule39 and when increasing local concentrations of toxins are produced, as they diffuse away from the bacterial body as soon as a bacterial cell settles in a given location in the tissue. The fine mechanisms of bacterial regulation are thus likely able to operate in the tissue environment that varies in a complex temporal way through the crosstalk between the bacteria and the host control mechanisms. The model system we describe here is therefore complementary to experimental models described in previous studies, using systemic injection of purified ET or LT,16,17 or infection with nonencapsulated9,18 or with an encapsulated laboratory B. anthracis.20,21 These experimental models preferentially explored the long-distance effects of significant amounts of circulating toxins, thus mimicking the terminal stage of anthrax. Depending on the presence of the capsule, LT was found to be required18 or not required21 for dissemination. Each toxin was found able to induce apoptosis/necrosis, cell depletion, and neutrophil recruitment in both red and white splenic pulp,16–18,20,21 which could be related to the significant levels of toxins that reach the spleen on a short time scale.
We were thus able to show that, even with encapsulated B. anthracis, the mouse model can be exploited to reveal the differential effects of the toxins. The bioluminescence technology was a key factor in this approach. It enabled access to previously elusive events, and in particular allowed real-time monitoring and detailed characterization of key steps of the infectious process within targeted organs at key time points.6–8 Another spin-off of our study is the evidence that the guinea pig is a highly informative model for anthrax infection and therapeutic development. Nontoxinogenic encapsulated B. anthracis, although attenuated, retains a significant level of virulence.23 The ET-alone and LT-alone secreting encapsulated strains were as virulent as the wild-type strain. All aspects of the anthrax toxi-infection could thus be studied and analyzed. The efficiency of vaccine regimens and formulae could easily be tested, whether they target toxin components, or spore or bacilli surface components.
This animal model should be of benefit for characterization of the in vivo efficiency of enzymatic inhibitors of LF40 and EF.29 Previous attempts have shown, for example, that neutralizing polyclonal41 and monoclonal42,43 antibodies are protective against edema toxin challenge, and suggest a modest benefit in vivo during murine infection with the Sterne strain.43 Clearly, the guinea pig model will help reassess the role of ET during infection. Inbred guinea pig strains are also available, facilitating immunological studies.44 Furthermore, microarrays have been developed that give an integrated view of the immune response.45 We show that inhibition of EF enzymatic activity could help control the infection. This in vivo proof of concept during infection with a highly virulent encapsulated strain paves the way for the development of new and more effective drugs targeting EF, thus contributing to better control of anthrax.
Acknowledgments
We thank Eddie Maranghi for help in the handling and therapeutic treatment of the guinea pigs, Michel Haustant for the construction of some of the B. anthracis mutant strains used in this study, Thomas Candela for the design of the corresponding genetic constructions, Craig Gibbs (Gilead Sciences, Foster City, CA) for kindly providing the EF inhibitor PMEA necessary for the in vivo therapeutic assays and for sharing his experience with us, David Govindin for the expert checking of the confinement boxes for BSL3 experiments, and Gilles Massoneau for the design of the electrical alimentation for the ventilation system. We give special thanks to Marie-Anne Nicola for her expertise and constant help in managing the bioluminescence system.
Footnotes
F.D. and G.J. contributed equally to the present work.
Current address of F.D., INRA, UMR1319 MICALIS, France.
References
- 1.Germain R.N., Miller M.J., Dustin M.L., Nussenzweig M.C. Dynamic imaging of the immune system: progress, pitfalls and promise. Nat Rev Immunol. 2006;6:497–507. doi: 10.1038/nri1884. [DOI] [PubMed] [Google Scholar]
- 2.Mock M., Fouet A. Anthrax. Annu Rev Microbiol. 2001;55:647–671. doi: 10.1146/annurev.micro.55.1.647. [DOI] [PubMed] [Google Scholar]
- 3.Candela T., Fouet A. Bacillus anthracis CapD, belonging to the gamma-glutamyltranspeptidase family, is required for the covalent anchoring of capsule to peptidoglycan. Mol Microbiol. 2005;57:717–726. doi: 10.1111/j.1365-2958.2005.04718.x. [DOI] [PubMed] [Google Scholar]
- 4.Moayeri M., Leppla S. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol Aspects Med. 2009;30:439–455. doi: 10.1016/j.mam.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tournier J.N., Rossi Paccani S., Quesnel-Hellmann A., Baldari C.T. Anthrax toxins: a weapon to systematically dismantle the host immune defenses. Mol Aspects Med. 2009;30:456–466. doi: 10.1016/j.mam.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Glomski I.J., Corre J.P., Mock M., Goossens P.L. Noncapsulated toxinogenic Bacillus anthracis presents a specific growth and dissemination pattern in naive and protective antigen-immune mice. Infect Immun. 2007;75:4754–4761. doi: 10.1128/IAI.00575-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glomski I.J., Dumetz F., Jouvion G., Huerre M.R., Mock M., Goossens P.L. Inhaled non-capsulated Bacillus anthracis in A/J mice: nasopharynx and alveolar space as dual portals of entry, delayed dissemination, and specific organ targeting. Microbes Infect. 2008;10:1398–1404. doi: 10.1016/j.micinf.2008.07.042. [DOI] [PubMed] [Google Scholar]
- 8.Glomski I.J., Piris-Gimenez A., Huerre M., Mock M., Goossens P.L. Primary involvement of pharynx and Peyer's patch in inhalational and intestinal anthrax. PLoS Pathog. 2007;3:e76. doi: 10.1371/journal.ppat.0030076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duong S., Chiaraviglio L., Kirby J.E. Histopathology in a murine model of anthrax. Int J Exp Pathol. 2006;87:131–137. doi: 10.1111/j.0959-9673.2006.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fritz D.L., Jaax N.K., Lawrence W.B., Davis K.J., Pitt M.L., Ezzell J.W., Friedlander A.M. Pathology of experimental inhalation anthrax in the rhesus monkey. Lab Invest. 1995;73:691–702. [PubMed] [Google Scholar]
- 11.Grinberg L.M., Abramova F.A., Yampolskaya O.V., Walker D.H., Smith J.H. Quantitative pathology of inhalational anthrax I: quantitative microscopic findings. Mod Pathol. 2001;14:482–495. doi: 10.1038/modpathol.3880337. [DOI] [PubMed] [Google Scholar]
- 12.Guarner J., Jernigan J.A., Shieh W.J., Tatti K., Flannagan L.M., Stephens D.S., Popovic T., Ashford D.A., Perkins B.A., Zaki S.R., Inhalational Anthrax Pathology Working Group Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am J Pathol. 2003;163:701–709. doi: 10.1016/S0002-9440(10)63697-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lever M.S., Stagg A.J., Nelson M., Pearce P., Stevens D.J., Scott E.A., Simpson A.J., Fulop M.J. Experimental respiratory anthrax infection in the common marmoset (Callithrix jacchus) Int J Exp Pathol. 2008;89:171–179. doi: 10.1111/j.1365-2613.2008.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shieh W.J., Guarner J., Paddock C., Greer P., Tatti K., Fischer M., Layton M., Philips M., Bresnitz E., Quinn C.P., Popovic T., Perkins B.A., Zaki S.R., Anthrax Bioterrorism Investigation Team The critical role of pathology in the investigation of bioterrorism-related cutaneous anthrax. Am J Pathol. 2003;163:1901–1910. doi: 10.1016/S0002-9440(10)63548-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vasconcelos D., Barnewall R., Babin M., Hunt R., Estep J., Nielsen C., Carnes R., Carney J. Pathology of inhalation anthrax in cynomolgus monkeys (Macaca fascicularis) Lab Invest. 2003;83:1201–1209. doi: 10.1097/01.lab.0000080599.43791.01. [DOI] [PubMed] [Google Scholar]
- 16.Firoved A.M., Miller G.F., Moayeri M., Kakkar R., Shen Y., Wiggins J.F., McNally E.M., Tang W.J., Leppla S.H. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am J Pathol. 2005;167:1309–1320. doi: 10.1016/S0002-9440(10)61218-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moayeri M., Haines D., Young H.A., Leppla S.H. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J Clin Invest. 2003;112:670–682. doi: 10.1172/JCI17991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loving C.L., Khurana T., Osorio M., Lee G.M., Kelly V.K., Stibitz S., Merkel T.J. Role of anthrax toxins in dissemination, disease progression, and induction of protective adaptive immunity in the mouse aerosol challenge model. Infect Immun. 2009;77:255–265. doi: 10.1128/IAI.00633-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pezard C., Berche P., Mock M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect Immun. 1991;59:3472–3477. doi: 10.1128/iai.59.10.3472-3477.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drysdale M., Olson G., Koehler T.M., Lipscomb M.F., Lyons C.R. Murine innate immune response to virulent toxigenic and nontoxigenic Bacillus anthracis strains. Infect Immun. 2007;75:1757–1764. doi: 10.1128/IAI.01712-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heninger S., Drysdale M., Lochia J., Hutt J., Lipscomb M.F., Koehler T.M., Lyons C.R. Toxin-deficient mutants of Bacillus anthracis are lethal in a murine model for pulmonary anthrax. Infect Immun. 2006;74:6067–6074. doi: 10.1128/IAI.00719-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan H.S., Drysdale M., Lochia J., Koehler T.M., Lipscomb M.F., Lyons C.R. Discriminating virulence mechanisms among Bacillus anthracis strains by using a murine subcutaneous infection model. Infect Immun. 2009;77:429–435. doi: 10.1128/IAI.00647-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brassier F., Levy M., Mock M. Anthrax spores make an essential contribution to vaccine efficacy. Infect Immun. 2002;70:661–664. doi: 10.1128/iai.70.2.661-664.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brassier F., Weber-Levy M., Mock M., Sicard J.C. Protective antigen-mediated antibody response against a heterologous protein produced in vivo by Bacillus anthracis. Infect Immun. 2000;68:5731–5734. doi: 10.1128/iai.68.10.5731-5734.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Remise V., Part G., Garrigue H., Guerdon J.L., Mock M. Identification and characterization of Bacillus anthracis by multiplex PCR analysis of sequences on plasmids pXO1 and pXO2 and chromosomal DNA. FEMS Microbiol Lett. 1996;145:9–16. doi: 10.1111/j.1574-6968.1996.tb08548.x. [DOI] [PubMed] [Google Scholar]
- 26.Gauthier Y.P., Tournier J.N., PaCOD J.C., Corre J.P., Mock M., Goossens P.L., Vidal D.R. Efficacy of a vaccine based on protective antigen and killed spores against experimental inhalational anthrax. Infect Immun. 2009;77:1197–1207. doi: 10.1128/IAI.01217-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reed L.J., Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 28.Keppie J., Smith H., Harris-Smith P.W. The chemical basis of the virulence of Bacillus anthracis: III. The role of the terminal bacteraemia in death of guinea-pigs from anthrax. Br J Exp Pathol. 1955;36:315–322. [PMC free article] [PubMed] [Google Scholar]
- 29.Shen Y., Zhukovskaya N.L., Zimmer M.I., Soelaiman S., Bergson P., Wang C.R., Gibbs C.S., Tang W.J. Selective inhibition of anthrax edema factor by adefovir, a drug for chronic hepatitis B virus infection. Proc Natl Acad Sci USA. 2004;101:3242–3247. doi: 10.1073/pnas.0306552101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khokhar A., Afdhal N.H. Therapeutic strategies for chronic hepatitis B virus infection in 2008. Am J Med. 2008;121(12 Suppl):S33–S44. doi: 10.1016/j.amjmed.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 31.Goossens P. Animal models of human anthrax: The quest for the holy grail. Mol Aspects Med. 2009;30:467–480. doi: 10.1016/j.mam.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 32.Maldonado-Arocho F.J., Fulcher J.A., Lee B., Bradley K.A. Anthrax oedema toxin induces anthrax toxin receptor expression in monocyte-derived cells. Mol Microbiol. 2006;61:324–337. doi: 10.1111/j.1365-2958.2006.05232.x. [DOI] [PubMed] [Google Scholar]
- 33.Kim C., Wilcox-Adelman S., Sano Y., Tang W.J., Collier R.J., Park J.M. Antiinflammatory cAMP signaling and cell migration genes co-opted by the anthrax bacillus. Proc Natl Acad Sci USA. 2008;105:6150–6155. doi: 10.1073/pnas.0800105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szarowicz S.E., During R.L., Li W., Quinn C.P., Tang W.J., Southwick F.S. Bacillus anthracis edema toxin impairs neutrophil actin-based motility. Infect Immun. 2009;77:2455–2464. doi: 10.1128/IAI.00839-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terra J.K., Cote C.K., France B., Jenkins A.L., Bozue J.A., Welkos S.L., LeVine S.M., Bradley K.A. Cutting edge: resistance to Bacillus anthracis infection mediated by a lethal toxin sensitive allele of Nalp1b/Nlrp1b. J Immunol. 2010;184:17–20. doi: 10.4049/jimmunol.0903114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Welkos S.L., Vietri N.J., Gibbs P.H. Non-toxigenic derivatives of the Ames strain of Bacillus anthracis are fully virulent for mice: role of plasmid pX02 and chromosome in strain-dependent virulence. Microb Pathog. 1993;14:381–388. doi: 10.1006/mpat.1993.1037. [DOI] [PubMed] [Google Scholar]
- 37.Salles I.I., Tucker A.E., Voth D.E., Ballard J.D. Toxin-induced resistance in Bacillus anthracis lethal toxin-treated macrophages. Proc Natl Acad Sci USA. 2003;100:12426–12431. doi: 10.1073/pnas.2134042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Firoved A.M., Moayeri M., Wiggins J.F., Shen Y., Tang W.J., Leppla S.H. Anthrax edema toxin sensitizes DBA/2J mice to lethal toxin. Infect Immun. 2007;75:2120–2125. doi: 10.1128/IAI.01781-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piris-Gimenez A., Corre J.P., Jouvion G., Candela T., Khun H., Goossens P.L. Encapsulated Bacillus anthracis interacts closely with liver endothelium. J Infect Dis. 2009;200:1381–1389. doi: 10.1086/644506. [DOI] [PubMed] [Google Scholar]
- 40.Tonello F., Seveso M., Marin O., Mock M., Montecucco C. Screening inhibitors of anthrax lethal factor. Nature. 2002;418:386. doi: 10.1038/418386a. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen M.L., Crowe S.R., Kurella S., Teryzan S., Cao B., Ballard J.D., James J.A., Farris A.D. Sequential B-cell epitopes of Bacillus anthracis lethal factor bind lethal toxin-neutralizing antibodies. Infect Immun. 2009;77:162–169. doi: 10.1128/IAI.00788-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Z., Moayeri M., Zhao H., Crown D., Leppla S.H., Purcell R.H. Potent neutralization of anthrax edema toxin by a humanized monoclonal antibody that competes with calmodulin for edema factor binding. Proc Natl Acad Sci USA. 2009;106:13487–13492. doi: 10.1073/pnas.0906581106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winterroth L., Rivera J., Nakouzi A.S., Dadachova E., Casadevall A. Neutralizing monoclonal antibody to edema toxin and its effect on murine anthrax. Infect Immun. 2010;78:2890–2898. doi: 10.1128/IAI.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Padilla-Carlin D.J., McMurray D.N., Hickey A.J. The guinea pig as a model of infectious diseases. Comp Med. 2008;58:324–340. [PMC free article] [PubMed] [Google Scholar]
- 45.Tree J.A., Elmore M.J., Javed S., Williams A., Marsh P.D. Development of a guinea pig immune response-related microarray and its use to define the host response following Mycobacterium bovis BCG vaccination. Infect Immun. 2006;74:1436–1441. doi: 10.1128/IAI.74.2.1436-1441.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]