

Abstract
Gap junction proteins (connexins) facilitate intercellular communication and serve several roles in regulation of tissue function and remodeling. To examine the physiologic effects of depleting two prominent endothelial connexins, Cx40 and Cx43, transgenic mice were generated by breeding Cx40-deficient mice (Cx40−/−) with a vascular endothelial cell (VEC)-specific Cx43-deficient mouse strain (VEC Cx43−/−) to produce double-connexin knockout mice (VEC Cx43−/−/Cx40−/−). The life span in VEC Cx43−/−/Cx40−/− mice was dramatically shortened, which correlated with severe spontaneous lung abnormalities as the mice aged including increased fibrosis, aberrant alveolar remodeling, and increased lung fibroblast content. Moreover, VEC Cx43−/−/Cx40−/− mice exhibited cardiac hypertrophy and hypertension. Because VEC Cx43−/−/Cx40−/− mice demonstrated phenotypic hallmarks that were remarkably similar to those in mice deficient in caveolin-1, pulmonary caveolin expression was examined. Lungs from VEC Cx43−/−/Cx40−/− mice demonstrated significantly decreased expression of caveolin-1 and caveolin-2. This suggests that expression of caveolin-1 may be linked to expression of Cx40 and endothelial Cx43. Moreover, the phenotype of caveolin-1−/− mice and VEC Cx43−/−/Cx40−/− mice may arise via a common mechanism.
Gap junctions are sites of direct cell-cell contact at the plasma membrane and contain arrays of channels that enable diffusion of soluble signaling molecules between cells as a means for intercellular communication. Gap junction channels are composed of proteins known as connexins.1,2 More than two dozen mammalian connexin genes have been identified,3 of which several isoforms are expressed differentially throughout the lung.4
Gap junction communication serves several functions in pulmonary physiology. For example, gap junction communication between type II alveolar epithelial (AT2) and type I alveolar epithelial (AT1) cells helps regulate secretion of pulmonary surfactant.5–7 This system enables AT1 cells to act as mechanosensors responsive to inflation because stretching AT1 cells causes an increase in intracellular calcium, which is subsequently transmitted via gap junctions to AT2 cells to stimulate secretion.6 Gap junction communication is not restricted to cells within an individual alveolus because calcium can also be transmitted between alveoli, even in the absence of mechanical stress.8
Functional communication between the pulmonary circulation and alveoli has been suggested in studies in which increased pulmonary vascular pressure stimulated surfactant secretion via a gap junction–dependent mechanism.9 Whether these signals are strictly due to mechanical distention of AT1 cells or whether gap junction communication from the endothelium to the alveoli is required is not known. Conversely, stimulation of the alveolar airspace with either tumor necrosis factor-α or ATP causes a calcium transient originating in the alveolar epithelium that is subsequently transmitted to the pulmonary endothelium.10,11 Although the role of gap junction communication in mediating these signals is not known, these findings demonstrate that signals that originate in the alveoli can be communicated to the endothelium to induce a physiologic response.
Connexin expression is frequently disrupted in response to lung disease. During the acute phase of lung injury, Cx43 expression in the alveoli is increased,5,12 whereas Cx40 expression at the whole-lung level is decreased.13 Moreover, lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis demonstrate reduced Cx43 expression and decreased intercellular communication.14 The consistent correlative changes in connexin expression with pulmonary disease suggest roles for gap junction communication in normal lung homeostasis and remodeling in addition to established roles for intercellular signaling.
Cx40 and Cx43 are two of the major connexins expressed by the pulmonary vasculature.15 Cx40-deficient mice are hypertensive16 due to misregulation of renin secretion by the kidney.17 Conducted vasodilation along the endothelium of Cx40−/− mice is attenuated,16 although Cx40 deficiency has little effect on large blood vessel architecture.18,19 In addition, there are also hints of blood leakage into the airspaces of lungs from Cx40−/− mice, although this has not been fully established.20 Because Cx43-deficient mice are not viable,21 roles for Cx43 in vascular function were examined using a tissue-targeted approach in which mice expressing Cre recombinase driven by a Tie-2 promoter were crossbred with mice containing a Cx43 gene flanked by loxP sites to generate endothelial-specific Cx43-deficient mice (VEC Cx43−/−).22,23 VEC Cx43−/− mice demonstrate decreased blood pressure,22 in contrast to Cx40−/− mice, which exhibit elevated blood pressure.16 This suggests distinct roles for these two connexins. Moreover, calcium wave propagation via Cx43 in the pulmonary vasculature was severely attenuated in VEC Cx43−/− mice, which indicates that Cx43 has a role in pulmonary function.24
To define the combined effect of depleting endothelial Cx43 and Cx40, double-connexin knockout mice were generated (VEC Cx43−/−/Cx40−/− mice). It was observed that, compared with either control, VEC Cx43−/− or Cx40−/− mice, VEC Cx43−/−/Cx40−/− mice demonstrated decreased viability as they aged. Based on functional and morphologic criteria, VEC Cx43−/−/Cx40−/− mice acquired severe spontaneous lung abnormalities as they aged, which included increased fibrosis and alveolar wall thickening, a phenotype that closely resembled caveolin-1–deficient mice. Consistent with this VEC Cx43−/−/Cx40−/− mice demonstrated significantly decreased caveolin-1 expression. Findings are discussed in the context of previous studies that documented that connexins and caveolins interact, and suggest that the lung pathologic findings in VEC Cx43−/−/Cx40−/− and caveolin-1−/− mice likely occur through a common pathway.
Materials and Methods
Animals
All mice were treated in accordance with animal protocols approved by the University of Virginia Animal Care and Use Committee, under animal protocol number 3648 (to B.E.I.). VEC-specific Cx43−/− mice, which are homozygous for Cx43 flanked by loxP sites and express Cre recombinase driven by the Tie-2 promoter, were generated as originally described.22 Cx40−/− mice were generated as previously described.16 VEC Cx43−/−/Cx40−/− mice were generated by first mating female VEC Cx43−/− mice with male Cx40−/− mice, followed by successive mating of progeny to produce a subset of mice homozygous for VEC Cx43−/− and Cx40−/−. Genomic DNA from tail clips confirmed the genotype of each animal studied. Kaplan-Meier analysis of strain viability was performed as described,25,26 using VassarStats (available at http://faculty.vassar.edu/lowry/VassarStats.html; last accessed December 30, 2010).
Arterial Blood pO2 Analysis
Mice were anesthetized using 60 to 90 mg/kg sodium pentobarbital and placed on a warming pad to prevent loss of body heat. The carotid artery was exposed, and 0.15 mL arterial blood was drawn into a heparinized syringe. Blood gas analysis was immediately performed on the sample using a 178 pH/Blood Gas Analyzer (Ciba Corning Diagnostics Corp., Medfield, MA).27 Results from four 32-week-old mice per genotype were averaged. Statistical significance was determined using one way analysis of variance.
Blood Pressure Measurement
Mean systolic blood pressure was obtained from tail cuff measurements in mice at designated times using the Visitech System (Physiological Research Instruments, Apex, NC), as described.22 Each group of knockout mice consisted of six to eight mice. At least two blood pressure recordings were obtained for each mouse on each day, and the procedure was repeated three or four times on different days. At least six measurements were obtained for each mouse, and data were pooled to obtain a single mean value for each mouse genotype.
Histologic Analysis
Aortas were processed as previously described.28 All lung sections used were taken from the distal left lobe of the mice. Lungs were perfused with saline solution and inflated with 1 mL 4% paraformaldehyde at a constant pressure of 30 cm H2O. Lungs were ligated at the trachea, removed en bloc, and immersed in 4% paraformaldehyde for 24 hours, at which time the immersion medium was changed to 70% alcohol, before paraffin embedding followed by sectioning to 8-μm thickness and H&E staining. Alternatively, Picro-sirius Red staining of paraffin-embedded sections to enhance collagen or the Verhoeff-Van Gieson procedure for staining elastin fibers was performed using standard techniques. Each image represents one of three mice analyzed from each genotype at each time point. Mean alveolar septal width was calculated by measuring the width of the alveolar wall between the pulmonary microcirculation and alveolar airspace in lung sections imaged at transmission electron microscopy.
Primary antibodies used were rabbit anti-Cx40 (ADI, Inc., San Antonio, TX), rabbit anti-Cx43 (Sigma-Aldrich Corp., St. Louis, MO), rabbit anti-prolyl 4-hydroxylase (anti-fibroblast; Abcam Inc., Cambridge, MA), and rabbit anti–caveolin-1 and rabbit anti–caveolin-2 (BD Biosciences, Franklin Lakes, NJ). A secondary antibody used was donkey anti-rabbit Alexa Fluor 594 [Molecular Probes, Eugene, OR (Invitrogen Corp., Carlsbad, CA)]. Immunofluorescence was performed as described.5,29,30 Deparaffinized thin sections were incubated for 30 minutes at room temperature with 1 mmol/L glycine in PBS to reduce nonspecific cross-linking and autofluorescence, washed with PBS, incubated for 10 minutes at room temperature with 1 mg/mL NaBH4 in PBS to further reduce autofluorescence, and washed with PBS plus 0.5% Triton X-100 and PBS plus 0.5% Triton X-100 plus 2% goat serum. The sections were incubated overnight at 4°C with primary antiserum diluted with PBS plus 0.5% Triton X-100 plus 2% goat serum, washed, and incubated with fluorescent goat anti-rabbit IgG (Roche Diagnostics Corp., Indianapolis, IN) for 2 hours at room temperature. Sections were imaged using a confocal microscope (FluoView; Olympus America, Center Valley, PA). Minimum and maximum intensity were adjusted for images in parallel so that the intensity scale remained linear to maximize dynamic range. To quantify the total immunofluorescence signal, total relative fluorescence units were determined from four blinded fields per condition, and were expressed as mean ± SE. Statistical significance was determined using Student's t-test.
Results
Previously, transgenic mice deficient in either Cx40 or Cx43 were used to identify roles for these connexins in the vasculature.22,31 To define the combined effect of depleting Cx43 and Cx40 expression by endothelium, double-connexin knockout mice (VEC Cx43−/−/Cx40−/− mice) were generated. Cx43 was a tissue-specific knockout using floxed Cx43 and Cre recombinase driven by the Tie-2 promoter, which is primarily expressed by vascular endothelium. VEC Cx43−/−/Cx40−/− mice were identified via genotyping of clipped tail samples. The relative expression of Cx40 and Cx43 in aortas of wild-type C57Bl/6 versus VEC Cx43−/−/Cx40−/− mice is shown in Figure 1. Confirming the extent of connexin deficiency, Cx40 was absent from the aortas in VEC Cx43−/−/Cx40−/− mice. In VEC Cx43−/−/Cx40−/− mice, Cx43 was specifically depleted from the endothelium but not the smooth muscle layer, consistent with a tissue-specific knockout.
Figure 1.
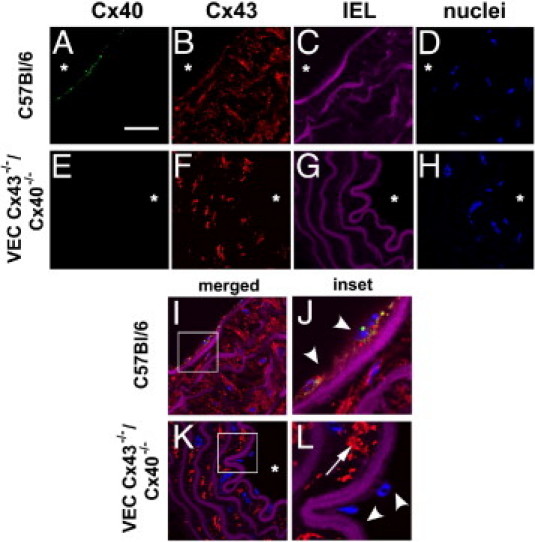
Cx40 and Cx43 expression in mouse aorta of control and VEC Cx43−/−/Cx40 −/− mice. Sections from C57Bl/6 mice (A–D, I, and J) and VEC Cx43−/−/Cx40 −/− mice (E–H, K, and L) were stained with anti-Cx40 [A and E (green)] and anti-Cx43 [B and F (red)]. C, G, and I–L: Violet areas represent autofluorescence of internal elastic lamina (IEL). D, H, and I–L: Nuclei were stained with Sytox Blue. I–L: Merged images. Note lack of Cx40 labeling in aortas from VEC Cx43−/−/Cx40 −/− mice; Cx43 was depleted from the endothelial layer in VEC Cx43−/−/Cx40 −/− mice (arrowheads) but remained present in the smooth muscle layer (arrows). J and L: Insets show threefold magnification of the regions of interest indicated in I and K. Scale bars = 100 μm.
VEC Cx43−/−/Cx40−/− mice died prematurely (Figure 2A). At 50 weeks, the 95% confidence interval for the probability of survival was 0.94 to 1.0 for C57Bl/6 mice (n = 70), 0.82 to 0.98 for Cx40−/− mice (n = 55), and 0.90 to 1.0 for VEC Cx43−/− mice (n = 44). In contrast, compared with the other strains, VEC Cx43−/−/Cx40−/− mice demonstrated significantly reduced viability at 50 weeks, with a probability of survival of <0.80 (95% confidence interval, 0.20 to 0.80; n = 10). Therefore, the VEC Cx43−/−/Cx40−/− mice were examined in greater detail. One clue that decreased lung function was a major contributor to premature death in VEC Cx43−/−/Cx40−/− mice was the appearance of the lungs at 32 weeks; they were enlarged and looked hemorrhagic, which suggested pulmonary dysfunction (Figure 2B). Consistent with impaired lung function at age 32 weeks, carotid arterial blood oxygenation was significantly reduced in VEC Cx43−/−/Cx40−/− mice compared with C57Bl/6, VEC Cx43−/−, or Cx40−/− mice (Figure 2C). It was also confirmed that Cx40 and Cx43 are present in the lungs of wild-type C57BL/6 mice and that VEC Cx43−/−/Cx40−/− mice lacked Cx40 expression (see Supplemental Figure S1 at http://ajp.amjpathol.org). VEC Cx43−/−/Cx40−/− mice retained some Cx43 expression in the lungs, consistent with diminished Cx43 expression by endothelium but preservation of Cx43 by other lung cells.32
Figure 2.
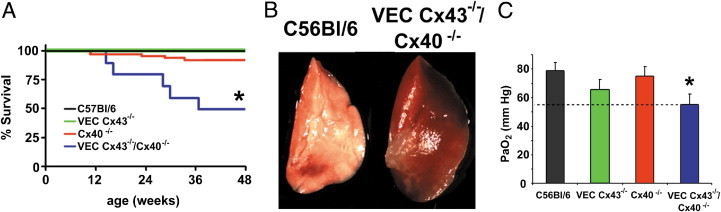
VEC Cx43−/−/Cx40−/− mice have a shortened life span and demonstrate lung injury and decreased blood oxygenation. A: C57Bl/6, Cx40−/−, and VEC Cx43−/− mice all had normal viability; however, nearly half of the VEC Cx43−/−/Cx40−/− mice died by age 32 weeks. *Significantly different from control and single-knockout mice based on efficient score method.25B: Gross anatomy of the left lobe of lungs from viable 32-week-old VEC Cx43−/−/Cx40−/− mice demonstrates a characteristic hemorrhagic appearance indicative of lung injury, as compared with healthy age-matched C57Bl/6 mice. C: Carotid arterial paO2 for C57Bl/6, VEC Cx43−/−, Cx40−/−, and VEC Cx43−/−/Cx40−/− mice measured at age 32 weeks (mean ± SE; n = 4). VEC Cx43−/−/Cx40−/− mice demonstrated significantly lower pO2 than did C57Bl/6 mice. *P < 0.05.
It was also visually apparent that the hearts of VEC Cx43−/−/Cx40−/− mice were larger than those of control mice. Hearts from VEC Cx43−/− or Cx40−/− mice were comparable to those of control mice (data not shown). In addition to cardiac hypertrophy, VEC Cx43−/−/Cx40−/− mice also demonstrated consistently elevated blood pressure at levels comparable to those of Cx40−/− mice (Figure 3B).31 Moreover, VEC Cx43−/− mice demonstrated decreased blood pressure,22 which suggested that loss of Cx40 was the predominant factor in determining the blood pressure of VEC Cx43−/−/Cx40−/− mice. Inasmuch as viability was significantly greater in Cx40−/− mice than in VEC Cx43−/−/Cx40−/− mice (Figure 2A), high blood pressure alone was not sufficient to shorten the life span of VEC Cx43−/−/Cx40−/− mice, and decreased viability was due to other confounding factors, most notably, decreased lung function.
Figure 3.
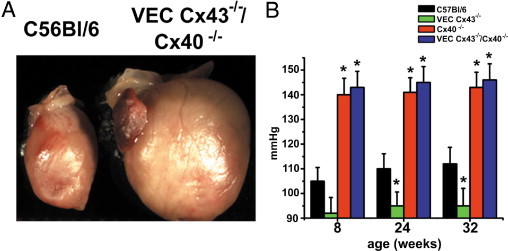
Compared with VEC Cx43−/−/Cx40−/− mice, Cx40−/− mice demonstrated increased blood pressure. A: Gross anatomy of hearts from 32-week-old VEC Cx43−/−/Cx40 −/− mice shows hypertrophy. B: Mean blood pressure measurements from C57Bl/6, VEC Cx43−/−, Cx40−/−, and VEC Cx43−/−/Cx40−/− mice measured at age 8, 24, or 32 weeks (mean ± SE; n = 4). Blood pressure in VEC Cx43−/− mice was significantly lower than C57Bl/6 mice at 24 and 32 weeks. *P < 0.05. Cx40−/− and VEC Cx43−/−/Cx40−/− mice had comparable blood pressure, which was significantly higher than C57Bl/6 mice. *P < 0.05.
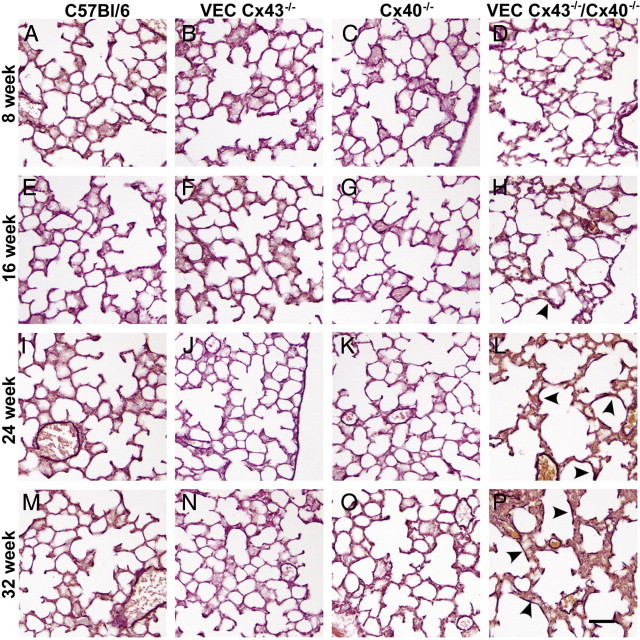
To further examine how the loss of endothelial Cx43 and Cx40 affects lung architecture as a function of age, H&E-stained lung sections were examined (Figure 4). As early as age 16 weeks, the lungs of VEC Cx43−/−/Cx40−/− mice began to exhibit disruption of the alveolar airspaces, which were larger than the alveoli in wild-type C57Bl/6 mice. The architecture of lungs from VEC Cx43−/−/Cx40−/− mice continued to deteriorate as the mice aged; at 32 weeks, morphologic features were comparable in all lungs from VEC Cx43−/−/Cx40−/− mice. In contrast, lungs from either VEC Cx43−/− or Cx40−/− mice appeared more normal.
Figure 4.
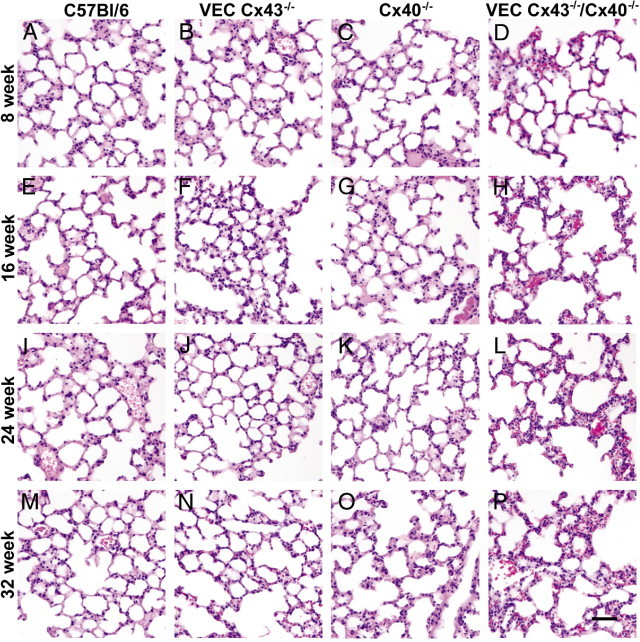
Altered architecture of lungs from VEC Cx43−/−/Cx40−/− mice. Paraffin-embedded distal lung sections from C57Bl/6 (A, E, I, and M), VEC Cx43−/− (B, F, J, and N), Cx40−/− (C, G, K, and O), and VEC Cx43−/−/Cx40−/− (D, H, L, and P) mice at age 8 weeks (A–D), 16 weeks (E–H), 24 weeks (I–L), or 32 weeks (M–P) were stained with H&E and imaged. Compared with the other strains examined, lungs from VEC Cx43−/−/Cx40−/− mice exhibited disrupted alveolar architecture and alveolar wall thickening as they aged. Scale bars = 60 μm.
Because the lungs of VEC Cx43−/−/Cx40−/− mice resembled fibrotic lung disease, elastin and collagen content were examined at histologic analysis. Compared with wild-type or single transgenic mice, lungs from VEC Cx43−/−/Cx40−/− mice were enriched in elastin, as determined using Verhoeff staining (Figure 5). Elastin deposition in lungs from VEC Cx43−/−/Cx40−/− mice seemed to lag behind the alterations in alveolar structure, in which the brown elastin labeling became prevalent beginning at age 24 weeks and persisted to age 32 weeks. By Picro-sirius Red staining, lungs from VEC Cx43−/−/Cx40−/− mice exhibited increased collagen deposition at age 32 weeks, although lungs from wild-type, VEC Cx43−/−, and Cx40−/− mice also demonstrated some localized enrichment with collagen using this approach (Figure 6). Given the changes in architecture and matrix deposition observed in lungs from VEC Cx43−/−/Cx40−/− mice, alveolar septal width was quantified by measuring tissue thickness between the pulmonary microvasculature and the alveolar epithelium. At age 32 weeks, alveolar septa of VEC Cx43−/−/Cx40−/− mice were roughly 25% thicker than septa of C57Bl/6 control mice (Figure 6E). Together, these results demonstrate that, compared with the other strains of mice examined, lungs from VEC Cx43−/−/Cx40−/− mice lost the ability to maintain proper lung architecture and instead were more sensitive to aging.
Figure 5.
Increased elastin deposition in lungs from VEC Cx43−/−/Cx40−/− mice. Paraffin-embedded distal lung sections from C57Bl/6 (A, E, I, and M), VEC Cx43−/− (B, F, J, and N), Cx40−/− (C, G, K, and O), and VEC Cx43−/−/Cx40−/− (D, H, L, and P) mice at age 8 weeks (A–D), 16 weeks (E–H), 24 weeks (I–L), or 32 weeks (M–P) were labeled using Verhoeff to stain elastin fibers dark brown, and were imaged. Compared with the other strains examined, lungs from VEC Cx43−/−/Cx40−/− mice showed increased elastin labeling (arrowheads). Scale bars = 60 μm.
Figure 6.
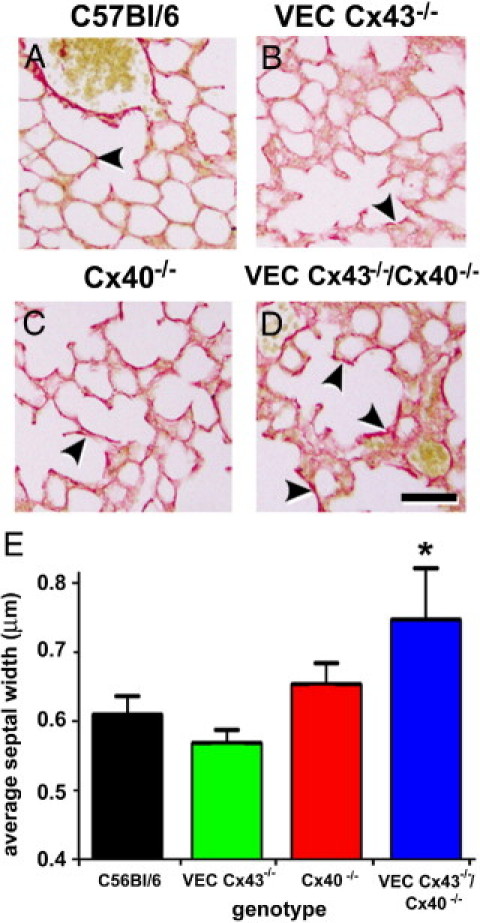
Collagen deposition in lungs from VEC Cx43−/−/Cx40−/− mice. Paraffin-embedded distal lung sections from C57Bl/6 (A), VEC Cx43−/− (B), Cx40−/− (C), and VEC Cx43−/−/Cx40−/− (D) mice at age 32 weeks were labeled using Picro-sirius Red to stain collagen, and were imaged. Compared with the other strains of mice examined, lungs from VEC Cx43−/−/Cx40−/− mice showed enhanced collagen staining (arrowheads). Scale bars = 60 μm. E: Paraffin-embedded distal lung sections from C57Bl/6, VEC Cx43−/−, Cx40−/−, and VEC Cx43−/−/Cx40−/− mice at age 32 weeks were analyzed using morphometric analysis. Compared with the other strains examined, VEC Cx43−/−/Cx40−/− mice demonstrated significantly thicker alveolar septal width between the pulmonary endothelium and the alveolar epithelium. *P < 0.05.
To determine whether the changes observed in VEC Cx43−/−/Cx40−/− mice were due to altered cell proliferation, Sytox Blue was used to label nuclei and scored for changes in lung cell number. Lungs from VEC Cx43−/−/Cx40−/− mice were hyperplastic, in particular at age 32 weeks (Figure 7A). Given the lung pathologic findings, these cells were likely fibroblasts. To confirm this, lung sections from 32-week-old mice were immunolabeled using the enzyme prolyl 4-hydroxylase as a fibroblast marker, which confirmed that there was a significant increase in fibroblasts in the lungs from VEC Cx43−/−/Cx40−/− mice (Figure 7, B–E). Quantitative image analysis of the sections demonstrated that VEC Cx43−/−/Cx40−/− mice had significantly higher fibroblast content than did lungs from the other strains examined (Figure 7F). Moreover, VEC Cx43−/− and Cx40−/− mice also had significantly higher fibroblast content than did wild-type mice, although they had fewer fibroblasts than lungs from double-knockout VEC Cx43−/−/Cx40−/− mice. This underscores the sensitivity of using prolyl 4-hydroxylase as a marker and suggests that the lungs depleted of either endothelial Cx43 or Cx40 demonstrate low levels of injury and/or fibrotic remodeling at age 32 weeks.
Figure 7.
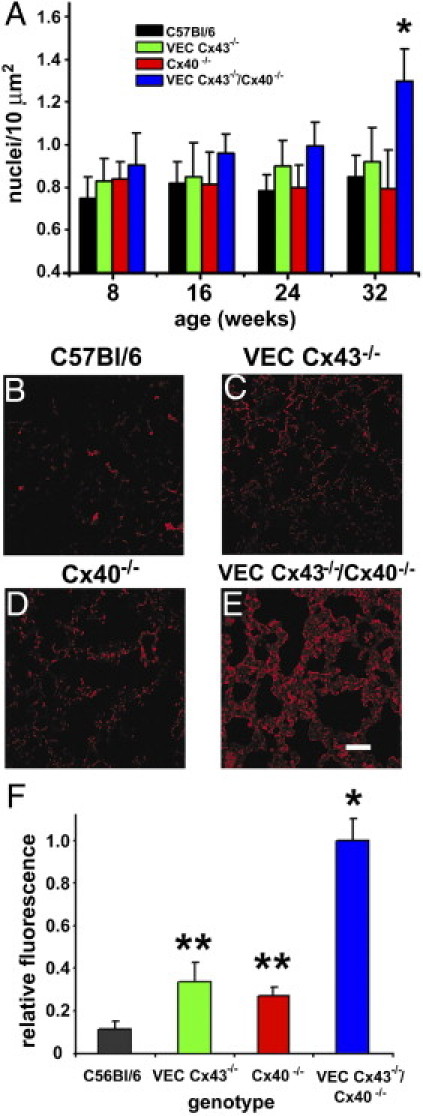
Lungs from VEC Cx43−/−/Cx40−/− mice are enriched in fibroblasts. A: Lung sections from mice at age 8, 16, 24, and 32 weeks were stained with Sytox Blue, and the number of nuclei per 10 μm2 were scored. At age 32 weeks, VEC Cx43−/−/Cx40−/− mice demonstrated significantly more nuclei than did the other strains examined, indicating enhanced cell proliferation (P < 0.05). B–E: Immunofluorescence analysis of paraffin-embedded lung sections from 32-week-old C57Bl/6 (B), VEC Cx43−/− (C), Cx40−/− (D), and VEC Cx43−/−/Cx40−/− (E) mice were immunostained for the fibroblast marker prolyl 4-hydroxylase. Scale bars = 60 μm. F: Quantification of fibroblast immunofluorescence from stained lung sections. Compared with the other strains examined, lungs from VEC Cx43−/−/Cx40−/− mice demonstrated significantly more fibroblasts at 32 weeks. *P < 0.01. The fibroblast content of lungs from single-knockout VEC Cx43−/− and Cx40−/− mice also had significantly more fibroblasts than did wild-type lungs at age 32 weeks. **P < 0.05.
The phenotype of VEC Cx43−/−/Cx40−/− mice was remarkably similar to the phenotype of caveolin-1 deficient mice, which are subject to premature death,33 alveolar disruption,34,35 alterations in lung extracellular cell matrix,36,37 cardiac hypertrophy,38 and hypertension.39 Thus, it was hypothesized that caveolin-1 expression is reduced in VEC Cx43−/−/Cx40−/− mice. Compared with lungs from wild-type, VEC Cx43−/−, and Cx40−/− mice, caveolin-1 was significantly reduced in lungs from VEC Cx43−/−/Cx40−/− mice as early as age 8 weeks (Figure 8; see also Supplemental Figure S2 at http://ajp.amjpathol.org). Moreover, compared with the other strains examined, caveolin-2 expression was also decreased in lungs from VEC Cx43−/−/Cx40−/− mice (Figure 8; see also Supplemental Figure S3 at http://ajp.amjpathol.org), consistent with previous findings that caveolin-2 expression is diminished in caveolin-1–deficient mice.34,35 Thus, the data suggest that expression of caveolin-1 and caveolin-2 is linked to expression of Cx43 and Cx40 and that the pulmonary damage observed in caveolin-1−/− mice and VEC Cx43−/−/Cx40−/− mice develops through a common pathway.
Figure 8.
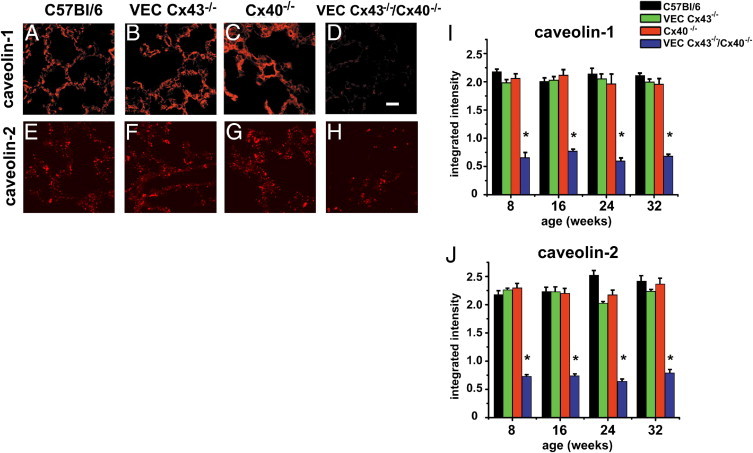
Decreased caveolin-1 and caveolin-2 expression by lungs from VEC Cx43−/−/Cx40−/− mice. Paraffin-embedded distal lung sections from C57Bl/6 (A and E), VEC Cx43−/− (B and F), Cx40−/− (C and G), and VEC Cx43−/−/Cx40−/− (D and H) mice at age 32 weeks were immunostained for caveolin-1 (A–D) or caveolin-2 (E–H) and imaged. Scale bars = 60 μm. Caveolin-1 (I) and Caveolin-2 (J) were quantified by immunofluorescence analysis of paraffin-embedded lung sections from C57Bl/6, VEC Cx43−/−, Cx40−/−, and VEC Cx43−/−/Cx40−/− mice at age 8, 16, 24, and 32 weeks (Figures S2 and S3). Compared with the other strains of mice examined, VEC Cx43−/−/Cx40−/− mice demonstrate decreased caveolin-1 and caveolin-2, as early as age 8 weeks. *P < 0.05.
Discussion
It was observed that as they aged, VEC Cx43−/−/Cx40−/− mice died prematurely, which correlated with severe spontaneous lung abnormalities. This included increased fibrosis and alveolar wall thickening, which had an adverse effect on blood oxygenation. Previous studies have associated decreased expression of Cx40 with acute lung injury,13 and of Cx43 with idiopathic pulmonary fibrosis.14 The present study provided evidence that reducing expression of these connexins in an intact mouse model is a causative factor in development of lung disease.
Deletion of vascular connexins had a deleterious effect on lung architecture and remodeling, which underscores the concept of coordinate regulation of pulmonary epithelial and vascular compartments to maintain proper lung architecture. Both sides of the alveolar-capillary unit are functionally coupled via transmission of calcium transients, which can originate on either the endothelial or epithelial side and, in some cases, require gap junction communication.9–11 By electron microscopy, human alveolar epithelium and pulmonary circulation are interconnected by a network of gap junctions passing through fibroblasts. This is disrupted in chronic obstructive pulmonary disease and further underscores the importance of intercellular communication in preventing lung disease.40,41
The phenotype of VEC Cx43−/−/Cx40−/− mice was remarkably comparable to that of caveolin-1–deficient mice.34,35 Although the similar pathologic findings in these two different mouse strains with distinct genotypes was surprising, it has been well established that several connexins directly interact with caveolins and contain caveolin-binding motifs.42–44 Moreover, mesenteric arteries from caveolin-1−/− mice exhibited defective vasodilation and, according to immunoblot analysis and morphologic criteria, demonstrated decreased expression of vascular Cx37, Cx40, and Cx43.32 This decrease in vascular connexin expression correlated with a blunted endothelium-derived hyperpolarizing factor response by caveolin-1−/−–deficient vessels, which underscores a role for caveolin-1 in proper targeting of connexins to myoendothelial junctions.32
The ability of connexins to bind to caveolin-1 and caveolin-2 also has been confirmed by co-immunoprecipitation using preparations isolated from cultured cells.42–44 Detailed analysis of caveolin/Cx43 interactions has revealed that these proteins initially associate at the trans-Golgi network to facilitate incorporation of connexin hemichannels into cholesterol-enriched membrane microdomains (“lipid rafts”).42 This in turn facilitates connexin transport to the plasma membrane. Connexins at the plasma membrane generally exhibit low levels of co-localization with caveolin-1,42,43,45–47 yet a significant fraction remains associated with membrane microdomains, based on co-localization with glycosphingolipids labeled with fluorescent cholera toxin.45,48,49 Moreover, connexins in mature gap junctions are not present in caveolae or glycolipid-enriched microdomains; rather, they are removed from rafts through the action of the cytoplasmic scaffold protein zonula occludens-1.50,51
Caveolar endocytosis is a prominent pathway for plasma membrane internalization by endothelial cells.52,53 However, Cx43 turnover does not depend on membrane microdomains.54 Rather, Cx43 is internalized by clathrin-coated pits and degraded by ubiquitinylation.55,56 Whether Cx40 is comparably processed has not been definitely established. Inasmuch as Cx40 and Cx43 hetero-oligomerize,57,58 Cx40 and Cx43 are likely co-internalized by clathrin-coated pits. Thus, the more prominent role for caveolins in controlling connexins is regulation of transport of newly synthesized connexins as opposed to a significant direct role in regulating connexin turnover.
Although caveolins and connexins interact directly, lungs from VEC Cx43−/−/Cx40−/− mice demonstrated a generalized decrease in caveolin-1 and caveolin-2 content beyond the pulmonary vasculature (Figure 8, and Figures S2 and S3). Thus, a model is favored in which initial disruption of endothelial connexins and caveolins in combination with stress during aging initiates a pathway that has a deleterious effect on overall lung caveolin content. As one possibility, idiopathic pulmonary fibrosis is associated with increased expression of transforming growth factor β (TGF-β) and decreased caveolin-1 content.59 Moreover, TGF-β–treated human lung fibroblasts have decreased caveolin-1 expression.60 A comparable increase in lung TGF-β and decrease in AT1 caveolin-1 expression was observed in a rodent model of radiation- induced pulmonary fibrosis.61 Although the decrease in caveolin-1 and caveolin-2 content in lungs from VEC Cx43−/−/Cx40−/− mice precedes overt disruption of lung architecture, it is possible that significant levels of TGF-β are present in young connexin-deficient mice, which might have a deleterious effect on caveolin expression. Consistent with this possibility, rats fed alcohol have morphologically normal lungs that contain fivefold more TGF-β than do control rats given normal feed. This increase in TGF-β in response to alcohol compromises lung barrier function and increases susceptibility to acute lung injury.62
Epithelial to mesenchyme transition has an important role in idiopathic pulmonary fibrosis.63,64 Recent evidence suggests that connexin expression inhibits epithelial to mesenchyme transition in several systems including mammary tumor cells, which revert to a normal cell phenotype when transfected to overexpress connexins,65 and epicardium, in which Cx43 is required for proper vessel development.66 A recent study by Langlois et al46 demonstrated that functional cooperation between Cx43 and caveolin protects normal keratinocytes from epithelial to mesenchyme transition induced by phorbol esters and epidermal growth factor. In a key set of experiments, overexpression of full-length Cx43 was protective; however, overexpression of a C-terminal truncated form of Cx43 that cannot bind to caveolin-1 was not, which suggests that cooperation between Cx43 and caveolin-1 was required to inhibit epithelial to mesenchyme transition.46 If a co-regulatory role for connexins and caveolins can be extended more generally to control of cell phenotype and migration, it can be speculated that connexin/caveolin complexes may modulate overexuberant vasculogenesis, which is associated with aberrant lung remodeling in response to injury.67–70 Whether this is the case remains to be determined.
A primary function of caveolin-1 is to provide a matrix that enables local enrichment of diverse proteins into an integrated platform that controls cell signaling. For example, endothelial nitric oxide synthase is associated with caveolin-enriched membrane microdomains71–73 and is hyperactivated in caveolin-1−/− mice, leading to hypertension.74 In addition, recent evidence suggests that gap junction communication across myoendothelial junctions is controlled by a cyclical nitrosylation/denitrosylation pathway.75 Whether endothelial nitric oxide synthase misregulation in caveolin-1−/− mice is due to disruption of normal cross-talk between nitrosylation and gap junction communication pathways remains to be determined. Nonetheless, by serving as part of a caveolin-signaling complex, connexins have the potential to expand an individual cell response into a multicellular response by enabling intracellular signaling to be coupled to direct intercellular gap junction communication.
Acknowledgments
We thank Susan I. Ramos, David N. Damon, and Katherine H. Day for initial experiments involved in generation of the mice, and Victor Laubach for assistance with blood pO2 measurements.
Footnotes
Supported by Emory Alcohol and Lung Biology Center/NIH grant P50-AA013757 (M.K.), NIH grants R01-HL083120 (M.K.), R01-HL053318 (B.R.D.), and R01-HL088554 (B.E.I.), an American Heart Association (AHA) Scientist Development Grant (B.E.I.), AHA Postdoctoral Fellowship Awards (M.B.), and an NIH T32 Fellowship (A.C.S.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.02.045.
Contributor Information
Michael Koval, Email: mhkoval@emory.edu.
Brant E. Isakson, Email: bei6n@virginia.edu.
Supplementary data
Cx40 and Cx43 expression in mouse lung. Immunofluorescence analysis of lung sections from 8-week-old C57Bl/6 (A and D) and VEC Cx43−/−/Cx40−/− (B, C, E, and F) mice shows the extent of Cx43 (A and B) and Cx40 (D and E) (arrowheads) present in lungs from healthy versus transgenic mice. C and F: Note partial depletion of Cx43 due to the VEC Cx43−/− genotype (B). Sections incubated with Cy3 goat anti-rabbit IgG alone (2°) show limited nonspecific labeling. Scale bars = 60 μm.
Decreased caveolin-1 expression by lungs of VEC Cx43−/−/Cx40−/− mice. Paraffin-embedded distal lung sections from C57Bl/6 (A, E, I, and M), VEC Cx43−/− (B, F, J, and N), Cx40−/− (C, G, K, and O), and VEC Cx43−/−/Cx40−/− (D, H, L, and P) mice at age 8 weeks (A–D), 16 weeks (E–H), 24 weeks (I–L), and 32 weeks (M–P) were immunostained for caveolin-1 and imaged. Scale bars = 60 μm. Compared with the other strains of mice examined, as early as age 8 weeks, VEC Cx43−/−/Cx40−/− mice demonstrated decreased caveolin-1. For quantification, see Figure 8A.
Decreased caveolin-2 expression by lungs from VEC Cx43−/−/Cx40−/− mice. Paraffin-embedded distal lung sections from C57Bl/6 (A, E, I, and M), VEC Cx43−/− (B, F, J, and N), 1 Cx40−/− (C, G, K, and O), and VEC Cx43−/−/Cx40−/− (D, H, L, and P) mice at age 8 weeks (A–D), 16 weeks (E–H), 24 weeks (I–L), and 32 weeks (M–P) were immunostained for caveolin-2 and imaged. Scale bars = 60 μm. Compared with the other strains of mice examined, as early as age 8 weeks, VEC Cx43−/−/Cx40−/− mice demonstrated decreased caveolin-2. For quantification, see Figure 8B.
References
- 1.Koval M. Pathways and control of connexin oligomerization. Trends Cell Biol. 2006;16:159–166. doi: 10.1016/j.tcb.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laird D.W. Life cycle of connexins in health and disease. Biochem J. 2006;394:527–543. doi: 10.1042/BJ20051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sohl G., Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res. 2004;62:228–232. doi: 10.1016/j.cardiores.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 4.Koval M. Sharing signals: connecting lung epithelial cells with gap junction channels. Am J Physiol Lung Cell Mol Physiol. 2002;283:L875–L893. doi: 10.1152/ajplung.00078.2002. [DOI] [PubMed] [Google Scholar]
- 5.Abraham V., Chou M.L., George P., Pooler P., Zaman A., Savani R.C., Koval M. Heterocellular gap junctional communication between alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1085–L1093. doi: 10.1152/ajplung.2001.280.6.L1085. [DOI] [PubMed] [Google Scholar]
- 6.Ashino Y., Ying X., Dobbs L.G., Bhattacharya J. [Ca(2+)](i) oscillations regulate type II cell exocytosis in the pulmonary alveolus. Am J Physiol. 2000;279:L5–L13. doi: 10.1152/ajplung.2000.279.1.L5. [DOI] [PubMed] [Google Scholar]
- 7.Guo Y., Martinez-Williams C., Yellowley C.E., Donahue H.J., Rannels D.E. Connexin expression by alveolar epithelial cells is regulated by extracellular matrix. Am J Physiol Lung Cell Mol Physiol. 2001;280:L191–L202. doi: 10.1152/ajplung.2001.280.2.L191. [DOI] [PubMed] [Google Scholar]
- 8.Ichimura H., Parthasarathi K., Lindert J., Bhattacharya J. Lung surfactant secretion by interalveolar Ca2+ signaling. Am J Physiol Lung Cell Mol Physiol. 2006;291:L596–L601. doi: 10.1152/ajplung.00036.2006. [DOI] [PubMed] [Google Scholar]
- 9.Wang P.M., Fujita E., Bhattacharya J. Vascular regulation of type II cell exocytosis. Am J Physiol Lung Cell Mol Physiol. 2002;282:L912–L916. doi: 10.1152/ajplung.00303.2001. [DOI] [PubMed] [Google Scholar]
- 10.Kiefmann R., Islam M.N., Lindert J., Parthasarathi K., Bhattacharya J. Paracrine purinergic signaling determines lung endothelial nitric oxide production. Am J Physiol Lung Cell Mol Physiol. 2009;296:L901–L910. doi: 10.1152/ajplung.90549.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuebler W.M., Parthasarathi K., Wang P.M., Bhattacharya J. A novel signaling mechanism between gas and blood compartments of the lung. J Clin Invest. 2000;105:905–913. doi: 10.1172/JCI8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasper M., Traub O., Reimann T., Bjermer L., Grossmann H., Muller M., Wenzel K.W. Upregulation of gap junction protein connexin43 in alveolar epithelial cells of rats with radiation-induced pulmonary fibrosis. Histochem Cell Biol. 1996;106:419–424. doi: 10.1007/BF02473301. [DOI] [PubMed] [Google Scholar]
- 13.Rignault S., Haefliger J.A., Waeber B., Liaudet L., Feihl F. Acute inflammation decreases the expression of connexin 40 in mouse lung. Shock. 2007;28:78–85. doi: 10.1097/shk.0b013e3180310bd1. [DOI] [PubMed] [Google Scholar]
- 14.Trovato-Salinaro A., Trovato-Salinaro E., Failla M., Mastruzzo C., Tomaselli V., Gili E., Crimi N., Condorelli D.F., Vancheri C. Altered intercellular communication in lung fibroblast cultures from patients with idiopathic pulmonary fibrosis. Respir Res. 2006;7:122. doi: 10.1186/1465-9921-7-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson L.N., Koval M. Cross-talk between pulmonary injury, oxidant stress, and gap junctional communication. Antiox Redox Signal. 2009;11:355–367. doi: 10.1089/ars.2008.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Figueroa X.F., Paul D.L., Simon A.M., Goodenough D.A., Day K.H., Damon D.N., Duling B.R. Central role of connexin40 in the propagation of electrically activated vasodilation in mouse cremasteric arterioles in vivo. Circ Res. 2003;92:793–800. doi: 10.1161/01.RES.0000065918.90271.9A. [DOI] [PubMed] [Google Scholar]
- 17.Wagner C., de Wit C., Kurtz L., Grunberger C., Kurtz A., Schweda F. Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ Res. 2007;100:556–563. doi: 10.1161/01.RES.0000258856.19922.45. [DOI] [PubMed] [Google Scholar]
- 18.Simon A.M., Goodenough D.A., Paul D.L. Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atrioventricular block and bundle branch block. Curr Biol. 1998;8:295–298. doi: 10.1016/s0960-9822(98)70113-7. [DOI] [PubMed] [Google Scholar]
- 19.Simon A.M., McWhorter A.R. Vascular abnormalities in mice lacking the endothelial gap junction proteins connexin37 and connexin40. Dev Biol. 2002;251:206–220. doi: 10.1006/dbio.2002.0826. [DOI] [PubMed] [Google Scholar]
- 20.Simon A.M., McWhorter A.R. Decreased intercellular dye-transfer and downregulation of non-ablated connexins in aortic endothelium deficient in connexin37 or connexin40. J Cell Sci. 2003;116:2223–2236. doi: 10.1242/jcs.00429. [DOI] [PubMed] [Google Scholar]
- 21.Reume A.G., deSousa P.A., Kulkarni S., Langille B.L., Zhu D., Davies T.C., Juneja S.C., Kidder G.M., Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 22.Liao Y., Day K.H., Damon D.N., Duling B.R. Endothelial cell-specific knockout of connexin 43 causes hypotension and bradycardia in mice. Proc Natl Acad Sci USA. 2001;98:9989–9994. doi: 10.1073/pnas.171305298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu Y., Bradley A. Engineering chromosomal rearrangements in mice. Nat Rev Genet. 2001;2:780–790. doi: 10.1038/35093564. [DOI] [PubMed] [Google Scholar]
- 24.Parthasarathi K., Ichimura H., Monma E., Lindert J., Quadri S., Issekutz A., Bhattacharya J. Connexin 43 mediates spread of Ca2+-dependent proinflammatory responses in lung capillaries. J Clin Invest. 2006;116:2193–2200. doi: 10.1172/JCI26605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newcombe R.G. Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat Med. 1998;17:857–872. doi: 10.1002/(sici)1097-0258(19980430)17:8<857::aid-sim777>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 26.Kaplan E.L., Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 27.Ross S.D., Kron I.L., Gangemi J.J., Shockey K.S., Stoler M., Kern J.A., Tribble C.G., Laubach V.E. Attenuation of lung reperfusion injury after transplantation using an inhibitor of nuclear factor-kappaB. Am J Physiol Lung Cell Mol Physiol. 2000;279:L528–L536. doi: 10.1152/ajplung.2000.279.3.L528. [DOI] [PubMed] [Google Scholar]
- 28.Isakson B.E., Damon D.N., Day K.H., Liao Y., Duling B.R. Connexin40 and connexin43 in mouse aortic endothelium: evidence for coordinated regulation. Am J Physiol Heart Circ Physiol. 2006;290:H1199–H1205. doi: 10.1152/ajpheart.00945.2005. [DOI] [PubMed] [Google Scholar]
- 29.Wang F., Daugherty B., Keise L.L., Wei Z., Foley J.P., Savani R.C., Koval M. Heterogeneity of claudin expression by alveolar epithelial cells. Am J Respir Cell Mol Biol. 2003;29:62–70. doi: 10.1165/rcmb.2002-0180OC. [DOI] [PubMed] [Google Scholar]
- 30.Isakson B.E., Olsen C.E., Boitano S. Laminin-332 alters connexin profile, dye coupling and intercellular Ca2+ waves in ciliated tracheal epithelial cells. Respir Res. 2006;7:105. doi: 10.1186/1465-9921-7-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Wit C., Roos F., Bolz S.S., Pohl U. Lack of vascular connexin 40 is associated with hypertension and irregular arteriolar vasomotion. Physiol Genomics. 2003;13:169–177. doi: 10.1152/physiolgenomics.00169.2002. [DOI] [PubMed] [Google Scholar]
- 32.Saliez J., Bouzin C., Rath G., Ghisdal P., Desjardins F., Rezzani R., Rodella L.F., Vriens J., Nilius B., Feron O., Balligand J.L., Dessy C. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor–mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation. 2008;117:1065–1074. doi: 10.1161/CIRCULATIONAHA.107.731679. [DOI] [PubMed] [Google Scholar]
- 33.Park D.S., Cohen A.W., Frank P.G., Razani B., Lee H., Williams T.M., Chandra M., Shirani J., De Souza A.P., Tang B., Jelicks L.A., Factor S.M., Weiss L.M., Tanowitz H.B., Lisanti M.P. Caveolin-1 null (−/−) mice show dramatic reductions in life span. Biochemistry. 2003;42:15124–15131. doi: 10.1021/bi0356348. [DOI] [PubMed] [Google Scholar]
- 34.Razani B., Engelman J.A., Wang X.B., Schubert W., Zhang X.L., Marks C.B., Macaluso F., Russell R.G., Li M., Pestell R.G., Di Vizio D., Hou H Jr, Kneitz B., Lagaud G., Christ G.J., Edelmann W., Lisanti M.P. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 35.Drab M., Verkade P., Elger M., Kasper M., Lohn M., Lauterbach B., Menne J., Lindschau C., Mende F., Luft F.C., Schedl A., Haller H., Kurzchalia T.V. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 36.Le Saux O., Teeters K., Miyasato S., Choi J., Nakamatsu G., Richardson J.A., Starcher B., Davis E.C., Tam E.K., Jourdan-Le Saux C. The role of caveolin-1 in pulmonary matrix remodeling and mechanical properties. Am J Physiol Lung Cell Mol Physiol. 2008;295:L1007–L1017. doi: 10.1152/ajplung.90207.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maniatis N.A., Shinin V., Schraufnagel D.E., Okada S., Vogel S.M., Malik A.B., Minshall R.D. Increased pulmonary vascular resistance and defective pulmonary artery filling in caveolin-1−/− mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L865–L873. doi: 10.1152/ajplung.00079.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen A.W., Park D.S., Woodman S.E., Williams T.M., Chandra M., Shirani J., Pereira de Souza A., Kitsis R.N., Russell R.G., Weiss L.M., Tang B., Jelicks L.A., Factor S.M., Shtutin V., Tanowitz H.B., Lisanti M.P. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am J Physiol Cell Physiol. 2003;284:C457–C474. doi: 10.1152/ajpcell.00380.2002. [DOI] [PubMed] [Google Scholar]
- 39.Zhao Y.Y., Liu Y., Stan R.V., Fan L., Gu Y., Dalton N., Chu P.H., Peterson K., Ross J Jr, Chien K.R. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA. 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sirianni F.E., Milaninezhad A., Chu F.S., Walker D.C. Alteration of fibroblast architecture and loss of Basal lamina apertures in human emphysematous lung. Am J Respir Crit Care Med. 2006;173:632–638. doi: 10.1164/rccm.200509-1434OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sirianni F.E., Chu F.S., Walker D.C. Human alveolar wall fibroblasts directly link epithelial type 2 cells to capillary endothelium. Am J Respir Crit Care Med. 2003;168:1532–1537. doi: 10.1164/rccm.200303-371OC. [DOI] [PubMed] [Google Scholar]
- 42.Langlois S., Cowan K.N., Shao Q., Cowan B.J., Laird D.W. Caveolin-1 and -2 interact with connexin43 and regulate gap junctional intercellular communication in keratinocytes. Mol Biol Cell. 2008;19:912–928. doi: 10.1091/mbc.E07-06-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schubert A.L., Schubert W., Spray D.C., Lisanti M.P. Connexin family members target to lipid raft domains and interact with caveolin-1. Biochemistry. 2002;41:5754–5764. doi: 10.1021/bi0121656. [DOI] [PubMed] [Google Scholar]
- 44.Lin D., Lobell S., Jewell A., Takemoto D.J. Differential phosphorylation of connexin46 and connexin50 by H2O2 activation of protein kinase Cgamma. Mol Vis. 2004;10:688–695. [PubMed] [Google Scholar]
- 45.Leaphart C.L., Dai S., Gribar S.C., Richardson W., Ozolek J., Shi X.H., Bruns J.R., Branca M., Li J., Weisz O.A., Sodhi C., Hackam D.J. Interferon-gamma inhibits enterocyte migration by reversibly displacing connexin43 from lipid rafts. Am J Physiol Gastrointest Liver Physiol. 2008;295:G559–G569. doi: 10.1152/ajpgi.90320.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langlois S., Cowan K.N., Shao Q., Cowan B.J., Laird D.W. The tumor-suppressive function of connexin43 in keratinocytes is mediated in part via interaction with caveolin-1. Cancer Res. 2010;70:4222–4232. doi: 10.1158/0008-5472.CAN-09-3281. [DOI] [PubMed] [Google Scholar]
- 47.Barth K., Gentsch M., Blasche R., Pfuller A., Parshyna I., Koslowski R., Barth G., Kasper M. Distribution of caveolin-1 and connexin43 in normal and injured alveolar epithelial R3/1 cells. Histochem Cell Biol. 2005;123:239–247. doi: 10.1007/s00418-004-0727-4. [DOI] [PubMed] [Google Scholar]
- 48.Sasseville M., Gagnon M.C., Guillemette C., Sullivan R., Gilchrist R.B., Richard F.J. Regulation of gap junctions in porcine cumulus-oocyte complexes: contributions of granulosa cell contact, gonadotropins, and lipid rafts. Mol Endocrinol. 2009;23:700–710. doi: 10.1210/me.2008-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Locke D., Liu J., Harris A.L. Lipid rafts prepared by different methods contain different connexin channels, but gap junctions are not lipid rafts. Biochemistry. 2005;44:13027–13042. doi: 10.1021/bi050495a. [DOI] [PubMed] [Google Scholar]
- 50.Laing J.G., Chou B.C., Steinberg T.H. ZO-1 alters the plasma membrane localization and function of Cx43 in osteoblastic cells. J Cell Sci. 2005;118:2167–2176. doi: 10.1242/jcs.02329. [DOI] [PubMed] [Google Scholar]
- 51.Laing J.G., Koval M., Steinberg T.H. Association with ZO-1 correlates with plasma membrane partitioning in truncated connexin45 mutants. J Membr Biol. 2005;207:45–53. doi: 10.1007/s00232-005-0803-2. [DOI] [PubMed] [Google Scholar]
- 52.Minshall R.D., Tiruppathi C., Vogel S.M., Niles W.D., Gilchrist A., Hamm H.E., Malik A.B. Endothelial cell-surface gp60 activates vesicle formation and trafficking via G(i)-coupled Src kinase signaling pathway. J Cell Biol. 2000;150:1057–1070. doi: 10.1083/jcb.150.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim K.J., Malik A.B. Protein transport across the lung epithelial barrier. Am J Physiol Lung Cell Mol Physiol. 2003;284:L247–L259. doi: 10.1152/ajplung.00235.2002. [DOI] [PubMed] [Google Scholar]
- 54.Mograbi B., Corcelle E., Defamie N., Samson M., Nebout M., Segretain D., Fenichel P., Pointis G. Aberrant connexin 43 endocytosis by the carcinogen lindane involves activation of the ERK/mitogen-activated protein kinase pathway. Carcinogenesis. 2003;24:1415–1423. doi: 10.1093/carcin/bgg093. [DOI] [PubMed] [Google Scholar]
- 55.Piehl M., Lehmann C., Gumpert A., Denizot J.P., Segretain D., Falk M.M. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell. 2007;18:337–347. doi: 10.1091/mbc.E06-06-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leithe E., Rivedal E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci. 2004;117:1211–1220. doi: 10.1242/jcs.00951. [DOI] [PubMed] [Google Scholar]
- 57.Stergiopoulos K., Alvarado J.L., Mastroianni M., Ek-Vitorin J.F., Taffet S.M., Delmar M. Hetero-domain interactions as a mechanism for the regulation of connexin channels. Circ Res. 1999;84:1144–1155. doi: 10.1161/01.res.84.10.1144. [DOI] [PubMed] [Google Scholar]
- 58.Cottrell G.T., Wu Y., Burt J.M. Cx40 and Cx43 expression ratio influences heteromeric/heterotypic gap junction channel properties. Am J Physiol Cell Physiol. 2002;282:C1469–C1482. doi: 10.1152/ajpcell.00484.2001. [DOI] [PubMed] [Google Scholar]
- 59.Broekelmann T.J., Limper A.H., Colby T.V., McDonald J.A. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc Natl Acad Sci USA. 1991;88:6642–6646. doi: 10.1073/pnas.88.15.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X.M., Zhang Y., Kim H.P., Zhou Z., Feghali-Bostwick C.A., Liu F., Ifedigbo E., Xu X., Oury T.D., Kaminski N., Choi A.M. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med. 2006;203:2895–2906. doi: 10.1084/jem.20061536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kasper M., Reimann T., Hempel U., Wenzel K.W., Bierhaus A., Schuh D., Dimmer V., Haroske G., Muller M. Loss of caveolin expression in type I pneumocytes as an indicator of subcellular alterations during lung fibrogenesis. Histochem Cell Biol. 1998;109:41–48. doi: 10.1007/s004180050200. [DOI] [PubMed] [Google Scholar]
- 62.Bechara R.I., Brown L.A., Roman J., Joshi P.C., Guidot D.M. Transforming growth factor beta1 expression and activation is increased in the alcoholic rat lung. Am J Respir Crit Care Med. 2004;170:188–194. doi: 10.1164/rccm.200304-478OC. [DOI] [PubMed] [Google Scholar]
- 63.Willis B.C., Liebler J.M., Luby-Phelps K., Nicholson A.G., Crandall E.D., du Bois R.M., Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim K.K., Kugler M.C., Wolters P.J., Robillard L., Galvez M.G., Brumwell A.N., Sheppard D., Chapman H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McLachlan E., Shao Q., Wang H.L., Langlois S., Laird D.W. Connexins act as tumor suppressors in three-dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res. 2006;66:9886–9894. doi: 10.1158/0008-5472.CAN-05-4302. [DOI] [PubMed] [Google Scholar]
- 66.Rhee D.Y., Zhao X.Q., Francis R.J., Huang G.Y., Mably J.D., Lo C.W. Connexin 43 regulates epicardial cell polarity and migration in coronary vascular development. Development. 2009;136:3185–3193. doi: 10.1242/dev.032334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tzouvelekis A., Anevlavis S., Bouros D. Angiogenesis in interstitial lung diseases: a pathogenetic hallmark or a bystander? Respir Res. 2006;7:82. doi: 10.1186/1465-9921-7-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Antoniou K.M., Tzouvelekis A., Alexandrakis M.G., Sfiridaki K., Tsiligianni I., Rachiotis G., Tzanakis N., Bouros D., Milic-Emili J., Siafakas N.M. Different angiogenic activity in pulmonary sarcoidosis and idiopathic pulmonary fibrosis. Chest. 2006;130:982–988. doi: 10.1378/chest.130.4.982. [DOI] [PubMed] [Google Scholar]
- 69.Burdick M.D., Murray L.A., Keane M.P., Xue Y.Y., Zisman D.A., Belperio J.A., Strieter R.M. CXCL11 attenuates bleomycin-induced pulmonary fibrosis via inhibition of vascular remodeling. Am J Respir Crit Care Med. 2005;171:261–268. doi: 10.1164/rccm.200409-1164OC. [DOI] [PubMed] [Google Scholar]
- 70.Hamada N., Kuwano K., Yamada M., Hagimoto N., Hiasa K., Egashira K., Nakashima N., Maeyama T., Yoshimi M., Nakanishi Y. Anti-vascular endothelial growth factor gene therapy attenuates lung injury and fibrosis in mice. J Immunol. 2005;175:1224–1231. doi: 10.4049/jimmunol.175.2.1224. [DOI] [PubMed] [Google Scholar]
- 71.Garcia-Cardena G., Oh P., Liu J., Schnitzer J.E., Sessa W.C. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci USA. 1996;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ghosh S., Gachhui R., Crooks C., Wu C., Lisanti M.P., Stuehr D.J. Interaction between caveolin-1 and the reductase domain of endothelial nitric-oxide synthase: consequences for catalysis. J Biol Chem. 1998;273:22267–22271. doi: 10.1074/jbc.273.35.22267. [DOI] [PubMed] [Google Scholar]
- 73.Ju H., Zou R., Venema V.J., Venema R.C. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem. 1997;272:18522–18525. doi: 10.1074/jbc.272.30.18522. [DOI] [PubMed] [Google Scholar]
- 74.Zhao Y.Y., Zhao Y.D., Mirza M.K., Huang J.H., Potula H.H., Vogel S.M., Brovkovych V., Yuan J.X., Wharton J., Malik A.B. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. 2009;119:2009–2018. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Straub A.C., Billaud M., Johnstone S.R., Best A.K., Yemen S., Dwyer S.T., Looft-Wilson R., Lysiak J.J., Gaston B., Palmer L., Isakson B.E. Compartmentalized connexin 43 S-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler Thromb Vasc Biol. 2011;31:399–407. doi: 10.1161/ATVBAHA.110.215939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cx40 and Cx43 expression in mouse lung. Immunofluorescence analysis of lung sections from 8-week-old C57Bl/6 (A and D) and VEC Cx43−/−/Cx40−/− (B, C, E, and F) mice shows the extent of Cx43 (A and B) and Cx40 (D and E) (arrowheads) present in lungs from healthy versus transgenic mice. C and F: Note partial depletion of Cx43 due to the VEC Cx43−/− genotype (B). Sections incubated with Cy3 goat anti-rabbit IgG alone (2°) show limited nonspecific labeling. Scale bars = 60 μm.
Decreased caveolin-1 expression by lungs of VEC Cx43−/−/Cx40−/− mice. Paraffin-embedded distal lung sections from C57Bl/6 (A, E, I, and M), VEC Cx43−/− (B, F, J, and N), Cx40−/− (C, G, K, and O), and VEC Cx43−/−/Cx40−/− (D, H, L, and P) mice at age 8 weeks (A–D), 16 weeks (E–H), 24 weeks (I–L), and 32 weeks (M–P) were immunostained for caveolin-1 and imaged. Scale bars = 60 μm. Compared with the other strains of mice examined, as early as age 8 weeks, VEC Cx43−/−/Cx40−/− mice demonstrated decreased caveolin-1. For quantification, see Figure 8A.
Decreased caveolin-2 expression by lungs from VEC Cx43−/−/Cx40−/− mice. Paraffin-embedded distal lung sections from C57Bl/6 (A, E, I, and M), VEC Cx43−/− (B, F, J, and N), 1 Cx40−/− (C, G, K, and O), and VEC Cx43−/−/Cx40−/− (D, H, L, and P) mice at age 8 weeks (A–D), 16 weeks (E–H), 24 weeks (I–L), and 32 weeks (M–P) were immunostained for caveolin-2 and imaged. Scale bars = 60 μm. Compared with the other strains of mice examined, as early as age 8 weeks, VEC Cx43−/−/Cx40−/− mice demonstrated decreased caveolin-2. For quantification, see Figure 8B.