

Abstract
Although the pathologic role of the prion protein in transmissible spongiform encephalopathic diseases has been widely investigated, the physiologic role of the cellular prion protein (PrPC) is not known. Among the many functions attributed to PrPC, there is increasing evidence that it is involved in cell survival and mediates neuroprotection. A potential role in the immune response has also been suggested. However, how these two functions interplay in autoimmune disease is unclear. To address this, autoimmune optic neuritis, a model of multiple sclerosis, was induced in C57Bl/6 mice, and up-regulation of PrPC was observed throughout the disease course. In addition, compared with wild-type mice, in PrPC-deficient mice and mice overexpressing PrPC, histopathologic analysis demonstrated that optic neuritis was exacerbated, as indicated by axonal degeneration, inflammatory infiltration, and demyelination. However, significant neuroprotection of retinal ganglion cells, the axons of which form the optic nerve, was observed in mice that overexpressed PrPC. Conversely, mice lacking PrPC demonstrated significantly more neurodegeneration. This suggests that PrPC may have a neuroprotective function independent of its role in regulating the immune response.
Cellular prion protein (PrPC) is a cell-surface copper-binding glycoprotein1 that is linked to the cellular membrane by a glycosylphosphatidylinositol anchor and is highly expressed in the central nervous system on the surface of both neuronal and glial cells.2,3 In a variety of prion disorders, also known as transmissible spongiform encephalopathic diseases including scrapie and bovine spongiform encephalopathy in animals and Creutzfeldt-Jakob disease in humans, it is believed that PrPC undergoes a conformational change into an abnormal protease-resistant isoform (PrPsc) that can form pathologic extracellular aggregates.4,5 However, to date, the normal physiologic function of PrPC remains unclear. It is thought to be involved in a wide range of cellular processes including neuronal adhesion,6 neuritogenesis,7 neurite outgrowth,8 and cell survival9,10 and has also been suggested to act as a putative receptor for a number of ligands including laminin, heparin, and a variety of synaptic proteins.11–13 Furthermore, it is required for long-term maintenance of myelin.14
Recently, much evidence has suggested that PrPC is anti-apoptotic and may promote neuronal survival. In in vitro experiments, it prevented neuronal apoptosis mediated by the pro-apoptotic protein Bax,9 and oxidative stress.15 Further evidence of a neuroprotective role for PrPC has been demonstrated in in vivo models of cerebral ischemia,16–21 contusion injury,22 axotomy,23 and epilepsy.24 In addition, it has been proposed that PrPC interacts with several signal transduction pathways involved in apoptosis and cell survival such as the phosphatidylinositol 3-kinase/Akt, protein kinase A, and mitogen-activated protein kinase pathways.25–27
It has been suggested that neurodegeneration during the disease course might not be caused by a toxic gain in function due to accumulation of pathologic PrPsc but by loss of the neuroprotective function of PrPC.28
PrPC is also expressed by a variety of nonneuronal cells including those of the immune system. It is expressed by T lymphocytes and cells of myeloid lineage,29–31 and is thought to have a role in many T-cell physiologic features including activation,29,30 antigen presentation,32 phagocytosis,33 differentiation, and survival.34 Collectively, the data suggest that PrPC may have multiple roles in autoimmune diseases such as multiple sclerosis. To investigate its potential role in both autoimmunity and neuroprotection, the present study used experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis that is associated with optic neuritis in more than 90% of animals. This was achieved via myelin oligodendrocyte glycoprotein (MOG) immunization of both PrPC-deficient mice and mice overexpressing PrPC compared with wild-type (WT) counterparts. Despite the inflammatory attack of the optic nerve being elevated in both genetically modified mice, there was a strong correlation between PrPC expression levels and neuronal survival.
Materials and Methods
Animals
Female mice aged 6 to 8 weeks were used in all experiments, and were kept under environmentally controlled conditions. Mice were used with a targeted disruption of the prnp gene, originally termed Zürich I,35 that had been back-crossed into a C57Bl/6 background for 10 generations. WT C57Bl/6N mice were purchased from Charles River Laboratories, Inc. (Sulzfeld, Germany). The Tg35 transgenic line of PrPC overexpressing mice was also used. These animals carry a cosmid transgene that encodes the mouse PrPC b allele, leading to a four- to fivefold increase in expression of PrPC.36 The WT animals for this strain were C57Bl/6 × 129/sv(ev) crossbreeds, as previously described.35
All experiments were performed in compliance with the relevant laws and institutional guidelines. The experiments were approved by the local authorities of the Saar-Pfalz Kreis, Germany.
Induction and Evaluation of EAE
Mice were immunized subcutaneously in the flanks with 300 μg MOG 35–55 in PBS emulsified in an equal volume of complete Freund's adjuvant (Sigma-Aldrich Corp., St. Louis, MO) supplemented with Mycobacterium tuberculosis H37RA (Difco Laboratories Inc., Detroit, MI) to a final concentration of 10 mg/mL. Immediately afterward and again 48 hours later, mice received intraperitoneal injections of 300 ng pertussis toxin (List Biological Laboratories, Inc., Campbell, CA). Every day, animals were weighed and disease severity assessed. Disease severity was scored on a scale of 0 to 5, where 0 indicated no clinical disease; 0.5, distal paresis of the tail; 1.0, complete paralysis of the tail; 1.5, paresis of the tail and slightly impaired righting reflex; 2.0, gait ataxia and severely reduced righting reflex; 2.5, bilateral severe hindlimb paresis; 3.0, complete bilateral hindlimb paralysis; 3.5, complete bilateral hindlimb paralysis and weakness of forelimbs; 4, paralysis of hindlimbs and paresis of forelimbs; 4.5, paralysis of hindlimbs and paralysis of forelimbs; and 5, moribund state or death. Mice sham-immunized with emulsion lacking MOG 35–55 served as controls.
Retrograde Labeling of Retinal Ganglion Cells
One week before immunization, all mice were anesthetized via intraperitoneal injection of 10% ketamine (150 mg/kg; Atarost GmbH and Co., Twistringen, Germany) and 2% xylazine (10 mg/kg; Albrecht GmbH & Co. KG, Aulendorf, Germany), the skin was excised mediosagitally, and holes were drilled in the skull above each superior colliculus (2.92 mm dorsal and 0.5 mm lateral from Bregma). Both superior colliculi were injected stereotactically with 1.5 μL fluorescent dye (Fluoro-Gold 5% in normal saline solution; Fluorochrome LLC, Denver, CO) at a rate of 0.75 μL/min.
Quantification of Retinal Ganglion Cell Density
Retinas were dissected and flat-mounted onto glass slides. Retinal ganglion cells (RGCs) were visualized via fluorescence microscopy (Axioplan 2; Carl Zeiss MicroImaging GmbH, Göttingen, Germany) using a UV filter (365/397 nm). RGC density was determined by counting labeled cells in three areas (62,500 μm2) per retinal quadrant at three different eccentricities (1/6, 3/6, and 5/6) of the retinal radius, in a minimum of eight animals at each time point (both retinas were considered individual data points). Cell counts were performed by two independent investigators who were blinded to the protocol.
Histopathologic Analysis
From each strain, a minimum of 10 mice on each of EAE days 1, 8, and 21 and four control mice received an overdose of ketamine/xylacine and were transcardially perfused with 4% paraformaldehyde in PBS. Retinas were removed, and optic nerves were processed for paraffin embedding. Histopathologic analysis was performed on 0.5-μm paraffin-embedded transverse sections of the optic nerve. Luxol fast blue staining and Bielschowsky's silver impregnation were performed to assess demyelination and axonal disease, respectively, as previously described.37 Immunohistochemistry (IHC) was performed on 0.5-μm paraffin-embedded sections. After antigen retrieval in citrate buffer and blocking, antibodies against Mac-3 (1:200; BD Biosciences Pharmingen Inc., San Diego, CA) to detect activated microglia or macrophages, CD3 (1:150; Dako A/S, Glostrup, Denmark) to detect T cells, and β-amyloid precursor protein (1:1000; Millipore Corp., Billerica, MA) to detect injured axons were diluted in the appropriate serum and incubated overnight at 4°C. After incubation in biotinylated secondary antibodies, avidin-biotin amplification was performed (ABC Elite kit; Vector Laboratories, Inc., Burlingame, CA). For all histopathologic analyses, a minimum of 10 sections from throughout the length of each optic nerve were quantified.
Analysis of Spinal Cord Lesions
Histologic analysis was performed on paraformaldehyde-fixed paraffin-embedded serial sections of spinal cords, in a minimum of eight mice from each strain, at EAE day 21. Demyelination was assessed using Luxol fast blue stain, and inflammation was analyzed by IHC using antibodies against Mac-3 and CD3 (see Histopathologic Analysis). Spinal cord histopathologic features were analyzed as previously described.37 The degree of demyelination was evaluated semiquantitatively using the following scoring system: 0.5, traces of perivascular or subpial demyelination; 1, marked perivascular or subpial demyelination; 2, confluent perivascular or subpial demyelination; 3, demyelination of half spinal cord cross section; and 4, transverse myelitis. Quantitative assessment of Mac-3– and CD3-positive cells and β-amyloid precursor protein–positive axons was achieved by determining the number of positive cells or axonal profiles in an average of 15 complete spinal cord cross sections. Values were converted to millimeters squared.
Western Blot Analysis
Animals were sacrificed using CO2, and retinas, brains, spinal cords, and optic nerves were removed. Samples were mechanically homogenized in ice-cold lysis buffer (50 mmol/L Tris HCl, 150 mmol/L NaCl, and 1% Triton X-100) containing inhibitors (complete protease inhibitor cocktail; Roche Diagnostics GmbH, Mannheim, Germany), and protein concentration was determined (BCA assay; Pierce Chemical Co., Rockford, IL). Lysates were separated by reducing SDS-PAGE, and protein was transferred to a polyvinylidene difluoride membrane and blocked with 5% skim milk powder in 0.1% Tween-20 in PBS. Membranes were incubated in the primary antibody [PrPC, 1:100 (Bio-Rad Laboratories GmbH, Munich, Germany); β-tubulin, 1:1000 (Sigma-Aldrich Corp.); Phospho-Stat1, 1:1000 Phospho-SAPK/JNK, 1:1000, and phospho-p44/42 MAPK, 1:1000 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 1:250 (Millipore Corp., Billerica, MA)] in either 5% skim milk powder (PrPC, phospho-p44/42 MAPK, and GAPDH) or 5% bovine serum albumin (Phospho-Stat1 and Phospho-SAPK/JNK) in 0.1% Tween-20, overnight at 4°C. Membranes were then washed and incubated with horseradish peroxidase–conjugated secondary antibodies (1:7000; GE Healthcare, Munich, Germany), and proteins were detected (ECL Plus reagent; GE Healthcare). Western blot analysis was repeated three times on lysates from four animals at each time measured.
Statistical Analysis
Student's t-tests were used for comparison. All data are given as mean ± SEM. P < 0.05 was considered statistically significant.
Results
PrPC Is Up-Regulated Throughout the Course of EAE
EAE was induced in C57Bl/6 mice via immunization with MOG 35–55, resulting in autoimmune optic neuritis, as characterized by inflammation and demyelination of the optic nerves, in 95% of mice (data not shown). The expression levels of PrPC were assessed using Western blot analysis in tissue of the optic system (retina and optic nerve) and central nervous system (brain and spinal cord). Although detectable levels were observed in all healthy samples, levels were up-regulated in all tissue examined on the first day of clinical symptoms (EAE day 1), and remained high throughout the course of the disease to day 21 after onset of clinical symptoms (EAE day 21), the last time examined (Figure 1, A and D).
Figure 1.

PrPC is up-regulated during EAE. A: PrPC expression was analyzed in WT C57Bl/6 mice in various tissue compartments throughout EAE. In all compartments analyzed (brain, spinal cord, retina, and optic nerves), PrPC expression was increased from the first day of symptoms of EAE and remained elevated until 21 days after onset of EAE, the last day of the experiment. B: PrPC levels were compared between brains from the prion-overexpressing Tg35 mouse line, the Prnp−/− knockout line, and their respective WT counterparts (WT-Tg35 mice, 129B6; and WT-Prnp, C57Bl/6). As expected, PrPC expression was greatly enhanced in the Tg35 line compared with the level in WT mice (Tg35 4.12-fold higher than WT), and was completely abolished in the Prnp−/− line. Two glycosylated forms (solid arrow) and one nonglycosylated form (open arrow) were visible. C: Quantification of PrPC expression in transgenic mice compared with their WT counterparts. D: Quantification of PrPC expression in various tissue compartments throughout EAE. *P < 0.05.
Both PrPC-Deficient Mice and Mice Overexpressing PrPC Have Increased EAE Disease Severity
Levels of PrPC expression were confirmed in brain lysates from Prnp−/− and Tg35 mice and their respective WT counterparts [WT-PrnP (C57Bl/6) and WT-Tg35 (129B6)]. As expected, and in accordance with previously published results,36 Tg35 mice demonstrated an approximate four- to fivefold higher level of PrPC expression, as compared with WT controls [4.12-fold (± 0.97-fold) higher; n = 4; Figure 1C], whereas Prnp−/− mice demonstrated undetectable levels of PrPC (Figure 1, B and C).
Subsequently, 15 animals of each strain were immunized as previously described and followed up until EAE day 21, during which period the clinical deficit was scored daily on a scale of 0 to 5 (Figure 2, A and B). The disease score was significantly elevated in both Tg35 and Prnp−/− mice throughout the disease course, as compared with WT control mice, and analysis of spinal cords from all strains was performed on EAE day 21 to investigate the histopathologic changes responsible for the observed increase in disease severity. Correspondingly, compared with WT control mice, both Prnp−/− and Tg35 mice demonstrated significantly more demyelination: Prnp−/−, 1.12 ± 0.077 versus WT, 1.46 ± 0.075 (P = 0.036); and Tg35, 1.28 ± 0.116 versus WT, 1.67 ± 0.1 (P = 0.026) (Figure 2C). Furthermore, Prnp−/− and Tg35 mice exhibited an increased number of Mac-3–positive activated microglia and macrophages in the spinal cord at EAE day 21: Prnp−/−, 281.74 ± 57.56 versus WT, 136.93 ± 28.47 (P = 0.049); and Tg35, 335.19 ± 31.77 versus WT, 155.03 ± 28.47 (P = 0.047) (Figure 2D). These data were mirrored by a significant increase in the number of CD3-positive T cells in both Prnp−/− and WT mice at EAE day 21: Prnp−/−, 244.37 ± 54.99 versus WT, 112.15 ± 35.46 (P = 0.031); and Tg35, 352.07 ± 89.79 versus WT, 96.71 ± 17.94 (P = 0.032) (Figure 2E).
Figure 2.
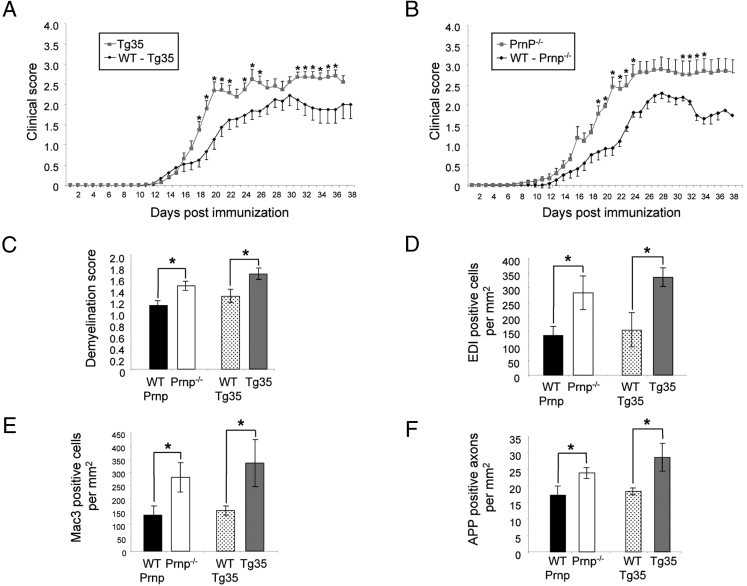
Clinical EAE symptoms and spinal cord lesions in Prnp transgenic mice. A: Clinical scores of Tg35 mice and their WT counterparts, 129B6 (WT-Tg35), were monitored after active immunization with MOG. Scores were significantly elevated in Tg35 mice. B: On comparing the clinical progression of EAE between Prnp−/− mice and their WT counterparts, C57Bl/6 (WT-Prnp) mice, disease severity was more severe in the Prnp−/− mice. Spinal cords were assessed by histopathologic analysis at EAE day 21, and demonstrated an increase in both transgenic strains (Prnp−/− and Tg35) compared with their WT counterparts with respect to demyelination (C), infiltration of Mac-3–positive cells (D) and CD3 positive cells (E), and axonal degeneration (F). *P < 0.05.
The extent of axonal degeneration in the spinal cord was also determined by IHC for β-amyloid precursor protein. At EAE day 21, compared with WT control mice, both Prnp−/− and Tg35 mice demonstrated a significant increase in the number of β-amyloid precursor protein–positive axonal profiles: Prnp−/−, 23.82 ± 1.63 versus WT, 17.17 ± 2.78 (P = 0.049); and Tg35, 28.59 ± 4.21 versus WT, 18.26 ± 1.02 (P = 0.047) (Figure 2F).
Tg35 and Prnp−/− Mice Have More Severe Optic Neuritis
To evaluate the extent of optic neuritis, infiltration of inflammatory cells was studied together with an assessment of the extent of demyelination of the optic nerves. The number of inflammatory cells in the optic nerves in immunized animals was assessed by IHC for activated microglia and macrophages and for T cells. Ten animals were assessed at each time point measured. As expected, in the WT mouse strains, infiltration of both CD3-positive and Mac-3–positive cells was observed in the optic nerves of mice, starting on EAE day 1 (Figure 3, E and J). However, the number of positive cells counted in optic nerve cross-sections from the genetically modified mice was significantly increased. Prnp−/− mice demonstrated significantly elevated numbers of CD3-positive cells starting on EAE day 1 [Prnp−/−, 1886.85 ± 397.46 versus WT, 1007.06 ± 128.33 (P = 0.011)] and continuing at both EAE day 8 [Prnp−/−, 1226.10 ± 106.74 versus WT, 1007.61 ± 163.04 (P = 0.00078)] and EAE day 21 [Prnp−/−, 1025.40 ± 81.61 versus WT, 615.97 ± 93.63 (P = 0.00078)]. Similarly, Tg35 mice demonstrated greater numbers of CD3-positive cells at EAE day 1 [Tg35, 3645.73 ± 521.51 versus WT, 1102.54 ± 201.65 (P = 0.000079)], EAE day 8 [Tg35, 2131.23 ± 581.65 versus WT, 60.87 ± 219.51 (P = 0.018)], and EAE day 21 [Tg35, 1364.40 ± 396.57 versus WT, 464.10 ± 85.09 (P = 0.0013)] (Figure 3, A–E).
Figure 3.
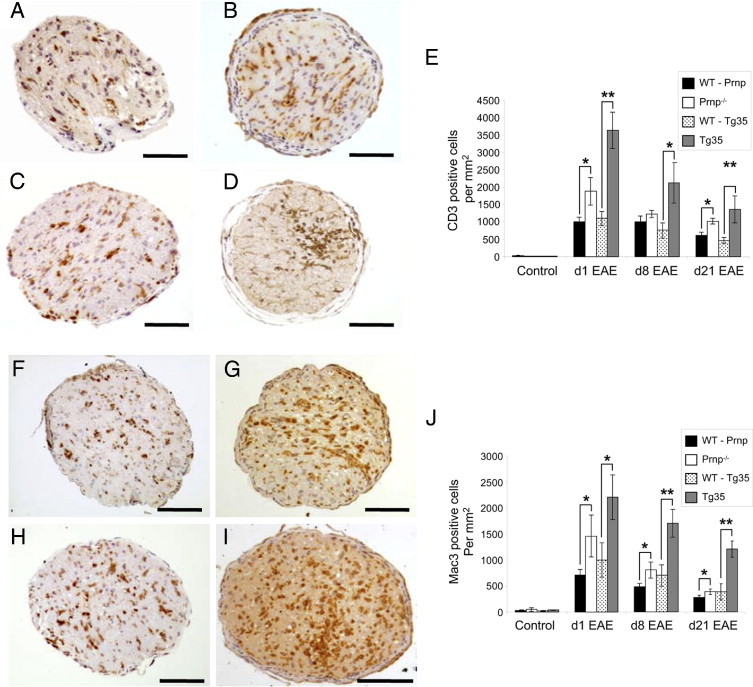
Inflammatory infiltration of optic nerves during EAE. A–D: Representative images of optic nerve cross-sections labeled with an antibody against CD3 at EAE day 1 in WT-Prnp (A), Prnp−/− (B), WT-Tg35 (C), and Tg35 (D) mice. E: Quantification of CD3-positive cells at various stages of EAE. Compared with WT mice, significantly greater numbers of CD3-positive cells were observed in optic nerves of Tg35 mice at all stages analyzed and Prnp−/− mice at days 1 and 21 of EAE. F–I: Representative images of optic nerve cross-sections labeled with an antibody against Mac-3 at EAE day 1 in WT-Prnp (F), Prnp−/− (G), WT-Tg35 (H), and Tg35 (I) mice. J: Quantification of infiltrating Mac-3–positive cells demonstrated increased numbers of Mac-3–positive cells at all times analyzed in both Prnp−/− and Tg35 mice, as compared with their WT counterparts. Scale bars: 100 μm (A–D and F–I). *P < 0.05. **P < 0.01.
The numbers of Mac-3–positive activated microglia and macrophages were also significantly increased at all time points measured in both the Prnp−/− and Tg35 mice (Figure 3, F–J). Compared with their WT counterparts, Prnp−/− mice demonstrated significantly more Mac-3–positive cells at EAE day 1 [Prnp−/−, 1464.60 ± 401.88 versus WT, 713.51 ± 108.51 (P = 0.027)], EAE day 8 [Prnp−/−, 545.29 ± 74.28 versus WT, 394.8 ± 56.94 (P = 0.032)], and EAE day 21 [Prnp−/−, 567.2 ± 109.28 versus WT, 338.87 ± 49.33 (P = 0.0092)]. Similarly, Tg35 mice demonstrated greater numbers of Mac-3–positive cells at EAE day 1 [Tg35, 2212.2 ± 428.75 versus WT, 541.35 ± 221.01 (P = 0.041)], EAE day 8 [Tg35, 1699.32 ± 217.01 versus WT, 541.35 ± 221.01 (P = 0.0019)], and EAE day 21 [Tg35, 1208.20 ± 192.43 versus WT, 498.98 ± 144.04 (P = 0.0039)].
In agreement with the data for inflammation, demyelination of the optic nerves was also more severe in both the Prnp−/− and Tg35 mice (Figure 4, F–J). Compared with their WT counterparts, Tg35 mice demonstrated increased demyelination starting on EAE day 1 [Tg35, 12.33% ± 2.13% versus WT, 4.28% ± 1.00% (P = 0.0056)] and continuing at both EAE day 8 [Tg35, 32.51% ± 7.20% versus WT, 6.37% ± 1.35% (P = 0.0035)] and EAE day 21 [Tg35, 48.05 ± 5.54% versus WT, 15.64% ± 1.68% (P = 0.00052)]. Prnp−/− mice demonstrated a trend toward increased demyelination; however, this reached significance only on EAE day 8 [Prnp−/−, 13.94% ± 2.94% versus Tg35, 7.47% ± 1.66% (P = 0.049)].
Figure 4.

Axonal degeneration and demyelination of optic nerves during EAE. A–D: Representative images of optic nerve cross-sections stained using Bielschowsky's silver impregnation to label axons at EAE day 21 in WT-Prnp (A), Prnp−/− (B), WT-Tg35 (C), and Tg35 (D) mice. B: Red arrow indicates an axon, and white arrow, a macrophage. E: Quantification of the number of surviving axons at various stages of EAE. Significantly decreased numbers of axons were present in Prnp−/− mice at EAE days 1 and 8, and in Tg35 mice at EAE days 1, 8, and 21, as compared with their respective WT counterparts. F–I: Representative images of optic nerve cross-sections at EAE day 21 stained using Luxol fast blue to label myelin in WT-Prnp (F), Prnp−/− (G), WT-Tg35 (H), and Tg35 (I) mice. J: Quantification of the percentage of demyelination of optic nerves showed significantly higher demyelination in Prnp−/− mice on EAE day 8 and in Tg35 mice at EAE days 1, 8, and 21. Scale bars: 20 μm (A–D); 100 μm (F–I). *P < 0.05. **P < 0.01.
In parallel with the increased inflammation and demyelination observed in the genetically modified mice, axonal numbers were also significantly decreased in these animals (Figure 4, A–E). As observed using Bielschowsky's silver impregnation method, Prnp−/− mice demonstrated significantly reduced numbers of axons at EAE day 1 [Prnp−/−, 71.1% ± 9.57% versus WT, 89.03% ± 1.90% (P = 0.00092)] and EAE day 8 [Prnp−/−, 63.95% ± 10.14% versus WT, 83.22% ± 1.94% (P = 0.013)]. Similarly, compared with WT mice, Tg35 mice demonstrated significantly reduced numbers of surviving axons at EAE day 1 [Tg35, 72.05 ± 5.02% versus WT, 91.17 ± 2.40% (P = 0.0014)], EAE day 8 [Tg35, 56.24 ± 6.88% versus WT, 82.05 ± 4.67% (P = 0.033)], and EAE day 21 [Tg35, 45.37 ± 4.49% versus WT, 63.43 ± 6.16% (P = 0.0256)].
These findings suggest that inflammation was greater in both Tg35 and Prnp−/− mice as compared with their WT controls and is presumed responsible for the greater levels of demyelination and axonal degeneration observed in these animals. This was in agreement with both the spinal cord analysis and clinical scores (Figure 2).
PrPC Expression Correlates with Neuroprotection of RGCs
To assess the neuroprotective effect of PrPC expression on RGC survival during autoimmune optic neuritis, RGCs were prelabeled via injection of the retrograde tracer Fluoro-Gold into the superior colliculi 1 week before immunization. After immunization, animals were kept until day 21 after onset of clinical symptoms before retinas were removed and flat-mounted for analysis. Healthy nonimmunized animals that were labeled with Fluoro-Gold but kept until the end of the EAE experiments (approximately 50 days after labeling) served as age-matched controls (n = 4 in each group). Each group comprised similar numbers of Fluoro-Gold–labeled RGCs (WT-Prnp, 2031.99 ± 73.52; Prnp−/−, 1983.93 ± 109.09; WT-Tg35, 2060 ± 69.09; and Tg35, 2215.52 ± 41.22; n > 6 per group), which demonstrated that age-related loss of RGCs did not interfere with the experiment; therefore, the observed loss of RGCs was due to MOG-induced EAE. Quantification of surviving RGCs revealed that in mice deficient in PrPC expression (Prnp−/−), significantly lower numbers of RGCs were present at EAE day 21 (757.49 ± 23.59, n = 10; Figure 5 E), as compared with their WT controls (1121.82 ± 109.22, n = 14, P = 0.036). In contrast, significantly greater numbers of RGCs were detected in Tg35 mice (1530.84 ± 142.59, n = 12), as compared with Tg35 WT controls (WT-Tg35, 1116.78 ± 142.59, n = 12, P = 0.026). In both WT strains, values were similar. Thus, the level of PrPC expressed correlated with the survival of RGCs at EAE day 21.
Figure 5.
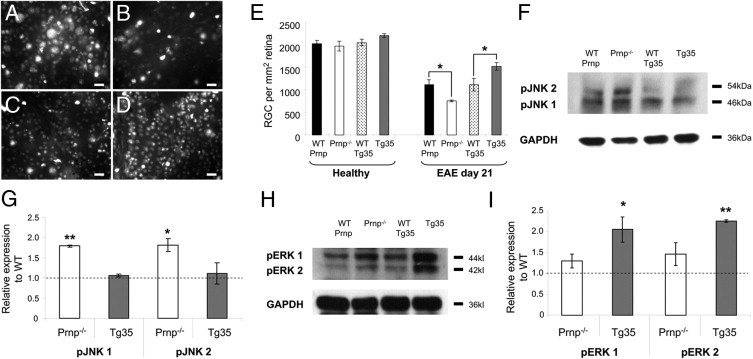
Neurodegeneration of RGCs at EAE day 21 and influences of PrPC on intracellular signaling pathways. A–D: Representative images of retinal whole mounts demonstrate Fluoro-Gold–labeled RGCs at EAE day 21, taken from WT-Prnp (A), Prnp−/− (B), WT-Tg35 (C), and Tg35 (D) mice. E: Quantification of the number of RGCs in healthy (nonimmunized) age-matched mice demonstrates no significant differences between all mice strains, and quantification of the number of surviving RGCs at EAE day 21 demonstrates that relative to their WT counterparts, a significant decrease in the number of surviving RGCs is observed in Prnp−/− mice, whereas a significant increase in the number of surviving RGCs is observed in Tg35 mice. F: Representative Western blots using antibodies against phosphorylated forms of JNK 1 and 2 (46 and 54 kDa, respectively). G: Quantification of pJNK 1 and 2 levels (relative to GAPDH levels) in genetically modified mice compared with their WT counterparts. H: Representative Western blots using antibodies against phosphorylated forms of ERK 1 and 2 (44 and 42 kDa, respectively). I: Quantification of pERK 1 and 2 levels (relative to GAPDH levels) in genetically modified mice as compared with their WT counterparts. Scale bars: 20 μm (A–D). *P < 0.05. **P < 0.01.
To assess signaling pathways affected in Prnp−/− and Tg35 mice, retinas from animals at EAE day 21 were analyzed using Western blot analysis for the levels of phosphorylated Jun-amino-terminal kinase (pJNK) 1 and 2, extracellular signal-regulated kinase (pERK) 1 and 2, and pStat1 because these proteins have been implicated in the effects of PrpC on neuronal survival.19,38 In retinal lysates obtained from animals on EAE day 21, significant elevation of pJNK 1 and 2 was observed in Prnp−/− mice as compared with their WT counterparts [pJNK 1, 1.791 ± 0.027 (P = 0.0084); pJNK 2, 1.725 ± 0.31 (P = 0.016)]; however, these were unchanged in Tg35 mice (Figure 5, F and G). Conversely, pERK 1 and 2 were both elevated in Tg35 mice as compared with WT mice [pERK 1, 2.055 ± 0.30 (P = 0.039), and pERK 2, 2.225 ± 0.03 (P = 0.0029)] but were not significantly changed in Prnp−/− mice (Figure 5, H and I). No significant changes were observed in pStat1 in either genetically modified mouse strain (data not shown).
Discussion
To assess the potential neuroprotective role of PrpC, in the present study, a knockout mouse strain deficient in PrpC (Prnp−/−) and a PrpC-overexpressing transgenic mouse strain (Tg35) were used. Autoimmune optic neuritis was induced in these animals and their WT counterparts via immunization using MOG 35–55. Analysis of the EAE disease course revealed that both genetically modified strains demonstrated an increased clinical score, associated increased demyelination, increased infiltration of T cells and activated microglia and macrophages in the spinal cord, and increased axonal degeneration. In the optic nerves, increased infiltration of inflammatory cells was also observed, correlating with increased demyelination and axonal loss that was also observed in both strains of mice as compared with their respective WT counterparts. In contrast, by analyzing the numbers of surviving RGCs at EAE day 21, a clear neuroprotective role of PrpC was demonstrated. In PrpC-deficient mice, there was a decrease in the number of surviving RGCs, whereas in PrpC-overexpressing mice, there was a significant increase in the number of surviving RGCs. These results confirm a potential neuroprotective role for PrpC, as has been suggested in previous studies, predominantly in models of ischemic brain injury,16–21 in which expression levels of PrpC correlated with infarct volume.18,19
It was not demonstrated that this neuroprotection is functional. Although PrpC overexpression resulted in preservation of RGC cell bodies, it did not protect axons from degeneration, presumably because of the concomitant increase in infiltration. Because of this reduced axonal survival, functional protection could not be demonstrated via measurement of pattern visual evoked potentials, which depend on maintenance of axonal integrity (data not shown).
Increased levels of PrpC were also observed throughout the central nervous system from onset of EAE and for the next 3 weeks. In other studies, PrpC was also observed to be up-regulated after both permanent and transient focal ischemia in mice.16,17,20 It could be hypothesized that PrpC is up-regulated as part of an adaptive neuronal survival mechanism, as supported by the observation of increased mitogen-activated protein kinase phosphorylation in PrPC-overexpressing mice or, alternatively, from decreased protein turnover. However, it is highly likely that much of the increased PrPC protein level observed was due to the infiltration of the central nervous system by both T cells and activated microglia and macrophages that occurs in EAE. Since cell surface expression of PrpC increases in T cells on activation,39 this provides an additional mechanism for PrpC up-regulation during the course of EAE.
Analysis of the EAE clinical scores demonstrated no significant changes in the day of onset of disease; however, in Prnp−/− mice, the course of disease was more severe, consistent with findings of other recent studies.34,39,40 In addition, the disease course in Tg35 mice was also more severe, in contrast to previously published data.34 The study by Hu et al34 used not only a different mode of EAE induction (adoptive T-cell transfer as opposed to active immunization with MOG) but also a different transgenic PrpC-overexpressing mouse line (Tga20 as opposed to Tg35), which may be crucial in explaining these varying results. Differences in immune cell phenotype have been reported between Tga20 mice and a further PrpC-overexpressing line, Tga19, possibly due to the positional genomic insertion of the Tga20 transgene near a region of the chromosome important for T-cell development.41
During EAE, increased inflammatory activity occurs in mice lacking prion protein.40 However, this study did not directly analyze the degeneration of neuronal cell bodies, and only axonal survival was quantified. Since increased inflammation was observed, it is difficult to determine whether the axonal loss observed resulted from loss of the neuroprotective role of PrpC or from the increased inflammation. Due to the higher disease score and greater infiltration observed in both strains, determination of the neuroprotective role of PrpC in an inflammatory model is necessary. To achieve this, the EAE-associated autoimmune optic neuritis model was used, which enables consecutive analysis of the inflammatory effects within the optic nerves, where RGC axons reside, as well as any neurodegenerative changes in the RGC cell bodies, which are compartmentalized in the retina.
In the Prnp−/− mice, neurodegeneration, as assessed by quantification of surviving RGCs, was elevated at the end of the disease course. It was presumed that this resulted from loss of the protective PrpC function, although it could have resulted from enhanced inflammatory activity in these mice. However, in the Tg35 mice, although increased axonal loss in the optic nerves was observed along with increased demyelination and inflammatory activity, this did not correlate with the numbers of cell bodies surviving in the retina when there was a distinct neuroprotective effect. To explain the increased inflammation observed in the Tg35 mice, it is possible that the anti-apoptotic effects of prion protein9,10,25 may also protect immune cells from apoptosis, resulting in an increase in their number. In contrast, the lack of prion protein in the Prnp−/− mice may have exacerbated the immune response by shifting the cytokine profile affecting the balance of Th1- to Th17-positive T cells, as hypothesized by Ingram et al.39 This has been suggested to result in increased proliferation and activation of T cells, which may also increase macrophage infiltration through active breakdown of the blood-brain barrier, and direct activation of macrophages.
Analysis of signaling pathways affected in the genetically modified mice strains provided further evidence in support of the neuroprotective role of PrPC. The increased neurodegeneration in Prnp−/− mice was accompanied by an increase in the activity of JNK, which is a key regulator of apoptosis.42 Correspondingly, the increased survival of RGCs in the Tg35 mice was accompanied by an increase in ERK 1 and ERK 2 activity, which is indicative of an elevation in endogenous neuroprotection, as previously documented in our autoimmune optic neuritis.43 Although previously, elevated ERK activity was reported in Prnp−/− mice, in contrast to PrpC-overexpressing mice,38 this was observed rapidly after acute damage induced by ischemia, and, therefore, may represent a rapid pro-apoptotic signaling arm associated with this model44 rather than a more sustained elevation representing its documented function as a mediator of cell survival.45,46 Considered together, JNK and ERK activity work antagonistically to determine whether a cell progresses to apoptosis or survival,47 and, thus, it is conceivable that one of the physiologic functions of PrpC may be to regulate the balance of signaling pathways that determine cell fate. The neuroprotection observed may also be mediated by an as yet unidentified pathway, and further studies are required to directly demonstrate the mechanism by which PrPC promotes neuronal survival in this model.
In conclusion, although PrPC has an important role in determining the immune response, it also independently regulates the susceptibility of neurons to degeneration. Further work to determine whether both functions are mediated by the same downstream signaling pathways may aid attempts to reduce immune responses during autoimmune diseases while concomitantly increasing the ability of neurons to withstand degeneration.
Acknowledgments
We thank Nadine Meyer, Ina Boger, and Marika Dienes for technical assistance.
Footnotes
Supported by the FP6 Program of the European Union (LSHM-CT-2005-018637; NeuroproMiSe).
References
- 1.Brown D.R., Qin K., Herms J.W., Madlung A., Manson J., Strome R., Fraser P.E., Kruck T., von Bohlen A., Schulz-Schaeffer W., Giese A., Westaway D., Kretzschmar H. The cellular prion protein binds copper in vivo. Nature. 1997;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- 2.Pruisner S. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sales N., Rodolfo K., Hassig R., Faucheux B., Di Giamberardino L., Moya K.L. Cellular prion protein localization in rodent and primate brain. Eur J Neurosci. 1998;10:2464–2471. doi: 10.1046/j.1460-9568.1998.00258.x. [DOI] [PubMed] [Google Scholar]
- 4.Caughey B., Chesebro B. Transmissible spongiform encephalopathies and prion protein interconversions. Adv Virus Res. 2001;56:277–311. doi: 10.1016/s0065-3527(01)56031-5. [DOI] [PubMed] [Google Scholar]
- 5.White A.R., Enever P., Taybei M., Mushens R., Linehan J., Brandner S., Anstee D., Collinge J., Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422:80–83. doi: 10.1038/nature01457. [DOI] [PubMed] [Google Scholar]
- 6.Mange A., Milhavet O., Umlauf D., Harris D., Lehmann S. PrP-dependent cell adhesion in N2a neuroblastoma cells. FEBS Lett. 2002;514:159–162. doi: 10.1016/s0014-5793(02)02338-4. [DOI] [PubMed] [Google Scholar]
- 7.Lopes M.H., Hajj G.N., Muras A.G., Mancini G.L., Castro R.M., Ribeiro K.C., Brentani R.R., Linden R., Martins V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J Neurosci. 2005;25:11330–11339. doi: 10.1523/JNEUROSCI.2313-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanaani J., Prusiner S.B., Diacovo J., Baekkeskov S., Legname G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J Neurochem. 2005;95:1373–1386. doi: 10.1111/j.1471-4159.2005.03469.x. [DOI] [PubMed] [Google Scholar]
- 9.Bounhar Y., Zhang Y., Goodyer C.G., LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem. 2001;276:39145–39149. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- 10.Brown D.R., Nicholas R.S., Canevari L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res. 2002;67:211–222. doi: 10.1002/jnr.10118. [DOI] [PubMed] [Google Scholar]
- 11.Gauczynski S., Peyrin J.M., Haïk S., Leucht C., Hundt C., Rieger R., Krasemann S., Deslys J.P., Dormont D., Lasmézas C.I., Weiss S. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001;20:5863–5865. doi: 10.1093/emboj/20.21.5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spielhaupter C., Schätzl H.M. PrPc directly interacts with proteins involved in signaling pathways. J Biol Chem. 2001;276:44604–44612. doi: 10.1074/jbc.M103289200. [DOI] [PubMed] [Google Scholar]
- 13.Pan T., Wong B.S., Liu T., Li R., Petersen R.B., Sy M.S. Cell-surface prion protein interacts with glycosaminoglycans. Biochem J. 2002;368:81–90. doi: 10.1042/BJ20020773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bremer J., Baumann F., Tiberi C., Wessig C., Fischer H., Schwarz P., Steele A.D., Toyka K.V., Nave K.-A., Weis J., Aguzzi A. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13:310–318. doi: 10.1038/nn.2483. [DOI] [PubMed] [Google Scholar]
- 15.Brown D.R., Schulz-Schaeffer W.J., Schmidt B., Kretzschmar H.A. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp Neurol. 1997;146:104–112. doi: 10.1006/exnr.1997.6505. [DOI] [PubMed] [Google Scholar]
- 16.McLennan N.F., Brennan P.M., McNeill A., Davies I., Fotheringham A., Rennison K.A., Ritchie D., Brannan F., Head M.W., Ironside J.W., Williams A., Bell J.E. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol. 2004;165:227–235. doi: 10.1016/S0002-9440(10)63291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weise J., Crome O., Sandau R., Schulz-Schaeffer W., Bähr M., Zerr I. Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci Lett. 2004;372:146–150. doi: 10.1016/j.neulet.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 18.Weise J., Sandau R., Schwarting S., Crome O., Wrede A., Schulz-Schaeffer W., Zerr I., Bähr M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke. 2006;37:1296–1300. doi: 10.1161/01.STR.0000217262.03192.d4. [DOI] [PubMed] [Google Scholar]
- 19.Weise J., Doeppner T.R., Müller T., Wrede A., Schulz-Schaeffer W., Zerr I., Witte O.W., Bähr M. Overexpression of cellular prion protein alters postischemic Erk1/2 phosphorylation but not Akt phosphorylation and protects against focal cerebral ischemia. Restor Neurol Neurosci. 2008;26:57–64. [PubMed] [Google Scholar]
- 20.Shyu W.C., Lin S.Z., Chiang M.F., Ding D.C., Li K.W., Chen S.F., Yang H.I., Li H. Overexpression of PrPc by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J Neurosci. 2005;25:8967–8977. doi: 10.1523/JNEUROSCI.1115-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitteregger G., Vosko M., Krebs B., Xiang W., Kohlmannsperger V., Nölting S., Hamann G.F., Kretzschmar H.A. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007;17:174–183. doi: 10.1111/j.1750-3639.2007.00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoshino S., Inoue K., Yokoyama T., Kobayashi S., Asakura T., Teramoto A., Itohara S. Prions prevent brain damage after experimental brain injury: a preliminary report. Acta Neurochir. 2003;86:297–299. doi: 10.1007/978-3-7091-0651-8_64. [DOI] [PubMed] [Google Scholar]
- 23.Couplier M., Messiaen S., Boucreaux D., Eloit M. Axotomy-induced motoneuron death is delayed in mice overexpressing PrpC. Neurosci. 2006;141:1827–1834. doi: 10.1016/j.neuroscience.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 24.Rangel A., Burgaya F., Gavín R., Soriano E., Aguzzi A., Del Río J.A. Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: role of AMPA/kainate receptors. J Neurosci Res. 2007;85:2741–2755. doi: 10.1002/jnr.21215. [DOI] [PubMed] [Google Scholar]
- 25.Chiarini L.B., Freitas A.R.O., Zanata S.M., Brentani R.R., Martins V.R., Linden R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002;21:3317–3326. doi: 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vassallo N., Herms J., Behrens C., Krebs B., Saeki K., Onodera T., Windl O., Kretzschmar H.A. Activation of phosphatidylinositol 3-kinase by cellular prion protein and its role in cell survival. Biochem Biophys Res Comm. 2005;332:75–82. doi: 10.1016/j.bbrc.2005.04.099. [DOI] [PubMed] [Google Scholar]
- 27.Krebs B., Dorner-Ciossek C., Schmalbauer R., Vassallo N., Herms J., Kretzschmar H.A. Prion protein induced signalling cascades in monocytes. Biochem Biophys Res Commun. 2006;340:13–22. doi: 10.1016/j.bbrc.2005.11.158. [DOI] [PubMed] [Google Scholar]
- 28.Harris D.A., True H.L. New insights into prion structure and toxicity. Neuron. 2006;50:353–357. doi: 10.1016/j.neuron.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 29.Cashman N.R., Loertscher R., Nalbantoglu J., Shaw I., Kascsak R.J., Bolton D.C., Bendheim P.E. Cellular isoform of the scrapie agent protein participates in lymphocyte activation. Cell. 1990;61:185–192. doi: 10.1016/0092-8674(90)90225-4. [DOI] [PubMed] [Google Scholar]
- 30.Mabbott N.A., Brown K.L., Manson J., Bruce M.E. T-lymphocyte activation and the cellular form of the prion protein. Immunology. 1997;92:161–165. doi: 10.1046/j.1365-2567.1997.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dürig J., Giese A., Schulz-Schaeffer W., Rosenthal C., Schmücker U., Bieschke J., Dührsen U., Kretzschmar H.A. Differential constitutive and activation-dependent expression of prion protein in human peripheral blood leucocytes. Br J Haematol. 2000;108:488–496. doi: 10.1046/j.1365-2141.2000.01881.x. [DOI] [PubMed] [Google Scholar]
- 32.Ballerini C., Gourdain P., Bachy V., Blanchard N., Levavasseur E., Grégoire S., Fontes P., Aucouturier P., Hivroz C., Carnaud C. Functional implication of cellular prion protein in antigen-driven interactions between T cells and dendritic cells. J Immunol. 2006;176:7254–7262. doi: 10.4049/jimmunol.176.12.7254. [DOI] [PubMed] [Google Scholar]
- 33.de Almeida C.J., Chiarini L.B., da Silva J.P., E Silva P.M., Martins M.A., Linden R. The cellular prion protein modulates phagocytosis and inflammatory response. J Leukocyte Biol. 2005;77:238–246. doi: 10.1189/jlb.1103531. [DOI] [PubMed] [Google Scholar]
- 34.Hu W., Nessler S., Hemmer B., Eagar T.N., Kane L.P., Rutger Leliveld S., Müller-Schiffmann A., Gocke A.R., Lovett-Racke, Ben L.-H., Hussain R.Z., Breil A., Elliott J.L., Puttaparthi K., Cravens P.D., Singh M.P., Petsch B., Stitz L., Racke M.K., Korth C., Stüve O. Pharmacological prion protein silencing accelerates central nervous system autoimmune disease via T cell receptor signalling. Brain. 2010;133:375–388. doi: 10.1093/brain/awp298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bueler H., Fischer M., Lang Y., Bluethmann H., Lipp H.-P., De Armond S.J., Pruisner S.B., Aguet M., Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 36.Fischer M., Rülicke T., Raeber A., Sailer A., Moser M., Oesch O., Brandner S., Aguzzi A., Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 37.Storch M.K., Stefferl A., Brehm U., Weissert R., WallströM E., Kerschensteiner M., Olsson T., Linington C., Lassmann H. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 1995;8:681–694. doi: 10.1111/j.1750-3639.1998.tb00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spudich A., Frigg R., Kilic E., Kilic U., Oesch B., Raeber A., Bassetti C.L., Hermann D.M. Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol Dis. 2005;20:442–449. doi: 10.1016/j.nbd.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Ingram R.J., Isaacs J.D., Kaur G., Lowther D.E., Reynolds C.J., Boyton R.J., Collinge J., Jackson G.S., Altmann D.M. A role of cellular prion protein in programming T-cell cytokine responses in disease. FASEB J. 2009;23:1672–1684. doi: 10.1096/fj.08-116087. [DOI] [PubMed] [Google Scholar]
- 40.Tsutsui S., Hahn J.N., Johnson T.A., Zenobia A., Jirik F.R. Absence of the cellular prion protein exacerbates and prolongs neuroinflammation in experimental autoimmune encephalomyelitis. Am J Pathol. 2008;173:1029–1041. doi: 10.2353/ajpath.2008.071062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zabel M., Greenwood C., Thackray A.M., Pulford B., Rens W., Bujdoso R. Perturbation of T-cell development by insertional mutation of a PrP transgene. Immunology. 2009;127:226–236. doi: 10.1111/j.1365-2567.2008.02944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsuruta F., Sunayama J., Mori Y., Hattori S., Shimizu S., Tsujimoto Y., Yoshioka K., Masuyama N., Gotoh Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004;23:1889–1899. doi: 10.1038/sj.emboj.7600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diem R., Hobom M., Maier K., Weissert R., Storch M.K., Meyer R., Bähr M. Methylprednisolone increases neuronal apoptosis during autoimmune CNS inflammation by inhibition of an endogenous neuroprotective pathway. J Neurosci. 2003;23:6993–7000. doi: 10.1523/JNEUROSCI.23-18-06993.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alessandrini A., Namura S., Moskowitz M.A., Bonventre J.V. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamada M., Tanabe K., Wada K., Shimoke K., Ishikawa Y., Ikeuchi T., Koizumi S., Hatanaka H. Differences in survival-promoting effects and intracellular signaling properties of BDNF and IGF-1 in cultured cerebral cortical neurons. J Neurochem. 2001;78:940–951. doi: 10.1046/j.1471-4159.2001.00497.x. [DOI] [PubMed] [Google Scholar]
- 46.Jin K., Mao X.O., Zhu Y., Greenberg D.A. MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. J Neurochem. 2002;80:119–125. doi: 10.1046/j.0022-3042.2001.00678.x. [DOI] [PubMed] [Google Scholar]
- 47.Xia Z., Dickens M., Raingeaud J., Davis R.J., Greenberg M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]