

SUMMARY
Combination therapy, in which two or more agents are applied, is more effective than single therapies for combating cancer. For this reason, combinations of chemotherapy with radiation are being explored in clinical trials, albeit with an empirical approach. We developed a screen to identify, from the onset, molecules that act in vivo in conjunction with radiation, using Drosophila as a model. Screens through two small molecule libraries from the NCI Developmental Therapeutics Program yielded microtubule poisons; this class of agents is known to enhance the effect of radiation in mammalian cancer models. Here we report an analysis of one microtubule depolymerizing agent, maytansinol isobutyrate (NSC292222; maytansinol), in Drosophila and in human cancer cells. We find that the effect of maytansinol is p53 dependent in Drosophila cells and human cancer cells, that maytansinol enhances the effect of radiation in both systems, and that the combinatorial effect of drug and radiation is additive. We also uncover a differential sensitivity to maytansinol between Drosophila cells and Drosophila larvae, which illustrates the value of studying cell behavior in the context of a whole organism. On the basis of these results, we propose that Drosophila might be a useful model for unbiased screens through new molecule libraries to find cancer drugs for combination therapy.
INTRODUCTION
No single mode of therapy presents a cure for cancer. Multi-modal therapy, in which more than one anti-cancer agent is applied in combination, has more promise and is being assessed in multiple clinical trials (www.clinicaltrials.gov). The precise efficacy and degree of tumor control exhibited by combination therapies, however, remains variable. Although the reasons for variability are unclear, discovery of additional novel drugs that synergize with an existing therapy, such as radiation, will allow multiple combinations to choose from, thereby increasing the likelihood of clinical success. How then can we identify drugs that work together with an existing agent to provide robust therapy against cancer? One solution would be to choose a suitable model and design screens that will allow co-discovery, from the onset, of anti-cancer agents that are effective in combination (as opposed to screening for an agent that has effect on its own and then testing whether it acts in combination with a second agent). Furthermore, a screening model that recapitulates the three-dimensional multicellular context of tumors might provide better predictive value than homogeneous single cell culture models.
We described previously a screen in Drosophila for chemical molecules that enhance the killing effect of ionizing radiation (IR) (Jaklevic et al., 2006). Briefly, third instar larvae were irradiated and cultured in food supplemented with different molecules. Survival to adulthood was quantified 10 days later. Molecules that produced a percent survival that was 2 standard deviations (s.d.) lower than the average survival for the cohort were considered potential hits and were further tested for reproducible effect. A pilot screen through a 1990-molecule library using Drosophila Chk1 (grapes) mutants yielded four molecules of interest (Jaklevic et al., 2006). Two of these are 20S-camptothecin and topotecan, a derivative of camptothecin. Camptothecin and its derivatives inhibit topoisomerase I. Topotecan is an FDA-approved anti-cancer agent that acts as a radiation sensitizer of mammalian tumors. These results serve as proof-of-concept that Drosophila can be a useful screening model for anti-cancer agents that can be used in combination therapy with a standard agent (i.e. radiation).
The pilot screen was performed on the Diversity Set library from the National Cancer Institute Developmental Therapeutics Program (NCI-DTP; http://dtp.nci.nih.gov/). Molecules in this library were chosen for structural diversity rather than biological activity, and the library contains both natural products and synthetic molecules. Of 1990 molecules in this library, approximately 2% are from natural products. Yet, all four hits in the pilot screen were of natural origin. This led us to explore the idea that natural products would be a good source of molecules that increase the effect of radiation. Therefore, we next screened through the Natural Products Set library from the NCI-DTP. This library consists of 235 molecules that were initially identified in extracts of plants or marine organisms. The active molecule in each extract was then identified, synthesized and supplied in a 96-well plate format at 10 mM concentration in DMSO by the DTP. The screen was done ‘blind’, i.e. without knowledge of the identity of molecules in the whole library or in each well of the coded plate.
The screen through the Natural Product Set (235 molecules) using Drosophila Chk1 and p53 mutants identified 13 molecules that reproducibly enhanced the effect of radiation (our unpublished data). These included three microtubule depolymerizing agents, a maytansine derivative (NSC292222), colchicine (NSC757) and vincristine (NSC67574). Colchicine was the drug we used in combination with radiation to design the Drosophila screen originally (Jaklevic et al., 2006). Its identification, therefore, was reassuring. Vincristine is an FDA-approved chemotherapy drug against several cancer types and is being tested in combination therapy with radiation in clinical trials (www.clinicaltrials.gov). It has also been used in experiments in Drosophila and was found to increase genetic instability in some assays (Clements et al., 1990; Tiburi et al., 2002). Maytansinol isobutyrate (NSC292222; hereafter referred to as maytansinol) was one of four hits in the pilot screen through the Diversity Set. Its repeat identification in the Natural Product screen attests to the robustness of the screening protocol. The effect of maytansinol and its derivatives in Drosophila has not been described before and forms the focus of this article.
Maytansine is an ansamycin antibiotic found in the Ethiopian shrub Maytenus (Kupchan et al., 1972). Maytansine inhibits microtubule assembly and induces microtubule disassembly in vitro, in cultured mammalian cells, and in embryos of sea urchins and clams (Ootsu et al., 1980; Remillard et al., 1975). Maytansine exhibits cytotoxicity against many tumor cell lines and inhibits tumor growth in vivo (e.g. Ootsu et al., 1980). Early clinical trials failed owing to toxicity at therapeutic levels. Currently there are multiple lines of effort to bypass toxicity by targeting cancer cells with maytansinoid-conjugated homing antibodies (e.g. Krop et al., 2010). Here we present a study of maytansinol in conjunction with IR in Drosophila larvae and human cell culture. The results support the idea that Drosophila larvae can be used to identify molecules that have efficacy in mammalian models of cancer. Furthermore, we report differences in the behavior of Drosophila cells and Drosophila larvae that illustrate the importance of studying cell behavior in a multicellular context in vivo.
RESULTS
We first confirmed that maytansinol depolymerizes microtubules in Drosophila. Wing imaginal discs were removed from third instar larvae, incubated in different concentrations of maytansinol in culture for 1 or 2 hours, fixed, and stained with an antibody to Tubulin. The discs were also stained with an antibody to phosphorylated Histone H3, a mitotic marker. Three lines of data indicate that maytansinol depolymerizes microtubules in Drosophila. First, mitotic spindles were visible in control discs but not in drug-treated discs (Fig. 1A–F, brackets). Second, different phases of mitosis were visible in control discs but not in drug-treated discs. Instead, mitotic cells in drug-treated discs displayed a prometaphase configuration (Fig. 1G,H). This was expected because microtubule depolymerization would trigger the spindle checkpoint and prevent mitotic exit. Third, mitotic index increased after exposure to maytansinol (Fig. 1I–K). Again, this is consistent with the activation of the spindle checkpoint. We conclude that maytansinol depolymerizes microtubules in Drosophila, as do its derivatives in vertebrate cells, an echinoderm and a mollusk (Ootsu et al., 1980; Remillard et al., 1975).
Fig. 1.

Maytansinol disrupts the mitotic spindle and prevents mitotic exit in Drosophila. (A–H) Wing imaginal discs from feeding-stage third instar larvae were incubated in PBS containing either DMSO (–M) or DMSO with 10 μM maytansinol (+M) for 1 hour, fixed and stained with antibodies to phosphorylated Histone H3 (red in A and B; also shown in C,D,G–J) and β-Tubulin (blue in A and B; also shown in E,F). (A–F) Mitotic spindles (brackets) are visible in control but not drug-treated discs. Scale bar: 6.3 μm. (G,H) Various phases of mitosis are visible in controls but not in drug-treated discs. pm, prometaphase; m, metaphase; a, anaphase; p, prophase. Scale bar: 10 μm. (I,J) Wing imaginal discs from feeding-stage third instar larvae were incubated in PBS containing either DMSO (–M) or DMSO with 2 μM maytansinol (+M) for 2 hours, fixed and stained with antibodies to phosphorylated Histone H3. The number of mitotic cells increased after incubation in maytansinol. (K) Quantification of the data for the experiment shown in I and J. ‘none’ represents discs treated with solvent (DMSO for maytansinol and 95% ethanol for colchicine; n=13 from three different experiments). ‘C’ represents discs incubated in 50 μg/ml colchicine for 2 hours (n=5). ‘M’ represents discs incubated with 2 μM maytansinol for 2 hours (n=8 from two different experiments). The differences between drug-treated discs and controls are statistically significant (Student’s t-test).
Maytansinol was identified in a screen for molecules that decreased the survival of irradiated larvae. We investigated whether it also decreased the survival of un-irradiated larvae. Third instar larvae were either irradiated or left un-irradiated (control) and cultured in food containing maytansinol at different concentrations. Survival to adulthood was quantified 10 days later by counting the percentage of pupal cases that were left empty as adult flies eclosed. We found that treatment with maytansinol alone (i.e. without radiation) decreased the survival of wild-type and p53 mutant larvae, but to different levels. The effect was greater for p53 mutants than for wild type at 0.5 and 1 μM maytansinol (Fig. 2). At 2 μM maytansinol, the effect was still greater for p53 mutants than for wild type on average but, when compiled over a number of experiments, was found to not be significant.
Fig. 2.
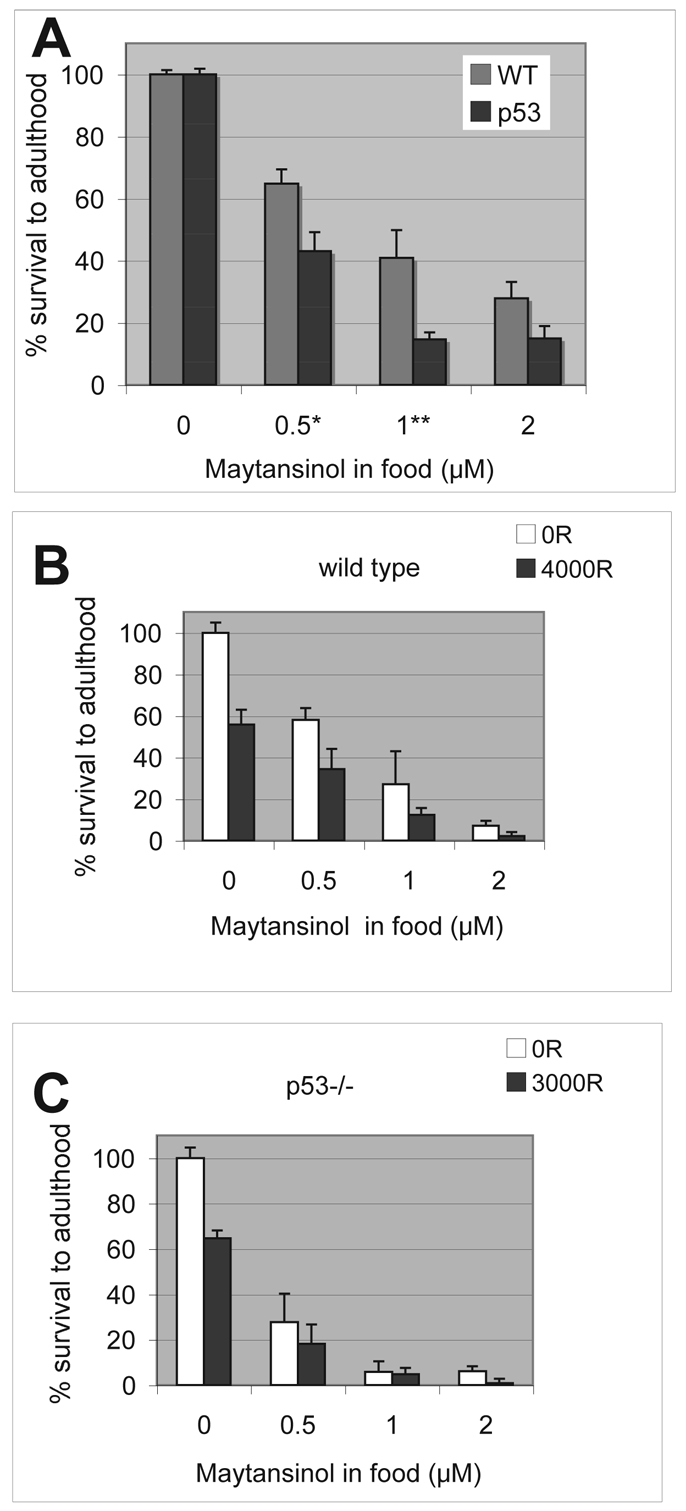
Sensitivity of Drosophila larvae to maytansinol and IR. Feeding-stage third instar larvae were placed in new food containing different concentrations of maytansinol. Survival was calculated as the fraction of pupae that eclosed as adult (empty pupa cases/total) and normalized to survival of solvent-treated controls for each respective genotype. Error bars extend beyond fractional survival of 1.0 for solvent controls (0 μM drug) simply because of normalization. WT, wild type; p53, p53 homozygous mutants. The data shown are averages from over 2500 pupae per genotype examined in four (wild type) or five (p53) different experiments. Error bar = 1 s.e.m. *P<0.05; **P<0.01 in Student’s t-test. (B,C) Feeding-stage third instar larvae were irradiated with doses of X-ray as shown and placed in new food containing different concentrations of maytansinol. The dose of X-ray was chosen to achieve approximately 50% killing for each genotype. Most, if not all, irradiated larvae formed pupae in these experiments. Survival was calculated as the fraction of pupae that eclose as adults from the pupae case. The data shown are from one set of experiments in which data from five biological replicates were averaged per genotype per treatment. These data sets were two of several used to compute averages show in A, which are higher than in these particular experiments but still within one s.d. Error bar = 1 s.d. Error bars extend beyond fractional survival of 1.0 for solvent controls (no drug) simply because of normalization. 100 R = 1 Gy.
Next we examined the combined effect of drug and radiation (Fig. 2B,C). The dose of X-rays that results in approximately 50% lethality for each genotype was used in these experiments. We found that the effect of drug and radiation was mostly additive. For example, for wild-type larvae, X-rays alone produced 55.8±7.2% survival and 0.5 nM of maytansinol alone produced 58.1±5.7% survival. The additive effect of drug and radiation was expected to produce 32.4±5.3% survival (55.8% of 58.1%; see Methods for computing the s.d. of a product). The observed survival under these conditions was 34.5±9.7%, which is not significantly different from the expected. Expected survival for additive action between drug and radiation is calculated for each dose of drug and shown in Table 1. For most conditions, the expected survival ± 1 s.d. and observed survival ± 1 s.d. overlap.
Table 1.
Expected and observed survival for Drosophila wild-type and p53 mutant larvae after exposure to a combination of maytansinol and IR
| Genotype | Drug dose (μ M) | Expected survival (%) | S.d. | Observed survival (%) | S.d. |
|---|---|---|---|---|---|
| Wild type | 0.5 | 32.4 | 5.3 | 34.5 | 9.7 |
| 1 | 15.2 | 9.1 | 12.4 | 3.3 | |
| 2 | 4.0 | 1.4 | 2.3 | 1.8 | |
| p53 mutant | 0.5 | 17.9 | 8.2 | 18.1 | 8.6 |
| 1 | 3.8 | 3.0 | 4.7 | 2.8 | |
| 2 | 3.9 | 1.4 | 0.8 | 1.9 |
The values for expected and observed survival are shown for three different doses of drug. Expected numbers are computed for the scenario in which drug and radiation act in an additive manner.
We noted experiment-to-experiment variability in survival after drug treatment (for example, compare the numbers for wild type in panels A and B of Fig. 2). But two trends were reproducible over multiple experiments: (1) sensitivity to maytansinol was similar or greater for p53 mutants than wild type, and (2) the combined effect of drug and radiation was mostly additive.
In humans, radiation exposure alters blood cell counts. The exact effect depends on cell type and radiation dose [pages 335–337 of Hall and Giaccia (Hall and Giaccia, 2006)]. In general, lymphocytes are highly radiation sensitive and are significantly depleted within a day after whole body radiation. Granulocytes, by contrast, increase in number initially, owing to mobilization of reserves, before declining in number. Red blood cells are the most resistant of all blood cell types and their counts remain unchanged for several days before declining. All cell types can recover but the time course and the extent of recovery depends on the radiation dose. Macrophages, which are derived from the same progenitor stem cells as lymphocytes, are more radiation resistant than lymphocytes and show little decline after radiation exposure in mammalian systems (Perkins et al., 1966). Hemocytes are circulating cells of invertebrates that have overlapping functions with macrophages in that both phagocytose damaged cells and pathogens. Hemocytes have been shown to behave more like macrophages than like lymphocytes in terms of radiation resistance; total hemocyte counts in insect and mollusk show little change after exposure to IR (Bezerra et al., 2003; Christensen et al., 1990). Drosophila S2 cells, which are derived from hemocytes, are also highly radiation resistant; we have seen no significant cell killing at doses up to 40 Gy (Jaklevic et al., 2006). Nonetheless, we addressed the possibility that changes in hemocyte count occur after exposure to radiation and maytansinol, and that these changes can explain the differential sensitivity of wild-type and p53 larvae. We examined total hemocyte counts in larvae at 4 and 24 hours after exposure to drug and radiation. We found no significant changes in either genotype in any of the treatments (supplementary material Fig. S1). Importantly, we did not see a correlation between hemocyte counts and survival into adulthood, suggesting that declining hemocyte numbers are not responsible for reduced survival (see Fig. 2). For example, drug treatment alone of wild type or p53 mutants resulted in no significant decline in hemocytes at either 4 or 24 hours after exposure (compare all –IR samples), even though survival was significantly reduced under these conditions. We conclude that differences in sensitivity to radiation and maytansinol between wild-type larvae and p53 mutants cannot be attributed to hemocyte numbers at these time points after treatment.
Although maytansine and its derivatives have been studied in several human cancer cell lines, the effect of p53 status has not been directly tested. Our findings in Drosophila suggest that p53 mutants might be more sensitive to maytansinol than wild type, at least at some drug concentrations. To investigate whether this is also true in mammalian cells, we used HCT116 colon cancer cells, which are available as isogenic lines with or without functional p53 (Bunz et al., 1998). We found that maytansinol reduced the growth and/or survival of HCT116 cells in a dose-dependent manner and that the effect was more severe for p53+/+ than for p53−/− cells at both low and high doses (Fig. 3). At intermediate doses, the effect was not significantly different on wild-type and p53 mutant cells. Although we do not know the basis for dose dependence, it is reproducible; the data shown depict the average of four independent experiments.
Fig. 3.

Maytansinol inhibits the growth of HCT116 human colon cancer cells. Isogenic p53 wild-type and mutant HCT116 cells were seeded in 96-well plates, allowed to grow for 24 hours and treated with drug at the final concentrations shown. Viable cells were quantified 6 days later using the MTT assay (see Methods). The difference between wild type and mutant was statistically significant at the concentrations indicated with an * (P<0.05 in Student’s t-test). Error bars = s.d. The data are from four different experiments.
The effect of p53 status on maytansinol sensitivity in HCT116 cells is different from what was found in Drosophila larvae, in which p53 mutants were more sensitive to maytansinol than wild type at least at some concentrations. To address the basis for this difference, we investigated whether Drosophila p53 mutant cells show different sensitivity to killing by maytansinol than do wild-type cells. Third instar larvae were transferred to food containing maytansinol or DMSO carrier (control). Imaginal discs were extirpated 1 day after the transfer and stained with Acridine Orange (AO), a vital dye that is excluded from live cells. In Drosophila, AO is specific for apoptotic cells and does not stain necrotic cells (Abrams et al., 1993). We found that maytansinol induces apoptosis in imaginal discs of wild-type larvae but not p53 mutant larvae (Fig. 4). This parallels the finding in human HCT116 cells, in which maytansinol was more effective when p53 was present, at least at some doses (Fig. 3).
Fig. 4.

Maytansinol induces cell death in wild-type but not p53 mutant Drosophila larvae. Feeding-stage third instar larvae were placed in food with maytansinol (+M) or DMSO (control; –M) for 24–26 hours. Wing imaginal discs were extirpated and stained with AO to visualize dead cells. Duplicate discs are shown to illustrate reproducibility. Nine drug-treated wild-type, 13 drug-treated p53 mutant and five DMSO-treated wild-type control wing imaginal discs were examined in two different experiments at 1 or 2 μM maytansinol, with similar results. Scale bar: 100 μm.
HCT116 cells also allowed us to investigate whether p53 status influenced the combinatorial effects of maytansinol with IR in human cells. The combined effects of two treatments were quantified in terms of Combination Index (CI; see Methods for the formula used) (Chou and Talalay, 1983). CI values of 1, >1 and <1 signify additive, antagonistic and synergistic effects, respectively, between two drugs. We found that adding 5 and 10 Gy of IR to drug treatment at concentrations shown in Fig. 3 produced CI values that are close to or above 1 for both p53+/+ and p53−/− isogenic HCT116 cells (Table 2), reflecting additive or antagonistic interaction between drug and radiation.
Table 2.
CI values for the combination of maytansinol and IR in HCT116 cells
| Genotype | Irradiation (Gy) | nM maytansinol | |||||
|---|---|---|---|---|---|---|---|
| 0.13 | 0.18 | 0.26 | 0.37 | 0.53 | 0.75 | ||
| p53+/+ | 5 | 1.934 | 0.78 | 0.912 | 1.373 | 1.446 | 1.479 |
| 10 | 1.481 | 1.432 | 1.045 | 1.359 | 1.761 | 2.044 | |
| p53−/− | 5 | 6.796 | 0.887 | 0.902 | 1.244 | 1.283 | 1.817 |
| 10 | 1.569 | 1.191 | 1.086 | 1.607 | 1.985 | 2.076 | |
The data for 5 and 10 Gy of radiation are shown. CI values are not measured values but are computed from measured values. The numbers reflect the extent of antagonism or synergy between two treatments and can be very large (severe antagonism), very small (strong synergy) or close to 1 (additive effect). 100 R = 1 Gy.
To investigate whether the combined effect of maytansinol and IR seen in HCT116 cells also applies to other cell lines, we measured CI values for this combination in eight head and neck cancer (HNC) and three non-small-cell lung cancer (NSCLC) cell lines. We found that, overall, the drug and IR combination acts in an additive to slightly antagonistic manner. The average CI values for three doses of IR and >100-fold change in drug concentration was 1.31±0.48. Cell lines included those with wild-type and mutant p53 (raw data, including Fa, fraction killed, are in supplementary material Table S1 and p53 status is in supplementary material Table S2). In these experiments, drug and IR were added on the same day (‘co-treatment’). To investigate whether an alternate schedule would change the interaction between drug and radiation, we administered maytansinol 24 hours prior to irradiation (‘pre-treatment’). The average CI value from this schedule was 1.35±0.63 [raw data, including Fa, are in supplementary material Table S3). Thus, although the average remained the same, the s.d. increased. This reflects the fact that, with pre-treatment, CI values decreased for some cell lines under some conditions but increased for others (supplementary material Table S3).
DISCUSSION
Irradiation of Drosophila larvae induces cell death in the diploid cells of imaginal discs; these cells are the precursors of adult organs. Successful production of an adult fly from a larva requires proper growth and differentiation of diploid imaginal discs. Following irradiation and induced cell death, remaining cells undergo compensatory proliferation to re-generate imaginal discs (Jaklevic and Su, 2004). Chemical molecules that interfere with compensatory proliferation and regeneration are expected to reduce the fraction of irradiated larva that survives to reach adulthood (Jaklevic et al., 2006). Microtubule depolymerizing agents such as colchicine, vincristine and maytansine would disrupt mitotic progression and thus interfere with compensatory cell proliferation. We speculate that this is the basis for their ability to act in combination with radiation to kill Drosophila larvae. This would explain why two independent screens we have performed for chemical molecules that enhance the killing effect of radiation in Drosophila yielded three microtubule poisons from a total of 2225 molecules (Jaklevic et al., 2006) (and this article). We acknowledge, however, that we cannot rule out the possibility that it is some other activity of this drug, rather than microtubule depolymerization, that increases the radiation sensitivity.
Drosophila larvae and human cancer cells behave similarly in the following ways: (1) maytansinol is more effective at killing p53+/+ than p53−/− cells in Drosophila imaginal discs (Fig. 4) and is more effective on p53+/+ than p53−/− human HCT116 cancer cells at certain concentrations (Fig. 3). (2) In both Drosophila larvae and human cancer cells, maytansinol generally increased the effect of radiation. The extent of this increase varied, however, reflecting a mostly additive interaction between drug and radiation in Drosophila larvae, and additive-to-antagonistic interactions in various cancer cell lines.
In addition to these similarities, Drosophila offers another dimension of experimental analysis, namely the ability to compare cells and multicellular tissues and/or organisms. For example, Drosophila p53 mutant cells, like human p53 mutant HCT116 cells, were more resistant to maytansinol. In the context of a whole organism, the situation seems to be reversed; p53-deficient larvae are more sensitive to maytansinol than were p53 wild-type larvae. This provides a clear example of how cells and multicellular organs or organisms can behave differently and illustrates the value of studying drug effects in vivo in metazoan models.
IR induces apoptosis in imaginal discs of wild-type larvae but not p53 mutant larvae at 4–6 hours after exposure to radiation. At longer times after irradiation, however, p53-independent apoptosis occurs (Wichmann et al., 2006). Maytansinol induced apoptosis in imaginal discs of wild-type larvae but not p53 mutant larvae at 24 hours after exposure to drug. It remains possible that maytansinol also induces p53-independent apoptosis at longer times after drug exposure. Although the difference in apoptosis induction between wild type and p53 mutants is seen at both 1 and 2 μM drug, the difference in eclosion is more pronounced and reproducible at 1μM drug. We do not know the basis for this but note that, although apoptosis is assayed 24 hours after drug exposure, eclosion is assayed 10 days after drug exposure. It is possible that longer-term effects of the drug, such as p53-independent apoptosis, contribute to these findings.
There is precedence for differential sensitivity between cells and organisms. Drosophila p53-deficient cells were more resistant than cells with wild-type p53 to killing by IR. Under the same conditions, Drosophila p53 mutant larvae are more sensitive to IR than larvae with wild-type p53 (Wichmann et al., 2006). But what is the basis for this difference? In both Drosophila and in mice, cells undergoing apoptosis signal to neighboring survivors and induce the latter to proliferate (Li et al., 2010; Ryoo et al., 2004). This process is important for the generation of new cells to replace those lost to apoptosis, for wound healing and for regeneration. In Drosophila, dying cells signal to neighbors through the Wnt (wingless) and JNK pathways (Ryoo et al., 2004). In mice, this phenomenon requires caspases 3 and 7, and operates through prostaglandin E(2), a promoter of stem or progenitor cell proliferation and tissue regeneration (Li et al., 2010). On the basis of these previous findings, we speculate that, in the presence of p53, the most severely damaged cells die by a p53-dependent mechanism but are replaced by healthier survivors that have been induced to proliferate. This allows for regeneration of damaged tissue and survival of the organism. In the absence of p53, damaged cells accumulate but are not replaced. Indeed, irradiated Drosophila p53 mutants acquire elevated genetic instability in loss-of-heterozygosity assays compared with wild type (Sogame et al., 2003). The resulting genetic instability might be too high to be compatible with organism survival, even though cells survive. This could explain the differences we see between larval survival and cell survival in p53 mutants.
Drosophila p53 displays a subset of activities attributed to mammalian p53. Mammalian p53 acts in cell cycle checkpoint control as well as in the induction of apoptosis in response to genotoxin exposure. Drosophila p53 has a proapoptotic function but is not needed for cell cycle checkpoints, at least after IR exposure (Brodsky et al., 2000; Jin et al., 2000; Ollmann et al., 2000; Sogame et al., 2003). Instead, Drosophila p53 is needed for compensatory proliferation after experimental induction of cell death (Wells et al., 2006). Thus, p53 mutants might be doubly disadvantaged after exposure to maytansinol. Not only are proliferative signals from dying cells reduced (because cells fail to undergo p53-dependent apoptosis), but surviving cells additionally have a reduced ability to undergo compensatory proliferation.
We note that the range of drug concentrations used in Drosophila and human cells are different. The experiments in HCT116 cells measure cumulative survival over 6 or 7 days; this protocol is typically used to measure the effect of IR and combination therapies. The range of maytansinol used in Drosophila (1–10 μM) obliterates human cancer cells completely, regardless of genotype (A.E. and T.T.S., unpublished data). Experiments in Drosophila imaginal discs were set up to measure the effect of the drug on mitosis within a cell cycle, and apoptosis shortly after. We also wanted to use the same drug concentrations used on larvae in these experiments so that we could directly compare cellular responses and organismal responses in the same system. Low (<1 nM) concentrations of drug that are effective on human cells do not affect larval survival regardless of genotype. In short, we used drug concentrations that show some effect but still allow survival in each system so that we can then assay for the effect of mutations or radiation on top of the drug effect.
In conclusion, these results support the idea that Drosophila can be used to identify anti-cancer drugs. Furthermore, Drosophila offers an opportunity to compare the effect of small molecules on cells and on multicellular structures. We report here the results of a screen to identify small molecules that increase the effect of radiation. We are adapting the screen to identify small molecules that increase the effect of other standard therapies. Drosophila models of neurodegeneration are currently being used for drug identification or testing, both in academic laboratories and in industry (e.g. Bortvedt et al., 2010; Rana et al., 2010; Sarantseva et al., 2009) (www.vitruvean.com). We hope that Drosophila models of human cancers will likewise become useful for identifying new anti-cancer drugs in the future. Expression of the Drosophila RET receptor tyrosine kinase, which has been mutated to mimic human oncogenic mutants, causes defects in the adult eye. Importantly, these defects can be suppressed by a known chemical inhibitor of human RET (Vidal et al., 2005). Such results support the idea that small molecule inhibitors found in Drosophila models have the potential to apply to human cancers.
METHODS
Fly stocks
Wild-type flies were of the Sevelin stock. p535A-1-4 results from targeted deletion of the gene (Rong et al., 2002).
Irradiation
Feeding-stage third instar larvae were irradiated as previously described (Jaklevic et al., 2006). Briefly, 120-hour-old larvae were rinsed to remove food and passed through sizing sieves to obtain animals of uniform size. Larvae were placed in a Petri dish and irradiated using a TORREX X-ray generator, set at 115 kV and 5 mA (producing 2.4 Rads/second). Irradiated larvae were then cultured on cornmeal-agar media (Jaklevic et al., 2006) containing drug or DMSO carrier. Human cells in 96-well plates were irradiated with a RS2000 Biological Irradiator (Rad Source Technologies) delivering 1 Gy/minute.
AO staining
Larvae were dissected in PBS. Imaginal discs were incubated for 5 minutes in PBS + 0.5 mM AO (Sigma) at room temperature, washed once with PBS, mounted in PBS, and imaged immediately using a Leica DMR fluorescence compound microscope, a Sensicam CCD camera and Slidebook software (Intelligent Imaging). Images were compiled using Photoshop software.
Antibody staining
To detect phosphorylated Histone H3, larval imaginal discs were extirpated in PBS, fixed for 10 minutes in PBT (PBS with 0.2% Tween) containing 10% formaldehyde and washed three times with PBT. Samples were incubated with primary antibodies in blocking solution, which is PBT + 3% normal goat serum, for 2 hours at room temperature or overnight at 4°C. Primary antibodies were rabbit polyclonal anti-phospho-Histone-H3 antibody (Upstate Biotechnology) diluted at 1:1000 and mouse monoclonal anti-β-Tubulin antibody (Developmental Hybridoma Bank) diluted at 1:100. Samples were then washed three times with PBT and incubated for 2–4 hours at room temperature with secondary antibody conjugated to rhodamine or fluorescein, diluted to 1:500 in blocking solution (Jackson ImmunoResearch). Samples were washed three times with PBT, stained with 10 μg/ml Hoechst 33258 in PBT for 2 minutes, and washed three times with PBT before mounting onto slides with Fluoromount G. Samples were imaged on a Leica DMR fluorescence microscope using a Sensicam CCD camera and Slidebook software (Intelligent Imaging). Images at different focal planes were combined using ImageJ (http://rsb.info.nih.gov/ij/), displayed using Photoshop software and the number of mitotic cells was counted manually.
Cell lines
The HNC and NSCLC cell lines were kindly provided by David Raben and Paul Bunn (University of Colorado Cancer Center, CO). These cell lines were maintained in RPMI media supplemented with 10% heat-inactivated fetal bovine serum (FBS; Hyclone, Logan, UT) in a humidified incubator with 5% CO2. HCT116 cells were kindly provided by Joaquin Espinosa (University of Colorado, Boulder, CO) and grown in McCoy’s medium with 10% FBS.
Cell growth assay
The growth inhibitory effects of maytansinol with radiation were evaluated using a modified tetrazolium salt (MTT) assay (Carmichael et al., 1988). In the MTT assay, 1000–2000 viable cells were plated in 100 μl of growth medium in 96-well plates (Corning, Ithaca, NY). Following an overnight incubation, maytansinol was added at varying concentrations and the plates were irradiated on the same day (co-treatment) or 24 hours later (pre-treatment) and incubated for 6–7 days. The tetrazolium salt was added at a concentration of 0.4 mg/ml to each well following the 6- to 7-day treatment. The plates were incubated with the salt for 4 hours at 37°C. At 4 hours, the medium was aspirated off, leaving the dark blue formazan product at the bottom of the wells. The reduced MTT product was solubilized by adding 100 μl of 0.2 N HCl in 75% isopropanol, 23% MilliQ water to each well. Thorough mixing was done using a Titertek multichannel pipetman. The absorbency of each well was measured using an automated plate reader (Molecular Devices, Sunnyvale, CA). All experiments were performed in triplicate.
Hemocyte counting
Hemolymph was collected by opening two larvae lengthwise in 20μl of PBS containing 10 μg/ml Hoechst 33342 (Molecular Probes), on a siliconized glass slide, using #5 forceps (Ted Pella). Larval carcasses were removed and hemolymph in PBS was incubated for 2 minutes in a humidified chamber to allow cell-permeable DNA dye to stain nuclei. 10 μl of stained cells in PBS was placed onto a hemocytometer and cells that were identified by UV illumination of Hoechst 33342 were counted. We used two larvae per sample to ensure that we had at least 10 μl of material left for counting after mixing and incubation; dissecting one larva in 10 μl of PBS resulted in less than 10 μl of recoverable material. ‘+IR’ was 4000 R for wild type and 3000 R for p53 for reasons explained in the Results.
We found that hemocytes are typically counted in one of two ways in published reports; total hemocytes per larva or hemocytes per unit volume of hemolymph. We used the former approach because of technical ease, but found that our numbers are in line with published counts from the latter approach. For example, we counted 725–5625 hemocytes per wild-type larva control (no IR, no drug, Sevelin strain). The amount of hemolymph that can be extracted from an individual Drosophila third instar larva ranges from 50 to 300 nl (Piyankarage et al., 2008). If we assume that our lower counts reflect a smaller volume of hemolymph extracted, our counts translate to 725/50 nl or 14.5×103 hemocytes per μl. At the high end, 5625/300 nl translate to 18.8×103 hemocytes per μl. The published values are similar [e.g. 7.8×103/μl for Canton S (Tokusumi et al., 2009) and ∼5×103/μl for a GAL4 control (Asha et al., 2003)]. The average hemocyte count per wild-type larva in our studies was ∼2500. This is in line with an average of ∼4000/animal measured for another wild-type strain, Oregon R (Sinenko et al., 2004).
Combination index
CI values are calculated according to the formula: CI=(D)1/(Dx)1 + (D)2/(Dx)2 (Chou and Talalay, 1983). (Dx)1 and (Dx)2 are the doses required to achieve a given effect level for each treatment, i.e. a specified value of fraction affected, or Fa. (D)1 and (D)2 are the doses of each treatment in a given combination which gives the same Fa.
Standard deviation
S.d. for expected fraction survival for co-treatment of drug and radiation is computed according to the formula for the s.d. of a product of two normally distributed variables. For two normally distributed variables with means m1 and m2 and s.d. s1 and s2, respectively, the product will have mean m1m2 and the s.d.=square root of (m12s22 + m22s12 + s12s22) [page 140 of Menzel (Menzel, 1960)]. Fractional survival after genotoxin treatment shows a normal (Gaussian) distribution around a mean [figure 4 of Jaklevic et al. (Jaklevic et al., 2006) and our unpublished observations].
Supplementary Material
Acknowledgments
The authors thank the laboratories of Joaquin Espinosa and Paul Bunn for the cell lines, NCI Developmental Therapeutics Program for small molecule libraries, and the Bloomington Stock Center for fly stocks. This work was funded by a University of Colorado Cancer Center summer fellowship and an HHMI undergraduate research fellowship to A.E., a post-doctoral fellowship from the Cancer League of Colorado to M.G., and grants from the NIH ( GM73973, GM87276 and DE017494) to T.T.S.
Footnotes
COMPETING INTERESTS
T.T.S. is a co-founder, D.R. is a member of the scientific advisory board and B.F. is a consultant for Suvica. The University of Colorado has an issued patent for the Drosophila screen described here (US Patent #7,695,899).
AUTHOR CONTRIBUTIONS
T.T.S., B.F. and D.R. conceived and designed the experiments. A.E., M.G. and P.Y. performed the experiments. A.E., M.G., P.Y. and T.T.S. analyzed the data. D.R. contributed reagents and materials. M.G. and T.T.S. wrote the paper.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.006486/-/DC1
REFERENCES
- Abrams J. M., White K., Fessler L. I., Steller H. (1993). Programmed cell death during Drosophila embryogenesis. Development 117, 29–43 [DOI] [PubMed] [Google Scholar]
- Asha H., Nagy I., Kovacs G., Stetson D., Ando I., Dearolf C. R. (2003). Analysis of Ras-induced overproliferation in Drosophila hemocytes. Genetics 163, 203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezerra F. S., Nogueira-Machado J. A., Martins-Souza R. L., Chaves M. M., Correa R. F., Coelho P. M. (2003). Effect of gamma radiation on the activity of hemocytes and on the course of Schistosoma mansoni infection in resistant Biomphalaria tenagophila snails. Mem. Inst. Oswaldo Cruz 98, 73–75 [DOI] [PubMed] [Google Scholar]
- Bortvedt S. F., McLear J. A., Messer A., Ahern-Rindell A. J., Wolfgang W. J. (2010). Cystamine and intrabody co-treatment confers additional benefits in a fly model of Huntington’s disease. Neurobiol. Dis. 40, 130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky M. H., Nordstrom W., Tsang G., Kwan E., Rubin G. M., Abrams J. M. (2000). Drosophila p53 binds a damage response element at the reaper locus. Cell 101, 103–113 [DOI] [PubMed] [Google Scholar]
- Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J. P., Sedivy J. M., Kinzler K. W., Vogelstein B. (1998). Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501 [DOI] [PubMed] [Google Scholar]
- Carmichael J., Mitchell J. B., DeGraff W. G., Gamson J., Gazdar A. F., Johnson B. E., Glatstein E., Minna J. D. (1988). Chemosensitivity testing of human lung cancer cell lines using the MTT assay. Br. J. Cancer 57, 540–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T. C., Talalay P. (1983). Analysis of combined drug effects: a new look at a very old problem. Trends Pharmacol. Sci. 4, 450–454 [Google Scholar]
- Christensen B. M., Huff B. M., Li J. (1990). Effect of gamma irradiation on the hemocyte-mediated immune response of Aedes aegypti against microfilariae. J. Invertebr. Pathol. 56, 123–127 [DOI] [PubMed] [Google Scholar]
- Clements J., Howe D., Lowry A., Phillips M. (1990). The effects of a range of anti-cancer drugs in the white-ivory somatic mutation test in Drosophila. Mutat. Res. 228, 171–176 [DOI] [PubMed] [Google Scholar]
- Hall E., Giaccia A. J. (2006). Radiobiology for the Radiologist. Philadelphia, PA: Lippincott Williams & Wilkins [Google Scholar]
- Jaklevic B., Uyetake L., Lemstra W., Chang J., Leary W., Edwards A., Vidwans S., Sibon O., Tin Su T. (2006). Contribution of growth and cell cycle checkpoints to radiation survival in Drosophila. Genetics 174, 1963–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaklevic B. R., Su T. T. (2004). Relative contribution of DNA repair, cell cycle checkpoints, and cell death to survival after DNA damage in Drosophila larvae. Curr. Biol. 14, 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S., Martinek S., Joo W. S., Wortman J. R., Mirkovic N., Sali A., Yandell M. D., Pavletich N. P., Young M. W., Levine A. J. (2000). Identification and characterization of a p53 homologue in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 97, 7301–7306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krop I. E., Beeram M., Modi S., Jones S. F., Holden S. N., Yu W., Girish S., Tibbitts J., Yi J. H., Sliwkowski M. X., et al. (2010). Phase I study of trastuzumab-DM1, a HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J. Clin. Oncol. 28, 2698–2704 [DOI] [PubMed] [Google Scholar]
- Kupchan S. M., Komoda Y., Court W. A., Thomas G. J., Smith R. M., Karim A., Gilmore C. J., Haltiwanger R. C., Bryan R. F. (1972). Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J. Am. Chem. Soc. 94, 1354–1356 [DOI] [PubMed] [Google Scholar]
- Li F., Huang Q., Chen J., Peng Y., Roop D. R., Bedford J. S., Li C. Y. (2010). Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 3, ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel D. H. (1960). Fundamental Formulas for Physics. Nineola, NY: Dover Publications, Inc [Google Scholar]
- Ollmann M., Young L. M., Di Como C. J., Karim F., Belvin M., Robertson S., Whittaker K., Demsky M., Fisher W. W., Buchman A., et al. (2000). Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 101, 91–101 [DOI] [PubMed] [Google Scholar]
- Ootsu K., Kozai Y., Takeuchi M., Ikeyama S., Igarashi K., Tsukamoto K., Sugino Y., Tashira T., Tsukagoshi S., Sakurai Y. (1980). Effects of new antimitotic antibiotics, ansamitocins, on the growth of murine tumors in vivo and on the assembly of microtubules in vitro. Cancer Res. 40, 1707–1717 [PubMed] [Google Scholar]
- Perkins E. H., Nettesheim P., Morita T. (1966). Radioresistance of the engulfing and degradative capacities of peritoneal phagocytes to kiloroentgen x-ray doses. J. Reticuloendothel. Soc. 3, 71–82 [PubMed] [Google Scholar]
- Piyankarage S. C., Augustin H., Grosjean Y., Featherstone D. E., Shippy S. A. (2008). Hemolymph amino acid analysis of individual Drosophila larvae. Anal. Chem. 80, 1201–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A., Seinen E., Siudeja K., Muntendam R., Srinivasan B., van der Want J. J., Hayflick S., Reijngoud D. J., Kayser O., Sibon O. C. (2010). Pantethine rescues a Drosophila model for pantothenate kinase-associated neurodegeneration. Proc. Natl. Acad. Sci. USA 107, 6988–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remillard S., Rebhun L. I., Howie G. A., Kupchan S. M. (1975). Antimitotic activity of the potent tumor inhibitor maytansine. Science 189, 1002–1005 [DOI] [PubMed] [Google Scholar]
- Rong Y. S., Titen S. W., Xie H. B., Golic M. M., Bastiani M., Bandyopadhyay P., Olivera B. M., Brodsky M., Rubin G. M., Golic K. G. (2002). Targeted mutagenesis by homologous recombination in D. melanogaster. Genes Dev. 16, 1568–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo H. D., Gorenc T., Steller H. (2004). Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell 7, 491–501 [DOI] [PubMed] [Google Scholar]
- Sarantseva S., Timoshenko S., Bolshakova O., Karaseva E., Rodin D., Schwarzman A. L., Vitek M. P. (2009). Apolipoprotein E-mimetics inhibit neurodegeneration and restore cognitive functions in a transgenic Drosophila model of Alzheimer’s disease. PLoS ONE 4, e8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinenko S. A., Kim E. K., Wynn R., Manfruelli P., Ando I., Wharton K. A., Perrimon N., MatheyPrevot B. (2004). Yantar, a conserved arginine-rich protein is involved in Drosophila hemocyte development. Dev. Biol. 273, 48–62 [DOI] [PubMed] [Google Scholar]
- Sogame N., Kim M., Abrams J. M. (2003). Drosophila p53 preserves genomic stability by regulating cell death. Proc. Natl. Acad. Sci. USA 100, 4696–4701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiburi M., Reguly M. L., Schwartsmann G., Cunha K. S., Lehmann M., Rodrigues de Andrade H. H. (2002). Comparative genotoxic effect of vincristine, vinblastine, and vinorelbine in somatic cells of Drosophila melanogaster. Mutat. Res. 519, 141–149 [DOI] [PubMed] [Google Scholar]
- Tokusumi T., Shoue D. A., Tokusumi Y., Stoller J. R., Schulz R. A. (2009). New hemocyte-specific enhancer-reporter transgenes for the analysis of hematopoiesis in Drosophila. Genesis 47, 771–774 [DOI] [PubMed] [Google Scholar]
- Vidal M., Wells S., Ryan A., Cagan R. (2005). ZD6474 suppresses oncogenic RET isoforms in a Drosophila model for type 2 multiple endocrine neoplasia syndromes and papillary thyroid carcinoma. Cancer Res. 65, 3538–3541 [DOI] [PubMed] [Google Scholar]
- Wells B. S., Yoshida E., Johnston L. A. (2006). Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr. Biol. 16, 1606–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann A., Jaklevic B., Su T. T. (2006). Ionizing radiation induces caspase-dependent but Chk2- and p53-independent cell death in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 103, 9952–9957 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.