

Abstract
Cytotoxic T lymphocytes (CTLs) that cause type 1 diabetes are activated in draining lymph nodes and become concentrated as fully active CTLs in inflamed pancreatic islets. It is unclear whether CTL function is driven by signals received in the lymph node or also in the inflamed tissue. We studied whether the development of cytotoxicity requires further activation in islets. Autoreactive CTLs found in the islets of diabetes-prone NOD mice had acquired much higher expression of the cytotoxic effector markers granzyme B, interferon γ, and CD107a than had those in the pancreatic lymph node (PLN). Increased expression seemed to result from stimulation in the islet itself. T cells held up from migrating from the PLN by administration of the sphingosine-1-phosphate agonist FTY720 did not increase expression of cytotoxic molecules in the PLN. Stimulation did not require antigen presentation or cytokine secretion by the target β cells because it was not affected by the absence of class I major histocompatibility complex expression or by the overexpression of suppressor of cytokine signaling-1. Activation of CD40-expressing cells stimulated increased CTL function and β-cell destruction, suggesting that signals derived from CD40-expressing cells promote the acquisition of cytotoxicity in the islet environment. These data provide in vivo evidence that stimulation of cytotoxic effector molecule expression occurs in inflamed islets and is independent of β cells.
Type 1 diabetes is an autoimmune disease in which the insulin-producing β cells of the pancreas are selectively destroyed. CD4+ and CD8+ T cells are required for efficient disease progression.1 The priming of naïve diabetogenic T cells is believed to occur in the pancreatic lymph node (PLN), where proteins derived from β cells are exposed to the immune system. T cells from β-cell–specific CD4+ TCR transgenic BDC2.5 mice proliferated in the PLN before their detection in the pancreas.2 Also, excision of the PLN from young NOD mice prevented the development of diabetes.3 Activation of lymphocytes in the regional secondary lymphoid tissue occurs not only in diabetes but generally in all immune responses. For example, in viral illness, such as influenza, activation of CD8+ cytotoxic T lymphocytes (CTLs) occurs in lymph nodes draining the site of infection.4–6 Activation results in extensive proliferation and differentiation before migration to infected tissue.6–8 Recent data from viral infection models have highlighted a role for signals in the target tissue as stimulating increased cytotoxicity by primed CTLs that promotes viral clearance.9–11
Extensive studies in the NOD mouse model, and indirect evidence from human diabetes, have identified CD8+ CTLs as the main cells that perform β-cell destruction.12,13 CTLs recognize peptide antigens presented on the β-cell surface by major histocompatibility complex (MHC) class I protein that consists of a polymorphic heavy chain and a constant light chain, β2-microglobulin. NOD mice deficient in β2-microglobulin lack MHC class I and CD8+ T cells and are protected from disease.14–17 Conditional deletion of MHC class I from the β cell alone resulted in a significant reduction in diabetes, and overexpression of adenovirus E19 protein in β cells inhibited MHC class I expression and prevented CTL lysis.18,19 Transgenic overexpression of suppressor of cytokine signaling 1 (SOCS1) in β cells blocked CTL killing of β cells by reducing MHC class I expression and antigen presentation. Therefore, a direct interaction between CTL and MHC class I on the β cell is required for β-cell destruction. It is possible that protection is seen because T cells do not achieve full activation with reduced β-cell antigen presentation or that they become fully armed but not able to target β cells adequately for killing to occur.
Although fully mature CTLs are concentrated in the islets of prediabetic NOD mice, it is unclear whether this is driven by signals received in the lymph node only or also in the inflamed tissue. The aim of this study is to explore the role of stimuli in islets in the development of diabetogenic CTLs. These data show that signals in the islet are required for the acquisition of molecules that indicate cytotoxic effector function.
Materials and Methods
Mice
All the mice were bred and maintained at the St. Vincent's Institute animal facility (Fitzroy, Australia). NOD/Lt mice were purchased from the animal breeding facility at the Walter and Eliza Hall Institute, Melbourne, VIC, Australia. The 8.3 mice expressing TCRαβ rearrangement of the H-2Kd-restricted, β-cell reactive, CD8+ T-cell clone NY8.3; NODRIP-SOCS1 mice expressing the SOCS1 transgene under the control of the rat insulin promoter; NODIGRP mice expressing islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) under the control of an MHC class II promoter (I-Eακ); and class I β-bald mice containing a conditional deletion of β2-microglobulin from β cells have all been described previously.18,20–22 Granzyme B (GzmB)–deficient mice were backcrossed to NOD for 10 generations and then were crossed with 8.3 to generate GzmB-deficient 8.3 mice (Z.U.M. and H.E.T., unpublished data).23 The Institutional Animal Ethics Committee approved all the experiments.
Antibodies and Peptides
Antibodies used for flow cytometric analysis were as follows: anti-CD8 [Ly2(53-6.7)] conjugated to phycoerythrin (PE) or allophycocyanin (BioLegend, San Diego, CA), anti–interferon (IFN) γ (XMG1.2) conjugated to PE (BD Pharmingen, San Diego, CA), and anti-mouse GzmB (I6G6) conjugated to PE (eBioscience Inc., San Diego, CA). Isotype controls were PE-conjugated rat IgG1 (R3-34) (BD Pharmingen) or PE-conjugated rat IgG2b (RTK4530) (BioLegend). The peptide IGRP206-214 (VYLKTNVFL) was purchased from Auspep (Louisville, KY).
CFSE Labeling and Adoptive Transfer
CD8+ T cells from 8.3 mice were labeled with carboxyfluorescein succinimidyl ester (CFSE) as previously described.21 Cells were resuspended at 2.5 × 107/mL in PBS, and 200 μL was injected i.v. into the tail vein of recipient mice. After 5 days, the mice were sacrificed, and the inguinal lymph node (ILN), PLN, and islets were harvested.
FTY720 and CD40 Agonist Treatments
The sphingosine-1-phosphate receptor-1 agonist FTY720 was purchased from Cayman Chemical Co., Ann Arbor, MI. Recipient NOD mice were treated i.p. with 3 mg/kg FTY720 in water on days 1 to 4 after adoptive transfer. CD40 agonist treatment was performed as previously described.24
Islet Isolation
Islets of Langerhans were isolated using collagenase P (Roche, Basel, Switzerland) and Histopaque-1077 density gradients (Sigma-Aldrich Corp, St. Louis, MO) as previously described.20,25
Flow Cytometry
Lymph nodes harvested from recipient mice were prepared as single-cell suspensions. Peripheral blood was collected by cardiac puncture, and lymphocytes were separated on a Ficoll gradient. Islets were dispersed to single cells with 0.1 mg/mL bovine trypsin (CalBiochem, San Diego, CA) and 2 mmol/L EDTA for 5 minutes at 37°C and gentle pipetting. Dispersed islets were washed in RPMI 1640 medium containing antibiotics, 2 mmol/L glutamine, nonessential amino acids, 50 μm of mercaptoethanol, and 10% fetal calf serum (complete RPMI; Gibco, Invitrogen Corp., Grand Island, NY) and were allowed to recover for 1 to 2 hours in complete RMPI at 37°C in 5% CO2. Cell surface markers were stained using standard procedures. Intracellular staining was performed according to the manufacturer's specifications using the Cytofix/Cytoperm Plus kit purchased from BD Biosciences. The specificity of staining was confirmed using isotype control antibodies. To ensure that collagenase digestion and trypsinization of islets did not alter staining of T cells, some PLN samples were treated as for islets, and expression of CFSE, CD8, and GzmB was analyzed. Pattern of expression was the same in the PLN with or without collagenase digestion and trypsinization. All analyses were performed using the FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ) and FlowJo analysis software (Tree Star Inc., Ashland, OR). Mean fluorescence intensity (MFI) was used to quantify and compare expression of markers of cytotoxicity where indicated.
CD107a Degranulation Assay
Unlabeled 8.3 T cells (5 × 106) in PBS were adoptively transferred into NOD recipients. After 5 days, the ILN, PLN, and islets were harvested, and single-cell suspensions were prepared. Degranulation was measured on transferred cells by staining with anti-CD8, IGRP206-214H-2Kd tetramer (made by ImmunoID Flow Cytometry Facility, Melbourne, Australia), and anti-CD107a (BD Pharmingen) and were analyzed by flow cytometry using standard methods.
Statistics
Analysis of data was performed using the GraphPad Prism program (GraphPad Software Inc., San Diego, CA), and the unpaired Student's t-test was used to assess statistical significance. Values are given as mean ± SEM throughout. Error bars on all graphs represent the SEM.
Results
Diabetogenic CTLs in Islets Display Increased Markers of Cytotoxicity
To determine whether restimulation in the islet environment is required for fully differentiated CTLs to develop, we adoptively transferred CFSE-labeled 8.3 CD8+ T cells into 8- to 9-week-old NOD recipients and analyzed CTL proliferation and expression of markers of cytotoxicity in the PLN versus the islets (Figure 1). The age of mice was carefully selected to be before the age where the PLN is dispensable for diabetes onset.3 In the PLN, the CFSE+CD8+ T cells underwent several rounds of cell division (divisions 1 to 3). In the islets, we detected further division that gave rise to a distinct pattern of proliferation in which most cells were detected in divisions 4 to 7 (Figure 1, A and B). This pattern suggests that initial proliferation occurs in the PLN in response to islet antigen presented on dendritic cells and that these cells migrate to the islets and undergo further rounds of division.
Figure 1.
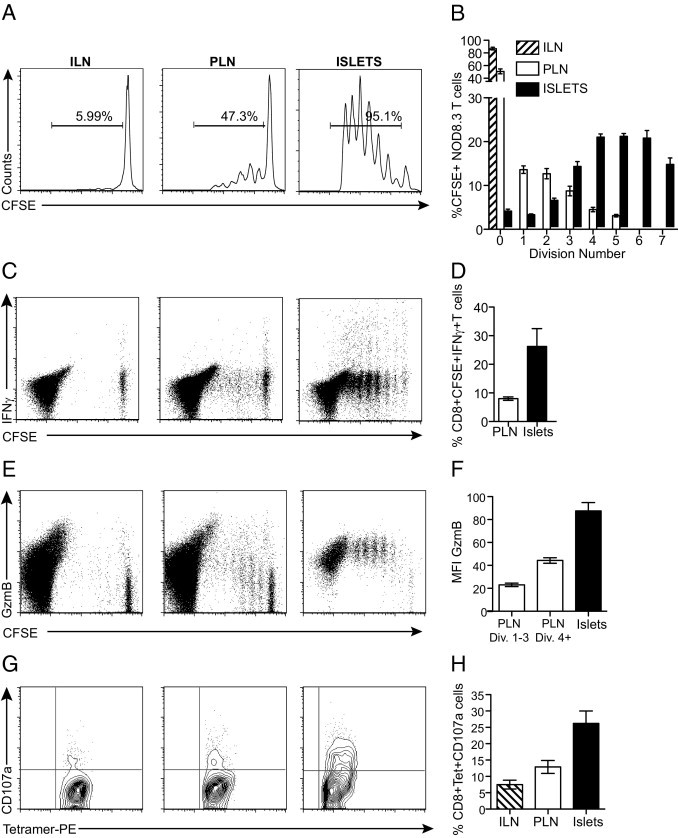
Proliferation, activation, and acquisition of cytotoxic effector function by diabetogenic CTLs. NOD recipients were adoptively transferred with 5 × 106 CFSE-labeled 8.3 T cells. Five days after transfer, the ILN, PLN, and islets were harvested and analyzed. A: Proliferation of 8.3 T cells. Representative histogram profiles are gated on CFSE-labeled CD8+ T cells. B: The percentage of CFSE-positive T cells per division (n = 7). C: Expression of IFNγ by proliferating 8.3 T cells. D: Percentage of CD8+CFSE+IFNγ+ cells in the PLN and islets (n = 5). E: Expression of GzmB. F: MFI for GzmB expression gated on all CD8+CFSE+ cells (n = 9). G: Expression of CD107a on transferred 8.3 T cells. H: Percentage of CD8+tetramer+CD107a+ T cells (n = 6). Data are given as the mean ± SEM (error bars).
We next examined the secretion of IFNγ and the expression of GzmB in dividing CFSE+CD8+ T cells. The secretion of IFNγ in response to antigen is often used as a marker of cytotoxic capacity in CD8+ T cells. Single-cell suspensions of the ILN, PLN, and islets were cultured in vitro for 6 hours with IGRP206-214 peptide, and capacity to secrete IFNγ was analyzed by intracellular cytokine stain (Figure 1C). The mean ± SEM percentage of proliferating CFSE+CD8+ T cells secreting IFNγ after transfer was higher in islets (26.2% ± 6.2%; mean ± SEM MFI = 1071 ± 86.2) compared with in the PLN (8.02% ± 0.61%; mean ± SEM MFI = 328.6 ± 47.04), and this did not correlate strongly with the number of cell divisions (P < 0.005) (Figure 1, C and D).
GzmB, a serine protease, is the most abundant component of the cytolytic granule and is a major part of the cytolytic mechanisms enabled by perforin.26 GzmB expression increased with each round of cell division of CFSE+CD8+ T cells in the PLN (Figure 1E). However, once the CD8+ T cells reached the islets, there was a further significant increase in GzmB expression. This was quantifiable by comparing the MFI for GzmB expression at each site (Figure 1F). In the PLN, the mean ± SEM MFI increased from 23.06 ± 1.53 in the early divisions to 44.3 ± 2.42 as the cells progressed to later divisions. Once in the islets, GzmB expression increased again (mean ± SEM MFI = 87.53 ± 7.36), and this was independent of cell cycle number (Figure 1F) (P < 0.0001, MFI for GzmB in islets compared with in the PLN). Expression in islets was comparable with GzmB expression after maximal in vitro stimulation (see Supplemental Figure S1 at http://ajp.amjpathol.org). To ensure that the detection of GzmB in 8.3 T cells was specific, we generated GzmB-deficient 8.3 mice and assessed expression in these T cells isolated from the ILN, PLN, and islets of NOD mice after adoptive transfer. Staining was absent in the GzmB-deficient 8.3 T cells (data not shown).
To assess cytotoxic function, we evaluated the degranulation of the 8.3 T cells in the PLN and islets, directly ex vivo (Figure 1, G and H). The mean ± SEM percentage of transferred 8.3 T cells expressing CD107a was increased on CD8+tetramer+ T cells from in the islets (26.2% ± 3.81%; mean ± SEM MFI = 32.65 ± 2.67) compared with in the PLN (12.8% ± 1.96%; mean ± SEM MFI = 18.3 ± 4.23) (P < 0.01). The expression of CD107a in islets was less than after in vitro stimulation, but the differences between the PLN and islets was similar (see Supplemental Figure S1 at http://ajp.amjpathol.org). The increased degranulation correlates with the increase in cytotoxic molecule expression in the islets. Most striking about all the variables measured were the substantial differences seen between the populations of cells isolated from islets compared with the PLN. Taken together, these data show that CTLs isolated from islets display increased cytotoxic ability, indicating that full activation was not apparent in the PLN.
Acquisition of Full Cytotoxic Effector Capacity Occurs in Islets
It is possible that the CD8+ T cells isolated from the islets acquired the features of cytotoxicity in the PLN and immediately migrated to islets, where they were detected. To address this possibility, we treated adoptive transfer recipients with the sphingosine-1-phosphate receptor 1 agonist FTY720, which prevents egress of lymphocytes from lymph nodes to the circulation.27 Recipient mice were treated on days 1 to 4 after transfer, and the PLN and islets were analyzed on day 5. FTY720-treated recipients had increased proliferation of 8.3 T cells in the PLN, indicating that FTY720 given after transfer did not prevent lymphocyte entrance into the lymph nodes or proliferation there but promoted accumulation and prevented migration to the islets (Figure 2, A and B). We then examined the expression of GzmB in the proliferating CD8+ T cells and found that in treated and untreated mice, dividing CD8+ T cells increased expression of GzmB to the same extent in the PLN (Figure 2B). There were no detectable CFSE-labeled CD8+ T cells in the islets of FTY720-treated mice.
Figure 2.
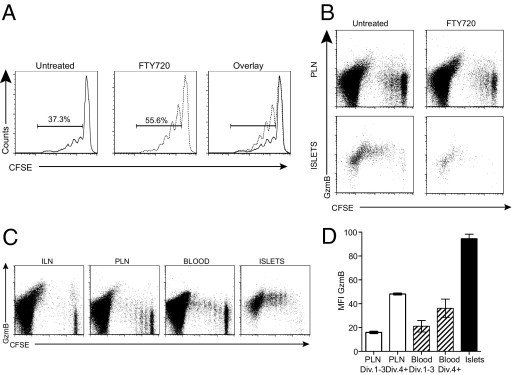
Full cytotoxic effector molecule expression does not occur in the PLN. A: Proliferation of CFSE-labeled 8.3 T cells in the PLNs of control and FTY720-treated mice. NOD mice were treated with 3 mg/kg FTY720 daily from the day of adoptive transfer until 5 days after transfer, when lymph nodes and islets were harvested (n = 8 for treated and untreated from four experiments). Histograms are gated on CFSE+CD8+ T cells, and the percentage of proliferating cells is indicated. B: Expression of GzmB in dividing CFSE-labeled CD8+ T cells isolated from the PLN and islets of untreated and FTY720-treated recipient NOD mice. Dot plots are gated on total CD8+ T cells. C: Flow cytometric analysis of GzmB expression in diabetogenic CTLs isolated from peripheral blood. Representative dot plots are gated on total CD8+ T cells. D: MFI for GzmB expression in the PLN, blood, and islets. For the PLN and blood, divisions (Div.) 1 to 3 and 4+ were gated and MFI was calculated (n = 4). Data are given as mean ± SEM (error bars).
An alternative interpretation of the differences between activation and cytotoxic capacity in the PLN and islets is that signaling for acquisition of cytotoxic effector function occurs in the PLN but expression is up-regulated only once the cells have egressed into the circulation. This could be because of negative signals, eg, regulatory T cells that inhibit expression in the PLN or because of positive signals delivered in the process of exiting the PLN. To assess this, we examined GzmB expression in CFSE-labeled CD8+ T cells in the peripheral blood (Figure 2, C and D). GzmB expression in proliferating cells was identical in the PLN and peripheral blood (Figure 2C). MFI for GzmB expression on cells in early division (1 to 3) and late division (4+) were calculated (Figure 2D). In the PLN and blood, the early divisions had a mean ± SEM MFI of 15.93 ± 1.82 and 21.2 ± 8.32, respectively, increasing to 48.13 ± 1.49 and 36.27 ± 13.19 in late division. Once the cells reached the islets, there was a further increase in GzmB expression reflected by a mean ± SEM MFI of 94.58 ± 7.364. These data demonstrate that acquisition of full cytotoxic capacity by diabetogenic CTLs is location dependent. Initial activation occurs in the PLN, resulting in the acquisition of some cytotoxic effector potential. These early CTLs then migrate to the islets and once in the islet environment undergo further stimulation to acquire full cytotoxic effector capacity.
Acquisition of Full Cytotoxic Effector Capacity Occurs Independently of Antigen Presentation by β Cells
We and others have previously shown that antigen presentation by β cells to CD8+ T cells is needed for the cytotoxic potential of β-cell–specific CTLs to be realized.18–20,28 To determine the role that antigen presentation by the β cell plays in increasing the expression of cytotoxic effector molecules in islets, we used class I β-bald mice. These mice contain a conditional deletion of β2-microglobulin from the β cells, resulting in complete elimination of MHC class I and, therefore, β-cell antigen presentation.18 There was no difference in cytotoxic effector molecule acquisition between 8.3 T cells transferred into NOD or class I β-bald recipients (Figure 3). A mean ± SEM of 26.27% ± 6.22% of CD8+CFSE+ CTLs isolated from the islets of NOD secreted IFNγ compared with 22.03% ± 4.68% isolated from class I β-bald mice (Figure 3, A and B). Similarly, the mean ± SEM MFI for GzmB expression was 19.67 ± 0.98 in CTLs from class I β-bald islets and 20.35 ± 1.57 in CTLs from NOD islets (Figure 3, C and D). These data demonstrate that the increase in cytotoxic capacity occurs in islets independently of β-cell antigen presentation. They also show that CTLs in β-bald mice seem to have identical cytotoxic capacity as CTLs in NOD islets but do not cause diabetes because they cannot directly recognize β cells.
Figure 3.
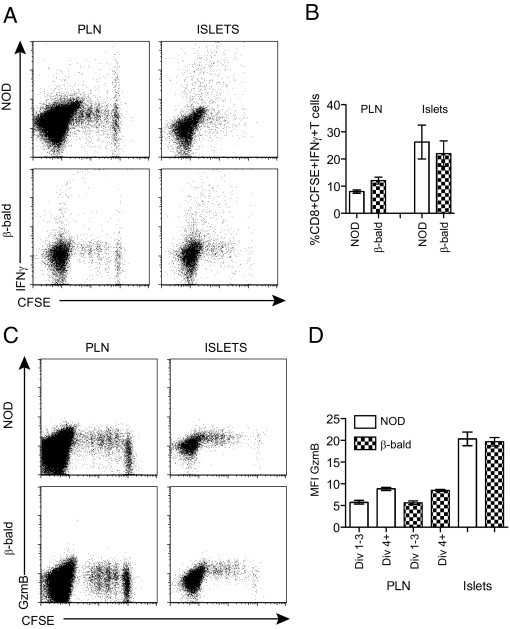
Complete elimination of β-cell antigen presentation does not prevent increased cytotoxic effector molecule expression in diabetogenic CTLs. Secretion of IFNγ (A and B) and expression of GzmB (C and D) in dividing 8.3 T cells isolated from the PLN and islets isolated from NOD and class I β-bald recipients. Dot plots (A and C) are gated on total CD8+ T cells and represent multiple experiments (n = 3 to 5). Data shown in the graphs (B and D) were calculated as described in Figure 1. Data are given as mean ± SEM (error bars).
Acquisition of Full Cytotoxic Effector Capacity Occurs Independently of Cytokine Effects on the β Cell
During the progression of diabetes, the β cell secretes cytokines in the islet environment in a feedback loop between the invading immune cells and the target tissue. For example, IFNγ is produced by infiltrating T cells that can act on the β cell not only to up-regulate MHC class I expression but also to secrete chemokines and cytokines. For example, IL-15 is produced by β cells, and this common γ-chain cytokine can drive T-cell activation and proliferation.20 To assess whether cytokine production by the β cell drives the increase in CTL effector molecule expression in islets, we compared CTLs isolated from NOD and NODRIP-SOCS1 recipients. NODRIP-SOCS1 have β cells unresponsive to multiple cytokines, including IFNγ, resulting in significant reductions in cytokine secretion, including IL-15, and prevention of MHC class I up-regulation.20 Analysis of CD8+CFSE+ T cells isolated from islets of NODRIP-SOCS1 and NOD mice showed similar secretion of IFNγ (Figure 4, A and B) and expression of GzmB (Figure 4, C and D), indicating that the cytokine secretion by the β cell has little effect on the increase in cytotoxicity observed in CTLs in islets.
Figure 4.
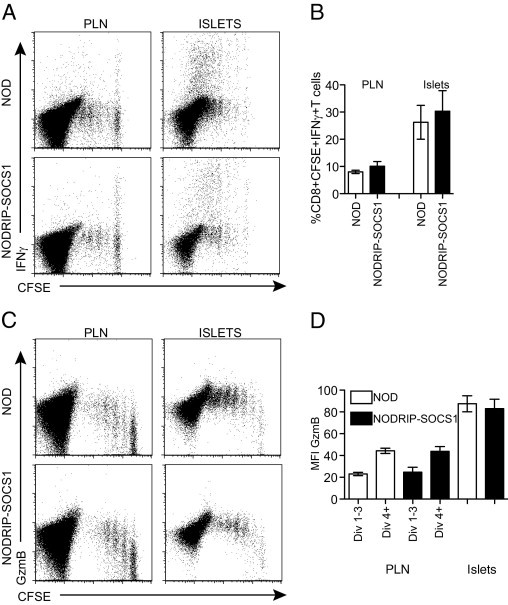
Reduced β-cell cytokine production does not prevent increased cytotoxic effector molecule expression in diabetogenic CTLs. Comparison of IFNγ secretion (A and B) and GzmB expression (C and D) in dividing 8.3 T cells from the PLN and islets isolated from NOD and NODRIP-SOCS1 recipients. Dot plots (A and C) are gated on total CD8+ T cells and represent multiple experiments. Percentage of IFNγ (γ) and MFI for GzmB expression (D) were calculated as described in Figure 1 (n = 4 to 5).
An Inflammatory Environment Promotes Acquisition of Full Cytotoxic Effector Capacity
The acquisition of full cytotoxic effector capacity is location dependent, occurring once the CTLs are in islets, but is independent of β-cell antigen presentation or cytokine production. This finding suggests that other stimuli from the inflamed islet environment are required to induce the increase in CTL cytotoxicity. To analyze this possibility, we examined whether activation of dendritic cells (DCs) and the creation of an inflammatory environment would result in fully differentiated CTLs outside the pancreas. NODIGRP mice overexpress IGRP206-214 under the control of the MHC class II promoter I-Eακ, resulting in IGRP expression in all DCs.21 We used these recipients as a source of specific antigen outside the islet and then treated with CD40 agonist antibody to stimulate up-regulation of co-stimulatory molecules and the production of proinflammatory cytokines, including IL-12.24,29 There was extensive proliferation of transferred cells in the spleen of all recipients consistent with the expression of IGRP in DCs with or without CD40 agonist, but recipient mice treated with CD40 agonist had enhanced proliferation (Figure 5A). The percentage of IFNγ+ cells was significantly increased in transferred cells isolated from recipient mice treated with CD40 agonist (Figure 5, A and B) (P = 0.03). Furthermore, diabetes resulted in recipients confirming within 14 days that the CTLs were capable of β-cell destruction. These data indicate that increasing inflammation by CD40-expressing cells can promote CD8+ T cells to acquire full cytotoxic capacity. They suggest that the acquisition of high levels of GzmB and other factors is physiologically important because this was associated with β-cell destruction and diabetes.
Figure 5.
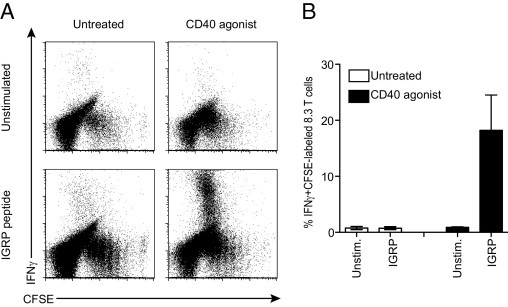
Increasing inflammation promotes acquisition of cytotoxic effector capacity. NODIGRP recipients were adoptively transferred with CFSE-labeled 8.3 T cells and were injected with CD40 agonist antibody (n = 5) or were left untreated (n = 5). A: Single-cell preparations of splenocytes were stimulated with IGRP peptide or were left unstimulated (Unstim.), and IFNγ secretion in dividing 8.3 T cells was measured. Dot plots are gated on total CD8+ T cells and represent multiple experiments. B: Percentage of IFNγ-secreting cells. Data are given as mean ± SEM (error bars).
Discussion
In this study, we demonstrated that the primary activation of β-cell–specific CTLs and the high level of expression of molecules associated with cytotoxicity may be distinct steps separated by location and driven by different stimuli. Initial proliferation and activation occurred in the PLN. However, CTLs then migrated to the islets and once in this environment acquired increased expression of GzmB and IFNγ and granule exocytosis measured by surface expression of CD107a, which are markers of cytotoxic capacity. Full expression of cytotoxic molecules could be mimicked by stimulation of antigen-presenting cells. Eliminating antigen presentation and reducing cytokine production by the β cell had no effect on the expression of cytotoxic markers. It is known that CTLs that are able to kill are concentrated in the islets, and the present data suggest that this increased capacity is acquired there.
Activation and differentiation of diabetogenic CTLs and the adaptive immune response to viral infections are analogous. T-cell activation occurs first in the lymph node that receives lymphatic drainage from an infected or antigen-expressing tissue.2,4–6,30 Activation in the lymph node has been thought to set in motion a sequence of events that results in homing to and immune attack on antigen-expressing cells and tissues. The present data and other recent data suggest that the contribution of further activation signals in the target tissue has been understated.11,31,32 Once in the islet environment, CTLs increased their cytotoxic effector capacity. This increase requires location-specific signals that are independent of the β cell.
Initially, we interpreted these data as suggesting that CTLs migrate to islets once they are fully activated, having lost the adhesion molecule and chemokine receptor expression that keep them in the PLN. Thus, markers of differentiated cytotoxic function observed on transferred CD8+ T cells in the islets might have actually been acquired in the PLN. They may not have been observed in the PLN because acquisition and CTL migration are closely coupled or because migration had occurred during the time required for expression of cytotoxic molecules. In addition, some CTLs that encounter antigen in the PLN have been observed to undergo apoptosis, a mechanism which may slow the progression of insulitis.33 Although not sufficient to completely prevent CTL differentiation and recruitment to islets, this apoptosis could contribute to the inability to detect fully activated CTLs in the PLN. Two experiments were designed to test these possibilities, and the results of both suggested that CTLs need to be in the inflamed islet for full differentiation. Administration of FTY720 held newly activated CTLs in the PLN rather than allowing them to migrate to islets, but this did not result in fully differentiated CTLs being increased in number in the PLN. Also, CTLs found in the peripheral blood that had evidence of previous activation due to CFSE dilution did not have increased CTL effector molecule expression, suggesting that these differentiation markers are not acquired immediately before exit from the PLN. An alternative explanation to CTLs increasing their expression of cytotoxic molecules in the islets is the possibility that the migration of activated CTLs to the islets is highly selective and that only CTLs with the highest level of expression gain access to the islets. Although this possibility is difficult to rule out, work is ongoing in our laboratory to identify whether this is the case.
There are several features of the islet environment that could contribute to the increase in cytotoxic effector molecule expression acquired once the CTLs are located there. The first feature is the increased expression of MHC class I on the surface of the β cell during insulitis progression.34 However, complete elimination of β-cell antigen presentation by conditional deletion of MHC class I had no effect on the increased expression of cytotoxic markers.18 This is in contrast to a recent study by Pang et al35 that concluded using influenza HA transgenic mice that β-cell antigen presentation is required for T-cell activation. However, the studies are not directly comparable owing to the different models studied.
Antigen presentation in the islet environment has been demonstrated as a requirement for the accumulation and retention of CTLs in islets.36,37 In the present experiments, antigen-specific CTLs accumulated in class I β-bald islets, indicating that the required antigen need not be presented by the β cell. It is likely that professional antigen-presenting cells in the islet could present specific antigen by cross presentation to CTLs. There is also evidence that vascular endothelial cells can present specific antigen to CD8+ T cells.38 Thus, either or both could act to present antigen to facilitate the retention of CTLs in the islet in class I β-bald mice.
The present data are consistent with a third signal being required for optimal activation and differentiation to fully functional CTLs. The third signal is provided once the cells have migrated to the islets. Candidates for the third signal identified in in vitro systems for CTL differentiation include the cytokines IFNα and IL-12 and the common γ-chain cytokines IL-2 and IL-21.29,39,40 IFNα has been associated with initiation of diabetes in NOD mice,41 and isolated macrophages and DCs have been shown to secrete increased levels of IL-12.42–44 NOD mice also have elevated levels of IL-21.45,46 We investigated two sources of these cytokines: β-cell– and CD40–expressing cells. Reducing cytokine secretion by the β cell through the overexpression of SOCS1 had no effect on the increased expression of cytotoxic molecules in the islets. This demonstrates that the β cell is not necessary to further activate CD8+ T cells in the islets. The ligation of CD40 on the surface of DCs promotes the secretion of inflammatory cytokines. Treatment with CD40 agonist promoted the development of CTLs with an islet signature of cytotoxicity, and these CTLs were able to cause diabetes. Therefore, inflammation and cytokine secretion may provide the third signal to stimulate acquisition of full cytotoxic effector capacity. Although this seems to implicate factors from DCs in increasing CTL expression of cytotoxic molecules, other cells that express CD40 might also be involved, including B lymphocytes and macrophages, both of which are abundant in the islet infiltrate.47–49
Two other features of the islet environment that could affect CTL differentiation are regulatory T cells and the formation of tertiary lymphoid organs (TLOs). The ratio of regulatory T cells (Tregs) to effector T cells is altered in islets due to a reduction in IL-2 and an increase in Treg apoptosis in the islet environment.50 This could allow the escape of CTLs from Treg suppression and promote the accumulation of fully activated CTLs in the islet. We addressed the possibility that Tregs may be involved by examining cells in the blood after they leave the regulated environment of the node and did not see increased effector molecule expression. Thus, it is unlikely that the reduction in islet Tregs affects CTL differentiation in islets. The extensive insulitis, including TLOs that form in NOD mouse pancreas, has been shown to directly promote the proliferation of autoreactive CD4+ T cells.51 However, these TLOs form in mice much older (≥15 weeks) than those used in the present study, making it unlikely that TLOs contribute to our observations.51,52 In addition, we did not detect CD8+CFSE+GzmB+ CTLs in the islets of FTY720-treated recipients, which indicates that direct pancreas priming does not occur in this model, ruling out TLOs as a requirement for the increased CTL cytotoxicty observed in islets.
These data provide new insights into the dynamics of CTL differentiation in diabetes and autoimmunity. We identified that cytotoxic function of the CTL becomes complete only after migration to the inflamed target tissue and probably depends on stimuli from antigen-presenting cells in the islet. Under the conditions of mild inflammation, as is generally seen in patients, it remains questionable to what degree these findings in the NOD mouse will translate to humans. However, there is evidence of inflammation in human islets, including the presence of immune cell subsets that include CD40-expressing cells (DCs, macrophages, and B cells), the widespread increased expression of MHC class I, the expression of IFNα, a strong candidate for the third signal to up-regulate cytotoxic molecule expression, and the vascular leakiness of infiltrated islets recently demonstrated using magnetic resonance imaging.12,13,51–55 A better understanding of CTL effector molecule expression may lead to an ability to reduce the damaging effects of CTLs in diabetes. The present data highlight the role that target tissue, not just the draining lymph node, plays during the generation of autoimmunity by facilitating the differentiation of autoreactive CD8+ T cells, which ultimately leads to tissue destruction and disease.
Acknowledgments
We thank Lorraine Elkerbout, Dip. Animal Technology, for her excellent animal care and technical assistance, Rochelle Ayala-Perez, BSc, for her technical assistance, Caroline Dobrzelak, BSc, for genotyping, and Anne Kelso, Ph.D., and Andrew Lew, Ph.D., for their helpful comments on the manuscript.
Footnotes
Supported by a program grant and a career development award from the National Health and Medical Research Council of Australia (H.E.T.); a program project grant (B.K.) and a postdoctoral fellowship (K.L.G.) from the Juvenile Diabetes Research Foundation; a Skip Martin Early Career Postdoctoral Fellowship from the Australian Diabetes Society (K.L.G.); and a Millennium Research Grant from Diabetes Australia (T.W.K.). P.S. is a scientist of the Alberta Heritage Foundation for Medical Research and is supported by the Canadian Institutes of Health Research and the Juvenile Diabetes Research Foundation. The Julia McFarlane Diabetes Research Centre is supported by the Diabetes Association (Foothills).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.02.015.
Supplementary data
Comparison of GzmB and CD107a expression in diabetogenic CTLs after in vitro stimulation. A: Expression of GzmB in unstimulated and IGRP-stimulated CFSE+CD8+ T cells. The graphs show the MFI for GzmB expression in unstimulated (B) and stimulated (C) cells (n = 4). Div., division. D: Expression of CD107a on transferred 8.3− T cells unstimulated or stimulated in vitro with PMA (25 ng/mL) and ionomycin (5 μg/mL). The graphs show the percentage of CD8+tetramer+CD107a+ T cells in unstimulated (E) and stimulated (F) cells (n = 6). Data are given as mean ± SEM (error bars).
References
- 1.Christianson S.W., Shultz L.D., Leiter E.H. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice: relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NODNON-Thy-1a donors. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 2.Hoglund P., Mintern J., Waltzinger C., Heath W., Benoist C., Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med. 1999;189:331–339. doi: 10.1084/jem.189.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gagnerault M.C., Luan J.J., Lotton C., Lepault F. Pancreatic lymph nodes are required for priming of β cell reactive T cells in NOD mice. J Exp Med. 2002;196:369–377. doi: 10.1084/jem.20011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coles R.M., Mueller S.N., Heath W.R., Carbone F.R., Brooks A.G. Progression of armed CTL from draining lymph node to spleen shortly after localized infection with herpes simplex virus 1. J Immunol. 2002;168:834–838. doi: 10.4049/jimmunol.168.2.834. [DOI] [PubMed] [Google Scholar]
- 5.Jones C.M., Cose S.C., Coles R.M., Winterhalter A.C., Brooks A.G., Heath W.R., Carbone F.R. Herpes simplex virus type 1-specific cytotoxic T-lymphocyte arming occurs within lymph nodes draining the site of cutaneous infection. J Virol. 2000;74:2414–2419. doi: 10.1128/jvi.74.5.2414-2419.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawrence C.W., Braciale T.J. Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J Immunol. 2004;173:1209–1218. doi: 10.4049/jimmunol.173.2.1209. [DOI] [PubMed] [Google Scholar]
- 7.Johnson B.J., Costelloe E.O., Fitzpatrick D.R., Haanen J.B., Schumacher T.N., Brown L.E., Kelso A. Single-cell perforin and granzyme expression reveals the anatomical localization of effector CD8+ T cells in influenza virus-infected mice. Proc Natl Acad Sci U S A. 2003;100:2657–2662. doi: 10.1073/pnas.0538056100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marshall D.R., Olivas E., Andreansky S., La Gruta N.L., Neale G.A., Gutierrez A., Wichlan D.G., Wingo S., Cheng C., Doherty P.C., Turner S.J. Effector CD8+ T cells recovered from an influenza pneumonia differentiate to a state of focused gene expression. Proc Natl Acad Sci U S A. 2005;102:6074–6079. doi: 10.1073/pnas.0501960102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohlmeier J.E., Cookenham T., Roberts A.D., Miller S.C., Woodland D.L. Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity. 2010;33:96–105. doi: 10.1016/j.immuni.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGill J., Van Rooijen N., Legge K.L. Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J Exp Med. 2008;205:1635–1646. doi: 10.1084/jem.20080314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wakim L.M., Gebhardt T., Heath W.R., Carbone F.R. Cutting edge: local recall responses by memory T cells newly recruited to peripheral nonlymphoid tissues. J Immunol. 2008;181:5837–5841. doi: 10.4049/jimmunol.181.9.5837. [DOI] [PubMed] [Google Scholar]
- 12.Bottazzo G.F., Dean B.M., McNally J.M., MacKay E.H., Swift P.G., Gamble D.R. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med. 1985;313:353–360. doi: 10.1056/NEJM198508083130604. [DOI] [PubMed] [Google Scholar]
- 13.Itoh N., Hanafusa T., Miyazaki A., Miyagawa J., Yamagata K., Yamamoto K., Waguri M., Imagawa A., Tamura S., Inada M. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92:2313–2322. doi: 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katz J., Benoist C., Mathis D. Major histocompatibility complex class I molecules are required for the development of insulitis in non-obese diabetic mice. Eur J Immunol. 1993;23:3358–3360. doi: 10.1002/eji.1830231244. [DOI] [PubMed] [Google Scholar]
- 15.Serreze D.V., Leiter E.H., Christianson G.J., Greiner D., Roopenian D.C. Major histocompatibility complex class I-deficient NOD-B2mnull mice are diabetes and insulitis resistant. Diabetes. 1994;43:505–509. doi: 10.2337/diab.43.3.505. [DOI] [PubMed] [Google Scholar]
- 16.Sumida T., Furukawa M., Sakamoto A., Namekawa T., Maeda T., Zijlstra M., Iwamoto I., Koike T., Yoshida S., Tomioka H. Prevention of insulitis and diabetes in β 2-microglobulin-deficient non-obese diabetic mice. Int Immunol. 1994;6:1445–1449. doi: 10.1093/intimm/6.9.1445. [DOI] [PubMed] [Google Scholar]
- 17.Wicker L.S., Leiter E.H., Todd J.A., Renjilian R.J., Peterson E., Fischer P.A., Podolin P.L., Zijlstra M., Jaenisch R., Peterson L.B. β 2-microglobulin-deficient NOD mice do not develop insulitis or diabetes. Diabetes. 1994;43:500–504. doi: 10.2337/diab.43.3.500. [DOI] [PubMed] [Google Scholar]
- 18.Hamilton-Williams E.E., Palmer S.E., Charlton B., Slattery R.M. β Cell MHC class I is a late requirement for diabetes. Proc Natl Acad Sci U S A. 2003;100:6688–6693. doi: 10.1073/pnas.1131954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamanouchi J., Verdaguer J., Han B., Amrani A., Serra P., Santamaria P. Cross-priming of diabetogenic T cells dissociated from CTL-induced shedding of β cell autoantigens. J Immunol. 2003;171:6900–6909. doi: 10.4049/jimmunol.171.12.6900. [DOI] [PubMed] [Google Scholar]
- 20.Chong M.M., Chen Y., Darwiche R., Dudek N.L., Irawaty W., Santamaria P., Allison J., Kay T.W., Thomas H.E. Suppressor of cytokine signaling-1 overexpression protects pancreatic β cells from CD8+ T cell-mediated autoimmune destruction. J Immunol. 2004;172:5714–5721. doi: 10.4049/jimmunol.172.9.5714. [DOI] [PubMed] [Google Scholar]
- 21.Krishnamurthy B., Dudek N.L., McKenzie M.D., Purcell A.W., Brooks A.G., Gellert S., Colman P.G., Harrison L.C., Lew A.M., Thomas H.E., Kay T.W. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest. 2006;116:3258–3265. doi: 10.1172/JCI29602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verdaguer J., Yoon J.W., Anderson B., Averill N., Utsugi T., Park B.J., Santamaria P. Acceleration of spontaneous diabetes in TCR-β-transgenic nonobese diabetic mice by β-cell cytotoxic CD8+ T cells expressing identical endogenous TCR-α chains. J Immunol. 1996;157:4726–4735. [PubMed] [Google Scholar]
- 23.Heusel J.W., Wesselschmidt R.L., Shresta S., Russell J.H., Ley T.J. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell. 1994;76:977–987. doi: 10.1016/0092-8674(94)90376-x. [DOI] [PubMed] [Google Scholar]
- 24.Krishnamurthy B., Mariana L., Gellert S.A., Colman P.G., Harrison L.C., Lew A.M., Santamaria P., Thomas H.E., Kay T.W. Autoimmunity to both proinsulin and IGRP is required for diabetes in nonobese diabetic 8.3 TCR transgenic mice. J Immunol. 2008;180:4458–4464. doi: 10.4049/jimmunol.180.7.4458. [DOI] [PubMed] [Google Scholar]
- 25.Liu M., Shapiro M.E. A new method for isolation of murine islets with markedly improved yields. Transplant Proc. 1995;27:3208–3210. [PubMed] [Google Scholar]
- 26.Smyth M.J., Kelly J.M., Sutton V.R., Davis J.E., Browne K.A., Sayers T.J., Trapani J.A. Unlocking the secrets of cytotoxic granule proteins. J Leukoc Biol. 2001;70:18–29. [PubMed] [Google Scholar]
- 27.Schwab S.R., Cyster J.G. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8:1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 28.Dudek N.L., Thomas H.E., Mariana L., Sutherland R.M., Allison J., Estella E., Angstetra E., Trapani J.A., Santamaria P., Lew A.M., Kay T.W. Cytotoxic T-cells from T-cell receptor transgenic NOD8.3 mice destroy beta-cells via the perforin and Fas pathways. Diabetes. 2006;55:2412–2418. doi: 10.2337/db06-0109. [DOI] [PubMed] [Google Scholar]
- 29.Casey K.A., Mescher M.F. IL-21 promotes differentiation of naive CD8 T cells to a unique effector phenotype. J Immunol. 2007;178:7640–7648. doi: 10.4049/jimmunol.178.12.7640. [DOI] [PubMed] [Google Scholar]
- 30.Jenkins M.R., Mintern J., La Gruta N.L., Kedzierska K., Doherty P.C., Turner S.J. Cell cycle-related acquisition of cytotoxic mediators defines the progressive differentiation to effector status for virus-specific CD8+ T cells. J Immunol. 2008;181:3818–3822. doi: 10.4049/jimmunol.181.6.3818. [DOI] [PubMed] [Google Scholar]
- 31.Feuerer M., Shen Y., Littman D.R., Benoist C., Mathis D. How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity. 2009;31:654–664. doi: 10.1016/j.immuni.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wakim L.M., Waithman J., van Rooijen N., Heath W.R., Carbone F.R. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science. 2008;319:198–202. doi: 10.1126/science.1151869. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y.G., Choisy-Rossi C.M., Holl T.M., Chapman H.D., Besra G.S., Porcelli S.A., Shaffer D.J., Roopenian D., Wilson S.B., Serreze D.V. Activated NKT cells inhibit autoimmune diabetes through tolerogenic recruitment of dendritic cells to pancreatic lymph nodes. J Immunol. 2005;174:1196–1204. doi: 10.4049/jimmunol.174.3.1196. [DOI] [PubMed] [Google Scholar]
- 34.Thomas H.E., Parker J.L., Schreiber R.D., Kay T.W. IFN-γ action on pancreatic β cells causes class I MHC upregulation but not diabetes. J Clin Invest. 1998;102:1249–1257. doi: 10.1172/JCI2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pang S., Zhang L., Wang H., Yi Z., Li L., Gao L., Zhao J., Tisch R., Katz J.D., Wang B. CD8(+) T cells specific for β cells encounter their cognate antigens in the islets of NOD mice. Eur J Immunol. 2009;39:2716–2724. doi: 10.1002/eji.200939408. [DOI] [PubMed] [Google Scholar]
- 36.Lennon G.P., Bettini M., Burton A.R., Vincent E., Arnold P.Y., Santamaria P., Vignali D.A. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity. 2009;31:643–653. doi: 10.1016/j.immuni.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Tsai S, Shameli A, Yamanouchi J, Alkemade G, Santamaria P: In situ recognition of autoantigen as an essential gatekeeper in autoimmune CD8+ T cell inflammation. Proc Natl Acad Sci U S A 107:9317–9322 [DOI] [PMC free article] [PubMed]
- 38.Savinov A.Y., Wong F.S., Stonebraker A.C., Chervonsky A.V. Presentation of antigen by endothelial cells and chemoattraction are required for homing of insulin-specific CD8+ T cells. J Exp Med. 2003;197:643–656. doi: 10.1084/jem.20021378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agarwal P., Raghavan A., Nandiwada S.L., Curtsinger J.M., Bohjanen P.R., Mueller D.L., Mescher M.F. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol. 2009;183:1695–1704. doi: 10.4049/jimmunol.0900592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janas M.L., Groves P., Kienzle N., Kelso A. IL-2 regulates perforin and granzyme gene expression in CD8+ T cells independently of its effects on survival and proliferation. J Immunol. 2005;175:8003–8010. doi: 10.4049/jimmunol.175.12.8003. [DOI] [PubMed] [Google Scholar]
- 41.Li Q., Xu B., Michie S.A., Rubins K.H., Schreriber R.D., McDevitt H.O. Interferon-α initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2008;105:12439–12444. doi: 10.1073/pnas.0806439105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poligone B., Weaver D.J., Jr., Sen P., Baldwin A.S., Jr., Tisch R. Elevated NF-κB activation in nonobese diabetic mouse dendritic cells results in enhanced APC function. J Immunol. 2002;168:188–196. doi: 10.4049/jimmunol.168.1.188. [DOI] [PubMed] [Google Scholar]
- 43.Sen P., Bhattacharyya S., Wallet M., Wong C.P., Poligone B., Sen M., Baldwin A.S., Jr., Tisch R. NF-κ B hyperactivation has differential effects on the APC function of nonobese diabetic mouse macrophages. J Immunol. 2003;170:1770–1780. doi: 10.4049/jimmunol.170.4.1770. [DOI] [PubMed] [Google Scholar]
- 44.Weaver D.J., Jr., Poligone B., Bui T., Abdel-Motal U.M., Baldwin A.S., Jr., Tisch R. Dendritic cells from nonobese diabetic mice exhibit a defect in NF-κ B regulation due to a hyperactive I κ B kinase. J Immunol. 2001;167:1461–1468. doi: 10.4049/jimmunol.167.3.1461. [DOI] [PubMed] [Google Scholar]
- 45.King C., Ilic A., Koelsch K., Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–277. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 46.McGuire H.M., Vogelzang A., Hill N., Flodstrom-Tullberg M., Sprent J., King C. Loss of parity between IL-2 and IL-21 in the NOD Idd3 locus. Proc Natl Acad Sci U S A. 2009;106:19438–19443. doi: 10.1073/pnas.0903561106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brodie G.M., Wallberg M., Santamaria P., Wong F.S., Green E.A. B-cells promote intra-islet CD8+ cytotoxic T-cell survival to enhance type 1 diabetes. Diabetes. 2008;57:909–917. doi: 10.2337/db07-1256. [DOI] [PubMed] [Google Scholar]
- 48.Hanninen A., Jalkanen S., Salmi M., Toikkanen S., Nikolakaros G., Simell O. Macrophages, T cell receptor usage, and endothelial cell activation in the pancreas at the onset of insulin-dependent diabetes mellitus. J Clin Invest. 1992;90:1901–1910. doi: 10.1172/JCI116067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wheat W., Kupfer R., Gutches D.G., Rayat G.R., Beilke J., Scheinman R.I., Wegmann D.R. Increased NF-κ B activity in B cells and bone marrow-derived dendritic cells from NOD mice. Eur J Immunol. 2004;34:1395–1404. doi: 10.1002/eji.200324490. [DOI] [PubMed] [Google Scholar]
- 50.Tang Q., Adams J.Y., Penaranda C., Melli K., Piaggio E., Sgouroudis E., Piccirillo C.A., Salomon B.L., Bluestone J.A. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Penaranda C., Tang Q., Ruddle N.H., Bluestone J.A. Prevention of diabetes by FTY720-mediated stabilization of peri-islet tertiary lymphoid organs. Diabetes. 2010;59:1461–1468. doi: 10.2337/db09-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Astorri E., Bombardieri M., Gabba S., Peakman M., Pozzilli P., Pitzalis C. Evolution of ectopic lymphoid neogenesis and in situ autoantibody production in autoimmune nonobese diabetic mice: cellular and molecular characterization of tertiary lymphoid structures in pancreatic islets. J Immunol. 2010;185:3359–3368. doi: 10.4049/jimmunol.1001836. [DOI] [PubMed] [Google Scholar]
- 53.Foulis A.K., Farquharson M.A., Meager A. Immunoreactive α-interferon in insulin-secreting β cells in type 1 diabetes mellitus. Lancet. 1987;2:1423–1427. doi: 10.1016/s0140-6736(87)91128-7. [DOI] [PubMed] [Google Scholar]
- 54.Gaglia J.L., Guimaraes A.R., Harisinghani M., Turvey S.E., Jackson R., Benoist C., Mathis D., Weissleder R. Noninvasive imaging of pancreatic islet inflammation in type 1A diabetes patients. J Clin Invest. 2011;121:442–445. doi: 10.1172/JCI44339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang X., Yuang J., Goddard A., Foulis A., James R.F., Lernmark A., Pujol-Borrell R., Rabinovitch A., Somoza N., Stewart T.A. Interferon expression in the pancreases of patients with type I diabetes. Diabetes. 1995;44:658–664. doi: 10.2337/diab.44.6.658. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of GzmB and CD107a expression in diabetogenic CTLs after in vitro stimulation. A: Expression of GzmB in unstimulated and IGRP-stimulated CFSE+CD8+ T cells. The graphs show the MFI for GzmB expression in unstimulated (B) and stimulated (C) cells (n = 4). Div., division. D: Expression of CD107a on transferred 8.3− T cells unstimulated or stimulated in vitro with PMA (25 ng/mL) and ionomycin (5 μg/mL). The graphs show the percentage of CD8+tetramer+CD107a+ T cells in unstimulated (E) and stimulated (F) cells (n = 6). Data are given as mean ± SEM (error bars).