

Abstract
An earlier study showed that a calix[4]arene could function as a central relay unit to form an ion conductance pathway through a phospholipid bilayer membrane. The present study expands the range of compounds from calix[4]arene to calix[6]arene and incorporates them either as central units or as headgroups, substituting one or more diaza-18-crown-6 residues in functioning hydraphiles. Ion release was assayed by detecting either Na+ or Cl− release from phospholipid vesicles. The ion transport activity for calix[4]arenes in either position is modest, but is almost non-existent when calix[6] residues were incorporated either as head groups or central relay units. The poor activity of the calix[6]arenes may result from an inability to penetrate to the midplane of the bilayer or pass entirely through it to form a conductance pathway. The transmembrane “flip-flop” may result from high polarity or steric bulk, or both. A hydraphile incorporating a single –NHCOC6H4OCH2CONH– as a central relay proved to be an excellent Na+ conductor, but less selective for Cl−. The fact that this new hydraphile molecule shows selectivity for Na + over Cl− transport and possesses two secondary amide residues in the central relay suggests a means to control ion selectivity in synthetic ion transporters.
Introduction
Less than a decade ago, no solid state structure had been reported for any protein channel molecule. In the years preceding the appearance of the Nobel prize winning1,2 structures,3,4 a number of synthetic ion channel mimics were devised in the hope that they could model channel function and reveal mechanistic details of transport. Several different channel designs emerged, some of which were based on the cation complexing and polar properties of crown ethers. The crown compounds served in the earliest model systems as headgroups5 and in others6,7 as what we have termed a “central relay.” The “central relay” that was designed into synthetic channels is equivalent to the “water and ion filled capsule” that was discovered in proteins8 when the first solid state structures were reported.
In considering channel design, there are several structural, steric, and polarity issues that must be considered. These include the span of the channel, the polarity of the headgroups, the presence and type of selectivity filter, the presence and type of central relay, and the general question of whether the channel will be flexible or rigid. These issues have been dealt with in a variety of ways by different investigators.9 Roks and Nolte,10 for example, used stacked isocyanocrown ethers as a rigid channel while Meillon and Voyer11 used crown ethers oriented into a pore by an α-helical peptide backbone. The span of synthetic unimolecular ion channels has varied, but has typically been between 35 and 45 Å. Where a distinguishable central relay was present, there was considerable variation as well. Thus Lehn and co-workers used β-cyclodextrin12 while we13,14 and Fyles et al.15 used crown ethers. The crown ethers often comprised both the selectivity filter and the polar head groups. Of course, entirely different, unique, and successful approaches have been reported.16–20
In addition to these important design variants, chemical characterization and documented function are critical. Demonstrating chemical purity is a routine matter for synthetic laboratories, but assaying ion transport is less common. Various methods have been used to detect ion transport. Probably the most informative technique is the use of planar bilayer voltage clamp methods.21 Although this method gives comprehensive information about channel behavior, it requires expensive instrumentation, considerable time, and operator expertise. Alternative methods use NMR,22 various fluorescent dyes,23 and ion selective electrodes (ISEs).24 Not all methods give identical information, but it is critical for claimed ion transport to be documented or a compound designed to be a channel may not be a transporter at all.
In a previous study, we used calixarenes as central units25 in a crown ether headgroup channel. In these studies, we compared synthetic systems in which the calixarenes were more or less sterically encumbered. Transport in these systems was assayed by using planar bilayer methods. In the work presented here, we have prepared ion transporters using calixarene headgroups in concert with our well-studied diaza-18-crown-6 central relay. The efficacy of these compounds as transporters was determined by using ISE methods to detect ion release from phospholipid vesicles.
Results and discussion
To our knowledge, only limited use has been made of the calixarene unit in ion transporters.26 In our previous report on25 calix[4]arenes as central relay units the structures had the form [PhCH2〈N18N〉(CH2)12]2calix[(CH2)12〈N18N〉CH2Ph]2, where 〈N18N〉 represents 4,13-diaza-18-crown-6.27 Three derivatives were prepared for the present study. One was a control compound in which the calix[4]arene was in the cone conformation. This means that all four of the –(CH2)12〈N18N〉CH2Ph chains emanating from the calixarene were oriented on the same side of that macrocycle. As such, there was insufficient overall length to span the bilayer, although all of the requisite structural elements were present.
Two 1,3-alternate calixarene derivatives were also prepared. These were intended to probe channel formation per se and the ability of a calix[4]arene to pass a cation through the center of the calix. Each arene's para position in the calix[4]arene was either hydrogen (calix-H) or tert-butyl (calix-Bu). The planar bilayer conductance evidence showed that the calix-H and calix-Bu channels were similar in ion transport behavior. Since the tert-butylated calixarene was too sterically hindered to pass Na+ through its center, it was concluded that the amphiphiles were forming pores that afforded Na+ a conductance pathway.25
Design criteria
The calixarene-derived channels reported here incorporate both calix[4]- and calix[6]arenes. The passage of Na+ through the calixarene's interior has been reported.28 To the extent this occurs, passage of Na+ must be more favorable through a calix[6]arene owing to its larger interior size. It is worth noting that determining a precise size for the hole of calixarenes is difficult owing to the conformational dynamics of these macrocycles. Even so, calix[4]arene and calix[6]arene residues were incorporated into putative channels as either head groups or central relays in the expectation that they might be effective but behave differently as transporters.
The compounds of the present study were designed based on the successful benzyl-terminated hydraphiles that are now well characterized. The best studied of these is PhCH2〈N18N〉(CH2)12〈N18N〉(CH2)12〈N18N〉CH2Ph, 1. The strategy was to replace either the two distal diaza-18-crown-6 macrocycles with calixarenes or to replace the central (medial) macrocycle of 1 by a single calixarene. By doing so, it would be possible to assess how the calixarenes compared to diazacrown ethers either as head groups or as central relay elements.
Compounds studied
Eight compounds are compared in the present study. Compounds 1 and 4 were prepared previously. Compound 1 is, as noted above, the best studied13,14 member of the hydraphile family. It is known to span the bilayer29 and to transport Na+ in a unimolecular fashion.30 Its transport efficacy constitutes the benchmark for the present study.
Compounds 2 and 3 were prepared to serve as controls. The 4-acetyloxybenzoic acid central relay of 2 is a fragment of the calixarene central units present in 5 and 6. Similarly, compound 3 is a control for 7 and 8, which are terminated by calix[4]arene or calix[6]arene head groups, respectively.
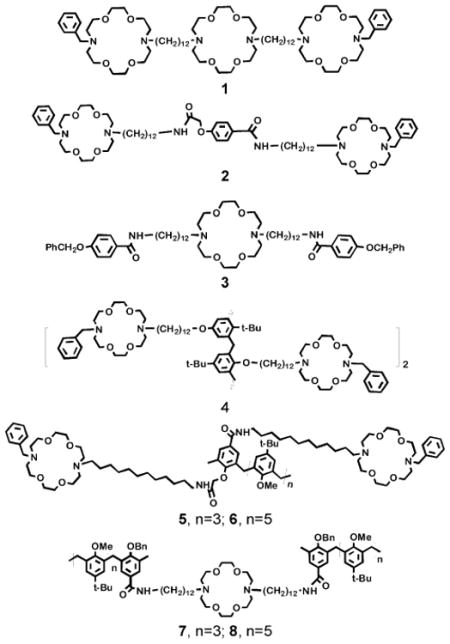
Compounds 5–8 are the major focus of this study. They incorporate calix[4]arene or calix[6]arene residues either as a central relay (5, 6) or as the amphiphile's polar head groups (7, 8). We anticipated that any free hydroxyl groups present on the calix might make these structures too polar to insert into the bilayer. A particular concern in this connection was that the calixes of 5 and 6 needed to reside at the midplane of the bilayer to serve as the central relay unit. The midplane is the least polar portion of the bilayer and a component of the channel having high polarity might simply disrupt the membrane.
An additional issue is that the formation of a conductance pathway requires, in principle at least, compounds 5–8 to extend through both leaflets of the bilayer membrane. If the calix is too large or too polar, it may be unable to penetrate the hydrocarbon regime of the bilayer and anchor on the opposite membrane surface. It is well known that the polar head groups of phospholipids do not readily pass through (“flip-flop”) the bilayer, even though they have high mobility within a single membrane leaflet.
Synthetic access
Compounds 2 and 3 incorporated a 4-hydroxybenzoic acid unit as a mimic of a calixarene, positioned either to function as the central relay (2) or head groups (3). Calix[4]- and calix[6]-arenes were incorporated as central units (5, 6) and as headgroups (7, 8). Compound 2 was prepared in three steps from ethyl 4-hydroxybenzoate. The ester was treated with methyl bromoacetate and K2CO3 in acetone followed by KOH hydrolysis to give 4-(carboxymethyloxy)benzoic acid, 9, and coupling with N-benzyl-N′-12-aminododecyl-4,13-diaza-18-crown-6 (12BDA)13 (63% overall yield). The preparation of 3 was also accomplished from ethyl 4-hydroxybenzoate, by O-benzylation, hydrolysis of the ethyl ester to give 4-benzyloxybenzoic acid, 10, and coupling with N,N′-bis(12-aminodecyl)-4,13-diaza-18-crown-6 (12DA12)13 (64% overall).
The preparation of calix channels 5–8 required the formation of the appropriate calixarene having one (7, 8) or two (5, 6) connection points on the same arene. Once the appropriately functionalized precursors were in hand, a carboxylic acid on the calixarene was coupled (using benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate PyBOP•PF6, see Experimental section) with the appropriate azacrown 12DA12 and 12BDA, respectively. The key calixarene structures are shown as 12,3115,3218, 19, 26 and 27. Compounds 12 and 15 are the starting materials for the calix[4]arene and calix[6]arene channels, respectively. Compounds 18 and 19 are the head groups for channels 7 and 8. The central units of 5 and 6 were prepared from 26 and 27, respectively.
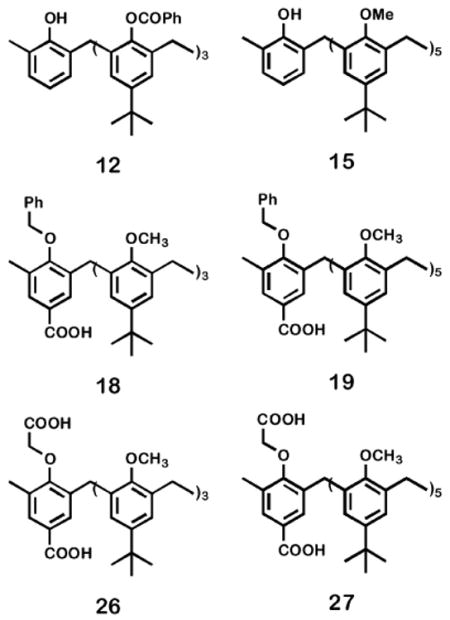
As with the control structures 2 and 3, the preparation of 5–8 required extensive functionalization of the calixarene starting materials (12, 15). Coupling of two 12BDA or one 12DA12 units on calixarenes 26–27 or 18–19, respectively gave 5–8 with good yields. Details of this effort are presented in the Experimental section. Therein, the conformational status of the calixarene derivatives are addressed. The final coupling step and the key calixarene derivatives are shown in Chart 1.
Chart 1.
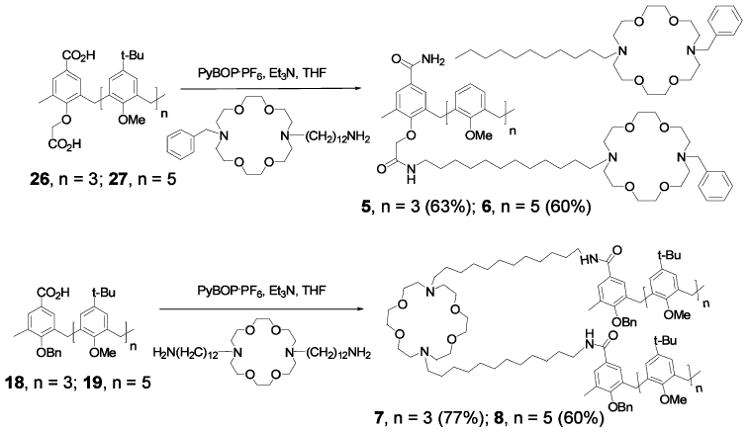
Final coupling steps to form calixarene derivatives 5, 6, 7 and 8.
Ion transport behavior
As noted above, the calixarene transporters were designed to incorporate the successful central relay system of the hydraphiles14 but bring to the system greater size, polarity and rigidity. The latter term may seem ambiguous considering the conformational flexibility of the calixarenes. The much larger surfaces of the arenes were expected to provide a better defined entry portal or central relay than do the crowns. It was known from previous studies that ion transport mediated by 4 did not permit passage of Na+ through the calixarene.25 It was anticipated that this would be the case for calix[4]arene compounds, whether the calix was incorporated as the central relay (5) or as headgroups (7). The calix[6]arene derivatives having larger central relay (6) or headgroup (8) elements were expected to readily pass cations.
The planar bilayer voltage clamp method gives extensive and detailed information concerning ion transport.21 Unfortunately, this technique is both tedious and time-consuming. Considering the number of compounds that were required to be assayed, we decided to use ion selective electrode methods to assess ion egress from liposomes.
The liposomes used in the studies reported here were prepared by the method of Szoka and Papahadjoupolos33 from 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC). In the Na+ release experiments, the internal buffer was 750 mM NaCl–15 mM HEPES buffer at pH 7.0. The liposomes were filtered through a 200 nm membrane and passed over a Sephadex G25 column and then equilibrated in sodium-free buffer (750 mM cholineCl–15 mM HEPES, pH 7.0). In the Cl− release experiments, liposomes were prepared similarly, but loaded with 600 mM KCl and 10 mM HEPES (pH = 7.0), then equilibrated with an external buffer (400 mM K2SO4−10 mM HEPES, pH = 7.0). The expected vesicle size (∼200 nm) was confirmed by light scattering measurements (particle analyzer). Ion efflux was measured by using either a pH/Na+ combination microelectrode24 or a chloride selective membrane electrode relative to the final value determined after vesicular lysis.
The results of the Na+ release studies for compounds 1–3 and 5–8 are shown in the upper panel of Fig. 1. Hydraphile 1 is equal or superior to all of the other compounds studied. It released ∼95% of the available sodium during our customary (arbitrary) 1500 s period of observation. We note that once a conductance pore is formed, ion release occurs nearly instantly. Thus, we presume that the curves shown in Fig. 1 reflect a combination of insertion dynamics and conformational adjustment required to establish an appropriate conductance pathway.
Fig. 1.
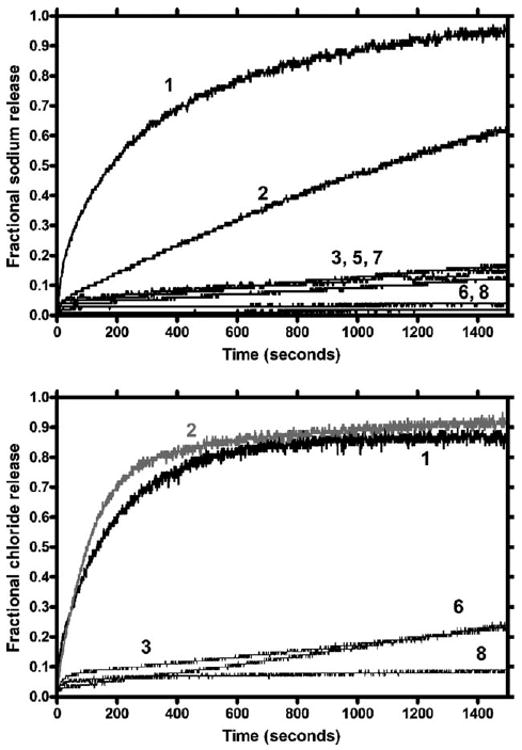
Sodium (upper panel) and chloride (lower panel) release from DOPC:DOPA liposomes mediated by synthetic ionophores.
Compound 2 is similar in overall structure to 1. It incorporates the –NHCOCH2OC6H4CONH– unit, a single element of the calixarene central relay present in compounds 5 and 6. In previous studies34 we noted that replacement of the diazacrown central relay of 1 by 4,4′-bipiperidyl gave a hydraphile that was 40% as active as 1. In this case, 2 is about 64% as active as 1 in releasing Na+ from liposomes. Compound 3 contains the diazacrown central relay but lacks the crown headgroups. Its distal residues were designed to mimic a single calixarene element of 7 and 8. The transport activity of 3 is measurable but too small to be of interest. Likewise, 5 and 7 show very low Na+ transport although it is above baseline. Only 6 and 8 parallel the baseline for the entire 1500 s observation period.
Benzyl channel 1 is an unusually effective Na+ transporter. In previous studies, only hydraphiles having crown or oligo(oxyethylene) central units approached its transport efficacy.14 Thus, the high release observed for 2 is particularly interesting. The central unit of 2, i.e., –NHCOCH2OC6H4CONH–, contains two carbonyl groups that can serve as H-bond acceptors from water and two NH residues that can donate hydrogen bonds to water. The carbonyl groups and the amide NH groups are both spaced about 9 Å apart. The carbonyl group spacing is too large for the carbonyl oxygens to directly ligate with a transient cation—an interaction that should dramatically slow transport. The activity of this compound supports our general understanding of the central relay unit, but is a very interesting alternative to crown ether relays.
The O–O or N–N spacing in diaza-18-crown-6 is typically only about 6 Å. In principle, any interaction with a transient cation should be stronger with a crown owing to this shorter distance. The carbonyl groups of 2 are farther apart but also more polar. It is interesting to note that the selectivity filter of the KcsA potassium selective channel uses carbonyl groups to replace the solvation shell of a transient K+ cation.1 In any event, sodium ion transport selectivity is apparent between 1 and 2.
The Na+ transport ability of 2 stands in contrast to that of 3, which contains a diaza-18-crown-6 central relay. In this case, the PhCH2OC6H4CONH– sidechains that replace the distal diaza-18-crown-6 macrocycles of 1 apparently lack sufficient polarity to function as headgroups or sufficient size to serve as cation entry portals. The presence of this molecule in the liposomal bilayer leads to Na+ release of barely 15% during 1500 s.
Ion transport by calixarene-containing amphiphiles
Compounds 5–8 incorporate calix[4]arenes (5, 7) or calix[6]-arenes (6, 8) as a central relay (5, 6) or as putative headgroups (7, 8). Transport of Na+ by 5 or 7 is modest. The overall transport behavior for 5 and 7 is indistinguishable from that found for 3, i.e., ∼15% at 1500 s. Compounds 6 and 8 show no evidence of Na+ transport during the 1500 s observation period. Taken together, these observations reveal two facts. First, and perhaps obviously, calix[6]arenes are bulkier and more polar than their calix[4]arene counterparts. Second, for the calix[4]arenes, it does not seem to matter whether a single calix is present as a central relay unit or two are incorporated as headgroups. Transport is poor suggesting that the formation of a stable conductance pathway is problematic.
Perhaps the most interesting observation is the inability of compound 8, which incorporates two distal calix[6]arene residues, to transport Na+ or Cl−, shown in the bottom panel of Fig. 1. In the Cl− transport case, a more limited sample of compounds was tested, but the results proved to be interesting. As with Na+ transport, compounds 3 and 6 proved to be only modest transporters, although both ultimately mediated the release of about 25% of the available Cl− ion.
The low transport activity of calixarenes amphiphiles
In order to evaluate the behavior of compounds 5–8, it is useful to consider the presumed active conformations. These are shown in Fig. 2. Diaza-18-crown-6 is represented by the ovals and the blocks represent the calixarenes.
Fig. 2.
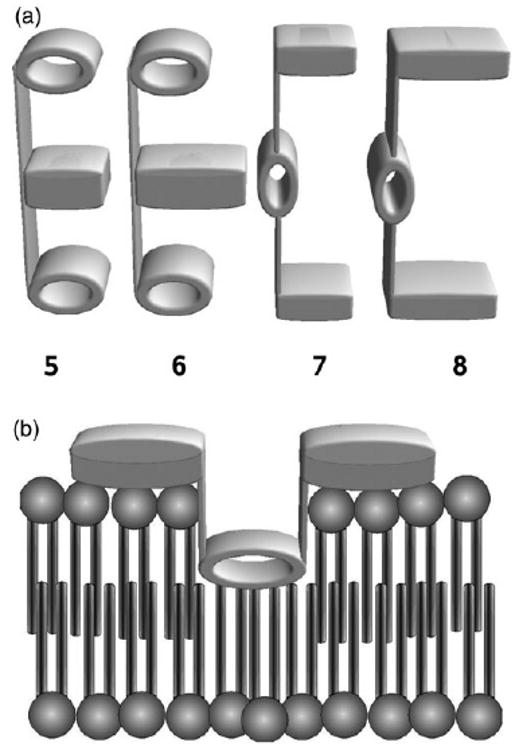
Schematic representations of compounds (left to right) 5, 6, 7 and 8. Postulated non-conducting structure for calixarene derivative 8.
In order for any of these molecules to form a conductance pathway, the molecule must insert into the upper leaflet of the bilayer and then unfold into the lower leaflet, ultimately passing half of the structure through the midplane of the bilayer to the opposite surface. Compounds 5–8 present a challenge in this respect because the calixarenes are both large and polar. The ability of the calixarene central units in either 5 or 6 to reside at the midplane of the bilayer is limited by their polarity and their ability to penetrate the upper leaflet's hydrophobic regime. The problem is exacerbated in 7 because one of the calix[4]arene units must pass all the way from the outer leaflet surface to the inner leaflet surface, i.e., transverse relaxation or “flip-flop”, a process that occurs very slowly. The problem is most severe for 8, in which the calix[6]arene is both large and polar. The data of Fig. 1 show that 8 is essentially inactive as a transporter either of cations or anions. Compound 8 might prove active if it was present and incorporated into the bilayer during liposome formation, but this experiment has not been attempted because insertion is critical for these compounds to be of biological relevance. Fig. 2 shows schematically how 7 or 8 might insert in the upper bilayer but fail to mediate ion transport.
Presumably, the diaza-18-crown-6 residues of 1, 2, 4, 5 and 6 have sufficient polarity to serve as headgroups and entry (or exit) portals, but they can still pass through the bilayer's hydrophobic regime to the opposite surface. This accounts, in part, for their efficacy as transporters, although transport mediated by 5 proved to be marginal.
Ion transport by compounds 1 and 2
The results obtained for 1 and 2 were especially interesting. Although there is a minor difference in line shape, probably reflecting small differences in insertion or conformational dynamics, both compounds release about 90% of the available Cl− ion by 1500 s. In previous studies of 1, we have focused on Na+ transport and the competition with other cations, such as K+ and Ag+. The fact that Cl− release is so effective is surprising and will merit additional study, which is beyond the scope of this work. Even so, it is interesting to compare 1, which effectively releases both Na+ and Cl−, with 2.
The central relay in 2 is –NHCOCH2OC6H4CONH– rather than 4,13-diaza-18-crown-6. Certainly either of these units can serve to organize water, which would comprise the requisite midplane polarity for transport. Both the amide carbonyl groups of 2 and the ether oxygens of 1 would show an affinity for alkali metal cations but only the amide NH groups of 2 should interact effectively with Cl−. Thus, it is not surprising that 2 shows a modest 1.5 : 1 transport selectivity for Na+ over Cl− (cf. upper and lower panels). The transport efficacies of the other compounds in this study are too low to determine selectivity with any confidence.
The results presented here show that replacement of the diazacrown central unit of 1 by a residue containing secondary amides alters the Na+/Cl− transport selectivity. In previous studies, we have prepared numerous amides,35 but in all cases prior to this one, they have been tertiary. Presumably the presence of a secondary amide alters the affinity of these molecules for Cl− compared to Na+ but this remains to be confirmed generally.
Conclusions
Previous work showed that a calix[4]arene could function effectively as a central unit to induce an ion conductance pathway in a phospholipid bilayer. In this case, each arene was substituted by a dodecylene chain terminated by a benzyldiazacrown residue. Structural modification showed that although a conductance path was formed, ions did not flow through the calix[4]arene. The present study expanded the calixarene size (4 and 6) and placed the calixarenes to function as either headgroups or central units. Only modest ion transport (cation or anion) was realized from compounds in which the calix[4]arene was present in either position. When the calixarene was placed to serve as a headgroup, low activity was observed for the four-arene macrocycle and no transport was apparent when the macrocycle was calix[6]arene. We speculate that the lack of transport activity reflects the inability of the large and polar calixarene to cross the bilayer and establish a conductance pathway. Ultimately, the most interesting and surprising observation relates to 1 and 2, which were included as control compounds. Compound 1, which has been extensively studied as a cation transporter, conducts Cl− as well as it does Na+. The fact that 2 favors Na+ over Cl− transport and possesses two secondary amide residues in the central relay suggests a strategy to incorporate a selective element in the design of more selective ion transporters.
Experimental
General methods
Unless otherwise indicated, reactions were conducted under dry and deoxygenated argon. Solvents were freshly distilled and dried by standard methods before use. Melting points are uncorrected and were measured in open capillaries on a Gallenkamp melting point apparatus. The NMR experiments (1H, 13C{1H}, COSY (H,H), HMQC and ROESY) were carried out at 500 MHz (1H) or 125 MHz (13C) in CDCl3 at ambient temperature, unless otherwise specified. Chemical shifts (δ, ppm) are externally referenced to solvent residual signal. Mass spectra were performed on a REFLEX spectrometer (MALDI-TOF) using ditranol (1,8-dihydroxyanthrone) as matrix and NaI as additive. Elemental analyses, performed on a LECO CHN 932 microanalyser and reported as percentage, indicated inclusion of solvent molecules for nearly all calixarene products and were supported by separate 1H NMR studies. TLC was performed on silica gel Alugram Sil G/UV254 (Macherey–Nagel).
Vesicle preparation and ion transport measurements
Liposomes were formed (20 s sonication) from 1,2-dioleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids, AL). The buffer captured within the liposomes was 750 mM NaCl–15 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.0. The liposomes were filtered (200 nm membrane) and passed over a Sephadex G25 column equilibrated in sodium-free buffer (750 mM cholineCl–15 mM HEPES, pH 7.0). Lipid concentration was measured as previously reported36 and vesicle size was confirmed by light scattering (submicron particle analyzer) to be ∼200 nm. Sodium efflux was measured by using a pH/Na+ combination microelectrode (Thermo-Orion). Aliquots of the amphiphile solution (15 μL, 5 mM in 2-PrOH, except 5, 5 mM in DMSO) were added into the system after the base line is stable. Final values were determined after vesicular lysis (n-octylglucose) and curve calibration.
Determination of chloride release by ISE methods
Liposomes were prepared as above, and loaded with 600 mM KCl and 10 mM (HEPES, pH = 7.0), then equilibrated with an external buffer (400 mM K2SO4, 10 mM HEPES, pH = 7.0). The pH of the buffers prepared was adjusted using 10% NaOH. An Accumet Chloride Combination Electrode was equilibrated (5 min) in external buffer (2.0 mL) and the vesicle solution was added. After recording the baseline voltage, aliquots of the amphiphile solution (5 mM in 2-PrOH, except 5, 5 mM in DMSO) were added. The amount of isopropyl alcohol was limited to 15 μL to avoid alcohol-induced leakage. Vesicles were lysed by addition of aqueous Triton X100 (100 μL, 2%). The lipid concentration used in the studies presented here was 0.40 mM.
Data analysis
The data were acquired in mV units, which were converted to concentration units by using the electrode calibration curve. After conversion from mV to concentration units, the release data were normalized to the total ion concentration, as determined by lysing the vesicles. Data manipulation was done using Origin Pro 7 software.
General procedure for azacrown-carboxylic acid coupling
To a solution of the carboxylic acid (1 mmol) was added benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP•PF6, 2.2 mmol), EtN3 (2.2 mmol), and the corresponding azacrown (2.2 mmol) in THF (10 mL). The mixture was stirred (under Ar) at rt for 24 h. The solvent was removed in vacuo and the residue was partitioned between CH2Cl2 and HCl (1 M). The organic soluble residue was purified by flash column chromatography over silica gel (solvent: 4% v/v MeOH in CH2Cl2).
N-Benzyldiaza-18-crown-6 (BDA, PhCH2〈N18N〉H) was prepared as previously reported.13
N-Benzyl-N′-12-aminododecyl-4,13-diaza-18-crown-6 (12BDA, PhCH2〈N18N〉(CH2)12NH2) was prepared as previously reported.13
N,N′-Bis(12-aminodecyl)-4,13-diaza-18-crown-6 (12DA12, H2N(CH2)12〈N18N〉(CH2)12NH2) was prepared as previously reported.13
N,N′-Bis[12-(N′ -benzyldiaza-18- crown-6) dodecyl]diaza-18-crown-6, [C6H5CH2〈N18N〉(CH2)12(N18N)(CH2)12〈N18N〉CH2C6H5], 1, was prepared as previously reported.13
C6H5CH2〈N18N〉(CH2)12 NHCOCH2OC6H4CONH (CH2)12-〈N18N〉CH2C6H5, 2
This was prepared by the coupling between 9 (44 mg, 0.22 mmol, general procedure), and 12BDA (250 mg, 0.54 mmol). Yield: 201 mg, 73%. 1H NMR: 1.25 [m, 36H, (CH2)9], 1.59 (m, 4H, CH2), 2.18 (m, 16H, CH2–N–CH2), 2.82 (m, 4H, CH2NHCO + CH2N), 3.67 (m, 32H, CH2OCH2-CH2OCH2), 3.76 (s, 4H, OCH2Ar), 6.11 (s, 2H, NH), 6.54 (s, 7.02 (d, 4H, J = 8.8 Hz, ArH), 7.38 (m, 4H, ArH), 7.46 (m, H, NH), 6.96 (d, 2H, J = 8.7, Hz, ArH), 7.25 (m, 4H, ArH), 7.32 (m, 6H, ArH), 7.77 (d, 2H, J = 8.7 Hz, ArH); 13C NMR: 27.1, 27.5, 28.9, 29.2, 29.3, 29.4 [(CH2)10], 39.2, 40.2 (CH2NH), 46.1, 46.3 (CH2N), 53.9, 54.0 (CH2NCH2), 68.0 (OCH2CO), 69.8, 69.9 (CH2OCH2CH2OCH2), 70.4 (OCH2Ar), 114.5, 126.9, 128.2, 128.7, 128.9, 129.0 (ArH), 127.1, 139.5, 159.5 (ArC), 166.6, 167.4 (CO); MALDI-TOF MS m/z (Ditranol + NaI), 1231.9 [M + H]+. Anal. (%). Calc. for C71H118N6O11: C, 69.23; H, 9.66; N, 6.82. Found: C, 69.55; H, 9.82; N, 6.95.
PhCH2OC6 H4 CONH(CH2)12 〈N18N〉(CH2)12 NHCOC6H4OCH2Ph, 3
This was prepared by the general procedure from 10 (100 mg, 0.44 mmol) and 12DA12 (207 mg, 0.51 mmol). Yield: 376 mg, 82%, mp 217 °C. 1H NMR: 1.30 [m, 36H, (CH2)9], 1.64 (q, 4H, J = 7.1 Hz, CH2), 2.72 (m, 8H, CH2–N–CH2), 3.05 (br s, 4H, CH2NHCO), 3.45 (c, 4H, J = 7.1 Hz, CH2NHCO), 3.65 (m, 16H, CH2OCH2CH2OCH2), 5.02 (s, 4H, OCH2Ar), 6.20 (s, 2H, NH), 7.02 (d, 4H, J = 8.8 Hz, ArH), 7.38 (m, 4H, ArH), 7.46 (m, 6H, ArH), 7.77 (d, 4H, J = 8.8 Hz, ArH); 13C NMR: 27.0, 29.3, 29.5, 29.7 [(CH2)10], 40.1 (CH2NH), 50.4 (CH2NCH2), 69.7, 69.9 (CH2OCH2-CH2OCH2), 70.1 (OCH2Ar), 114.6, 127.5, 128.2, 128.7 (ArH), 126.5, 136.5, 162.4 (ArC), 167.5 (CO); MALDI-TOF MS m/z (Ditranol + NaI), 1049.4 [M + H]+, 1072.4 [M + Na]+. Anal. (%): Calc. for C64H96N4O8: C, 73.25; H, 9.22; N, 5.34. Found: C, 73.30; H, 9.25; N, 5.41.
Calix[4]arene channel 4
This has been previously reported.25
Calix[4]arene channel 5
This was prepared from 26 (50 mg, 0.068 mmol) by coupling (general procedure) with 12BDA (91 mg, 0.17 mmol). Yield: 82 mg, 69%. Asymmetric partial cone:1H NMR: 0.84 [s, 9H, C(CH3)3], 1.12 [s, 9H, C(CH3)3], 1.25 [s, 9H, C(CH3)3], 1.32 [m, 36H, (CH2)9], 2.69 (m, 4H, CH2–N–CH2), 2.75 (s, 3H, OCH3), 3.02 (br s, 4H, CH2NHCO), 3.15–3.25 (m, 2H, ArCH2Ar), 3.62 (s, 3H, OCH3), 3.68 (m, 8H, CH2OCH2CH2OCH2), 3.74 (s, 3H, OCH3), 3.70–3.80 (m, 4H, ArCH2Ar), 4.17 (m, 1H, ArCH2-Ar), 4.23 (m, 1H, ArCH2Ar), 4.35 (s, 2H, OCH2CO), 5.70 (1H, NH), 5.75 (br s, 1H, NH), 6.34 (d, 1H, J = 2.0 Hz, ArH), 6.92 (d, 1H, J = 2.0 Hz, ArH), 7.13 (2H, ArH), 7.18 (s, 1H, J = 1.9 Hz, ArH), 7.34 (2H, ArH), 7.68 (d, 1H, J = 1.9 Hz, ArH); 13C NMR: 27.0, 29.3, 29.5, 29.7 [(CH2)10], 30.8, 31.6, 31.7 [C(CH3)3], 30.9, 31.2, 37.7, 38.4 (ArCH2Ar), 33.4, 33.8, 34.2 [C(CH3)3], 40.1 (CH2NH), 50.4 (CH2NCH2), 58.4, 59.8, 60.6 (OCH3), 69.7, 69.9 (CH2OCH2 CH2OCH2), 70.4 (OCH2CO), 124.9, 126.2, 126.4, 127.3, 128.2, 128.3, 128.6, 130.4, 131.8 (ArH), 132.0, 132.7, 132.8, 134.6, 134.8, 135.8, 136.4, 137.3, 143.5, 144.2, 145.4, 151.6, 154.8, 155.3, 155.7 (Ar), 169.0, 170.5 (CO). Cone:1H NMR: 0.72 [s, 9H, C(CH3)3], 1.32 [m, 36H, (CH2)9], 1.35 [s, 18H, C(CH3)3], 2.69 (m, 4H, CH2–N–CH2), 3.02 (br s, 4H, CH2NHCO), 3.27 (m, 2H, ArCH2Ar), 3.29 (m, 2H, ArCH2Ar), 3.61 (s, 3H, OCH3), 3.68 (m, 8H, CH2OCH2-CH2OCH2), 3.73 (s, 6H, OCH3), 4.40 (d, 2H, J = 9.8 Hz, ArCH2Ar), 4.42 (d, 2H, J = 10.0 Hz, ArCH2Ar), 4.62 (s, 2H, OCH2CO), 5.70 (br s, 1H, NH), 5.75 (br s, 1H, NH), 6.26 (s, 2H, ArH), 7.02 (s, 2H, ArH), 7.14 (s, 2H, ArH), 7.24 (s, 2H, ArH); 13C NMR: 30.7, 31.4 [C(CH3)3], 30.9, 31.1 (ArCH2Ar), 33.8, 34.2 [C(CH3)3], 51.4, 51.9 (CO2CH3), 58.3, 60.5 (OCH3), 71.2 (OCH2CO), 169.0, 170.5 (CO); MALDI-TOF MS m/z (Ditranol + NaI), 1722.5 [M]+, 1795.5 [M + Na]+. Anal. (%): Calc. for C108H166N6O14: C, 73.18; H, 9.44; N, 4.74. Found: C, 73.41; H, 9.57; N, 4.87.
Calix[6]arene channel, 6
This was prepared by general procedure, from 27 (30 mg, 0.028 mmol) and (12BDA, 37 mg, 0.07 mmol). Yield: 36 mg, 60%. 1H NMR (C2D2Cl4, 388 K (115 °C): 0.95 [s, 9H, C(CH3)3], 1.00 [s, 18H, C(CH3)3], 1.22 [s, 18H, C(CH3)3], 1.32 [m, 36H, (CH2)9], 1.61 (m, 4H, CH2), 2.69 (m, 4H, CH2–N–CH2), 3.02 (br s, 4H, CH2NHCO), 3.16 (s, 2H, OCH2CO2), 3.26 (s, 6H, OCH3), 3.41 (s, 6H, OCH3), 3.46 (s, 3H, OCH3), 3.68 (m, 8H, CH2OCH2CH2OCH2), 3.92 (s, 8H, ArCH2Ar), 3.98 (s, 4H, ArCH2Ar), 5.02 (s, 4H, OCH2Ar), 5.70 (br s, 1H, NH), 5.75 (1H, NH), 6.71 (s, 2H, ArH), 6.73 (s, 2H, ArH), 6.85 (s, 2H, ArH), 7.04 (s, 4H, ArH), 7.22 (s, 2H, ArH), 729–735 (m, 10H, ArH); 13C NMR: 27.0, 29.3, 29.5, 29.7 [(CH2)10], 30.1, 30.2, 30.7 (ArCH2Ar), 30.8, 30.9, 31.3 [C(CH3)3], 33.4, 33.43, 33.5 [C(CH3)3], 40.1 (CH2NH), 50.4 (CH2NCH2), 59.5, 59.53, 59.9 (OCH3), 68.8 (OCH2CO2), 69.7, 69.9 (CH2OCH2CH2OCH2), 74.8 (OCH2Ar), 123.8, 124.2, 124.5, 126.5, 126.7, 132.0 (ArH) 123.9, 131.5, 132.3, 132.4, 132.7, 132.9, 134.5, 145.2, 145.6, 145.65, 153.0, 153.2 (ArC), 159.8, 167.1 (CO); MALDI-TOF MS m/z (Ditranol + NaI), 2126.5 [M + H]+, 2148.0 [M + Na]+. Anal. (%): Calc. for C132H198N6O16: C, 74.71; H, 9.39; N, 3.95. Found: C, 75.01; H, 9.57; N, 4.10.
Calix[4]arene channel 7
This was prepared by the general procedure, from 18 (50 mg, 0.065 mmol) and 12DA12 (31 mg, 0.15 mmol). Yield: 107 mg, 77% (oil). Asymmetric partial cone:1H NMR: 0.99 [s, 9H, C(CH3)3], 1.22 [s, 9H, C(CH3)3], 1.30 [m, 36H, (CH2)9], 1.41 [s, 9H, C(CH3)3)], 1.64 (m, 4H, CH2), 2.69 (m, 8H, CH2–N–CH2), 2.73 (s, 8H, OCH3 + ArCH2Ar), 3.02 (s, 3H, OCH3), 3.13 (d, 1H, J = 13.5 Hz, ArCH2Ar), 3.15 (d, 1H, J = 13.3 Hz, ArCH2Ar), 3.71–3.79 (m, 4H, ArCH2Ar), 3.72 (s, 3H, OCH3), 3.74 (s, 3H, OCH3), 4.08 (d, 1H, J = 13.5 Hz, ArCH2Ar), 4.17 (d, 1H, J = 13.3 Hz, ArCH2Ar), 4.78 (d, 1H, J = 8.0 Hz, OCH2Ar), 4.85 (d, 1H, J = 8.0 Hz, OCH2Ar), 5.81 (br s, 2H, NH), 6.33 (d, 1H, J = 2.1 Hz, ArH), 6.92 (d, 1H, J = 2.1 Hz, ArH), 7.07 (d, 1H, J = 2.2 Hz, ArH), 7.13 (d, 1H, J = 2.2 Hz, ArH), 7.15 (d, 1H, J = 2.1 Hz, ArH), 7.27–7.28 (m, 2H, ArH), 7.30–7.38 (m, 3H, ArH), 7.41–7.45 (m, 2H, ArH), 7.80 (d, 1H, J = 2.1 Hz, ArH); 13C NMR: 27.0, 29.3, 29.5, 29.7 [(CH2)10], 30.9, 31.2, 36.5, 36.9 (ArCH2Ar), 31.1, 31.5, 31.7 [C(CH3)3], 33.4, 33.8, 34.3 [C(CH3)3], 40.1 (CH2NH), 50.4 (CH2NCH2), 58.4, 59.8, 60.6 (OCH3), 69.7, 69.9 (CH2OCH2CH2OCH2), 76.7 (OCH2Ar), 124.9, 125.2, 126.2, 126.4, 127.3, 128.2, 128.3, 128.6, 128.61, 130.4, 131.8 (ArH), 132.0, 132.7, 132.8, 133.2, 134.6, 134.8, 135.8, 136.4, 137.3, 143.5, 144.2, 145.4, 151.6, 154.8, 155.3, 155.7 (Ar), 161.0 (CO). Cone:1H NMR: 0.69 [s, 9H, C(CH3)3], 1.30 [m, 36H, (CH2)9], 1.43 [s, 18H, C(CH3)3], 1.64 (m, 4H, CH2), 2.69 (m, 8H, CH2–N–CH2), 3.18 (d, 2H, J = 13.1 Hz, ArCH2Ar), 3.20 (d, 2H, J = 13.0 Hz, ArCH2Ar), 3.73 (s, 3H, OCH3), 3.85 (s, 6H, OCH3), 4.38 (d, 2H, J = 13.1 Hz, ArCH2Ar), 4.41 (d, 2H, J = 13.0 Hz, ArCH2Ar), 4.86 (s, 2H, OCH2Ar), 5.81 (2H, NH), 6.24 (s, 2H, ArH), 7.21 (br s, 6H, ArH), 7.27–7.28 (m, 2H, ArH), 7.41–7.45 (m, 3H, ArH); 13C NMR: 29.7, 30.0 (ArCH2Ar), 30.8, 31.3 [C(CH3)3], 34.2, 34.3 [C(CH3)3], 60.6, 62.4 (OCH3), 76.8 (OCH2Ar), 160.0 (CO); MALDI-TOF MS m/z (Ditranol + NaI), 2131.0 [M]+, 2154.0 [M + Na]+. Anal. (%): Calc. for C138H192N4O14: C, 77.78; H, 9.08; N, 2.63. Found: C, 77.53; H, 9.19; N, 2.83.
Calix[6]arene channel, 8
This was prepared by general the procedure, from 19 (100 mg, 0.089 mmol) and 12DA12 (42 mg, 0.067 mmol). Yield: 189 mg, 75%, mp 254 °C; 1H NMR: 1.07 [s, 18H, C(CH3)3], 1.13 [s, 9H, C(CH3)3], 1.26 [s, 18H, C(CH3)3], 1.30 [m, 36H, (CH2)9], 1.64 (m, 4H, CH2), 2.69 (m, 8H, CH2–N–CH2), 2.73 (s, 8H, OCH3 + ArCH2Ar), 3.04 (s, 3H, OCH3), 3.07 (4H, CH2NHCO), 3.29 (s, 6H, OCH3), 3.38 (m, 8H, CH2NHCO + ArCH2Ar), 3.64 (m, 16H, CH2OCH2CH2OCH2), 3.83 (m, 4H, ArCH2Ar), 4.36 (m, 2H, ArCH2Ar), 4.91 (s, 2H, OCH2Ar), 5.81 (2H, NH), 6.81 (d, J = 2.4 Hz, 2H, ArH), 6.98 (d, J = 2.4 Hz, 2H, ArH), 7.02 (s, 2H, ArH), 7.18 (d, J = 2.4 Hz, 2H, ArH), 7.26 (d, J = 2.4 Hz, 2H, ArH), 7.31–7.40 (m, 3H, ArH), 7.47–7.49 (m, 2H, ArH), 7.48 (s, 2H, ArH); 13C NMR: 27.0, 29.3, 29.5, 29.7 [(CH2)10], 29.7, 30.2, 31.2 (ArCH2Ar), 31.3, 31.5, 31.6 [C(CH3)3], 34.0, 34.1, 34.2 [C(CH3)3], 40.1 (CH2NH), 50.4 (CH2NCH2), 59.9, 58.0, 60.0 (OCH3), 69.7, 69.9 (CH2OCH2-CH2OCH2), 74.8 (OCH2Ar), 124.8, 124.9, 126.8, 127.2, 127.7, 127.8, 128.0, 128.2, 128.5, 128.6, 129.7 (ArH), 132.2, 133.4, 133.5, 133.8, 135.9, 145.8, 146.2, 153.4, 154.3, 158.8 (ArC), 168.5 (CO); MALDI-TOF MS m/z (Ditranol + NaI), 2836.0 [M + H]+, 2859.0 [M + Na]+. Anal. (%): Calc. for C186H256N4O18: C, 78.77; H, 9.10; N, 1.98. Found: C, 78.95; H, 9.29; N, 2.09.
4-Carboxymethyloxybenzoic acid, 9
A suspension of ethyl 4-hydroxybenzoate (300 mg, 1.81 mmol) and K2CO3 (500 mg, 3.61 mmol) in Me2CO (25 mL) was stirred at rt (1 h) and methyl bromoacetate (0.25 mL, 2.72 mmol) was added. After stirring an additional 4 h, the solvent was removed in vacuo and the residue was partitioned between CH2Cl2 and HCl (1 M). The organic layer was washed with H2O, dried (MgSO4) and evaporated. The residue was dissolved in THF, then KOH (50 mg) was added, and the mixture was heated under reflux for 24 h. The solvent was evaporated, the residue was partitioned between CH2Cl2 and HCl (1 M), and the organic layer was washed (H2O), dried (MgSO4), and evaporated. The residue was purified by flash column chromatography with (hexane–THF 9 : 1) to give 9 (340 mg, 83%) as a colorless solid, mp 97 °C. 1H NMR (DMSO-d6): 4.77 [s, 2H, OCH2CO), 7.02 (d, 2H, J = 7.5 Hz, Ar), 8.00 (d, 2H, J = 7.5 Hz, Ar); 13C NMR (DMSO-d6): 64.4 (OCH2CO), 113.9, 131.4 (ArH), 123.5, 161.9 (Ar), 168.2, 170.7 (CO); electrospray MS m/z, 196.0 [M]+. Anal. (%): Calc. for C9H8O5: C, 55.11; H, 4.11. Found: C, 55.07; H, 4.08.
4-Benzyloxybenzoic acid (10)
A suspension of ethyl 4-hydroxybenzoate (300 mg, 1.81 mmol) and NaH 60% (87 mg, 3.61 mmol) in DMF (20 mL) was stirred at rt for 1 h and benzyl bromide (0.32 mL, 2.72 mmol) was added. The mixture was stirred for 4 h, evaporated, and the residue was partitioned between CH2Cl2 and HCl (1 M). The organic layer was washed (H2O), dried (MgSO4), and evaporated. The residue was dissolved in THF, KOH (50 mg) was added, and the mixture was heated under reflux for 24 h. The solvent was evaporated, the residue was partitioned between CH2Cl2 and HCl (1 M), the organic layer was washed with H2O, dried (MgSO4) and evaporated. The residue chromatographed (flash, hexane–THF 9 : 1) to give 10, (320 mg, 78%) as a colorless solid, mp 89 °C. 1H NMR: 5.14 [s, 2H, OCH2Ar), 7.02 (d, 2H, J = 7.8 Hz, Ar), 7.36–7.44 (m, 5H, Ar), 8.06 (d, 2H, J = 7.8 Hz, Ar); 13C NMR: 70.8 (OCH2Ar), 114.2, 127.3, 127.5, 128.8, 131.9 (ArH), 121.9, 136.4, 162.3 (Ar), 169.3 (CO); electrospray MS m/z, 229.2 [M]+. Anal. (%): Calc. for C14H12O3: C, 73.67; H, 5.30. Found: C, 73.62; H, 5.27.
26-Benzyloxy-11,17,23-tri-tert-butyl-25,27,28-trihydroxycalix[4]arene, 11 (cone)
A suspension of 25,27,28-tribenzoyloxy-11,17,23-tri-tert-butyl-26-hydroxycalix[4]arene 12,31 (0.5 mg, 0.55 mmol) and NaH 60% (27 mg, 0.66 mmol) in DMF (200 mL) was stirred for 1 h at rt, benzyl bromide (0.79 mL, 0.66 mmol) was added, and the mixture was stirred for 4 h. After evaporation of the solvent, the residue was partitioned between CH2Cl2 and HCl (1 M), the organic layer was washed (H2O), dried (MgSO4) and evaporated. The residue was dissolved in THF, KOH (100 mg) was added, and the mixture was heated (reflux) for 24 h. The solvent was evaporated, the residue was partitioned between CH2Cl2 and HCl (1 M), the organic layer was washed (H2O) dried (MgSO4), and evaporated. Chromatography (flash, hexane–THF 9 : 1) gave 11 (cone), (350 mg, 93%) as a colorless solid, mp 221 °C. 1H NMR: 1.23 [s, 9H, C(CH3)3)], 1.26 [s, 18H, C(CH3)3)], 3.43 (d, 2H, J = 13.7 Hz, ArCH2Ar), 3.45 (d, 2H, J = 13.1 Hz, ArCH2Ar), 4.24 (d, 2H, J = 13.7 Hz, ArCH2Ar), 4.38 (d, 2H, J = 13.1 Hz, ArCH2Ar), 5.23 (s, 2H, OCH2Ar), 6.95 (t, 1H, J = 7.5 Hz, ArH), 7.02 (s, 2H, ArH), 7.04 (d, 2H, J = 2.4 Hz, ArH), 7.07 (d, 2H, J = 2.4 Hz, ArH), 7.14 (s, 1H, ArH), 7.16 (s, 1H, ArH), 7.49–7.56 (m, 3H, ArH), 7.73–7.75 (m, 2H, ArH), 9.28 (s, 2H, OH), 9.69 (s, 1H, OH). 13C NMR: 31.4, 31.5 [C(CH3)3], 32.1, 32.8 (ArCH2Ar), 33.9 [C(CH3)3], 79.3 (OCH2Ar), 125.2, 125.7, 125.9, 126.2, 128.95, 129.0, 129.1, 129.3 (ArH), 127.4, 127.9, 128.1, 134.7, 135.6, 143.1, 143.8, 147.0, 148.6, 151.5 (Ar); MALDI-TOF MS m/z (Ditranol + NaI), 682.4 [M]+, 705.4 [M + Na]+. Anal. (%): Calc. for C47H54O4: C, 82.66; H, 7.97. Found: C, 82.34; H, 7.98.
25,27,28-Tribenzoyloxy-11,17,23-tri-tert-butyl-26-hydroxycalix[4]arene 12
This was prepared as previously reported.31
26-Benzyloxy-11, 17, 23-tri-tert-butyl-25, 27, 28-trimethoxycalix[4]arene, 13 (asymmetric partial cone : cone 3 : 1)
A suspension of 11 (200 mg, 2.9 mmol) and NaH 60% (70 mg, 1.74 mmol) in DMF (15 mL) was stirred for 20 min at rt, CH3I (0.11 mL, 1.74 mmol) was added, and the mixture was stirred for 2 h. The solvent was evaporated, the residue was partitioned between CH2Cl2 and HCl (1 M), the organic layer was washed (H2O), dried (MgSO4) and evaporated. Chromatography (flash, hexane–THF 95 : 5) gave 13, (233 mg, 90%). Asymmetric partial cone:1H NMR: 1.07 [s, 9H, C(CH3)3)], 1.19 [s, 9H, C(CH3)3)], 1.38 [s, 9H, C(CH3)3)], 3.04 (s, 3H, OCH3), 3.05 (d, 1H, J = 13.3 Hz, ArCH2Ar), 3.10 (d, 1H, J = 13.8 Hz, ArCH2Ar), 3.65–3.75 (m, 4H, ArCH2Ar), 3.65 (s, 3H, OCH3), 3.70 (s, 3H, OCH3), 4.06 (d, 1H, J = 13.8 Hz, ArCH2Ar), 4.17 (d, 1H, J = 13.3 Hz, ArCH2Ar), 4.73 (d, 1H, J = 10.7 Hz, OCH2Ar), 4.82 (d, 1H, J = 10.7 Hz, OCH2Ar), 6.23 (s, 2H, ArH), 6.34 (d, 1H, J = 7.2 Hz, ArH), 6.44 (t, 1H, J = 7.2 Hz, ArH), 6.81 (d, 1H, J = 2.4 Hz, ArH), 6.88 (d, 1H, J = 7.2 Hz, ArH), 6.94 (d, 1H, J = 2.4 Hz, ArH), 7.01 (d, 1H, J = 2.4 Hz, ArH), 7.13 (d, 1H, J = 2.4 Hz, ArH), 7.36–7.44 (m, 3H, ArH), 7.51–7.53 (m, 2H, ArH). 13C NMR: 30.7, 31.1, 36.2, 36.6 (ArCH2Ar), 31.1, 31.2, 31.4 [C(CH3)3], 34.0, 34.2, 34.3 [C(CH3)3], 58.7, 59.7, 60.3 (OCH3), 76.0 (OCH2Ar), 122.0, 124.5, 124.9, 126.2, 126.3, 127.9, 128.4. 128.5, 130.7, 131.2 (ArH), 131.9, 132.5, 133.1, 134.7, 135.9, 136.2, 136.7, 137.6, 144.2, 144.5, 145.2, 154.9, 155.5 (Ar). Cone:1H NMR: 0.77 [s, 9H, C(CH3)3)], 1.41 [s, 18H, C(CH3)3)], 3.18–3.19 (m, 4H, ArCH2Ar), 3.75 (s, 3H, OCH3), 3.84 (s, 6H, OCH3), 4.36 (d, 2H, J = 14.0 Hz, ArCH2Ar), 4.42 (d, 2H, J = 14.0 Hz, ArCH2Ar), 4.81 (s, 2H, OCH2Ar), 6.10 (s, 2H, ArH), 6.98 (m, 3H, ArH), 7.01 (s, 2H, ArH), 7.04 (s, 1H ArH), 7.08 (s, 1H ArH), 7.36–7,37 (m, 3H, ArH), 7.48–7.49 (m, 2H, ArH). 13C NMR: 31.4, 31.5 [C(CH3)3], 32.1, 32.7 (ArCH2Ar), 34.2, 34.3 [C(CH3)3], 60.5, 62.3 (OCH3), 79.8 (OCH2Ar), 125.3, 125.7, 125.8, 126.3, 128.8, 129.0, 129.1, 129.3 (ArH), 127.4, 127.8, 128.3, 134.9, 135.4, 143.2, 143.7, 146.8, 148.4, 151.5 (Ar); MALDI-TOF MS m/z (Ditranol + NaI), 724.4 [M]+, 747.4 [M + Na]+. Anal. (%): Calc. for C50H60O4: C, 82.83; H, 8.34. Found: C, 82.99; H, 8.20.
5-Bromo-11,17, 23, 29, 35-penta-tert- butyl-38-hydroxy-37,39,-40,41,42-pentamethoxycalix[6]arene, 14
Br2 (0.13 mL, 2.5 mmol) was added dropwise to a 0 °C solution of 5, 11, 17, 23, 29-penta-tert- butyl-37-hydroxy-38, 39, 40, 41,42-pentamethoxycalix[6]arene, 15, (500 mg, 0.50 mmol) in CHCl3 (50 mL). The solution was allowed to warm to ambient temperature while stirring for 1 h, whereupon 10% aq. NaHSO3 (10 mL) was added to quench the reaction. The organic layer was washed with water, dried (MgSO4), and evaporated. The residue was triturated with MeOH to give 14, (490 mg, 92%) as a colorless solid, mp 190 °C. 1H NMR: 0.95 [s, 9H, C(CH3)3], 1.07 [s, 18H, C(CH3)3], 1.11 [s, 18H, C(CH3)3], 2.93 (s, 6H, OCH3), 3.14 (s, 6H, OCH3), 3.41 (s, 3H, OCH3), 3.69 (br s, 4H, ArCH2Ar), 3.85 (br s, 8H, ArCH2Ar), 6.83 (s, 2H, ArH), 6.86 (d, J = 2.4 Hz, 2H, ArH), 6.91 (s, 2H, ArH), 6.94 (s, 4H, ArH), 6.99 (d, J = 2.4 Hz, 2H, ArH), 7.73 (s, 1H, OH). MALDI-TOF MS m/z (Ditranol + NaI), 1064.6 [M + H]+, 1087.6 [M + Na]+.
5, 11, 17, 23, 29-Penta-tert-butyl-37-hydroxy-38,39,40,41,42-pentamethoxycalix[6]arene, 15
This was prepared as previously reported.32
26-Benzyloxy-5-bromo-11, 17, 23-tri-tert-butyl-25, 27, 28-tri-methoxycalix[4]arene, 16 (asymmetric partial cone : cone 5 : 1)
Br2 (0.13 mL, 2.5 mmol) was added dropwise to a cooled solution of 13 (500 mg, 0.67 mmol) in CHCl3 (50 mL) at 0 °C and stirring was continued for 1 h. NaHSO3 (20 mL, 10% aq.) was added to quench the reaction. The organic layer was washed (H2O), dried (MgSO4), and evaporated. The residue was precipitated from CHCl3–MeOH (3 : 1) to give 16 (498 mg, 90%). Asymmetric partial cone:1H NMR: 1.07 [s, 9H, C(CH3)3)], 1.10 [s, 9H, C(CH3)3)], 1.33 [s, 9H, [C(CH3)3)], 2.94 (s, 3H, OCH3), 3.05 (d, 1H, J = 13.4 Hz, ArCH2Ar), 3.09 (d, 1H, J = 13.4 Hz, ArCH2Ar), 3.62–3.64 (m, 4H, ArCH2Ar), 3.63 (s, 3H, OCH3), 3.64 (s, 3H, OCH3), 4.01 (d, 1H, J = 13.4 Hz, ArCH2Ar), 4.05 (d, 1H, J = 13.4 Hz, ArCH2Ar), 4.66 (d, 1H, J = 10.8 Hz, OCH2Ar), 4.73 (d, 1H, J = 10.8 Hz, OCH2Ar), 6.38 (d, 1H, J = 2.4 Hz, ArH), 6.58 (d, 1H, J = 2.6 Hz, ArH), 6.93 (d, 1H, J = 2.4 Hz, ArH), 6.97 (d, 1H, J = 2.6 Hz, ArH), 7.03 (d, 1H, J = 2.4 Hz, ArH), 7.08 (d, 1H, J = 2.4 Hz, ArH), 7.13 (d, 1H, J = 2.4 Hz, ArH), 7.24 (d, 1H, J = 2.4 Hz, ArH), 7.36–7.44 (m, 3H, ArH), 7.51–7.53 (m, 2H, ArH). 13C NMR: 30.9, 31.1, 36.3, 36.8 (ArCH2Ar), 31.2, 31.3, 31.5 [C(CH3)3], 34.1, 34.2, 34.4 [C(CH3)3], 58.6, 60.0, 60.8 (OCH3), 76.5 (OCH2Ar), 124.8, 125.2, 126.2, 126.5, 127.9, 128.4, 128.5, 130.2, 131.6 (ArH), 114.3, 131.9, 132.7, 133.1, 134.6, 134.9, 136.2, 136.3, 137.6, 143.2, 144.0, 145.2, 154.9, 155.3 (Ar). Cone:1H NMR: 0.89 [s, 9H, C(CH3)3)], 1.31 [s, 18H, C(CH3)3)], 3.14 (d, 2H, J = 7.8 Hz, ArCH2Ar), 3.18 (d, 2H, J = 8.0 Hz, ArCH2Ar), 3.77 (s, 3H, OCH3), 3.80 (s, 6H, OCH3), 4.29 (d, 2H, J = 8.0 Hz, ArCH2Ar), 4.31 (d, 2H, J = 8.00 Hz, ArCH2Ar), 4.75 (s, 2H, OCH2Ar), 6.50 (s, 2H, ArH), 6.69 (s, 2H, ArH), 7.08 (d, 2H, J = 2.4 Hz, ArH), 7.18 (d, 2H, J = 2.4 Hz, ArH), 7.36–7.44 (m, 3H, ArH), 7.58–7.60 (m, 2H, ArH). 13C NMR: 29.9, 30.1 (ArCH2Ar), 31.2, 31.5 [C(CH3)3], 34.2, 34.3 [C(CH3)3], 60.5, 62.7 (OCH3), 77.8 (OCH2Ar), 124.6, 124.7, 127.2, 127.7, 129.0, 131.5 (ArH), 115.0, 128.1, 130.7, 141.2, 143.3, 152.2, 153.1 (Ar); MALDI-TOF MS m/z (Ditranol + NaI), 802.4 [M]+, 825.3 [M + Na]+. Anal. (%): Calc. for C50H59O4; C, 74.70; H, 7.40. Found: C, 74.79; H, 7.43.
38- Benzyloxy- 5- bromo-11, 17, 23, 29, 35- penta-tert- butyl-37,39,40,41,42-pentamethoxycalix[6]arene, 17
A suspension of 14 (630 mg, 0.59 mmol) and NaH 60% (36 mg, 0.88 mmol) in THF (40 mL) was heated at 80 °C for 1 h. Benzyl bromide (0.11 mL, 0.88 mmol) was added and the mixture was refluxed for 5 h. The solvent was evaporated and the residue was partitioned between CH2Cl2 and HCl (1 M). The organic layer was washed (H2O), dried (MgSO4) and evaporated. The residue was precipitated from MeOH to give 17 (439 mg, 65%), mp 205 °C. 1H NMR: 1.06 [s, 18H, C(CH3)3], 1.28 [s, 9H, C(CH3)3], 1.32 [s, 18H, C(CH3)3], 2.63 (s, 6H, OCH3), 2.77 (s, 3H, OCH3), 3.41 (s, 6H, OCH3), 3.53 (br s, 6H, ArCH2Ar), 3.86–4.00 (br s, 6H, ArCH2Ar), 4.97 (s, 2H, OCH2Ar), 6.77 (d, J = 2.2 Hz, 2H, ArH), 6.87 (br s, 2H, ArH), 6.91 (d, J = 2.2 Hz, 2H, ArH), 7.15 (s, 2H, ArH), 7.17 (d, J = 2.2 Hz, 2H, ArH), 7.20 (d, J = 2.2 Hz, 2H, ArH), 7.38–7.44 (m, 3H, ArH), 7.51–7.55 (m, 2H, ArH). 13C NMR: 30.9 (ArCH2Ar), 31.3, 31.5 [C(CH3)3], 34.1, 34.2 [C(CH3)3], 59.9, 60.0 (OCH3), 74.7 (OCH2Ar), 124.9, 126.6, 127.7, 127.9, 128.1, 128.6, 130.2 (ArH), 132.5, 133.4, 133.60, 133.64, 137.7, 145.8, 146.2, 153.4, 154.2 (ArC). MALDI-TOF MS m/z (Ditranol + NaI), 1155.5 [M + H]+, 1177.4 [M + Na]+. Anal. (%): Calc. for C74H91BrO6•MeOH: C, 75.80; H, 8.06. Found: C, 75.92; H, 8.14.
26-Benzyloxy-11,17,23-tri-tert-butyl-5-hydroxycarbonyl-25,27,-28-trimethoxycalix[4]arene, 18,(asymmetric partial cone : cone 9 : 1)
tert-Butyllithium (1.5 M solution in pentane) (0.96 mL, 1.3 mmol) was added dropwise to a −78 °C solution of 16 (0.5 mg, 0.63 mmol) in dry THF (10 mL). The resulting yellow solution was stirred at −78 °C for 20 min. Cooling was continued at 0 °C and CO2 gas was bubbled in during 30 min. The solvent was evaporated and the residue was partitioned between CH2Cl2 and HCl (1 M). The organic layer was washed (H2O), dried (MgSO4) and evaporated. The residue chromatographed (flash, hexane–THF 8 : 1) to give 18 (395 mg, 82%). Asymmetric partial cone:1H NMR: 0.99 [s, 9H, C(CH3)3)], 1.22 [s, 9H, C(CH3)3)], 1.41 [s, 9H, C(CH3)3)], 3.02 (s, 3H, OCH3), 3.13 (d, 1H, J = 13.5 Hz, ArCH2Ar), 3.15 (d, 1H, J = 13.3 Hz, ArCH2Ar), 3.71–3.79 (m, 4H, ArCH2Ar), 3.72 (s, 3H, OCH3), 3.74 (s, 3H, OCH3), 4.08 (d, 1H, J = 13.5 Hz, ArCH2Ar), 4.17 (d, 1H, J = 13.3 Hz, ArCH2Ar), 4.78 (d, 1H, J = 8.0 Hz, OCH2Ar), 4.85 (d, 1H, J = 8.0 Hz, OCH2Ar), 6.33 (d, 1H, J = 2.1 Hz, ArH), 6.92 (d, 1H, J = 2.1 Hz, ArH), 7.07 (d, 1H, J = 2.2 Hz, ArH), 7.13 (d, 1H, J = 2.2 Hz, ArH), 7.15 (d, 1H, J = 2.1 Hz, ArH), 7.27–7.28 (m, 2H, ArH), 7.30–7.38 (m, 3H, ArH), 7.41–7.45 (m, 2H, ArH), 7.80 (d, 1H, J = 2.1 Hz, ArH). 13C NMR: 30.9, 31.2, 36.5, 36.9 (ArCH2Ar), 31.1, 31.5, 31.7 [C(CH3)3], 33.4, 33.8, 34.3 [C(CH3)3], 58.4, 59.8, 60.6 (OCH3), 76.7 (OCH2Ar), 124.9, 125.2, 126.2, 126.4, 127.3, 128.2, 128.3, 128.6, 128.61, 130.4, 131.8 (ArH), 132.0, 132.7, 132.8, 133.2, 134.6, 134.8, 135.8, 136.4, 137.3, 143.5, 144.2, 145.4, 151.6, 154.8, 155.3, 155.7 (Ar), 161.0 (CO). Cone:1H NMR: 0.69 [s, 9H, C(CH3)3)], 1.43 [s, 18H, C(CH3)3)], 3.18 (d, 2H, J = 13.1 Hz, ArCH2Ar), 3.20 (d, 2H, J = 13.0 Hz, ArCH2Ar), 3.73 (s, 3H, OCH3), 3.85 (s, 6H, OCH3), 4.38 (d, 2H, J = 13.1 Hz, ArCH2Ar), 4.41 (d, 2H, J = 13.0 Hz, ArCH2Ar), 4.86 (s, 2H, OCH2Ar), 6.24 (s, 2H, ArH), 7.21 (br s, 6H, ArH), 7.27–7.28 (m, 2H, ArH), 7.41–7.45 (m, 3H, ArH). 13C NMR: 29.7, 30.0 (ArCH2Ar), 30.8, 31.3 [C(CH3)3], 34.2, 34.3 [C(CH3)3], 60.6, 62.4 (OCH3), 76.8 (OCH2Ar), 160.0 (CO); MALDI-TOF MS m/z (Ditranol + NaI), 768.4 [M]+, 791.4 [M + Na]+. Anal. (%): Calc. for C51H60O6: C, 79.65; H, 7.86. Found: C, 79.05; H, 7.62.
38-Benzyloxy-11,17,23,29,35-penta-tert-butyl-5-hydroxycarbonyl-37,39,40,41,42-pentamethoxycalix[6]arene, 19
tert-Butyllithium (0.15 mL, 0.16 mmol) was added dropwise at −78 °C to a solution of 17 (150 mg, 0.13 mmol) in dry THF (10 mL). The resulting yellow solution was stirred at −78 °C for 20 min, cooling was changed to an ice bath, and CO2 gas was bubbled in during 30 min (0 °C). The solvent was evaporated and the residue was partitioned between CH2Cl2 and HCl (1 M). The organic layer was washed (H2O), dried (MgSO4) and evaporated. The residue was chromatographed (flash, hexane–THF 8 : 1) to give 19 (110 mg, 76%) as a white solid, mp 220° C. 1H NMR (C2D2Cl4, 403 K (130 °C)): 0.99 [s, 18H, C(CH3)3], 1.20 [s, 9H, C(CH3)3], 1.25 [s, 18H, C(CH3)3], 2.69 (s, 6H, OCH3), 2.88 (s, 3H, OCH3), 3.29 (s, 6H, OCH3), 3.90 (s, 12H, ArCH2Ar), 4.97 (s, 2H, OCH2Ar), 6.72 (d, J = 2.4 Hz, 2H, ArH), 6.85 (d, J = 2.4 Hz, 2H, ArH), 7.04 (s, 2H, ArH), 7.07 (d, J = 2.4 Hz, 2H, ArH), 7.15 (d, J = 2.4 Hz, 2H, ArH), 7.31–7.40 (m, 3H, ArH), 7.47–7.49 (m, 2H, ArH), 7.50 (s, 2H, ArH). 13C NMR: 29.7, 30.2, 31.2 (ArCH2Ar), 31.3, 31.5, 31.6 [C(CH3)3], 34.0, 34.1, 34.2 [C(CH3)3], 59.9, 59.96, 60.01 (OCH3), 74.8 (OCH2Ar), 124.8, 124.9, 126.8, 127.2, 127.7, 127.8, 128.0, 128.2, 128.5, 128.6, 129.7 (ArH), 132.4, 133.4, 133.5, 133.6, 135.7, 145.8, 146.2, 153.4, 154.3, 158.9 (ArC), 166.2 (CO). MALDI-TOF MS m/z (Ditranol + NaI), 1143.8 [M + Na]+. Anal. (%): Calc. for C75H92O8: C, 80.32; H, 8.27. Found: C, 80.21; H, 8.49.
26-Benzyloxy-11,17,23-tri-tert-butyl-25,27,28-trimethoxy-5-methoxycarbonylcalix[4]arene, 20,(asymmetric partial cone : cone 9 : 1)
A suspension of 18 (250 mg, 0.33 mmol), Cs2CO3 (530 mg, 1.63 mmol) and CH3I (1 mL, 16.3 mmol) in THF (25 mL) was stirred for 12 hours at rt, the mixture was evaporated, and the residue was partitioned between CH2Cl2 and HCl (1M). The organic layer was washed (H2O), dried (MgSO4) and evaporated. The residue was chromatographed (flash, hexane–THF 95 : 5) to give 20 (200 mg, 81%). Asymmetric partial cone:1H NMR: 0.86 [s, 9H, C(CH3)3], 1.10 [s, 9H, C(CH3)3], 1.28 [s, 9H, C(CH3)3], 2.91 (s, 3H, OCH3), 2.98–3.03 (m, 2H, ArCH2Ar), 3.55–3.64 (m, 4H, ArCH2Ar), 3.58 (s, 3H, OCH3), 3.61 (s, 3H, OCH3), 3.66 (s, 3H, CO2CH3), 3.96 (d, 1H, J = 13.9 Hz, ArCH2Ar), 4.04 (d, 1H, J = 13.9 Hz, ArCH2Ar), 4.64 (d, 1H, J = 11.1 Hz, OCH2Ar), 4.72 (d, 1H, J = 11.0 Hz, OCH2Ar), 6.22 (d, 1H, J = 2.0 Hz, ArH), 6.79 (d, 1H, J = 2.0 Hz, ArH), 6.96 (d, 1H, J = 2.2 Hz, ArH), 7.01 (d, 1H, J = 2.2 Hz, ArH), 7.02 (d, 1H, J = 2.1 Hz, ArH), 7.08 (s, 1H, ArH), 7.15 (s, 1H, ArH), 7.29–7.31 (m, 3H, ArH), 7.39–7.42 (m, 2H, ArH), 7.60 (d, 1H, J = 2.1 Hz, ArH). 13C NMR: 30.9, 31.5, 31.7 [C(CH3)3], 30.9, 31.2, 36.6, 37.0 (ArCH2Ar), 33.4, 33.8, 34.2 [C(CH3)3], 51.4 (CO2CH3), 58.3, 59.8, 60.6 (OCH3), 76.6 (OCH2Ar), 124.9, 125.2, 126.2, 126.4, 127.3, 128.2, 128.3, 128.6, 128.61, 130.4, 131.8 (ArH), 132.0, 132.7, 132.8, 133.2, 134.6, 134.8, 135.8, 136.4, 137.3, 143.5, 144.2, 145.4, 151.6, 154.8, 155.3, 155.7 (Ar), 160.2 (CO). Cone:1H NMR: 0.56 [s, 9H, C(CH3)3)], 1.31 [s, 18H, C(CH3)3)], 3.06–3.09 (m, 4H, ArCH2Ar), 3.51 (s, 3H, CO2CH3), 3.64 (s, 3H, OCH3), 3.73 (s, 6H, OCH3), 4.24 (d, 2H, J = 9.8 Hz, ArCH2Ar), 4.29 (d, 2H, J = 9.6 Hz, ArCH2Ar), 4.73 (s, 2H, OCH2Ar), 6.12 (s, 2H, ArH), 7.01 (s, 2H, ArH), 7.08 (s, 4H, ArH), 7.26–7.31 (m, 3H, ArH), 7.39–7.41 (m, 2H, ArH). 13C NMR: 30.7, 31.4 [C(CH3)3], 30.9, 31.0 (ArCH2Ar), 33.8, 34.2 [C(CH3)3], 51.3 (CO2CH3), 58.3, 60.5 (OCH3), 76.8 (OCH2Ar), 160.2 (CO); MALDI-TOF MS m/z (Ditranol + NaI) 783.4 [M + H]+, 805.4 [M + Na]+. Anal. (%): Calc. for C52H62O6: C, 79.76; H, 7.98. Found: C, 79.55; H, 7.72.
38-Benzyloxy-11, 17, 23, 29, 35-penta-tert-butyl-37, 39,40,41,42-pentamethoxy-5-methoxycarbonylcalix[6]arene, 21
A solution of 19 (230 mg, 0.21 mmol) and DCC (64 mg, 0.31 mmol) was stirred at rt (5 h) in dry MeOH (5 mL), the solvent was evaporated, and the residue was partitioned between CH2Cl2 and HCl (1 M). The organic layer was washed (H2O) dried (MgSO4), evaporated, and chromatographed (flash, hexane–THF 9 : 1) to give 21 (175 mg, 75%) as a white solid, mp 190 °C. 1H NMR: 0.98 [s, 18H, C(CH3)3], 1.27 [s, 9H, C(CH3)3], 1.33 [s, 18H, C(CH3)3], 2.55 (s, 6H, OCH3), 2.78 (s, 3H, OCH3), 3.42 (s, 6H, OCH3), 3.52 (s, J = 16.0 Hz, 2H, ArCH2Ar), 3.71 (s, 3H, CO2Me), 3.76 (s, J = 15.6 Hz, 2H, ArCH2Ar), 3.80 (s, J = 14.0 Hz, 2H, ArCH2Ar), 4.17 (s, J = 14.0 Hz, 2H, ArCH2Ar), 4.20 (s, J = 15.6 Hz, 2H, ArCH2Ar), 4.50 (s, J = 16.0 Hz, 2H, ArCH2Ar), 5.00 (s, 2H, OCH2Ar), 6.71 (d, J = 2.3 Hz, 2H, ArH), 6.87 (d, J = 2.3 Hz, 2H, ArH), 7.15 (s, 2H, ArH), 7.16 (d, J = 2.5 Hz, 2H, ArH), 7.26 (d, J = 2.5 Hz, 2H, ArH), 7.35–7.44 (m, 5H, ArH), 7.54 (s, 2H, ArH). 13C NMR: 29.7, 30.5, 31.1 (ArCH2Ar), 30.2, 30.8, 31.5 [C(CH3)3], 34.0, 34.15, 34.2 [C(CH3)3], 51.7 (CO2Me) 59.9, 60.0 (OCH3), 74.8 (OCH2Ar), 124.7, 124.9, 126.8, 127.3, 128.0, 128.1 (ArH), 125.4, 128.6, 132.5, 133.4, 133.6, 135.4, 137.1, 145.7, 146.1, 153.4, 154.3 (ArC), 166.0 (CO). MALDI-TOF MS m/z (Ditranol + NaI), 1157.6 [M + Na]+, 1233.6 [M + Na]+. Anal. (%): Calc. for C76H94O8: C, 80.38; H, 8.34. Found: C, 80.84; H, 8.12.
11, 17, 23- Tri-tert- butyl-26-hydroxy-25,27,28-trimethoxy-5-methoxycarbonylcalix[4]arene, 22 (cone : symmetric partial cone 10 : 1)
Hydrogen was bubbled into a suspension of 20 (0.5 g, 0.38 mmol), Pd/C (50 mg) in THF (50 mL) during 30 min. The mixture was filtered (celite), evaporated, and the solid was triturated with EtOH, filtered, and dried to give 22 (262 mg, 100%). Cone:1H NMR: 0.87 [s, 18H, C(CH3)3], 1.39 [s, 9H, C(CH3)3], 3.28 (d, 2H, J = 12.9 Hz, ArCH2Ar), 3.44 (d, 2H, J = 13.6 Hz, ArCH2Ar), 3.90 (s, 6H, OCH3), 3.96 (s, 3H, CO2Me), 4.01 (s, 3H, OCH3), 4.33 (d, 2H, J = 13.6 Hz, ArCH2Ar), 4.35 (d, 2H, J = 12.9 Hz, ArCH2Ar), 6.56 (d, 2H, J = 2.5 Hz, ArH), 6.63 (d, 2H, J = 2.5 Hz, ArH), 7.21 (s, 2H, ArH), 7.30 (s, 1H, OH), 7.87 (s, 2H, ArH). 13C NMR: 31.1, 31.7 [C(CH3)3], 31.2, 31.3 (ArCH2Ar), 33.8, 34.2 [C(CH3)3], 51.7 (CO2CH3), 60.4, 63.0 (OCH3), 124.7, 125.4, 125.5, 130.3 (ArH), 120.2, 129.1, 130.8, 132.3, 135.7, 146.0, 152.9, 155.3, 158.3 (Ar), 167.7 (CO). Symmetric partial cone:1H NMR: 1.14 [s, 18H, C(CH3)3)], 1.47 [s, 9H, C(CH3)3)], 2.59 (s, 3H, OCH3), 3.39 (d, 2H, J = 13.9 Hz, ArCH2Ar), 3.77 (s, 6H, OCH3), 3.89 (s, 4H, ArCH2Ar), 3.95 (s, 3H, CO2Me), 4.06 (d, 2H, J = 13.9 Hz, ArCH2Ar), 6.83 (d, 2H, J = 2.5 Hz, ArH), 7.08 (d, 2H, J = 2.5 Hz, ArH), 7.33 (s, 2H, ArH), 7.82 (s, 1H, OH), 7.84 (s, 2H, ArH). 13C NMR: 31.1, 31.7 [C(CH3)3], 31.1, 38.4 (ArCH2-Ar), 33.9, 34.2 [C(CH3)3], 50.9 (CO2CH3), 58.2, 60.8 (OCH3), 125.6, 127.0, 127.2, 130.3 (ArH), 120.2, 128.3, 131.7, 133.1, 133.5, 146.1, 146.3, 153.4 (Ar), 167.7 (CO); MALDI-TOF MS m/z (Ditranol + NaI), 693.4 [M + H]+, 715.4 [M + Na]+. Anal. (%): Calc. for C45H56O6: C, 78.00; H, 8.15. Found: C, 77.89; H, 8.03.
5, 11, 17, 23, 29-Penta-tert-butyl-38-hydroxy-37,39,40,41,42-pentamethoxy-5-methoxycarbonylcalix[6]arene, 23
Hydrogen was bubbled into a suspension of 21 (200 mg, 0.18 mmol), Pd/C (20 mg) in THF (20 mL) during 30 min. The mixture was filtered (celite), evaporated, and the solid was triturated with EtOH. The mixture was filtered to give 23 (187 mg, 100%) as a white solid, mp 193–196 °C. 1H NMR: 1.06 [s, 9H, C(CH3)3], 1.17 [s, 18H, C(CH3)3], 1.19 [s, 18H, C(CH3)3], 3.02 (s, 6H, OCH3), 3.32 (s, 6H, OCH3), 3.57 (s, 3H, OCH3), 3.83 (s, 3H, CO2Me), 3.87 (s, 4H, ArCH2Ar), 3.98 (s, 8H, ArCH2Ar), 6.94 (s, 2H, ArH), 6.96 (d, J = 2.4 Hz, 2H, ArH), 7.02 (d, J = 2.5, 2H, ArH), 7.06 (d, J = 2.5 Hz, 2H, ArH), 7.11 (d, J = 2.4 Hz, 2H, ArH), 7.71 (s, 2H, ArH), 8.41 (s, 1H, OH). 13C NMR: 29.7, 30.9 (ArCH2Ar), 30.4, 31.3, 31.4 [C(CH3)3], 34.0, 34.1, 34.2 [C(CH3)3], 51.6 (CO2Me), 60.4, 60.42, 60.8 (OCH3), 125.4, 125.5, 125.9, 126.2, 126.5, 133.3 (ArH) 126.2, 130.3, 131.8, 132.9, 133.6, 133.7, 145.3, 145.7, 146.9, 152.8, 154.0, 154.4, 156.8 (ArC) 167.3 (CO). MALDI-TOF MS m/z (Ditranol + NaI), 1067.6 [M + Na]+. Anal. (%): Calc. for C69H88O8: C, 79.27; H, 8.48. Found: C, 79.57; H, 8.89.
11,17,23-Tri-tert-butyl-25, 27, 28-trimethoxy-5-methoxycarbonyl-26-methoxycarbonylmethyloxycalix[4]arene, 24 (asymmetric partial cone : cone 9 : 1)
A suspension of 22 (300 mg, 0.43 mmol) and K2CO3 (120 mg, 0.87 mmol) in CH3CN (50 mL) was heated at 80 °C for 1 h. Methyl bromoacetate (0.8 mL, 0.87 mmol) was added and heating continued for 3 h. The solvent was evaporated, the residue was partitioned between CH2Cl2 and HCl (1 M), the organic layer was washed (H2O), dried (MgSO4), evaporated, and the residue was chromatographed (flash, hexane–THF 8 : 2) to give 24 (190 mg, 69%). Asymmetric partial cone:1H NMR: 0.87 [s, 9H, C(CH3)3)], 1.08 [s, 9H, C(CH3)3)], 1.28 [s, 9H, C(CH3)3)], 2.71 (s, 3H, OCH3), 3.12–3.15 (m, 1H, ArCH2Ar), 3.16–3.21 (m, 1H, ArCH2Ar), 3.62 (s, 3H, OCH3), 3.65 (s, 3H, OCH3), 3.61 (s, 3H, CO2CH3), 3.70–3.80 (m, 4H, ArCH2Ar), 3.72 (s, 3H, CO2CH3), 4.11 (d, 1H, J = 13.9 Hz, ArCH2Ar), 4.19 (d, 1H, J = 13.9 Hz, ArCH2Ar), 4.35 (d, 1H, J = 11.1 Hz, OCH2CO), 4.57 (d, 1H, J = 11.0 Hz, OCH2CO), 6.34 (d, 1H, J = 2.0 Hz, ArH), 6.92 (d, 1H, J = 2.0 Hz, ArH), 7.13 (br s, 2H, ArH), 7.18 (s, 1H, J = 1.9 Hz, ArH), 7.34 (br s, 2H, ArH), 7.68 (d, 1H, J = 1.9 Hz, ArH). 13C NMR: 30.8, 31.6, 31.7 [C(CH3)3], 30.9, 31.2, 36.3, 36.9 (ArCH2Ar), 33.4, 33.8, 34.2 [C(CH3)3], 51.5, 51.9 (CO2CH3), 58.4, 59.8, 60.6 (OCH3), 70.8 (OCH2CO), 124.9, 126.2, 126.4, 127.3, 128.2, 128.3, 128.6, 130.4, 131.8 (ArH), 132.0, 132.7, 132.8, 134.6, 134.8, 135.8, 136.4, 137.3, 143.5, 144.2, 145.4, 151.6, 154.8, 155.3, 155.7 (Ar), 159.8, 166.9 (CO). Cone:1H NMR: 0.66 [s, 9H, C(CH3)3)], 1.31 [s, 18H, C(CH3)3)], 3.20 (d, 2H, J = 10.0 Hz, ArCH2Ar), 3.23 (d, 2H, J = 9.8 Hz, ArCH2Ar), 3.19 (s, 3H, CO2CH3), 3.61 (s, 3H, OCH3), 3.73 (s, 6H, OCH3), 3.79 (s, 3H, CO2CH3), 4.40 (d, 2H, J = 9.8 Hz, ArCH2Ar), 4.42 (d, 2H, J = 10.0 Hz, ArCH2Ar), 4.49 (s, 2H, OCH2CO), 6.26 (s, 2H, ArH), 7.02 (s, 2H, ArH), 7.14 (s, 2H, ArH), 7.24 (s, 2H, ArH). 13C NMR: 30.7, 31.4 [C(CH3)3], 30.9, 31.1 (ArCH2Ar), 33.8, 34.2 [C(CH3)3], 51.4, 51.9 (CO2CH3), 58.3, 60.5 (OCH3), 71.3 (OCH2CO), 159.8, 166.9 (CO); MALDI-TOF MS m/z (Ditranol + NaI), 764.5 [M + H]+, 787.5 [M + Na]+. Anal. (%): Calc. for C52H62O6: C, 75.36; H, 7.91. Found: C, 75.56; H, 7.99.
11,17,23,29,35-Penta-tert-butyl-37,39,40,41,42-pentamethoxy-5- methoxycarbonyl- 38- methoxycarbonylmethyloxycalix[6]arene, 25
A suspension of 23 (150 mg, 0.14 mmol) and K2CO3 (29 mg, 0.21 mmol) in Me2CO (20 mL) was heated at 80 °C for 1 h. Methyl bromoacetate (19 μL, 0.21 mmol) was added and heating continued for 5 h. The solvent was evaporated, the residue was partitioned between CH2Cl2 and HCl (1 M), the organic layer was washed (H2O), dried (MgSO4), evaporated, and the residue was precipitated from MeOH to give 25 (135 mg, 87%) as a white solid mp 197 °C; 1H NMR: 1.03 [s, 18H, C(CH3)3], 1.26 [s, 9H, C(CH3)3], 1.33 [s, 18H, C(CH3)3], 2.72 (s, 6H, OCH3), 2.84 (s, 3H, OCH3), 3.33 (s, 6H, OCH3), 3.74 (s, 3H, CO2Me), 3.79 (s, 3H, CO2Me), 3.98 (br s, 12H, ArCH2Ar), 4.39 (s, 2H, OCH2CO2), 6.77 (s, 2H, ArH), 6.93 (s, 2H, ArH), 7.13 (s, 2H, ArH), 7.14 (s, 2H, ArH), 7.26 (s, 2H, ArH), 7.58 (s, 2H, ArH). 13C NMR: 29.7, 30.1, 30.9 (ArCH2-Ar), 29.5, 30.2, 31.4 [C(CH3)3], 34.0, 34.2, 34.2 [C(CH3)3], 51.8, 52.0 (CO2Me), 59.9, 60.0 (OCH3), 69.2 (OCH2CO2), 125.1, 126.6, 127.0, 127.5, 129.7 (ArH), 125.2, 125.8, 132.4, 133.3, 133.5, 133.6, 135.1, 145.6, 145.7, 146.3, 153.6, 154.2, 154.3, 157.7 (ArC), 166.3, 169.2 (CO); MALDI-TOF MS m/z (Ditranol + NaI), 1139.7 [M + Na]+. Anal. (%): Calc. for C72H92O10: C, 77.38; H, 8.30. Found: C, 77.05; H, 8.15.
11,17,23-Tri-tert-butyl-5-hydroxycarbonyl-26-hydroxycarbonylmethyloxy-25,27,28-trimethoxycalix[4]arene, 26 (asymmetric partial cone : cone 9 : 1)
A suspension of 24 (200 mg, 0.26 mmol) and KOH (50 mg) in THF (50 mL) was heated at 80 °C for 5 h. The solvent was evaporated, the residue was partitioned between CH2Cl2 and HCl (1 M), the organic layer was washed (H2O), dried (MgSO4), evaporated, and then precipitated from MeOH to give 26 (175 mg, 92%). Asymmetric partial cone:1H NMR: 0.84 [s, 9H, C(CH3)3)], 1.12 [s, 9H, C(CH3)3)], 1.25 [s, 9H, C(CH3)3)], 2.75 (s, 3H, OCH3), 3.15–3.25 (m, 2H, ArCH2Ar), 3.62 (s, 3H, OCH3), 3.74 (s, 3H, OCH3), 3.70–3.80 (m, 4H, ArCH2Ar), 4.17 (m, 1H, ArCH2Ar), 4.23 (m, 1H, ArCH2Ar), 4.35 (s, 2H, OCH2-CO), 6.34 (d, 1H, J = 2.0 Hz, ArH), 6.92 (d, 1H, J = 2.0 Hz, ArH), 7.13 (br s, 2H, ArH), 7.18 (s, 1H, J = 1.9 Hz, ArH), 7.34 (br s, 2H, ArH), 7.68 (d, 1H, J = 1.9 Hz, ArH). 13C NMR: 30.8, 31.6, 31.7 [C(CH3)3], 30.9, 31.2, 37.7, 38.4 (ArCH2Ar), 33.4, 33.8, 34.2 [C(CH3)3], 58.4, 59.8, 60.6 (OCH3), 70.4 (OCH2CO), 124.9, 126.2, 126.4, 127.3, 128.2, 128.3, 128.6, 130.4, 131.8 (ArH), 132.0, 132.7, 132.8, 134.6, 134.8, 135.8, 136.4, 137.3, 143.5, 144.2, 145.4, 151.6, 154.8, 155.3, 155.7 (Ar), 169.0, 170.5 (CO). Cone:1H NMR: 0.72 [s, 9H, C(CH3)3)], 1.35 [s, 18H, C(CH3)3)], 3.27 (m, 2H, ArCH2-Ar), 3.29 (m, 2H, ArCH2Ar), 3.61 (s, 3H, OCH3), 3.73 (s, 6H, OCH3), 4.40 (d, 2H, J = 9.8 Hz, ArCH2Ar), 4.42 (d, 2H, J = 10.0 Hz, ArCH2Ar), 4.62 (s, 2H, OCH2CO), 6.26 (s, 2H, ArH), 7.02 (s, 2H, ArH), 7.14 (s, 2H, ArH), 7.24 (s, 2H, ArH). 13C NMR: 30.7, 31.4 [C(CH3)3], 30.9, 31.1 (ArCH2Ar), 33.8, 34.2 [C(CH3)3], 51.4, 51.9 (CO2CH3), 58.3, 60.5 (OCH3), 71.2 (OCH2CO), 169.0, 170.5 (CO). MALDI-TOF MS m/z (Ditranol + NaI), 736.5 [M]+, 759.4 [M + Na]+. Anal. (%): Calc. for C46H56O8: C, 74.97; H, 7.66. Found: C, 74.80; H, 7.51.
11,17,23,29,35-Penta(tert-butyl-5-hydroxycarbonyl-38-hydroxycarbonylmethyloxy-37,39,40,41,42-pentamethoxycalix[6]arene, 27
A suspension of 25 (150 mg, 0.13 mmol) and KOH (200 mg) in THF (20 mL) was heated at 80 °C for 5 h, the solvent was evaporated, the residue was partitioned between CH2Cl2 and HCl (1 M), the organic layer was washed (H2O), dried (MgSO4), evaporated, and the residue was precipitated from MeOH to give 27 (140 mg, 99%) as a white solid, mp 220 °C. 1H NMR (C2D2Cl4, 388 K (115 °C)): 0.95 [s, 9H, C(CH3)3], 1.00 [s, 18H, C(CH3)3], 1.22 [s, 18H, C(CH3)3], 3.16 (s, 2H, OCH2CO2), 3.26 (s, 6H, OCH3), 3.41 (s, 6H, OCH3), 3.46 (s, 3H, OCH3), 3.92 (s, 8H, ArCH2Ar), 3.98 (s, 4H, ArCH2Ar), 6.71 (s, 2H, ArH), 6.73 (s, 2H, ArH), 6.85 (s, 2H, ArH), 7.04 (s, 4H, ArH), 7.84 (s, 2H, ArH). 13C NMR: 30.1, 30.2, 30.7 (ArCH2Ar), 30.8, 30.9, 31.3 [C(CH3)3], 33.4, 33.43, 33.5 [C(CH3)3], 59.5, 59.53, 59.9 (OCH3), 68.8 (OCH2-CO2), 123.8, 124.2, 124.5, 126.5, 126.7, 132.0 (ArH), 123.9, 131.5, 132.3, 132.4, 132.7, 132.9, 134.5, 145.2, 145.6, 145.65, 153.0, 153.2 (ArC), 159.8, 167.1 (CO). MALDI-TOF MS m/z (Ditranol + NaI), 1111.7 [M + Na]+. Anal. (%): Calc. for C70H88O10: C, 77.17; H, 8.14. Found: C, 77.32; H, 8.41.
Acknowledgments
G. W. G. thanks the NIH for grants (GM 36262, GM 63190) and P. P. and J. dM. thank the Ministero de Educación y Ciencia for grant (CTQ2005-08948) that supported this work.
Footnotes
Dedicated to Professor Jerry Atwood on the occasion of his 65th birthday.
References
- 1.MacKinnon R. Angew Chem Int Ed. 2004;43:4265–4277. doi: 10.1002/anie.200400662. [DOI] [PubMed] [Google Scholar]
- 2.Agre P. Angew Chem Int Ed. 2004;43:4278–4290. doi: 10.1002/anie.200460804. [DOI] [PubMed] [Google Scholar]
- 3.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 4.Yasui M, Hazama A, Kwon TH, Nielsen S, Guggino WB, Agre P. Nature. 1999;402:184–187. doi: 10.1038/46045. [DOI] [PubMed] [Google Scholar]
- 5.Nakano A, Xie Q, Mallen JV, Echegoyen L, Gokel GW. J Am Chem Soc. 1990;112:1287–1289. [Google Scholar]
- 6.Jullien L, Lehn JM. Tetrahedron Lett. 1988:3803–3806. [Google Scholar]
- 7.Carmichael VE, Dutton PJ, Fyles FM, James TD, Swan JA, Zojaji M. J Am Chem Soc. 1989;111:767–769. [Google Scholar]
- 8.Roux B, MacKinnon R. Science. 1999;285:100–102. doi: 10.1126/science.285.5424.100. [DOI] [PubMed] [Google Scholar]
- 9.Fyles TM. Chem Soc Rev. 2007;36:335–347. doi: 10.1039/b603256g. [DOI] [PubMed] [Google Scholar]
- 10.Roks MFM, Nolte RJM. Macromolecules. 1992;25:5398–5407. [Google Scholar]
- 11.Meillon JC, Voyer N. Angew Chem Int Ed Engl. 1997;36:967–969. [Google Scholar]
- 12.Canceill J, Jullien L, Lacombe L, Lehn JM. Helv Chim Acta. 1992;75:791–812. [Google Scholar]
- 13.Murillo O, Watanabe S, Nakano A, Gokel GW. J Am Chem Soc. 1995;117:7665–7679. [Google Scholar]
- 14.Gokel GW. Chem Commun. 2000:1–9. [Google Scholar]
- 15.Fyles TM, James TD, Kaye KC. J Am Chem Soc. 1993;115:12315–12321. [Google Scholar]
- 16.Ghadiri MR, Granja JR, Buehler LK. Nature. 1994;369:301–304. doi: 10.1038/369301a0. [DOI] [PubMed] [Google Scholar]
- 17.Matile S. J Am Chem Soc. 1997;119:8726–8727. [Google Scholar]
- 18.Stadler E, Dedek P, Yamashita K, Regen SL. J Am Chem Soc. 1994;116:6677–6682. [Google Scholar]
- 19.Åkerfeldt KS, Lear JD, Wasserman ZR, Chung LA, DeGrado WF. Acc Chem Res. 1993;26:191–197. [Google Scholar]
- 20.Montal M. Annu Rev Biophys Biomol Struct. 1995;24:31–57. doi: 10.1146/annurev.bb.24.060195.000335. [DOI] [PubMed] [Google Scholar]
- 21.Sakmann B, Neher E. Single-channel Recording. Kluwer Academic Publishers; Dordrecht: 1995. [Google Scholar]
- 22.Riddell FG, Arumugam S. Biochim Biophys Acta. 1989;984:6–10. [Google Scholar]
- 23.Hervé M, Cybulska B, Gary-Bobo CM. Eur Biophys J. 1985;12:121–128. [Google Scholar]
- 24.Weber ME, Schlesinger PH, Gokel GW. J Am Chem Soc. 2005;126:636–642. doi: 10.1021/ja044936+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Mendoza J, Cuevas F, Prados P, Meadows ES, Gokel GW. Angew Chem Int Ed. 1998;37:1534–1537. doi: 10.1002/(SICI)1521-3773(19980619)37:11<1534::AID-ANIE1534>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 26.Iqbal KS, Cragg PJ. Dalton Trans. 2007:26–32. doi: 10.1039/b613867p. [DOI] [PubMed] [Google Scholar]
- 27.Hernandez JC, Trafton JE, Gokel GW. Tetrahedron Lett. 1991:6269–6272. [Google Scholar]
- 28.(a) Ikeda A, Shinkai S. J Am Chem Soc. 1994;116:3102–3110. [Google Scholar]; (b) Koh KN, Araki K, Shinkai S, Asfari Z, Vicens J. Tetrahedron Lett. 1995;36:6095–6098. [Google Scholar]
- 29.Leevy WM, Weber ME, Schlesinger PH, Gokel GW. Chem Commun. 2005:89–91. doi: 10.1039/b413588a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abel E, Maguire GEM, Murillo O, Suzuki I, Gokel GW. J Am Chem Soc. 1999;121:9043–9052. [Google Scholar]
- 31.Berthalon S, Motta-Viola L, Regnouf-de-Vains JB, Lamartine R, Lecocq S, Perrin M. Eur J Org Chem. 1999:2269–2274. [Google Scholar]
- 32.de Mendoza J, Carramolino M, Cuevas F, Manuel Nieto P, Prados P, Reinhoudt DN, Verboom W, Ungaro R, Casnati A. Synthesis. 1994:47–50. [Google Scholar]
- 33.Szoka F, Papahadjopoulos D. Proc Natl Acad Sci USA. 1978;75:4194–4198. doi: 10.1073/pnas.75.9.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murray CL, Shabany H, Gokel GW. Chem Commun. 2000:2371–2372. [Google Scholar]
- 35.Weber ME, Wang W, Steinhardt SE, Gokel MR, Leevy WM, Gokel GW. New J Chem. 2006;30:177–184. doi: 10.1039/b510863m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart JC. Anal Biochem. 1980;104:10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]