

Abstract
The Fas death receptor (CD95) is expressed on macrophages, smooth muscle cells, and T cells within atherosclerotic lesions. Given the dual roles of Fas in both apoptotic and nonapoptotic signaling, the aim of the present study was to test the effect of hematopoietic Fas deficiency on experimental atherosclerosis in low-density lipoprotein receptor-null mice (Ldlr−/−). Bone marrow from Fas−/− mice was used to reconstitute irradiated Ldlr−/− mice as a model for atherosclerosis. After 16 weeks on an 0.5% cholesterol diet, no differences were noted in brachiocephalic artery lesion size, cellularity, or vessel wall apoptosis. However, Ldlr−/− mice reconstituted with Fas−/− hematopoietic cells had elevated hyperlipidemia [80% increase, relative to wild-type (WT) controls; P < 0.001] and showed marked elevation of plasma levels of CXCL1/KC, CCL2/MCP-1, IL-6, IL-10, IL-12 subunit p70, and soluble Fas ligand (P < 0.01), as well as systemic microvascular inflammation. It was not possible to assess later stages of atherosclerosis because of increased mortality in Fas−/− bone marrow recipients. Our data indicate that hematopoietic Fas deficiency does not affect early atherosclerotic lesion development in Ldlr−/− mice.
Complications from atherosclerosis remain major causes of morbidity and mortality worldwide.1 Atherosclerosis is widely accepted as an inflammatory process.2,3 The development and composition of atherosclerotic lesions reflect a balance between accumulation, proliferation, and apoptosis of cellular components of the vessel wall, including macrophages, T lymphocytes, endothelial cells, and smooth muscle cells.
Fas (Apo-1, CD95), a 45-kDa member of the death receptor family, is known to induce apoptosis in susceptible cells on binding Fas ligand (FasL, CD178).4 Fas expression has been demonstrated on macrophages, T lymphocytes, and smooth muscle cells within human atherosclerotic lesions.5 In contrast to lymphocytes and smooth muscle cells, macrophages are relatively resistant to Fas-mediated apoptosis.6 This correlates with increased expression of cFLIP during monocyte to macrophage differentiation.7 However, macrophage resistance to Fas-mediated apoptosis can be reversed in vitro by Toll-like receptor ligands, including lipopolysaccharide, lipid A, zymosan, poly(I:C), and CpG DNA.8 Furthermore, there is increasing recognition of nonapoptotic responses to Fas ligation, including cellular proliferation, differentiation, and NF-κB activation in several cell types.9,10 In macrophages, Fas engagement also triggers proinflammatory cytokine production.6,11
Mouse models of Fas or FasL deficiency develop progressive lymphadenopathy and splenomegaly due to dysregulated lymphocyte proliferation.12,13 Fas-deficient mouse models include Faslpr/lpr mice (lymphoproliferation, lpr), which have an inactivating point mutation in the cytoplasmic domain,14 and Fas−/− mice, which are complete deficient.12 When lpr mice are crossed with atherosclerosis-prone Apoe−/− mice, the animals developed accelerated atherosclerosis accompanied by lymphoproliferation and a lupus-like syndrome.15 Autoimmune disorders such as systemic lupus erythematosus, recognized as an independent risk factor for atherosclerosis, are associated with an approximately 7.5-fold increase in cardiovascular complications.16 Thus, in the previous model,15 development of autoimmunity may have confounded the relationship between Fas deficiency and atherogenesis.
Here, we describe the effect of Fas expression in bone marrow-derived cells, using the atherosclerosis-prone low-density lipoprotein receptor-deficient (Ldlr−/−) mouse (human ortholog, LDLR). We find excess mortality with hematopoietic Fas deficiency in this atherosclerosis model, with no evidence of lymphoproliferation. Absence of lymphoproliferation is unexpected, because Fas−/− mice develop increased splenomegaly and lymphadenopathy at an earlier age than do lpr mice.17 Surprisingly, we found no significant effect on atherosclerotic lesion development or vascular wall apoptosis at an early stage of lesion development, despite an enhanced atherogenic state characterized by increased plasma lipids and systemic cytokine and chemokine levels.
Materials and Methods
Mice
Ldlr−/− and Fas−/− mice (B6.129P2-Fastm1Osa/J, Stock ID 003233)17 on the C57Bl/6J background were obtained from Jackson Laboratories (Bar Harbor, ME). Female Ldlr−/− mice, 12 weeks old, received 11 Gy irradiation for marrow ablation. The next day, bone marrow cells obtained from male WT C57Bl/6J (n = 16) or Fas−/− (n = 16) mice were administered to irradiated recipients via retro-orbital injection. Following a 4-week recovery period, mice were placed on an 0.5% cholesterol diet (Harlan Teklad RD-97234; Harlan Sprague Dawley, Indianapolis, IN) for a planned duration of 24 weeks.18 Mice were maintained under specific pathogen-free conditions. Blood was obtained by cardiac puncture at the time of sacrifice. Individual complete blood counts were performed on a Hemavet 950 veterinary hematology analyzer (Drew Scientific, Dallas, TX). All experimental procedures were performed with approval from the Animal Care Committee of the University of Washington.
Assessment of Atherosclerotic Lesions
At the time of sacrifice, mice were perfused via the left ventricle with 10 mL PBS containing 1 mmol/L EDTA, followed by 30 mL fixative solution (PBS, 4% paraformaldehyde in PBS). The heart including the aortic root, thoracic aorta, and its branching vessels was dissected and fixed overnight. The brachiocephalic artery from the branch off the aortic arch to the bifurcation of the right subclavian artery and common carotid artery was dissected, embedded in paraffin, and sectioned into 100 5-μm sections. For quantitation of lesion area, six H&E-stained sections (50-μm intervals) per mouse were analyzed using ImageJ software version 1.42 (NIH, Bethesda, MD). The internal and external elastic lamellae and lesion borders were traced and used to derive lesion areas.19 Terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) staining was performed according to the manufacturer's instructions (In Situ Cell Death Detection Kit; Roche Applied Science, Indianapolis, IN) with DAPI as a nuclear counterstain. DAPI-positive and TUNEL-positive cells were counted under fluorescence microscopy (Nikon Eclipse TE300), using 6 to 10 sections of brachiocephalic artery per mouse. Three independent researchers (X.Z., J.T., A.I.) without knowledge of the tissue source quantified lesion area and TUNEL analysis.
Immunostaining of Tissues
Tissue sections from liver, lung, spleen, and kidney were stained with H&E to evaluate histological changes, and adjacent sections were stained with antibodies to macrophages, (Mac-2; ATCC, Manassas, VA), B lymphocytes (B220; BD Biosciences, San Jose, CA), and T lymphocytes (CD3; Dako, Carpinteria, CA), as well as control rabbit and rat IgG.
Cell Isolation and Flow Cytometry
Spleens were harvested, weighed, and dispersed, and cells were passed through a 70-μm strainer. Peripheral blood leukocytes and isolated splenocytes were analyzed after red-cell lysis with multicolor flow cytometry on a FACScan system (BD Biosciences) with CellQuest software version 3.4. For leukocyte characterization the following antibodies were used: Thy1.2, B220, Ly6G, CD11b, CD95 (Fas), and CD178 (FasL). Fluorochrome-conjugated monoclonal antibodies were all purchased from BD Biosciences.
Chemokine and Cytokine Analysis
The plasma concentrations of CXCL1/KC, CXCL2/MIP-2, CCL2/MCP-1, IL-6, IL-10, IL-12, TNF-α, and IFN-γ were determined using a fluorokine MAP multiplex mouse cytokine panel (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. The plasma level of soluble FasL was determined using a commercially available enzyme-linked immunosorbent assay kit (R&D Systems).
Plasma Cholesterol and Lipid Profiles
Plasma cholesterol levels were measured at the Northwest Lipid Research Laboratories (Seattle, WA). The lipoprotein cholesterol profiles were determined for three mice per group on mouse plasma separated by fast protein liquid chromatography20 performed by the University of Washington Clinical Nutrition Research Unit.
Statistical Analysis
Data are reported as means ± SD, unless stated otherwise. Data were analyzed using the GraphPad Prism version 5 software (GraphPad Software, La Jolla, CA), and significance was set at P < 0.05. Unpaired two-tailed Student's t-test was conducted if variance was normally distributed; otherwise, the Mann-Whitney U-test was used. Survival analysis was performed using Kaplan-Meier method with log-rank test to determine differences in survival.
Results
Hematopoietic Fas−/−→Ldlr−/− Chimeric Mice Show Normal Hematopoietic Reconstitution and No Evidence of Lymphoproliferation
Bone marrow transplantation was used to generate mice that lacked hematopoietic Fas expression. 12-week-old female Ldlr−/− mice were reconstituted with bone marrow from male donor Fas−/− mice (n = 16) or their respective WT controls (n = 16). After a 4-week recovery period to allow for macrophage repopulation, the mice were then started on an 0.5% cholesterol diet. At 20 weeks after transplantation (16 weeks on the 0.5% cholesterol diet), excess mortality was noted in the Fas−/− bone marrow recipients, relative to WT bone marrow recipients: 5 of 16 dead in Fas−/−→Ldlr−/− versus 1 of 16 dead in WT→Ldlr−/− (P = 0.07, log-rank test). The remaining mice were then sacrificed for analysis at 21 weeks after transplantation, 3 weeks ahead of the planned schedule.
Hematopoietic recovery was similar between the two groups. Hematocrit, hemoglobin, total leukocyte, lymphocyte, and monocyte counts were within normal limits and were not statistically different between the two groups (data not shown). Platelet counts were lower in the Fas−/−→Ldlr−/− group (541 ± 141, versus 657 ± 89 K/μL in the WT group; P = 0.02) and absolute neutrophil count was higher in the Fas−/−→Ldlr−/− group (2.2 ± 1.0, versus 1.3 ± 0.6 K/μL in the WT group; P = 0.01). Flow cytometry of peripheral blood leukocytes confirmed expected phenotypes after bone marrow transplantation (Figure 1A).
Figure 1.
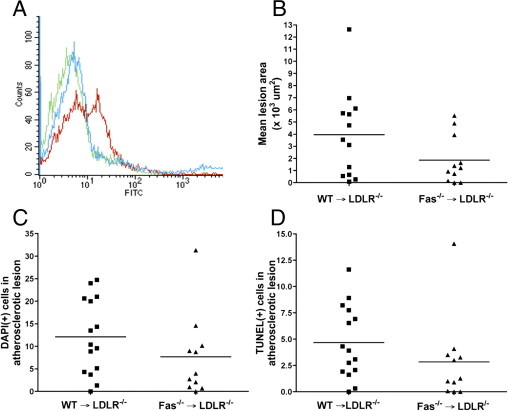
Hematopoietic Fas−/−→Ldlr−/− chimeric mice do not show changes in atherosclerotic lesion area, cellularity, or apoptosis. WT→Ldlr−/− (n = 15) and Fas−/−→Ldlr−/− mice (n = 11) were generated and fed with an 0.5% cholesterol diet for 16 weeks. A: Flow cytometry was performed on peripheral blood of the bone-marrow chimeric mice at the time of sacrifice. Representative profiles are shown. Green indicates background staining with no antibody in Fas−/−→Ldlr−/− mice; red indicates positive staining with CD95 antibody in WT→Ldlr−/− chimeric mice; and blue indicates lack of staining with CD95 (Fas) antibody in Fas−/−→Ldlr−/− chimeric mice. B: Atherosclerotic lesion area was determined from H&E-stained brachiocephalic artery sections (6 per mouse, 50-μm intervals) using ImageJ software. DAPI-positive cells (C) and TUNEL-positive cells (D) were counted from brachiocephalic artery sections (6 to 10 sections per mouse), using fluorescence microscopy. Each symbol represents an individual mouse; horizontal lines indicate the mean value. P > 0.05, Mann-Whitney U-test (B, C, and D).
Although lymphoproliferation has previously been well described in Fas-deficient mice and bone marrow chimeras using Fas-deficient bone marrow donors,21 there was no evidence of splenomegaly or lymphadenopathy in the Fas−/−→Ldlr−/− or WT→Ldlr−/− mice (Table 1).
Table 1.
Absence of Lymphoproliferation in Hematopoietic Fas−/−→Ldlr−/− Chimeric Mice
| P | |||
|---|---|---|---|
| WT→Ldlr−/− | Fas−/−→Ldlr−/− | value | |
| Body weight (g) | 19.7 ± 1.3 | 20.2 ± 0.6 | NS |
| Spleen weight (mg) | 108.9 ± 28.3 | 108.9 ± 39.8 | NS |
| Spleen/body weight (mg/g) | 5.6 ± 1.3 | 5.4 ± 1.9 | NS |
| Lymphadenopathy | Not detected | Not detected |
Lymphoproliferation parameters were assessed in WT→Ldlr−/− (n = 15) and Fas−/−→Ldlr−/− (n = 11) chimeric mice after 16 weeks on an 0.5% cholesterol diet. Data are reported as means ± SD; NS, not significant.
Hematopoietic Fas−/−→Ldlr−/− Chimeric Mice Have Hypercholesterolemia, but No Differences Are Observed in Lesion Size, Cellularity, or Apoptosis
Analysis of brachiocephalic artery atherosclerotic lesions showed no significant difference in lesion area between WT→Ldlr−/− and Fas−/−→Ldlr−/− mice (Figure 1B). There were no significant differences in lesion cellularity or apoptosis as assessed by DAPI-positive and TUNEL-positive cells, respectively (Figure 1, C and D).
Total plasma cholesterol levels were increased in Fas−/−→Ldlr−/− versus WT→Ldlr−/− chimeric mice (593 ± 129, versus 330 ± 38 mg/dL in WT chimeric; P < 0.001) (Figure 2A). Lipoprotein profile analysis on pooled samples showed increased very low density lipoprotein (VLDL) and low density lipoprotein (LDL) levels in Fas−/−→Ldlr−/− chimeric mice, compared with WT→Ldlr−/− mice (Figure 2B).
Figure 2.
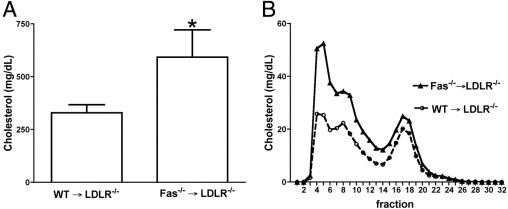
Hypercholesterolemia is increased in hematopoietic Fas−/−→Ldlr−/− chimeric mice. WT→Ldlr−/− (n = 15) and Fas−/−→Ldlr−/− mice (n = 11) were generated and fed with an 0.5% cholesterol diet for 16 weeks. A: Total plasma cholesterol levels. *P < 0.001, Student's t-test. Data are reported as means ± SD. B: Fast protein liquid chromatography profiles were performed on pooled plasma samples from WT→Ldlr−/− and Fas−/−→Ldlr−/− mice.
Markers of Systemic Inflammation Are Elevated in Hematopoietic Fas−/−→Ldlr−/− Chimeric Mice
Plasma levels of CXCL1/KC, CCL2/MCP-1, IL-6, IL-10, IL-12 subunit p70 (IL-12p70), and soluble FasL were significantly elevated in Fas−/−→Ldlr−/− versus WT→Ldlr−/− mice (Table 2). No significant differences between the two groups were observed for CXCL2/MIP-2, TNF-α, and IFN-γ. Plasma levels of CXLCL1/KC, CCL2/MCP-1, and IL-12p70 were also determined in Fas−/− (n = 4) and WT (n = 3) mice, which were not subjected to transplantation. In these untreated mice, IL-12p70 was below limits of detection in both Fas−/− and WT, in contrast to the marked elevation observed in the Fas−/−→Ldlr−/− chimeric mice (Table 2). Similarly, levels of CXCL1/KC and CCL2/MCP-1 were not significantly different between untreated Fas−/− and WT mice (CXCL1/KC: 791 ± 103 pg/mL in Fas−/− versus 1275 ± 229 pg/mL in WT; CCL2/MCP-1: 105 ± 53 pg/mL in Fas−/− versus 142 ± 38 in WT; means ± SEM), whereas levels of these two chemokines were markedly elevated in Fas−/−→Ldlr−/− versus WT→Ldlr−/− chimeric mice (Table 2).
Table 2.
Hematopoietic Fas−/−→Ldlr−/− Chimeric Mice Have Elevated Systemic Cytokine and Chemokine Levels
| WT→>Ldlr−/− (pg/mL) | Fas−/−→Ldlr−/− (pg/mL) | P value⁎ | Lower limit of detection (pg/mL) | |
|---|---|---|---|---|
| CXCL1/KC | 541 ± 311 | 1434 ± 832 | <0.01 | 10.8 |
| CXCL2/MIP-2 | 46 ± 17 | 37 ± 6 | NS | 40.4 |
| CCL2/MCP-1 | Not detected | 2112 ± 1153 | <0.01 | 148.4 |
| IL-6 | Not detected | 119 ± 86 | <0.01 | 4.2 |
| IL-10 | Not detected | 262 ± 209 | <0.01 | 5.1 |
| IL-12 subunit p70 | Not detected | 4701 ± 3541 | <0.01 | 89.7 |
| TNF-α | Not detected | Not detected | NS | 5.6 |
| IFN-γ | Not detected | Not detected | NS | 30.4 |
| Fas ligand | 13 ± 7 | 71 ± 51 | <0.01 | 7.2 |
Plasma cytokines, chemokines, and FasL levels were determined in WT→Ldlr−/− (n = 15) and Fas−/−→Ldlr−/− (n = 11) chimeric mice after 16 weeks on an 0.5% cholesterol diet. WT, wild type. Data are reported as means ± SD.
P values were determined by Student's t-test (MIP-2, FasL) or Mann-Whitney U-test (remainder). NS, not significant.
The H&E stained sections of lungs, kidneys, liver, and spleen showed inflammatory cell infiltrates surrounding parenchymal microvasculature in the Fas−/−→Ldlr−/− group. As shown by immunocytochemistry analysis, the cellular infiltrate was rich in T lymphocytes and B lymphocytes, as well as macrophages (Figure 3). Similar foci of microvascular inflammation were also observed in the spleen and kidney (data not shown).
Figure 3.
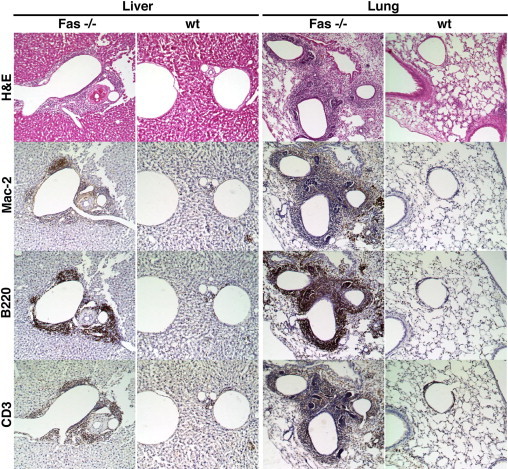
Microvascular inflammation is observed in hematopoietic Fas−/−→Ldlr−/− chimeric mice. Liver and lung tissue from Fas−/− and WT hematopoietic chimeras generated on Ldlr−/− background were evaluated after 16 weeks on an 0.5% cholesterol diet. Adjacent sections were stained with H&E, the macrophage antibody Mac-2, the B lymphocyte antibody B220, or the T lymphocyte antibody CD3. Original micrographs, ×20.
Discussion
In the present study, mice were generated to examine the effect of hematopoietic Fas deficiency on atherosclerotic lesion development. Whether the apoptotic or the nonapoptotic function of macrophage Fas expression predominates in vivo is unknown. No differences were found in brachiocephalic artery lesion area, cellularity, or vessel wall apoptosis between Fas−/−→Ldlr−/− versus WT→Ldlr−/− chimeric mice. We focused our analysis on brachiocephalic artery lesions, because the brachiocephalic artery is a highly reproducible site of lesion formation throughout various stages of lesion development.22 Although many studies use the aortic root or en face staining with Oil Red O, both of these approaches have major limitations. A site with comparable flow disturbance to the aortic root of the mouse does not exist in humans, and effects of gene deletion on lesions in the aortic root are often distinct from other aortic sites.23 Also, en face staining of the aorta with Oil Red O is limited to evaluation of lipid accumulation and does not allow evaluation of cell type distribution within lesions or other potential molecular mediators. Most importantly, the brachiocephalic vessel has been shown to model the characteristics of advanced atherosclerotic lesions in humans.24
Although it would have been desirable to analyze lesions at later time points, it was not possible to keep Fas−/−→Ldlr−/− chimeric mice on the atherogenic diet for more than 16 weeks, because of excess mortality in this group. In the original description of Fas−/− mice, Adachi et al12 reported 50% mortality by 5 months, in keeping with the present results. Thus, the increased mortality we observed is unrelated to the transplantation procedure or the Ldlr−/− background, but rather is intrinsic to complete absence of Fas. The reasons for the increased mortality in the Fas−/− mice are not understood.
Our findings extend the results of previous studies examining the role of dysregulated FasL/Fas signaling in atherosclerosis (Table 3).15,25–27 Global deficiency of Fas signaling leads to a modest increase in aortic root lesion area in Apoe−/− mice, and ineffective apoptosis was the hypothesized mechanism for accelerated atherogenesis.15 Another study showed an approximately 20% increase in aortic root lesion size (cross-sectional) and en face lesion area.25 Both of these studies used lpr mice, which carry a leaky Fas mutation that may result in expression of small amounts of intact Fas mRNA or protein.28,29 In the present study, we used the Fas−/− strain generated by Adachi et al,12 and we confirmed complete absence of hematopoietic Fas expression in the hematopoietic Fas−/−→Ldlr−/− chimeric mice by flow cytometry.
Table 3.
Comparison of Global and Hematopoietic Fas-Deficient and FasL-Deficient Models and Their Effect in Mouse Models of Atherosclerosis
| Fas−/−→Ldlr−/− hematopoietic Fas-deficiency | lpr/Apoe−/− global Fas-deficiency | gld→Ldlr−/− hematopoietic FasL-deficiency | gld/Apoe−/− global FasL-deficiency | |
|---|---|---|---|---|
| Atherosclerotic lesion area | Unchanged | Up | Up | Up |
| Site of lesion analysis | Brachiocephalic artery cross-section | Aortic root cross-section (Refs. 17 and 25) and en face aorta (Ref. 25) | Aortic root cross-section | en face aorta |
| Age of lesion (weeks) | 16 | 20 (Ref. 17) to 24 (Ref. 25) | 12 | 12 |
| Diet | 0.5% cholesterol | Normal chow | Western | Western |
| Cholesterol levels, compared with control | Up | Down | Unchanged | Down |
| Lymphoproliferation (splenomegaly, adenopathy) | None | +/+ | +/Not reported | +/+ |
| Systemic chemokine and cytokine levels | Up | Not reported | Down | Not reported |
| References | Present study | Adachi et al17; Ma et al25 | Gautier et al26 | Aprahamian et al27 |
Unchanged, unchanged compared to control; up, increased compared to control; down, decreased compared to control.
Faslgld/gld mice (generalized lymphoproliferative disease; gld) carry a point mutation near the carboxy terminal of the FasL gene. This results in expression of a nonfunctional protein, incapable of inducing Fas-mediated apoptosis.30 The effect of nonfunctional FasL on atherosclerosis was previously examined by generation of gld→Apoe−/− chimeric mice; en face plaque area in the aorta of gld→Apoe−/− mice was increased threefold, relative to Apoe−/− controls.27 Similar findings were noted when gld expression was restricted to the hematopoietic compartment using bone marrow transplants into Ldlr−/− recipient mice; approximately a 60% increase in lesion area was observed in the aortic root.26 The use of Western-type diets for both the gld→Apoe−/−27 and gld→Ldlr−/− models26 should be noted, because results may be confounded by effects of metabolic syndrome and TH2 versus TH1 immune response.31 The high-cholesterol diet used in the present study had been shown to induce hypercholesterolemia and extensive atherosclerosis in Ldlr−/− mice without the features of metabolic syndrome (obesity, hyperglycemia, hypertriglyceridemia, insulin resistance).18 In the present study, restriction of Fas−/− deficiency to hematopoietic elements was not associated with enhanced atherosclerotic lesion formation. Nonetheless, differences in mouse models, diet, lesion age, and site of lesion analysis among the relevant studies (Table 3) limit the extent to which study results can be directly compared.
Fas-mutant or FasL-mutant chimeric mice have also been used to study other models of inflammation, with varying outcomes. In an experimental autoimmune uveitis model, WT mice reconstituted with lpr or gld bone marrow had improved ocular inflammation scores.32 In a model of Fas-mediated acute lung injury, WT mice reconstituted with lpr bone marrow were not protected from alveolar damage, as evidenced by increases in tissue caspase 3 activation, total lung neutrophil content, and alveolar permeability.33
A notable feature in our model is the marked increase in the level of several circulating proatherogenic and antiatherogenic cytokines or chemokines in the Fas−/−→Ldlr−/− chimeric mice, compared with WT→Ldlr−/− chimeric mice. We tested several of the mediators that were markedly elevated in the Fas−/−→Ldlr−/− chimeric mice (CXCL1/KC, CCL2/MCP-1, and IL-12p70) and found no difference in levels between Fas−/− mice and WT mice that had not been subjected to transplantation, suggesting that the elevation of cytokines and chemokines in the Fas−/−→Ldlr−/− chimeric mice is a response to transplantation into the Ldlr−/− background. There is ample evidence on the proatherogenic effects (CXCL1/KC,34 CCL2/MCP-1,35,36 and IL-1237) and antiatherogenic effects (IL-638,39 and IL-1040) of these mediators, as assessed by knockout mouse models. Soluble FasL is capable of activating NF-κB and promotes autoimmunity and tumorigenesis through nonapoptotic mechanisms.41,42
As already noted, the excess mortality in Fas−/−→Ldlr−/− group is consistent with observations by Adachi et al,12 who reported a 50% mortality rate at 5 months in Fas−/− mice. Because no other causes for excess mortality in Fas−/−→Ldlr−/− group were found (no graft failure, infection, or lymphoproliferation), the increased mortality in the Fas−/−→Ldlr−/− mice is likely due to this intrinsic effect of Fas deficiency, compounded by the proinflammatory milieu generated in the Ldlr−/− background, as evidenced by the development of microvascular inflammation, neutrophilia, and thrombocytopenia in this group. The absence of lymphadenopathy or splenomegaly in the Fas−/−→Ldlr−/− mice, compared with the Fas−/− mice, is also notable. The most likely explanation is that the altered immune milieu with elevation of multiple cytokines and chemokines acts to suppress lymphoproliferation.
In summary, hematopoietic Fas deficiency is not associated with enhanced early atherosclerosis, despite worse hypercholesterolemia and systemic inflammation. Differences in mouse strains (Fas−/− versus lpr), site of lesion analysis, lymphoproliferation, and systemic inflammation may explain the discord between the present results and previously published findings of worse atherosclerosis associated with dysregulated FasL/Fas signaling. Recognition of these confounding factors increases our understanding of the role of FasL/Fas signaling in atherosclerotic lesion development.
Footnotes
Supported in part by grants from the NIH (HL087165 to A.R.deC., HL018645 to E.W.R. and J.M.H., and HL080623 to J.M.H.).
R.A.deC. and X.Z. contributed equally to the present work.
Current address of R.A.deC.: Food and Drug Administration, Washington, DC.
References
- 1.Lopez A.D., Mathers C.D., Ezzati M., Jamison D.T., Murray C.J. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367:1747–1757. doi: 10.1016/S0140-6736(06)68770-9. [DOI] [PubMed] [Google Scholar]
- 2.Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 3.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 4.Nagata S., Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 5.Cai W., Devaux B., Schaper W., Schaper J. The role of Fas/APO 1 and apoptosis in the development of human atherosclerotic lesions. Atherosclerosis. 1997;131:177–186. doi: 10.1016/s0021-9150(97)06099-1. [DOI] [PubMed] [Google Scholar]
- 6.Park D.R., Thomsen A.R., Frevert C.W., Pham U., Skerrett S.J., Kiener P.A., Liles W.C. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J Immunol. 2003;170:6209–6216. doi: 10.4049/jimmunol.170.12.6209. [DOI] [PubMed] [Google Scholar]
- 7.Perlman H., Pagliari L.J., Georganas C., Mano T., Walsh K., Pope R.M. FLICE-inhibitory protein expression during macrophage differentiation confers resistance to fas-mediated apoptosis. J Exp Med. 1999;190:1679–1688. doi: 10.1084/jem.190.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukui M., Imamura R., Umemura M., Kawabe T., Suda T. Pathogen-associated molecular patterns sensitize macrophages to Fas ligand-induced apoptosis and IL-1 beta release. J Immunol. 2003;171:1868–1874. doi: 10.4049/jimmunol.171.4.1868. [DOI] [PubMed] [Google Scholar]
- 9.Wajant H., Pfizenmaier K., Scheurich P. Non-apoptotic Fas signaling. Cytokine Growth Factor Rev. 2003;14:53–66. doi: 10.1016/s1359-6101(02)00072-2. [DOI] [PubMed] [Google Scholar]
- 10.Park S.M., Schickel R., Peter M.E. Nonapoptotic functions of FADD-binding death receptors and their signaling molecules. Curr Opin Cell Biol. 2005;17:610–616. doi: 10.1016/j.ceb.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Altemeier W.A., Zhu X., Berrington W.R., Harlan J.M., Liles W.C. Fas (CD95) induces macrophage proinflammatory chemokine production via a MyD88-dependent, caspase-independent pathway. J Leukoc Biol. 2007;82:721–728. doi: 10.1189/jlb.1006652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adachi M., Suematsu S., Kondo T., Ogasawara J., Tanaka T., Yoshida N., Nagata S. Targeted mutation in the Fas gene causes hyperplasia in peripheral lymphoid organs and liver. Nat Genet. 1995;11:294–300. doi: 10.1038/ng1195-294. [DOI] [PubMed] [Google Scholar]
- 13.Cohen P.L., Eisenberg R.A. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9:243–269. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe-Fukunaga R., Brannan C.I., Copeland N.G., Jenkins N.A., Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 15.Feng X., Li H., Rumbin A.A., Wang X., La Cava A., Brechtelsbauer K., Castellani L.W., Witztum J.L., Lusis A.J., Tsao B.P. ApoE-/-Fas-/- C57BL/6 mice: a novel murine model simultaneously exhibits lupus nephritis, atherosclerosis, and osteopenia. J Lipid Res. 2007;48:794–805. doi: 10.1194/jlr.M600512-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Esdaile J.M., Abrahamowicz M., Grodzicky T., Li Y., Panaritis C., du Berger R., Cote R., Grover S.A., Fortin P.R., Clarke A.E., Senécal J.L. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001;44:2331–2337. doi: 10.1002/1529-0131(200110)44:10<2331::aid-art395>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 17.Adachi M., Suematsu S., Suda T., Watanabe D., Fukuyama H., Ogasawara J., Tanaka T., Yoshida N., Nagata S. Enhanced and accelerated lymphoproliferation in Fas-null mice. Proc Natl Acad Sci USA. 1996;93:2131–2136. doi: 10.1073/pnas.93.5.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartvigsen K., Binder C.J., Hansen L.F., Rafia A., Juliano J., Horkko S., Steinberg D., Palinski W., Witztum J.L., Li A.C. A diet-induced hypercholesterolemic murine model to study atherogenesis without obesity and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2007;27:878–885. doi: 10.1161/01.ATV.0000258790.35810.02. [DOI] [PubMed] [Google Scholar]
- 19.Gough P.J., Gomez I.G., Wille P.T., Raines E.W. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ordovas J.M., Osgood D. Preparative isolation of plasma lipoproteins using fast protein liquid chromatography (FPLC) Methods Mol Biol. 1998;110:105–111. doi: 10.1385/1-59259-582-0:105. [DOI] [PubMed] [Google Scholar]
- 21.Montecino-Rodriguez E.M., Loor F. Haematopoietic cell transfers between C57BL/6 mice differing at the lpr or gld locus. Immunology. 1991;74:127–131. [PMC free article] [PubMed] [Google Scholar]
- 22.Nakashima Y., Plump A.S., Raines E.W., Breslow J.L., Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 23.VanderLaan P.A., Reardon C.A., Getz G.S. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004;24:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0. [DOI] [PubMed] [Google Scholar]
- 24.Rosenfeld M.E., Polinsky P., Virmani R., Kauser K., Rubanyi G., Schwartz S.M. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20:2587–2592. doi: 10.1161/01.atv.20.12.2587. [DOI] [PubMed] [Google Scholar]
- 25.Ma Z., Choudhury A., Kang S.A., Monestier M., Cohen P.L., Eisenberg R.A. Accelerated atherosclerosis in ApoE deficient lupus mouse models. Clin Immunol. 2008;127:168–175. doi: 10.1016/j.clim.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gautier E.L., Huby T., Ouzilleau B., Doucet C., Saint-Charles F., Gremy G., Chapman M.J., Lesnik P. Enhanced immune system activation and arterial inflammation accelerates atherosclerosis in lupus-prone mice. Arterioscler Thromb Vasc Biol. 2007;27:1625–1631. doi: 10.1161/ATVBAHA.107.142430. [DOI] [PubMed] [Google Scholar]
- 27.Aprahamian T., Rifkin I., Bonegio R., Hugel B., Freyssinet J.M., Sato K., Castellot J.J., Jr, Walsh K. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med. 2004;199:1121–1131. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi S., Hirano T., Kakinuma M., Uede T. Transcriptional repression and differential splicing of Fas mRNA by early transposon (ETn) insertion in autoimmune lpr mice. Biochem Biophys Res Commun. 1993;191:617–624. doi: 10.1006/bbrc.1993.1262. [DOI] [PubMed] [Google Scholar]
- 29.Mariani S.M., Matiba B., Armandola E.A., Krammer P.H. The APO-1/Fas (CD95) receptor is expressed in homozygous MRL/lpr mice. Eur J Immunol. 1994;24:3119–3123. doi: 10.1002/eji.1830241231. [DOI] [PubMed] [Google Scholar]
- 30.Lynch D.H., Watson M.L., Alderson M.R., Baum P.R., Miller R.E., Tough T., Gibson M., Davis-Smith T., Smith C.A., Hunter K., Bhat D., Din W., Goodwin R.G., Seldin M.F. The mouse Fas-ligand gene is mutated in gld mice and is part of a TNF family gene cluster. Immunity. 1994;1:131–136. doi: 10.1016/1074-7613(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 31.Zhou X., Paulsson G., Stemme S., Hansson G.K. Hypercholesterolemia is associated with a T helper (Th) 1/Th2 switch of the autoimmune response in atherosclerotic apo E-knockout mice. J Clin Invest. 1998;101:1717–1725. doi: 10.1172/JCI1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wahlsten J.L., Gitchell H.L., Chan C.C., Wiggert B., Caspi R.R. Fas and Fas ligand expressed on cells of the immune system, not on the target tissue, control induction of experimental autoimmune uveitis. J Immunol. 2000;165:5480–5486. doi: 10.4049/jimmunol.165.10.5480. [DOI] [PubMed] [Google Scholar]
- 33.Matute-Bello G., Lee J.S., Liles W.C., Frevert C.W., Mongovin S., Wong V., Ballman K., Sutlief S., Martin T.R. Fas-mediated acute lung injury requires fas expression on nonmyeloid cells of the lung. J Immunol. 2005;175:4069–4075. doi: 10.4049/jimmunol.175.6.4069. [DOI] [PubMed] [Google Scholar]
- 34.Boisvert W.A., Rose D.M., Johnson K.A., Fuentes M.E., Lira S.A., Curtiss L.K., Terkeltaub R.A. Up-regulated expression of the CXCR2 ligand KC/GRO-alpha in atherosclerotic lesions plays a central role in macrophage accumulation and lesion progression. Am J Pathol. 2006;168:1385–1395. doi: 10.2353/ajpath.2006.040748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu L., Okada Y., Clinton S.K., Gerard C., Sukhova G.K., Libby P., Rollins B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 36.Aiello R.J., Bourassa P.A., Lindsey S., Weng W., Natoli E., Rollins B.J., Milos P.M. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1518–1525. doi: 10.1161/01.atv.19.6.1518. [DOI] [PubMed] [Google Scholar]
- 37.Davenport P., Tipping P.G. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117–1125. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schieffer B., Selle T., Hilfiker A., Hilfiker-Kleiner D., Grote K., Tietge U.J., Trautwein C., Luchtefeld M., Schmittkamp C., Heeneman S., Daemen M.J., Drexler H. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation. 2004;110:3493–3500. doi: 10.1161/01.CIR.0000148135.08582.97. [DOI] [PubMed] [Google Scholar]
- 39.Madan M., Bishayi B., Hoge M., Amar S. Atheroprotective role of interleukin-6 in diet- and/or pathogen-associated atherosclerosis using an ApoE heterozygote murine model. Atherosclerosis. 2008;197:504–514. doi: 10.1016/j.atherosclerosis.2007.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caligiuri G., Rudling M., Ollivier V., Jacob M.P., Michel J.B., Hansson G.K., Nicoletti A. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med. 2003;9:10–17. [PMC free article] [PubMed] [Google Scholar]
- 41.Ahn J.H., Park S.M., Cho H.S., Lee M.S., Yoon J.B., Vilcek J., Lee T.H. Non-apoptotic signaling pathways activated by soluble Fas ligand in serum-starved human fibroblasts: Mitogen-activated protein kinases and NF-kappaB-dependent gene expression. J Biol Chem. 2001;276:47100–47106. doi: 10.1074/jbc.M107385200. [DOI] [PubMed] [Google Scholar]
- 42.O'Reilly L.A., Tai L., Lee L., Kruse E.A., Grabow S., Fairlie W.D., Haynes N.M., Tarlinton D.M., Zhang J.G., Belz G.T., Smyth M.J., Bouillet P., Robb L., Strasser A. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature. 2009;461:659–663. doi: 10.1038/nature08402. [DOI] [PMC free article] [PubMed] [Google Scholar]