

SUMMARY
The human fibroblast growth factor receptor 3 (FGFR3) gene is frequently mutated in superficial urothelial cell carcinoma (UCC). To test the functional significance of FGFR3 activating mutations as a ‘driver’ of UCC, we targeted the expression of mutated Fgfr3 to the murine urothelium using Cre-loxP recombination driven by the uroplakin II promoter. The introduction of the Fgfr3 mutations resulted in no obvious effect on tumorigenesis up to 18 months of age. Furthermore, even when the Fgfr3 mutations were introduced together with K-Ras or β-catenin (Ctnnb1) activating mutations, no urothelial dysplasia or UCC was observed. Interestingly, however, owing to a sporadic ectopic Cre recombinase expression in the skin and lung of these mice, Fgfr3 mutation caused papilloma and promoted lung tumorigenesis in cooperation with K-Ras and β-catenin activation, respectively. These results indicate that activation of FGFR3 can cooperate with other mutations to drive tumorigenesis in a context-dependent manner, and support the hypothesis that activation of FGFR3 signaling contributes to human cancer.
INTRODUCTION
Urothelial cell carcinoma (UCC) of the bladder is the fifth commonest cancer, with 357,000 new cases diagnosed yearly on a world-wide basis (Parkin et al., 2005). The majority (75%) of these tumors are noninvasive and well differentiated, and can be controlled by transurethral resection of the tumor lesions. However, up to 70% of the patients with a superficial UCC will have recurrences after its removal, and 10–15% will progress to invasive UCC (http://info.cancerresearchuk.org/cancerstats/). Even in cases of no progression, regular surveillance by cystoscopy is required, making bladder cancer one of the most expensive and labor-intensive cancers to manage.
A number of genetic and epigenetic alterations have been identified in bladder tumorigenesis, including activating mutations in fibroblast growth factor receptor 3 (FGFR3) and RAS family genes, amplification of ERBB2, and loss of the TP53, RB1 and PTEN tumor suppressors (Schulz, 2006; Luis et al., 2007; Cordon-Cardo, 2008; Diaz et al., 2008). Among these, somatic mutations in FGFR3 have been identified at a high frequency in cases of superficial UCC (60–80%) (Knowles, 2008b). FGFR3 is a receptor tyrosine kinase that is known to mediate the effects of fibroblast growth factors (FGFs). When these activating mutations occur in the germ line, they are known to cause several autosomal dominant human skeletal dysplasia syndromes (Muenke and Schell, 1995). Several studies have shown that somatic mutations of FGFR3 are strongly associated with bladder cancer of a low tumor grade and stage (Billerey et al., 2001; Jebar et al., 2005; Lamy et al., 2006; Lindgren et al., 2006).
Although some previous studies strongly suggest the importance of FGFR3 mutations in tumor formation (Chesi et al., 2001; Logie et al., 2005), thus far they have been uninformative on the precise mechanistic role of FGFR3 mutations in tumor formation and progression. The role of FGF signaling in bladder cancer is not well understood. Generation of a relevant mouse model is essential not only for the investigation of mechanism but also for testing potential therapeutic approaches. In this study, the role of two potent activating mutations of FGFR3 that are found in UCC was assessed in the initiation and development of UCC as a sole driver in vivo, and in synergy with β-catenin (Ctnnb1) and K-Ras mutations.
RESULTS
Targeting of the Fgfr3 mutations in the bladder
In order to achieve urothelium-specific conditional expression, mice expressing Cre recombinase driven by the mouse uroplakin II (UroII) promoter were selected. UroII is expressed throughout the urothelial layers in mice (Mo et al., 2005). The UroIICre promoter has been reported to successfully drive the expression of proteins that lead to bladder tumor formation, including SV40 large T antigen and H-Ras (Zhang et al., 2001). To demonstrate Cre-driven recombination in the urothelium, we crossed the UroIICre+ line with Z/EG reporter mice (Novak et al., 2000). Visualization with the Olympus OV100 whole-mouse fluorescent imaging system showed that recombination had occurred in the urothelial lining of the bladder, as well as in the ureters (supplementary material Fig. S1A).
We then bred UroIICre+ mice to lines carrying Fgfr3 K644E and Fgfr3 K644M mutations (murine equivalent of human K652E and K652M, respectively) (Iwata et al., 2000; Iwata et al., 2001). Both of these mutations have been found in human UCC (Knowles, 2008a) and are known to highly activate the kinase activity of the receptor (Iwata et al., 2001). Both lines carry a neomycin resistant gene (neo) flanked by loxP sites in the intron prior to the exon with the K644 mutation. This neo insertion is known to suppress the expression of the Fgfr3 mutant allele in the absence of Cre recombination. In the presence of Cre, the neo gene is excised, allowing expression of the mutant Fgfr3 protein. In the offspring (UroIICre+Fgfr3+/K644E and UroIICre+Fgfr3+/K644M), the pattern and approximate levels of Fgfr3 protein expression were similar to those of the age-matched wild-type mice (supplementary material Fig. S1B,C,F).
Fgfr3 mutation alone does not drive tumorigenesis of the bladder
To investigate the role of Fgfr3 mutations as a driver of UCC, we aged the UroIICre+Fgfr3+/K644E and UroIICre+Fgfr3+/K644M mutant mice to 18 months (tumor phenotype is summarized in supplementary material Table S1). Neither of these lines developed urothelial hyperplasia, dysplasia or carcinoma (Fig. 1E,I). After 2 hours of in vivo incorporation of 5-bromo-2-deoxyuridine (BrdU), no apparent positivity was observed, indicating that little or no cell proliferation took place in the urothelium either in the wild-type or in the mutant cohorts (Fig. 1F,J). We next examined some of the well-known core signaling pathways downstream of FGF. A strong upregulation of expression of phosphorylated extracellular-signal-regulated kinases 1 and 2 (pERK1/2) was observed in both UroIICre+Fgfr3+/K644E and UroIICre+Fgfr3+/K644M mutants compared with wild type (Fig. 1G,K and supplementary material Fig. S2). This was accompanied by an upregulation of Sprouty2 levels (Fig. 1H,L and supplementary material Fig. S2). No significant pAKT(Ser473) or p-mTOR staining was present in the Fgfr3 mutant cohorts, similar to wild type (supplementary material Fig. S3).
Fig. 1.

Fgfr3 mutation is not the sole driver of tumorigenesis in the bladder. The samples are from 12-month-old wild-type (A–D), UroIICre+Fgfr3+/K644E (E–H), UroIICre+Fgfr3+/K644M (I–L), UroIICre+Fgfr3+/K644EK-RasG12D/+ (M–P) and UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ (Q–T) mice. Hematoxylin and eosin (H&E) staining showed no apparent lesions in the urothelium in all Fgfr3 mutants (A,E,I,M) except for the compound model with β-cateninexon3/+ (Q), in which hyperplasia was observed (red arrowhead; also in S,T). No significant cell proliferation was observed (B,F,J,N) except for the area of hyperplastic lesion in UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice (black arrowhead) (R). Upregulation of pERK1/2 (G,K,O,S) and Sprouty2 (H,L,P,T) was observed in Fgfr3 mutant mice, compared with wild type (C,D, respectively). Scale bar: 200 μm.
Next, we addressed whether Fgfr3 mutations require other mutations for the formation of UCC. FGFR3 and H-RAS mutations are reported to be mutually exclusive events in human UCC (Jebar et al., 2005). Previous studies that have examined K-RAS mutations in a mixture of human bladder cancer have suggested a wide variation in frequency (4–29%) (Uchida et al., 1995; Olderoy et al., 1998; Ayan et al., 2001). In order to address how much of the FGFR3 signaling activities are augmented by additional upregulation of the ERK-MAPK pathway, we examined the role of oncogenic K-Ras G12D (Jackson et al., 2001) in UroIICre+Fgfr3+/K644EK-RasG12D/+ mice (n=23) (supplementary material Table S1). Upon aging to 12 months, no UroIICre+Fgfr3+/K644EK-RasG12D/+ mice developed urothelial hyperplasia, dysplasia or carcinoma (Fig. 1M) and no apparent changes in BrdU incorporation were observed (Fig. 1N). Similar to the single mutants, a strong upregulation of pERK1/2 (Fig. 1O) as well as an accompanying upregulation of Sprouty2 (Fig. 1P) was observed. Comparable levels of upregulation of pERK1/2 and Sprouty2 were observed in the UroIICre+K-RasG12D/+ urothelium (data not shown). Minimal levels of upregulation of pAKT(Ser473) and p-mTOR were observed in the UroIICre+Fgfr3+/K644EK-RasG12D/+ cohorts (supplementary material Fig. S3).
Mounting evidence is present for a role of Wnt pathway activation in human bladder cancer development, including an association between the accumulation of nuclear β-catenin and reduced patient survival (Kastritis et al., 2009). We recently reported the first in vivo evidence of β-catenin activation leading to the formation of UCC when combined with other tumor suppressor mutations, such as a loss of Pten (Ahmad et al., 2011a). In addition, we have also shown that the β-catenin mutation can cooperate with H-Ras mutation to drive superficial bladder cancer (Ahmad et al., 2011b). In order to test whether there is cooperation between the FGF and Wnt pathways in bladder tumorigenesis, we examined the role of β-catenin activating mutations (Harada et al., 1999) in UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice (n=27) (supplementary material Table S1).
In order to drive deregulated Wnt signaling, we used mice that carry a dominant allele of the β-catenin gene in which exon 3 is flanked by loxP sequences (Harada et al., 1999). In these mice, Cre recombinase deletes exon 3, which contains the residues that are phosphorylated by GSK3β. Thus, β-catenin will accumulate in the nucleus and drive Wnt signaling (Moon et al., 2004). Although areas of hyperproliferation were observed in the bladders of UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice from approximately 3 months of age in 100% of mice (n=20), equivalent lesions were also found in the UroIICre+β-cateninexon3/+ urothelium (Ahmad et al., 2011a). In both groups these lesions incorporated BrdU (Fig. 1R); however, they never progressed further (examination up to 12 months) (Fig. 1Q). Once again, upregulation of pERK1/2 and Sprouty2 was observed in this model (Fig. 1S,T). The areas of hyperproliferation that were observed in UroIICre+β-cateninexon3/+ mice showed comparable levels of upregulation of BrdU, pERK1/2 and Sprouty2 in the lesions (data not shown), indicating that Fgfr3 mutations are not contributing to urothelial hyperplasia in UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice. No upregulation of pAKT(Ser473) or p-mTOR was observed in this model (supplementary material Fig. S3). Taken together, these data suggest that K-Ras and β-catenin activating mutations do not cooperate with Fgfr3 mutation to drive UCC or to enhance signaling.
Skin papilloma formation in UroIICre+Fgfr3+/K644EK-RasG12D/+ mice
In contrast to observations in the urothelium, by 1 year of age, 44% (9/23) of the UroIICre+Fgfr3+/K644EK-RasG12D/+ cohort developed papilloma (Fig. 2A; supplementary material Table S1, Fig. S4). The tumors reached 1 cm in diameter by a median of 220 days (mean 249 days). Upon crossing of the UroIICre+Fgfr3+/K644EK-RasG12D/+ mice to the Z/EG reporter line, a strong GFP signal was observed within papillomas, indicating that the tumor formation was due to sporadic Cre recombination in the tumor (Fig. 2G). By contrast, no papilloma was observed in the UroIICre+K-RasG12D/+ cohort aged up to 18 months (n=20), indicating that Fgfr3 mutation cooperates with K-Ras mutation to drive papilloma formation.
Fig. 2.
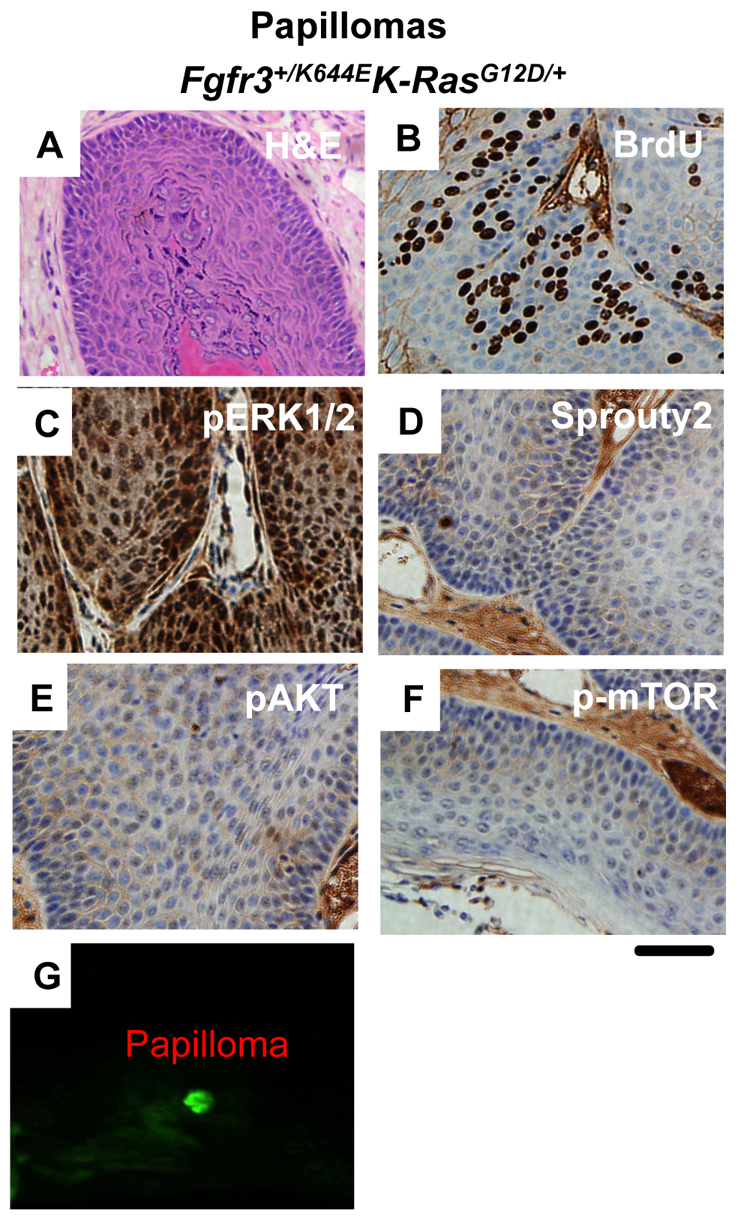
Formation of papilloma lesions in the UroIICre+Fgfr3+/K644EK-RasG12D/+ model. Papilloma samples are from 12-month-old UroIICre+Fgfr3+/K644EK-RasG12D/+ mice. H&E staining shows the formation of a papilloma (A), in which BrdU immunoreactivity demonstrated cell proliferation (B). Upregulation of pERK1/2 was observed (C), as was observed in the urothelium (Fig. 1O). However, upregulation of Sprouty2 was not present in UroIICre+Fgfr3+/K644EK-RasG12D/+ mice (D). Neither pAKT(Ser473) (E) nor p-mTOR (F) was upregulated in papillomas formed in UroIICre+Fgfr3+/K644EK-RasG12D/+ mice. GFP imaging indicated Cre-recombined cells in the papilloma (G). 14× magnification was used. Scale bar: 200 μm in all panels except for G.
The papillomas in the UroIICre+Fgfr3+/K644EK-RasG12D/+ cohort incorporated BrdU (Fig. 2B). In terms of signaling, tumor formation was associated with a robust activation of pERK1/2 expression in the absence of Sprouty2 upregulation (Fig. 2C,D). This is distinct from the observation in the urothelium of these mice (Fig. 1P), where Sprouty2 upregulation was seen (Fig. 2D), suggesting that the negative feedback counteracting the oncogenic ERK-MAPK pathway has not occurred in this line. Levels of pAKT(Ser473) and p-mTOR were unchanged (Fig. 2E,F). We also compared the signaling profiles observed in these papillomas of Fgfr3 mutants with those of papillomas from UroIICre+K-RasG12D/+Ptenfl/+ mice, which regularly develop papillomas (8/19; 42%) (supplementary material Fig. S5A). We found a similar increase of BrdU incorporation (supplementary material Fig. S5B). However, in contrast to UroIICre+Fgfr3+/K644EK-RasG12D/+ papillomas, only mild upregulation of nuclear and cytoplasmic pERK1/2 was observed in the lesion, verified by the Histoscore quantification (n=3, P<0.001 and 0.05, respectively) (supplementary material Fig. S5C,I,J). Sprouty2 was also mildly upregulated (supplementary material Fig. S5D). Furthermore, the ERK downstream effectors pElk and Pea3 were downregulated (supplementary material Fig. S5E,F), indicating that overall ERK was not very high in this model. Conversely, we found much higher levels of pAKT(Ser473) staining in papillomas of the UroIICre+K-RasG12D/+Ptenfl/+ mice in comparison to UroIICre+Fgfr3+/K644EK-RasG12D/+ mice (n=3, P<0.001; supplementary material Fig. S5G,H,K,L).
Formation of lung tumors in UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice
Although there was no papilloma formation, 36% (10/27) of the UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice developed lung tumors by 1 year of age (supplementary material Table S1 and Fig. S4). Imaging with the OV100 microscope showed a strong GFP signal in the lung tumor, indicating that the tumor formation was due to sporadic Cre recombination in the tumor (supplementary material Fig. S6A). These tumors resembled most closely solitary fibrous tumors (SFTs) of the lung (supplementary material Fig. S6B). SFTs were mostly termed hemangiopericytomas (HPCs) in the past. According to the World Health Organization (WHO), these tumors are mesenchymal neoplasms of subendothelial origin that can be found mostly in the pleura but also in extraserosal sites, such as lung, mediastinum, liver, head and neck, and deep soft tissues of the extremities. Most SFTs behave as slowly growing, painless masses (Kouki et al., 2008). No lung tumors were observed in the UroIICre+β-cateninexon3/+ mice aged up to 18 months (n=20; supplementary material Table S1). Therefore, in this instance, Fgfr3 mutation cooperates with β-catenin activation to drive lung tumorigenesis. In these lung tumors formed in UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice, strong BrdU positivity indicated rapid cell proliferation (supplementary material Fig. S6C). In addition, tumors showed high levels of nuclear β-catenin, consistent with the activation of β-catenin owing to Cre-mediated excision of exon 3 (supplementary material Fig. S6D). In humans, nuclear β-catenin is found in up to a third of SFTs (Yamaguchi et al., 2004; Ng et al., 2005; Rakheja et al., 2005; Andino et al., 2006; Carlson and Fletcher, 2007).
In terms of downstream signaling, increased levels of pAKT(Ser473) and p-mTOR were observed (supplementary material Fig. S6E,F), suggesting the involvement of the AKT pathway in tumorigenesis. By contrast, we found minimal upregulation of pERK1/2 and Sprouty2 (supplementary material Fig. S6G,H).
We compared this phenotype with lung tumors found in UroIICre+β-cateninexon3/+K-RasG12D/+ mice (n=17) (Ahmad et al., 2011b). In these tumors, we found little upregulation of pAKT (supplementary material Fig. S6I), but strong upregulation of pERK1/2 with a corresponding fall in Sprouty2 expression (supplementary material Fig. S6K,L). Upon quantification of the staining intensity using a weighted Histoscore technique, we found statistically significant increases in both nuclear and cytoplasmic pERK1/2 staining in the UroIICre+β-cateninexon3/+K-RasG12D/+ cohort compared with the UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice (n=3, P<0.001; supplementary material Fig. S6M,N). Conversely, we found much higher levels of pAKT(Ser473) staining in lung tumors of the UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice in comparison to UroIICre+β-cateninexon3/+K-RasG12D/+ mice (n=3, P<0.001; supplementary material Fig. S6O,P). Consistent with this, less p-mTOR staining was present in UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice (supplementary material Fig. S6J).
Taken together, these results suggest that activation of the AKT pathway might contribute to tumorigenesis in the lung of UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ mice and papilloma of UroIICre+K-RasG12D/+Ptenfl/+ mice. This differs to the signaling observed in papillomas of UroIICre+Fgfr3+/K644EK-RasG12D/+ mice, and in bladder tumors of UroIICre+H-RasQ61L and UroIICre+β-cateninexon3/exon3H-RasQ61L mice (Ahmad et al., 2011b), in which elevation of the ERK-MAPK pathway might play a major role in tumorigenesis. In summary, we hypothesize that there are two independent pathways, activation of ERK-MAPK and that of AKT, that could play a role in tumorigenesis in the presence of Fgfr3 mutations. This hypothesis is presented as a model in supplementary material Fig. S7.
Elevation of ERK downstream signaling is associated with tumor formation in the bladder and in papilloma
Given the difficulty of assigning ERK pathway activity in vivo, we next examined the expression of two known targets of ERK: Pea and pELK (O’Hagan et al., 1996; Cruzalegui et al., 1999). In the bladder of UroIICre+Fgfr3+/K644E mice, the levels of Pea3 and pElk1 were mildly upregulated (Fig. 3C,D) when compared with wild-type bladders (Fig. 3A,B). By contrast, in papillomas from UroIICre+Fgfr3+/K644EK-RasG12D/+ mice, Pea3 and pElk1 expression was highly upregulated (Fig. 3E,F). Quantitative reverse-transcriptase PCR (RT-PCR) experiments showed that, in UroIICre+Fgfr3+/K644EK-RasG12D/+ papillomas (n=2), the relative expression level of the Pea3 transcript normalized by actin was increased by 120-fold compared with that in wild-type bladders (n=4), with the average threshold cycle (Ct) values of 9.37 and 16.28±0.88 in papilloma and wild-type bladders, respectively. By contrast, a difference in the expression levels of Pea3 between the bladders of UroIICre+Fgfr3+/K644E (n=3) and wild type was not detected at the transcript level (average Ct = 15.76±0.97 in UroIICre+Fgfr3+/K644E; statistically not significant). This is in accordance with our hypothesis that the upregulation of pERK1/2 in the absence of Sprouty2 leads to an overall increase in ERK signaling in papillomas of UroIICre+Fgfr3+/K644EK-RasG12D/+ mice (Fig. 2C,D). By contrast, we suggest that Sprouty2 upregulation is sufficient to suppress the overall ERK signaling levels in the bladders of UroIICre+Fgfr3+/K644E mice (Fig. 1G,H). We have also examined Pea3 and pELK1 levels in two established bladder tumor models, UroIICre+H-RasQ61L and UroIICre+β-cateninexon3/exon3H-RasQ61L (Ahmad et al., 2011b). In these models, both Pea3 and pElk1 levels are highly upregulated in the bladder tumors (Fig. 3G–J), indicating that strong activation of the ERK-MAPK pathway might be one of the pathways that drives bladder tumor formation.
Fig. 3.

Elevation of ERK downstream signaling is associated with tumor formation in the bladder and in papilloma formation. Activation of overall ERK-MAPK signaling was examined in the bladders of wild-type (A,B) and UroIICre+Fgfr3+/K644E (C,D) mice, in papilloma of UroIICre+Fgfr3+/K644EK-RasG12D/+ mice (E,F), and in bladder tumors of UroIICre+H-RasQ61L (G,H) and UroIICre+β-cateninexon3/exon3H-RasQ61L (I,J) mice by assessing expression of Pea3 (A,C,E,G,I) and pElk1 (B,D,F,H,J). Upregulation of both Pea3 and pElk1 was observed in the bladder of UroIICre+Fgfr3+/K644E mice compared with that of wild-type, and further upregulation was observed in papilloma of UroIICre+Fgfr3+/K644EK-RasG12D/+ mice, as well as in bladder tumor models with H-RasQ61L mutation. Scale bar: 200 μm.
Finally, we have examined the ERK-MAPK signaling pathway in human UCC by using a tissue microarray (TMA) that contains 60 UCC and 20 benign controls (Folio Biosciences, OH). Using the Histoscore technique, we have quantified the staining intensities of pERK1/2, pElk1 and Sprouty2 (Fig. 4A). A significant correlation was observed in the upregulation of pERK1/2 and that of pElk1 (Pearson correlation coefficient=0.6082, P<0.0001, two-tailed) (MiniTab v15) (Fig. 4B), indicating that an elevation of overall ERK signaling is associated with human UCC. Furthermore, a significant correlation was also observed between the upregulation of pERK1/2 and the downregulation of Sprouty2 (Pearson correlation coefficient=0.6596, P<0.0001, two-tailed) (data not shown), indicating that the absence of Sprouty2 upregulation might underlie the mechanism leading to UCC formation in humans. In addition, using Oncomine (https://www.oncomine.org), Lindgren and colleagues demonstrated that, in FGFR3 mutant bladder cancer, there is a downregulation of Sprouty2 at the mRNA level (P=0.015) (Lindgren et al., 2006). In the same study, Sprouty2 mRNA levels were found to be downregulated as the disease progresses from Grade 1 to 3 (P=0.015).
Fig. 4.

Correlation between pERK1/2 and downstream effectors in human UCC TMA. The downstream signaling of the ERK-MAPK pathway was examined using a commercially available TMA containing 60 UCC and 20 benign bladder tumors. (A) An example of UCC samples, showing strong immunohistochemical staining of pERK1/2 and pElk1, and a weak staining of Sprouty2. Each core size is 1.5 mm in diameter. (B) Scatterplot demonstrating a correlation between the upregulation of pERK1/2 with that of pElk1.
DISCUSSION
We demonstrate here that activating mutations of FGFR3 are unlikely to be the sole initiating factor for UCC and that neitherβ-cateninexon3/+ nor K-RasG12D/+ mutation cooperates with FGFR3 to drive UCC. FGFR3 mutations are thought to be mutually exclusive with H-RAS mutation (Jebar et al., 2005), and FGFR3 is only rarely mutated with p53 (<5%) (Bakkar et al., 2003). Therefore, generation of mouse models that recapitulate superficial UCCs that have FGFR3 mutations might be difficult and will require greater knowledge of the genetic changes that accompany FGFR3 mutations (Jebar et al., 2005; Wu, 2005; Puzio-Kuter et al., 2009).
The study, however, showed that somatic Fgfr3 mutations caused a modest upregulation of the ERK-MAPK pathway in the urothelium, which was accompanied by upregulation of Sprouty2, one of the feedback inhibitors of the ERK-MAPK pathway (Fig. 1). We speculate that upregulation of negative-feedback genes, such as Sprouty2, could be one of the mechanisms by which UCC is normally prevented. In papillomas in UroIICRE+Fgfr3+/K644EK-RasG12D/+ mice, no concomitant upregulation of Sprouty2 had occurred, potentially leading to uncontrolled activation of the ERK-MAPK pathway in the absence of a negative-feedback mechanism (Fig. 2). In the case of lung tumors in the UroIICre+Fgfr3+/K644Eβ-cateninexon3/+ cohort, a strong upregulation of the PI3K-pAKT and Wnt–β-catenin pathways might underlie tumorigenesis (supplementary material Fig. S6). It is tempting to propose that these differential downstream signaling profiles are the underlying drivers of tumorigenesis, with the different oncogenic mutations cooperating with FGFR3 mutation in a context-dependent fashion (supplementary material Fig. S7). It would be interesting to assess in the future whether downregulation of Sprouty2 in our mouse system results in UCC formation. Tissue-specific effects of Fgfr3 mutation could also be caused by differential kinetics of Fgfr3 protein turnover in the bladder, skin and lung. Such a possibility could be tested by the generation of a mouse model that allows expression of a tagged Fgfr3 protein.
Fgfr3 K652E and K652M mutations, which we described here, are found in human UCC but are much less frequent (3%) than the S249C mutation, which accounts for 67% of the FGFR3 mutations in bladder cancer (Knowles, 2008a). Thus, it is possible that S249C mutation is more oncogenic in the bladder than are K652E or K652M. In vitro, both S249C and K652E or K653M mutations are shown to constitutively increase the FGFR3 kinase activity, albeit with different mechanisms (Naski et al., 1996; d’Avis et al., 1998). S249C is known to cause ligand-independent dimerization, whereas K652E or K652M promotes a conformational change that favors autophosphorylation, and its kinase activity can be further enhanced by additional ligand stimulation (Naski et al., 1996; d’Avis et al., 1998). Upon measuring transcription-inducing activity by the fos-luciferase reporter assay, K644E mutant protein was found to be twice as active as S249C mutant in the absence of ligand (d’Avis et al., 1998). Preliminary data suggest that transgenic mice with a human FGFR3IIIb isoform with S249C mutation did not show any tumors in the bladder up to 1 year of age (Margaret Knowles, Leeds Institute for Molecular Medicine, UK, personal communication).
The skin papilloma and lung tumors observed in our study with Fgfr3 mutations are relevant in human cancers (Woenckhaus et al., 2006; Hafner et al., 2007; Cortese et al., 2008). Overexpression of the FGFR3 S249C mutation from the K5 promoter in transgenic mice resulted in the formation of a benign skin tumor (Logie et al., 2005). Our study is consistent with this previous report.
Recent work has elegantly demonstrated that knockdown of FGFR3 expression suppresses the growth of bladder cell lines and mouse xenograft models of bladder cancer (Qing et al., 2009). Furthermore, FGFR3-specific monoclonal antibody (R3Mab) significantly inhibits growth and progression of xenograft tumors that harbor the S249C and K650E mutations (Qing et al., 2009). Further studies of the FGFR3 signaling pathway, which is frequently deregulated in UCC, is therefore essential, firstly to allow one to stratify patients according to risk of progression and/or recurrence, and secondly to aid in patient selection for either the single agent or combination therapy with small-molecule- and monoclonal-antibody-based treatments in the future (Black et al., 2007; Knowles, 2008a; Qing et al., 2009). Moreover, identifying the cooperating molecular events that occur alongside FGFR3 mutation to drive UCC will aid the development of genetic models of UCC to test combinatorial therapies.
METHODS
Mice
Uroplakin II Cre mice (UroIICre+) (Zhang et al., 1999) were intercrossed with mice harboring Fgfr3 K644E and Fgfr3 K644M (Fgfr3+/K644Eneo and Fgfr3+/K644Mneo) (Iwata et al., 2000; Iwata et al., 2001), β-cateninexon3/+ (Harada et al., 1999), and K-RasG12D/+ (Jackson et al., 2001) mutations. Mice were genotyped by PCR as previously described (Harada et al., 1999; Zhang et al., 1999; Iwata et al., 2000; Iwata et al., 2001; Jackson et al., 2001). Mice were of a mixed background and littermates were used as control mice. All experiments were carried out in accordance with the Project Licence under Home Office Animal (Scientific Procedures) Act 1986 in the UK.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on formalin-fixed, paraffin-embedded samples. Upon harvesting, the bladders were emptied of urine before being placed in formalin for overnight fixation and were paraffin embedded. All bladders were processed and cut in the same manner by a single histology technician to aid standardization. For each genotype we stained at least three samples from different mice and took representative images. We used antibodies against: FGFR3 (C-15, Santa Cruz Biotechnology, 1:20, no antigen retrieval), BrdU (347580, BD Biosciences, 1:500, citrate buffer and microwave antigen retrieval), pERK1/2 (#9101, Cell Signaling, 1:100, citrate buffer and microwave antigen retrieval), Sprouty2 (ab60719, AbCam, 1:300, citrate buffer and microwave antigen retrieval), pAKT(Ser473) (#3787, Cell Signaling, 1:50, citrate buffer and microwave antigen retrieval), β-catenin (C19220, Transduction Labs, 1:50, Tris/EDTA water bath antigen retrieval with 50 minutes at 99°C), p-mTOR(Ser2448) (#2976, Cell Signaling, 1:100, citrate buffer and microwave antigen retrieval), pElk1(Ser389) (ab28818, Abcam, 1:50, citrate buffer and microwave antigen retrieval) and Pea3 (ab70425, Abcam, 1:200, citrate buffer and microwave antigen retrieval).
Quantification of IHC staining
For each tissue section (n=5), the percentage of positive immunoreactivity in the nucleus and cytoplasm was evaluated with 40× magnification objectives. Staining intensity was categorized into three categories: 0, 1, 2 or 3, denoting negative, weak, moderate or strong staining, respectively. The final Histoscore was calculated from the sum of (1 × % weakly positive tumor cells) + (2 × % moderately positive tumor cells) + (3 × % strongly positive tumor cells), with a maximum Histoscore of 300 (Kirkegaard et al., 2006). Statistics were performed using the Student’s t-test (Minitab).
Human TMA
The TMA (Folio Biosciences, OH) consisted of 60 UCC and 20 benign bladder cores with data that consisted of patient sex, age and tumor grade. Slides were scanned using the Aperio slide scanner.
RNA isolation and quantitative RT-PCR
Bladders and papilloma tissues were collected in RNAlater RNA stabilization reagent and RNA was isolated using the RNeasy Mini Kit (Qiagen). DNase digest was performed using the DNAfree Kit (Ambion). Reverse transcription was carried out using the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen). Finally, quantitative PCR was carried out using DyNAmo SYBR Green qPCR Kit (Finnzymes) on a Chromo4 Real-Time PCR cycler (BioRad). PCR primers for actin and Pea3 were obtained from Qiagen (Quantitect qPCR Primer Assay).
Supplementary Material
Acknowledgments
This work was funded by Cancer Research UK and an MRC fellowship to I.A. We thank BICR Services, Biological Services Unit, and Colin Nixon and his histology department. We thank the ‘Think Pink’ charity for the purchase of the Aperio slide scanner and the Slidepath software.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
I.A., L.B.S., M.F. and C.-A.M. performed the experiments, M.M.T. and X.-R.W. supplied reagents, I.A., O.J.S. and T.I. wrote the paper. H.Y.L. supervised I.A.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.006874/-/DC1
REFERENCES
- Ahmad I., Morton J. P., Singh L. B., Radulescu S. M., Ridgway R. A., Patel S., Woodgett J., Winton D. J., Taketo M. M., Wu X. R., et al. (2011a). beta-Catenin activation synergizes with PTEN loss to cause bladder cancer formation. Oncogene 30, 178–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad I., Patel R., Liu Y., Singh L. B., Taketo M. M., Wu X. R., Leung H. Y., Sansom O. J. (2011b). Ras mutation cooperates with β-catenin activation to drive bladder tumourigenesis. Cell Death Dis. 2, e124; doi: 10.1038/cddis.2011.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andino L., Cagle P. T., Murer B., Lu L., Popper H. H., Galateau-Salle F., Sienko A. E., Barrios R., Zander D. S. (2006). Pleuropulmonary desmoid tumors: immunohistochemical comparison with solitary fibrous tumors and assessment of beta-catenin and cyclin D1 expression. Arch. Pathol. Lab. Med. 130, 1503–1509 [DOI] [PubMed] [Google Scholar]
- Ayan S., Gokce G., Kilicarslan H., Ozdemir O., Yildiz E., Gultekin E. Y. (2001). K-RAS mutation in transitional cell carcinoma of urinary bladder. Int. Urol. Nephrol. 33, 363–367 [DOI] [PubMed] [Google Scholar]
- Bakkar A. A., Wallerand H., Radvanyi F., Lahaye J. B., Pissard S., Lecerf L., Kouyoumdjian J. C., Abbou C. C., Pairon J. C., Jaurand M. C., et al. (2003). FGFR3 and TP53 gene mutations define two distinct pathways in urothelial cell carcinoma of the bladder. Cancer Res. 63, 8108–8112 [PubMed] [Google Scholar]
- Billerey C., Chopin D., Aubriot-Lorton M. H., Ricol D., Diez de Medina S. G., Van Rhijn B., Bralet M. P., Lefrere-Belda M. A., Lahaye J. B., Abbou C. C., et al. (2001). Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am. J. Pathol. 158, 1955–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black P. C., Agarwal P. K., Dinney C. P. (2007). Targeted therapies in bladder cancer-an update. Urol. Oncol. 25, 433–438 [DOI] [PubMed] [Google Scholar]
- Carlson J. W., Fletcher C. D. (2007). Immunohistochemistry for beta-catenin in the differential diagnosis of spindle cell lesions: analysis of a series and review of the literature. Histopathology 51, 509–514 [DOI] [PubMed] [Google Scholar]
- Chesi M., Brents L. A., Ely S. A., Bais C., Robbiani D. F., Mesri E. A., Kuehl W. M., Bergsagel P. L. (2001). Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood 97, 729–736 [DOI] [PubMed] [Google Scholar]
- Cordon-Cardo C. (2008). Molecular alterations associated with bladder cancer initiation and progression. Scand. J. Urol. Nephrol. Suppl. 42,154–165 [DOI] [PubMed] [Google Scholar]
- Cortese R., Hartmann O., Berlin K., Eckhardt F. (2008). Correlative gene expression and DNA methylation profiling in lung development nominate new biomarkers in lung cancer. Int. J. Biochem. Cell Biol. 40, 1494–1508 [DOI] [PubMed] [Google Scholar]
- Cruzalegui F. H., Cano E., Treisman R. (1999). ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene 18, 7948–7957 [DOI] [PubMed] [Google Scholar]
- d’Avis P. Y., Robertson S. C., Meyer A. N., Bardwell W. M., Webster M. K., Donoghue D. J. (1998). Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth Differ. 9, 71–78 [PubMed] [Google Scholar]
- Diaz D. S., Segersten U., Malmstrom P. U. (2008). Molecular genetics of bladder cancer: an update. Minerva Urol. Nefrol. 60, 205–216 [PubMed] [Google Scholar]
- Hafner C., Lopez-Knowles E., Luis N. M., Toll A., Baselga E., Fernandez-Casado A., Hernandez S., Ribe A., Mentzel T., Stoehr R., et al. (2007). Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc. Natl. Acad. Sci. USA 104, 13450–13454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N., Tamai Y., Ishikawa T., Sauer B., Takaku K., Oshima M., Taketo M. M. (1999). Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 18, 5931–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata T., Chen L., Li C., Ovchinnikov D. A., Behringer R. R., Francomano C. A., Deng C. X. (2000). A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum. Mol. Genet. 9, 1603–1613 [DOI] [PubMed] [Google Scholar]
- Iwata T., Li C. L., Deng C. X., Francomano C. A. (2001). Highly activated Fgfr3 with the K644M mutation causes prolonged survival in severe dwarf mice. Hum. Mol. Genet. 10, 1255–1264 [DOI] [PubMed] [Google Scholar]
- Jackson E. L., Willis N., Mercer K., Bronson R. T., Crowley D., Montoya R., Jacks T., Tuveson D. A. (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jebar A. H., Hurst C. D., Tomlinson D. C., Johnston C., Taylor C. F., Knowles M. A. (2005). FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene 24, 5218–5225 [DOI] [PubMed] [Google Scholar]
- Kastritis E., Murray S., Kyriakou F., Horti M., Tamvakis N., Kavantzas N., Patsouris E. S., Noni A., Legaki S., Dimopoulos M. A., et al. (2009). Somatic mutations of adenomatous polyposis coli gene and nuclear b-catenin accumulation have prognostic significance in invasive urothelial carcinomas: evidence for Wnt pathway implication. Int. J. Cancer 124, 103–108 [DOI] [PubMed] [Google Scholar]
- Kirkegaard T., Edwards J., Tovey S., McGlynn L. M., Krishna S. N., Mukherjee R., Tam L., Munro A. F., Dunne B., Bartlett J. M. S. (2006). Observer variation in immunohistochemical analysis of protein expression, time for a change? Histopathology 48, 787–794 [DOI] [PubMed] [Google Scholar]
- Knowles M. A. (2008a). Novel therapeutic targets in bladder cancer: mutation and expression of FGF receptors. Future Oncol. 4, 71–83 [DOI] [PubMed] [Google Scholar]
- Knowles M. A. (2008b). Molecular pathogenesis of bladder cancer. Int. J. Clin. Oncol. 13, 287–297 [DOI] [PubMed] [Google Scholar]
- Kouki H. S., Koletsis E. N., Zolota V., Prokakis C., Apostolakis E., Dougenis D. (2008). Solitary fibrous tumor of the lung. Gen. Thorac. Cardiovasc. Surg. 56, 249–251 [DOI] [PubMed] [Google Scholar]
- Lamy A., Gobet F., Laurent M., Blanchard F., Varin C., Moulin C., Andreou A., Frebourg T., Pfister C. (2006). Molecular profiling of bladder tumors based on the detection of FGFR3 and TP53 mutations. J. Urol. 176, 2686–2689 [DOI] [PubMed] [Google Scholar]
- Lindgren D., Liedberg F., Andersson A., Chebil G., Gudjonsson S., Borg A., Mansson W., Fioretos T., Hoglund M. (2006). Molecular characterization of early-stage bladder carcinomas by expression profiles, FGFR3 mutation status, and loss of 9q. Oncogene 25, 2685–2696 [DOI] [PubMed] [Google Scholar]
- Logie A., Dunois-Larde C., Rosty C., Levrel O., Blanche M., Ribeiro A., Gasc J. M., Jorcano J., Werner S., Sastre-Garau X., et al. (2005). Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum. Mol. Genet. 14, 1153–1160 [DOI] [PubMed] [Google Scholar]
- Luis N. M., Lopez-Knowles E., Real F. X. (2007). Molecular biology of bladder cancer. Clin. Transl. Oncol. 9, 5–12 [DOI] [PubMed] [Google Scholar]
- Mo L., Cheng J., Lee E. Y., Sun T. T., Wu X. R. (2005). Gene deletion in urothelium by specific expression of Cre recombinase. Am. J. Physiol. Renal Physiol. 289, F562–F568 [DOI] [PubMed] [Google Scholar]
- Moon R. T., Kohn A. D., De Ferrari G. V., Kaykas A. (2004). WNT and beta-catenin signalling: diseases and therapies. Nat. Rev. Genet. 5, 691–701 [DOI] [PubMed] [Google Scholar]
- Muenke M., Schell U. (1995). Fibroblast-growth-factor receptor mutations in human skeletal disorders. Trends Genet. 11, 308–313 [DOI] [PubMed] [Google Scholar]
- Naski M. C., Wang Q., Xu J., Ornitz D. M. (1996). Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat. Genet. 13, 233–237 [DOI] [PubMed] [Google Scholar]
- Ng T. L., Gown A. M., Barry T. S., Cheang M. C., Chan A. K., Turbin D. A., Hsu F. D., West R. B., Nielsen T. O. (2005). Nuclear beta-catenin in mesenchymal tumors. Mod. Pathol. 18, 68–74 [DOI] [PubMed] [Google Scholar]
- Novak A., Guo C., Yang W., Nagy A., Lobe C. G. (2000). Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis 28, 147–155 [PubMed] [Google Scholar]
- O’Hagan R. C., Tozer R. G., Symons M., McCormick F., Hassell J. A. (1996). The activity of the Ets transcription factor PEA3 is regulated by two distinct MAPK cascades. Oncogene 13, 1323–1333 [PubMed] [Google Scholar]
- Olderoy G., Daehlin L., Ogreid D. (1998). Low-frequency mutation of Ha-ras and Ki-ras oncogenes in transitional cell carcinoma of the bladder. Anticancer Res. 18, 2675–2678 [PubMed] [Google Scholar]
- Parkin D. M., Bray F., Ferlay J., Pisani P. (2005). Global cancer statistics, 2002. CA Cancer J. Clin. 55, 74–108 [DOI] [PubMed] [Google Scholar]
- Puzio-Kuter A. M., Castillo-Martin M., Kinkade C. W., Wang X., Shen T. H., Matos T., Shen M. M., Cordon-Cardo C., Abate-Shen C. (2009). Inactivation of p53 and Pten promotes invasive bladder cancer. Genes Dev. 23, 675–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing J., Du X., Chen Y., Chan P., Li H., Wu P., Marsters S., Stawicki S., Tien J., Totpal K., et al. (2009). Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. J. Clin. Invest. 119, 1216–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakheja D., Molberg K. H., Roberts C. A., Jaiswal V. R. (2005). Immunohistochemical expression of beta-catenin in solitary fibrous tumors. Arch. Pathol. Lab. Med. 129, 776–779 [DOI] [PubMed] [Google Scholar]
- Schulz W. A. (2006). Understanding urothelial carcinoma through cancer pathways. Int. J. Cancer 119, 1513–1518 [DOI] [PubMed] [Google Scholar]
- Uchida T., Wada C., Ishida H., Egawa S., Ao T., Yokoyama E., Koshiba K. (1995). Infrequent involvement of mutations on neurofibromatosis type 1, H-ras, K-ras and N-ras in urothelial tumors. Urol. Int. 55, 63–67 [DOI] [PubMed] [Google Scholar]
- Woenckhaus M., Klein-Hitpass L., Grepmeier U., Merk J., Pfeifer M., Wild P., Bettstetter M., Wuensch P., Blaszyk H., Hartmann A., et al. (2006). Smoking and cancer-related gene expression in bronchial epithelium and non-small-cell lung cancers. J. Pathol. 210, 192–204 [DOI] [PubMed] [Google Scholar]
- Wu X. R. (2005). Urothelial tumorigenesis: a tale of divergent pathways. Nat. Rev. Cancer 5, 713–725 [DOI] [PubMed] [Google Scholar]
- Yamaguchi U., Hasegawa T., Masuda T., Sekine S., Kawai A., Chuman H., Shimoda T. (2004). Differential diagnosis of gastrointestinal stromal tumor and other spindle cell tumors in the gastrointestinal tract based on immunohistochemical analysis. Virchows Arch. 445, 142–150 [DOI] [PubMed] [Google Scholar]
- Zhang Z. T., Pak J., Shapiro E., Sun T. T., Wu X. R. (1999). Urothelium-specific expression of an oncogene in transgenic mice induced the formation of carcinoma in situ and invasive transitional cell carcinoma. Cancer Res. 59, 3512–3517 [PubMed] [Google Scholar]
- Zhang Z. T., Pak J., Huang H. Y., Shapiro E., Sun T. T., Pellicer A., Wu X. R. (2001). Role of Ha-ras activation in superficial papillary pathway of urothelial tumor formation. Oncogene 20, 1973–1980 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}