

Abstract
In this study, we elucidated the mechanism by which adiponectin modulates hepatic stellate cell activation and fibrogenesis. Adiponectin-overexpressing transgenic mice receiving thioacetamide were resistant to fibrosis, compared with controls. In contrast, adiponectin-null animals developed severe fibrosis. Expression of collagen α1(I) and α-smooth muscle actin (α-SMA) mRNAs were significantly lower in adiponectin-overexpressing mice, compared with controls. In wild-type stellate cells exposed to a lentivirus encoding adiponectin, expression of peroxisome proliferator-activated receptor-γ (PPARγ), SREBP1c, and CEBPα mRNAs was significantly increased (3.2-, 4.1-, and 2.2-fold, respectively; n = 3; P < 0.05, adiponectin virus versus control), consistent with possible activation of an adipogenic transcriptional program. Troglitazone, a PPARγ agonist, strongly suppressed up-regulation of collagen α1(I) and α-SMA mRNA in stellate cells isolated from wild-type mice; however, stellate cells from adiponectin-null animals failed to respond to troglitazone. Furthermore, in isolated stellate cells in which PPARγ was depleted using an adenovirus-Cre-recombinase system and in which adiponectin was also overexpressed, collagen α1(I) and α-SMA were significantly inhibited. We conclude that the PPARγ effect on stellate cell activation and the fibrogenic cascade appears to be adiponectin-dependent; however, the inhibitory effect of adiponectin on stellate cell activation was not dependent on PPARγ, suggesting the presence of PPARγ-dependent as well as independent pathways in stellate cells.
Altered hepatic pathology, function, or both are among the most common sequelae of the metabolic syndrome.1,2 Steatosis is generally the first step in this process, which may progress to development of inflammation, fibrosis, and even end-stage liver failure.3 The determinants of liver disease in metabolic syndrome are under intense investigation. A number of factors likely contribute significantly to the process, including dysregulated metabolic pathways, oxidative stress, and chronic inflammation.3
Activated stellate cells (myofibroblasts) are key effectors of the fibrogenic response in the liver.4 Recent evidence has emphasized an adipogenic transcriptional program in stellate cells that is regulated by typical transcription factors in this pathway, including peroxisome proliferator-activated receptor-γ (PPARγ), SREBP1c, and CEBPα; this program appears to promote maintenance of stellate cells in a quiescent state.5,6 During activation, stellate cells lose retinoids and transform into a myofibroblast-like appearance.5,6 A critical corollary to the morphological transition is a set of remarkable functional changes that include production of extracellular matrix, as well as profibrotic mediators such as tissue inhibitors of matrix metalloproteinases (TIMPs).4
Adiponectin has been suggested to play an important role in the pathogenesis of liver fibrosis.7–15 Notably, in addition to adipose tissue, adiponectin is expressed in stellate cells.16 Previous studies have shown that adiponectin has important effects specifically on stellate cells, and the mechanism of these effects remains an area of active investigation.6,17 Here, we have hypothesized that the relationship between adiponectin and adipogenic signaling partners, and in particular the classic transcription factor PPARγ, is critical in modulating hepatic stellate cell activation and fibrogenesis. We examined the role of adiponectin in modulating hepatic stellate cell function in isolated primary cells and in genetic models that lack or overexpress adiponectin. Our findings highlight a complicated relationship between PPARγ and adiponectin in stellate cell activation.
Materials and Methods
Animals and Models of Liver Injury
Homozygous adiponectin-deficient and heterozygous adiponectin-overexpressing mice (using aP2 as a promoter) and corresponding controls on FVB background were generated as described previously.18,19 Male mice between the ages of 12 and 16 weeks were used for this study. All animals were maintained under temperature-controlled conditions (22 ± 2°C) in 12-hour light/dark cycles with unlimited access to food and water. All animals received humane care in compliance with University of Texas Southwestern Medical Center Institutional Animal Care and Use Guidelines. To induce liver fibrosis, the mice were given i.p. injections of 0.2 g/kg body weight of thioacetamide (TAA) dissolved in saline two times per week for 8 weeks as described previously.20 At the end of the experimental period, liver samples were collected for histological and biochemical examinations. Animal protocols used to induce injury and fibrogenesis were approved by the University of Texas Southwestern Medical Center Animal Care and Use Committee. Mice containing PPARγ LoxP sites on a C57BL/6J mixed background (4 to 5 weeks of age) were kindly provided by Dr. Bruce Spiegelman (Dana Farber Cancer Institute, Boston, MA).21
Cell Isolation and Culture
Hepatic stellate cells were isolated by digestion of the liver with pronase (Roche Applied Science, Indianapolis, IN) followed by collagenase (Crescent Chemical, Hauppauge, NY). In brief, stellate cells were separated from other liver nonparenchymal cells by ultracentrifugation over gradients of 8.2% and 15.6% cell separation medium (Accident medium; Accurate Chemical & Scientific, Westbury, NY). The resulting upper layer consisted of >95% stellate cells. Cells were placed on uncoated plastic and were maintained in standard stellate cell growth medium (modified 199OR containing 10% fetal bovine serum and 10% calf serum) as described previously.22 Isolated stellate cells were seeded at a density of 3 × 102 cells/mm2. Cultures were incubated at 37°C in a humidified incubator (containing 95% O2 and 2.5% CO2), and the medium was changed every 24 hours. Cell viability was >90% in all cultures used. The cells were considered to be quiescent at 24 hours after plating (day 1). Cells at day 7 were considered activated, with >95% staining positive for α-SMA.
Adenovirus
Recombinant adenovirus expressing green fluorescent protein (Ad-GFP) was prepared as described previously.23 Adenovirus encoding Cre recombinase (Ad-Cre) was purchased from Vector Biolabs (Philadelphia, PA). Viruses were amplified and titered according to the manufacturer's instructions (BD Biosciences, San Jose, CA). Stellate cells were isolated as described above, plated on 35-mm dishes at approximately 85% confluency and at the specified stage of culture were routinely infected as described previously.23 Infection efficiency was monitored by the expression of GFP and typically reached 80% to 90% within 48 hours.
Lentivirus
Lentivirus vectors were kindly provided by Dr. Zhao Wang (UT Southwestern Medical Center, Dallas, TX). Stellate cells were isolated as above, plated on 35-mm dishes at approximately 85% confluency, and at the specified stage of culture, were routinely infected with lentivirus at a multiplicity of infection (MOI) of 100 or 350.
RT-PCR
Total RNA was extracted using TRIZOL reagent according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). The reverse-transcription reaction was performed by using 1 μg of RNA that was reverse transcribed using oligo(dt) primers and SuperScript (Invitrogen) reverse transcriptase. Amplification reactions were performed using SYBR Green PCR master mix (Applied Biosystems, Foster City, CA). Five microliters of diluted cDNA samples (1:5 dilution) were used for quantitative two-step PCR (a 10-minute step at 95°C followed by 50 cycles of 15 seconds of 95°C and 1 second at 65°C) in the presence of 400 nmol/L specific forward and reverse primers and SYBR Green PCR master mix. Each sample was analyzed in triplicate. As negative controls, water was used as a template for each reaction. Primer sequences were as follows: type I collagen (COL1α1) forward, 5′-TTCCCTGGACCTAAGGGTACT-3′ and reverse, 5′-TTGAGCTCCAGCTTCGCC-3′; α-SMA forward, 5′-GTGGATCACCAAGCAGGAGT-3′ and reverse, 5′-CATAGCACGATGGTCGAT-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward, 5′-ACCCAGAAGACTGTGGATGG-3′ and reverse, 5′-CATCGAAGGTGGAAGAGTGG-3′; CEBPα forward, 5′-AAGAAGTCGGTGGATAAGAACAG-3′ and reverse, 5′-GTTGCGCTGTTTGGCTTTATCTC-3′; PPARγ forward, 5′-CCTGAAGCTCCAAGAATACCAAA-3′ and reverse, 5′-AGAGTTTTTCAGAATAATAAGG-3′; and SREBP1c forward, 5′-AGCTGTCGGGGTAGCGTCTG-3′ and reverse, 5′-GAGAGTTGGCACCTGGGCTG-3′.
Immunoblotting
Cell lysates were prepared in buffer containing 1% Triton X-100, 150 mmol/L NaCl, 20 mmol/L Tris pH 7.5, 1 mmol/L EDTA, 50 mmol/L NaF, 50 mmol/L sodium-2-glycerophosphate, 0.05 mmol/L Na3VO4, 10 μg leupeptin, 10% glycerol, and 100 mmol/L phenylmethylsulfonyl fluoride. Samples containing 50 g of total protein were subjected to SDS-PAGE, after which proteins were transferred to nitrocellulose membranes (Schleicher & Schuell Bioscience, Keene, NH). Membranes were incubated for 1 hour at room temperature in blocking buffer (10 mmol/L sodium phosphate, 0.5 mol/L NaCl, 0.05% Tween 20, and 2.5% dried milk) and then with primary antibody (1:1000) overnight at 4°C. Next, membranes were washed of excess primary antibody at room temperature in a phosphate-buffered saline Tween buffer (TBST: 10 mol/L 0.05% Tris pH 8, 0.9% sodium chloride, and Tween 20 0.05%) and then incubated for 1 hour at room temperature with secondary antibody. After the washing, specific signals were visualized using enhanced chemiluminescence detection according to the manufacturer's instructions (Thermo Fisher Scientific, Rockford. IL). Specific bands were scanned and data were collected over a narrow range of X-ray film (Eastman Kodak Co., Rochester, NY) linearity and then were quantitated by scanning densitometry.
Morphometry
Livers were fixed in 10% phosphate-buffered formalin for 48 hours at 4°C, washed twice with water, stored in 70% ethanol at 4°C for 24 hours, and then embedded in paraffin. Sections 5-μm thick were then dehydrated and stained with 0.1% Sirius Red F3B in saturated picric acid and counterstained with Fast Green FCF (all from Sigma-Aldrich, St. Louis, MO). The proportion of tissue stained with picrosirus red was assessed by morphometric analysis using MetaView software (Universal Imaging, Downingtown, PA) as described previously.22
Statistical Analysis
Data are reported as means ± SE. Significance was established using the Student's t-test and analysis of variance when appropriate. Differences were considered significant at P < 0.05.
Results
Stellate Cells from Adiponectin-Deficient Mice Exhibit an Accelerated Activation Phenotype
We initially characterized stellate cells from adiponectin-deficient and adiponectin-overexpressing mice. At early time points in culture, hepatic stellate cells from wild-type mice exhibited typical characteristics of quiescent cells, including abundant perinuclear retinoid, and a relatively rounded appearance (Figure 1A). Cells from adiponectin-deficient mice tended to spread more rapidly and had comparatively reduced amounts of retinoid droplets, consistent with the morphological appearance of activated stellate cells, whereas those from adiponectin-overexpressing mice retained features of quiescence. The differences were readily visible at 12, 24, and 48 hours (Figure 1A). At day 3 in culture, we quantified expression of activation and fibrosis markers in wild-type and adiponectin-null stellate cells (Figure 1, B–D) and found that both mRNA and protein levels of these indicators were significantly higher in stellate cells from adiponectin-null animals, compared with controls. These results suggest that lack of adiponectin accelerates the activation process and could explain the susceptibility of adiponectin-null mice to hepatic fibrosis.
Figure 1.
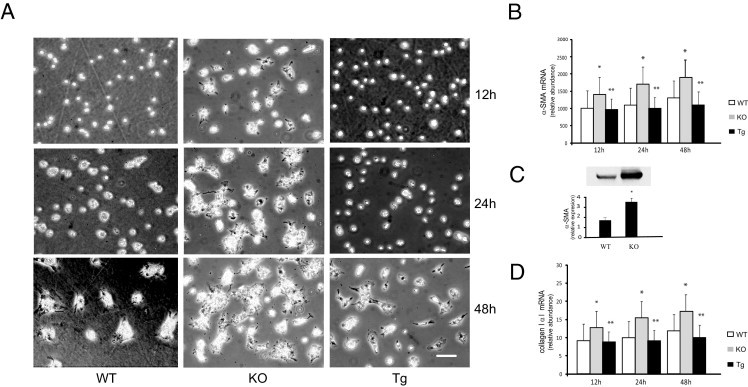
Characterization of stellate cells derived from adiponectin-deficient and transgenic mice. A: Stellate cells were isolated from wild type (WT), adiponectin-knockout (KO), and adiponectin-transgenic (Tg) mice and were grown in 20% serum-containing medium for up to 2 days. Representative images are shown (n > 10). Scale bar = 10 μm. B and D: mRNA levels of α-SMA (B) and collagen α1(I) (D) were measured after 7 days in culture (n = 4). *P < 0.05 versus WT; **P < 0.05 versus KO. C: Immunoblotting was performed to detect α-SMA and data were quantitated (means ± SE; n = 4). *P < 0.05 versus WT.
Mice Lacking Adiponectin Have Low-Grade Fibrosis and Are More Susceptible to TAA-Induced Fibrogenesis
Next, we induced liver fibrosis in mice with TAA. Adiponectin-deficient animals weighed less than either wild-type or adiponectin-overexpressing mice, although the differences were not statistically significant (Table 1). After TAA administration, all mice lost weight, and the loss in weight was greatest in adiponectin-deficient mice. The exposure to TAA led to injury and inflammation, as expected. There were increases in alanine transaminase levels in each of the groups of mice; relative increases in alanine transaminase were similar among all groups (Table 1). After 8 weeks of exposure to TAA, liver sections stained with H&E revealed hepatocellular necrosis, which was focused predominantly in pericentral and intralobular areas and was associated with a mixed inflammatory infiltrate (Figure 2 and Table 2). When inflammatory changes were quantitated using the Knodell scoring system,24 there was slightly more necrosis in the knockout mice and slightly less necrosis in the transgenic mice, compared with wild-type mice. These data suggest that there was no biochemical evidence of differential injury to hepatocytes caused by TAA among the different groups, but that there were small differences in the degree of inflammation among the groups.
Table 1.
Body Weight and Serum ALT and ALP Levels in Control and TAA-Injected Mice
| Variable | WT | KO | Tg | WT+TAA | KO+TAA | Tg+TAA |
|---|---|---|---|---|---|---|
| Body weight (g) | 32.9 ± 2.9 | 30.4 ± 1.5 | 32.7 ± 3.2 | 32.7 ± 2.3 | 29.8 ± 1.7 | 32.5 ± 1.5 |
| Serum ALT (IU/L) | 45 ± 8 | 54 ± 8⁎ | 43 ± 9 | 62 ± 12† | 77 ± 14†‡ | 58 ± 9† |
| Serum ALP (IU/L) | 34 ± 7 | 37 ± 5 | 32 ± 7 | 37 ± 6 | 45 ± 7 | 36 ± 8 |
Data are reported as means ± SE . n = 5 to 7 per group.
ALT, alanine transaminase; ALP, alkaline phosphatase; KO, adiponectin knockout mice; TAA, thioacetamide; Tg, adiponectin transgenic mice; WT, wild type.
P < 0.05 versus WT or Tg;
P < 0.05 versus untreated animals;
P < 0.001 versus WT+TAA or Tg+TAA mice.
Figure 2.
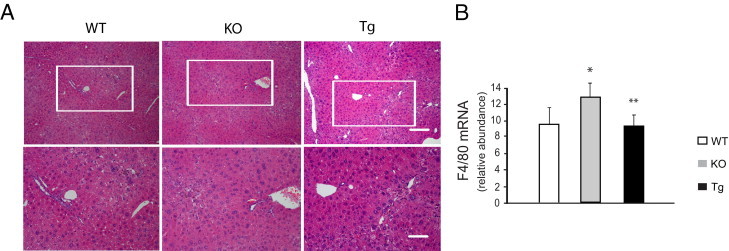
Liver morphology and inflammation in adiponectin-deficient and transgenic mice. Liver fibrosis was induced with repetitive intraperitoneal injection of 0.2 g/kg body weight of thioacetamide. At the end of the experimental period, liver samples were collected for histological and biochemical assays. A: Representative images of liver sections stained with H&E. Areas in insets are shown in higher magnification in the bottom row. Scale bars = 50 μm. B: mRNA was isolated from liver tissue, and relative abundance of F4/80 mRNA levels was measured by RT-PCR (means ± SE; n = 4). *P < 0.05 versus WT; **P < 0.05 versus KO.
Table 2.
Liver Histological Findings
| Variable | WT+TAA | KO+TAA | Tg+TAA |
|---|---|---|---|
| Periportal–bridge necrosis | 0.3 ± 0.2 | 0.4 ± 0.2 | 0.2 ± 0.1 |
| Intralobular degeneration and focal necrosis | 2 ± 0.3 | 3 ± 0.5 | 1 ± 0.2 |
| Portal inflammation | 0.2 ± 0.1 | 0.3 ± 0.2 | 0.1 ± 0.1 |
n = 5 to 7 per group.
KO, adiponectin knockout mice; TAA, thioacetamide; Tg, adiponectin transgenic mice; WT, wild type.
Notably, adiponectin-deficient mice exhibited greater fibrosis at baseline than did controls (Figure 3, A and B). Additionally, after exposure to TAA, adiponectin-deficient mice exhibited greater fibrosis than did matched controls (Figure 3, A and B). We further measured collagen α1(I) and α-SMA mRNA levels in these animals, and found that each was significantly up-regulated, compared with controls (Figure 3, C and D). We also explored a gain-of-function model in which adiponectin overexpression is driven by the aP2 promoter.19 After TAA-induced injury, collagen production in adiponectin-overexpressing mice was reduced, compared with controls (Figure 3, C and D). Additionally, expression of collagen α1(I) and α-SMA mRNA was significantly lower in adiponectin-overexpressing animals (Figure 3, C and D). Finally, we evaluated PPAR expression in the in vivo injury models (Figure 4). PPARγ was down-regulated in adiponectin-deficient and up-regulated in adiponectin-overexpressing mice. TAA appeared to blunt expression of PPARγ, which remained reduced in knockout mice and increased in overexpressing mice. The changes in PPARα mRNA expression were similar in their trends (Figure 4B).
Figure 3.
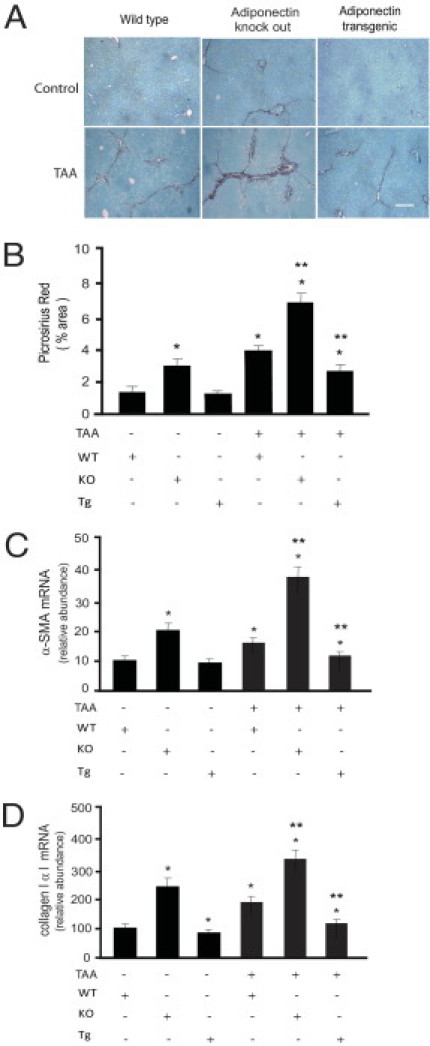
Mice lacking adiponectin are susceptible to fibrosis and mice overexpressing adiponectin are resistant to fibrosis. Liver fibrosis was induced by repetitive intraperitoneal injection of 0.2 g/kg body weight of thioacetamide (TAA). At the end of the experimental period, liver samples were collected for histological and biochemical examinations. A: Liver sections were stained with picrosirius red. Scale bar = 50 μm. B: Histomorphometric analysis was performed on random picrosirius red-stained liver sections (means ± SE; n = 10 fields/liver and 10 livers/group). *P < 0.05 versus WT alone; **P < 0.05 versus TAA alone. C and D: Livers were harvested, total RNA was extracted, and real-time PCR was performed to detect mRNA expression of α-SMA (C) and collagen α1(I) (D) (means ± SE; n = 6). *P < 0.05 versus WT control; **P < 0.05 versus WT and TAA.
Figure 4.
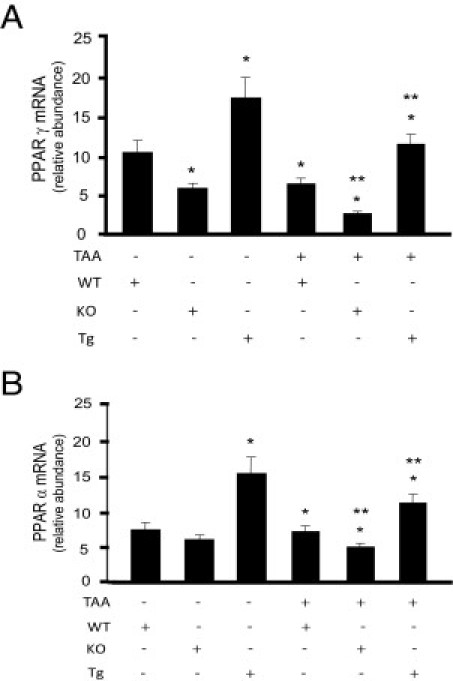
PPAR expression in adiponectin-deficient and transgenic mice. Liver fibrosis was induced by repetitive intraperitoneal injection of 0.2 g/kg body weight of thioacetamide (TAA). Livers were harvested, total RNA was extracted, and real-time PCR was performed to detect mRNA expression of PPARγ (A) or PPARα (B) (means ± SE; n = 6). *P < 0.05 versus WT; **P < 0.05 versus WT and TAA.
Adiponectin Overexpression Decreases Stellate Cell Activation and Stimulates an Adipokine Phenotype in Culture
Given that earlier studies showed that activation of stellate cells in culture is associated with a decline in expression of adiponectin,25 we asked whether activation could be reversed by re-expression of adiponectin in cultured cells. First, we demonstrated that infection of stellate cells with a lentivirus containing an adiponectin construct led to increased production of adiponectin (Figure 5A); additionally, a time-course experiment revealed that expression of adiponectin increased from 12 to 48 hours after infection (Figure 5B). Next, we found that exposure of stellate cells to this virus led to a reduction in α-SMA and collagen α1(I) mRNA, compared with control (Figure 6, A and B), consistent with retaining a more quiescent phenotype relative to controls.
Figure 5.
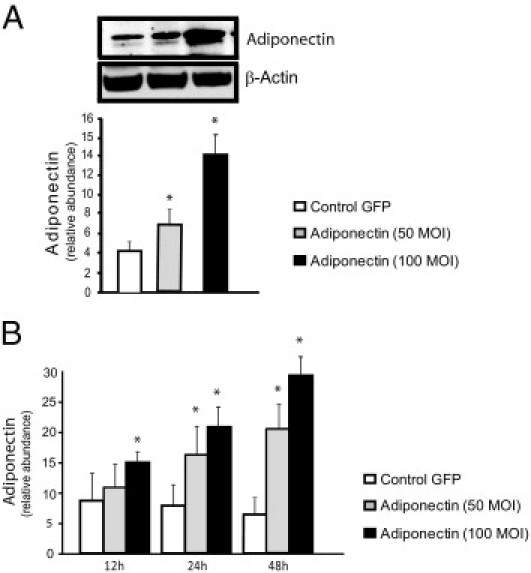
Quantification of adiponectin protein expression in hepatic stellate cells. Stellate cells from WT mice on FVB background were isolated, cultured for up to 48 hours, and then infected with a lentivirus expressing GFP, or adiponectin at 50 and 100 MOI. Cell lysates were harvested at 48 hours (A) or at 12, 24, and 48 hours (B) and were subjected to immunoblotting. A representative immunoblot is shown (A). Subsequently, specific bands from repeated experiments were quantified and normalized to the signal for β-actin (A and B) (means ± SE; n = 3). *P < 0.05 versus control GFP.
Figure 6.
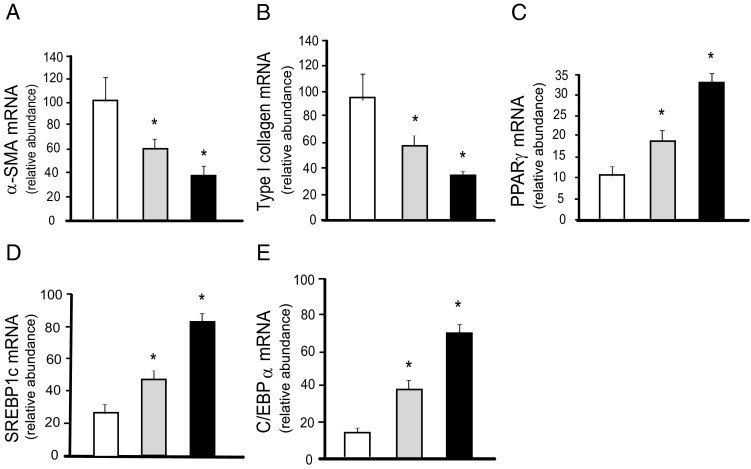
Effects of adiponectin overexpression on stellate cell activation. Stellate cells from normal rats were isolated, plated at equivalent density, and allowed to undergo culture-induced activation for 3 days. Cells were transduced with a control lentivirus expressing GFP or lentivirus overexpressing adiponectin at 50 and 100 MOI (n = 3/group). Total RNA was harvested 72 hours later and was subjected to RT-PCR to detect mRNA expression of α-SMA (A), collagen α1(I) (B), PPARγ (C), SREBP1c (D), and CEBPα (E) (means ± SE; n = 3). White bars indicate control GFP; gray bars indicate adiponectin (50 MOI); black bars indicate adiponectin (100 MOI). *P < 0.05 versus control GFP.
Activation of hepatic stellate cells is associated with enhanced expression of the nuclear receptor PPARβ/δ, recognized for its role in energy homeostasis, particularly lipid oxidation,26 and with reduced expression of PPARγ, known for its role in regulation of adipocyte differentiation.6,27 We next examined whether adiponectin overexpression modulated the expression of specific members of the adipogenic program in stellate cells. PPARγ, SREBP1c, and CEBPα were all up-regulated after overexpression of adiponectin in stellate cells (Figure 6, C–E).
PPARγ Ligands Are Ineffective in Reversing Stellate Cell Activation in the Absence of Adiponectin
Given that stellate cell activation is associated with significant decline in PPARγ and that forced expression of PPARγ leads to reversal of stellate cell activation,27,28 we asked whether the effect of PPARγ is dependent on adiponectin. Primary stellate cells from adiponectin-deficient animals and controls were exposed to the PPARγ ligand troglitazone (5 μg/mL) for 5 days. Troglitazone strongly suppressed the up-regulation of α-SMA and collagen α1(I) mRNA levels in stellate cells isolated from wild-type mice (Figure 7, A and B); however, stellate cells from adiponectin-null animals did not respond to troglitazone. Additionally, CEBPα, SREBP1c, and PPARγ mRNAs were each reduced in stellate cells from adiponectin knockout animals, and their response to troglitazone was blunted (Figure 7, C–E). These results suggest that abrogation of stellate cell fibrogenesis and activation, as well as transcriptional activation of adipogenic partners mediated by PPARγ ligands, may be adiponectin dependent.
Figure 7.
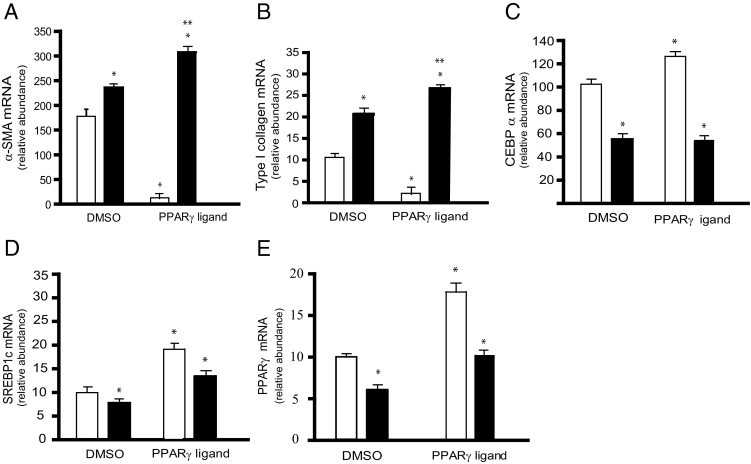
Adiponectin is required for PPARγ ligands to be effective in inhibiting stellate cell activation. Stellate cells isolated from WT and adiponectin knockout (ADNKO) mice were cultured in the presence of serum-containing medium for 24 hours. Cells were exposed to 5 μg/mL of the PPARγ ligand troglitazone, or an equivalent volume of dimethyl sulfoxide (vehicle) for 4 days, after which total RNA was extracted from lysates and mRNA expression of α-SMA (A), collagen α1(I) (B), CEBPα (C), SREBP1c (D), and PPARγ (E) was quantified by real-time PCR (means ± SE; n = 3). White bars indicate WT; black bars indicate ADNKO. *P < 0.05 versus WT cells exposed to vehicle alone; **P < 0.05 versus WT cells exposed to troglitazone.
Antifibrotic Actions of Adiponectin Are Not Dependent on PPARγ
PPARγ ligands are known to prevent stellate cell activation27 (Figure 7, A and B). Furthermore, our data suggest that the effect of PPARγ ligands on stellate cells is adiponectin dependent. We therefore asked whether the effect of adiponectin on stellate cells might be PPARγ ligand dependent. To this end, we developed a system in which PPARγ could be conditionally deleted, using PPARγ Cre/Lox conditional knockout mice and a Cre-expressing adenovirus. We isolated primary stellate cells from these knockout mice, and on day 2 we transduced these cells with a Cre-expressing adenovirus (to knock down PPARγ) and/or the lentivirus encoding adiponectin (to overexpress adiponectin). We initially demonstrated that, using this system, PPARγ expression could be significantly, although not altogether, reduced (Figure 8). Deletion of PPARγ significantly enhanced the stellate cell activation and fibrogenesis, as evidenced by the increased expression of the respective activation markers, α-SMA and collagen α1(I) (Figure 9, A and B). This finding was consistent with the finding that troglitazone inhibited expression of α-SMA and collagen α1(I) in activated stellate cells (Figure 7, A and B). Notably, adiponectin overexpression in PPARγ-null cells suppressed the expression of α-SMA and collagen α1(I) even in the absence of PPARγ (Figure 9, A and B). Furthermore, CEBPα and SREBP1c mRNAs were each reduced in PPARγ-null stellate cells, but these mRNAs were stimulated after exposure to adiponectin (Figure 9, C and D).
Figure 8.
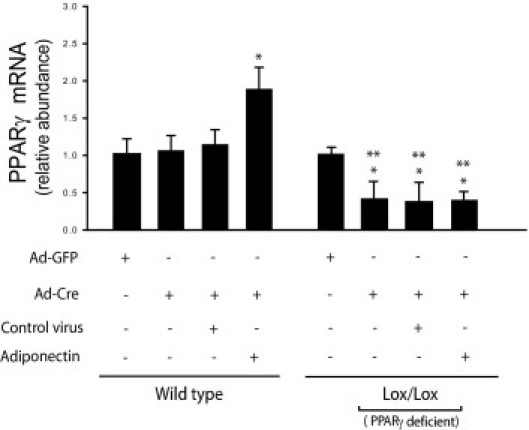
Deletion of PPARγ with a Cre-expressing adenovirus. Stellate cells were isolated from WT and PPARγ Cre/Lox mice and grown in culture. After 48 hours, cells were infected with adenovirus expressing Cre and/or lentivirus encoding adiponectin or corresponding control for 4 days. Total cellular RNA was isolated and mRNA expression of PPARγ was quantified by RT-PCR (means ± SE; n = 3). *P < 0.05 versus WT cells exposed to adenovirus expressing GFP alone; **P < 0.05 versus Lox/Lox cells exposed to adenovirus expressing GFP alone.
Figure 9.
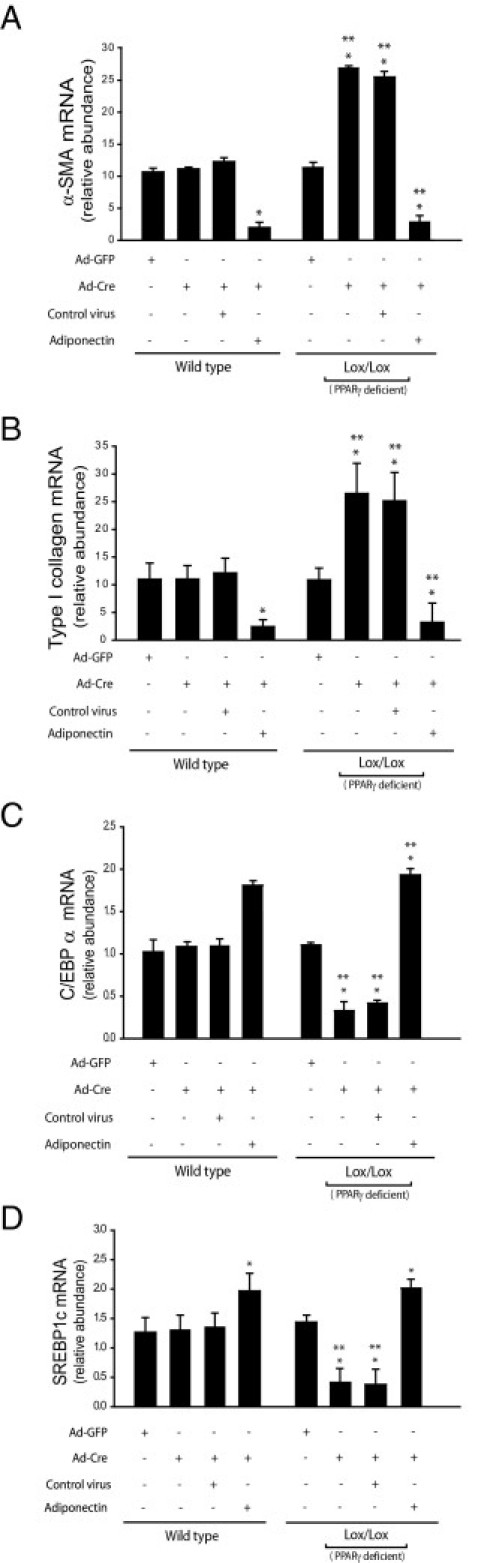
The antiactivating and antifibrotic actions of adiponectin are independent of PPARγ. Stellate cells were isolated from WT and PPARγ Cre/Lox mice and grown in culture. After 48 hours, cells were infected with Cre-expressing adenovirus and/or lentivirus encoding adiponectin or corresponding control for four days. Total cellular RNA was isolated and mRNA expression of α-SMA (A), collagen α1(I) (B), CEBPα (C), and SREBP1c (D) was quantified by RT-PCR (means ± SE; n = 3). *P < 0.05 versus WT cells exposed to adenovirus expressing GFP alone; **P < 0.05 versus Lox/Lox cells exposed to adenovirus expressing GFP alone.
Discussion
This work further highlights the importance of adiponectin in hepatic fibrosis. In the genetic models used, we demonstrated that adiponectin-overexpressing animals were significantly less susceptible to chemically induced fibrosis. Conversely, adiponectin-deficient animals were more sensitive to fibrosis than were littermate controls. Perhaps the most important advance emerging from this work is the idea that the antifibrotic effect of adiponectin may be based on the ability to prevent stellate cell activation. Indeed, it appeared that stellate cells isolated from adiponectin knockout mice were significantly more activated, compared with control. On the other hand, increasing the expression of adiponectin in primary stellate cells prevented and/or reversed the myofibroblastic transformation of stellate cells in culture. A highly novel finding was that the inhibitory effect of adiponectin on stellate cell activation was not dependent on PPARγ. However, the PPARγ ligand troglitazone was unable to halt stellate cell activation in primary stellate cells genetically lacking adiponectin, suggesting that PPARγ agonist-mediated antifibrotic actions may be dependent on adiponectin.
A noteworthy and important finding in this study was that the PPARγ agonist troglitazone did not maintain stellate cell quiescence in the absence of adiponectin. Additionally, adiponectin promoted stellate cell quiescence independent of PPARγ. Consistent with these data is the finding that PPARγ ligands are less effective in improving the metabolic syndrome in adiponectin-null mice.18 Given the similarities between PPARγ and adiponectin effects, we think that adiponectin may have acted in a PPARγ-dependent manner. We have clearly shown that adiponectin is a major effector for PPARγ actions, but the reverse is not true. Further work will be needed to help elucidate the mechanism underlying the pathways responsible for these observations.
The mechanism underlying the effects of adiponectin on stellate cells remains to be determined. Certainly, the data presented here suggest a direct affect of adiponectin on stellate cells. For example, adiponectin appears to signal through adenosine monophosphate-activated protein kinase,29 which in turn may suppress reactive oxygen species and AKT activation. Another possibility is that the antisteatotic and/or anti-inflammatory activity of adiponectin could be important in regulation of stellate cell function. Recent studies in our laboratory suggest that adiponectin tightly limits systemic availability of free fatty acids, presumably by enhancing glyceroneogenesis and re-esterification of free fatty acids in the subcutaneous adipose depot.30 Such a lowering effect on plasma free fatty acids not only protects the liver from steatosis and subsequent degeneration, but also reduces free fatty acid-mediated inflammation. Thus, the antifibrotic actions of adiponectin may be a combination of the direct effects of adiponectin on stellate cells but may also be a result of reduction of other systemic stimuli and risk factors that initiate or mediate liver injury.
One potential mechanism underlying the effect of adiponectin in the liver in vivo could be related to the effects on mitochondrial integrity and oxidative stress. One study revealed that genetic loss of adiponectin led to reduced activity of the respiratory chain, which in turn could set the stage for oxidative stress and generation of lipid peroxides, perhaps facilitating liver injury; the authors further attributed mitochondrial dysfunction to decreased expression of UCP-2 and increased steatosis.31 In contrast, adiponectin overexpression results in significantly higher cellular levels of mitochondria.30 Another study suggested an antioxidant role for adiponectin during liver injury, in addition to its effect on Kupffer cell recruitment.11 We recognize that the detailed mechanism of action by which adiponectin exerts its potent protective effects on stellate cells remains to be determined.
Another compound important in metabolism and fibrogenesis is leptin. Although we did not study leptin in the current study, abundant evidence indicates that leptin has an important role in the fibrogenic response. Indeed, it appears to enhance expression of inflammatory mediators and to contribute to hepatic fibrogenesis.32 Notably, there appears to be an antagonistic interaction between leptin and adiponectin; for example, based on our data and those of others, adiponectin and leptin play opposite roles in modulating liver fibrosis.16 Additionally, loss of leptin increases metabolic risk, manifested as obesity and diabetes. Studies from our laboratory have shown that overexpression of adiponectin in a leptin-deficient mouse model improves the metabolic phenotype but at the expense of a twofold increase in body weight and massive enlargement of the subcutaneous adipose mass.33 Although it is very likely that adiponectin inhibits the profibrotic effects of leptin, the mechanisms underlying this process are not clear.
An intriguing notion that emerges from the finding that adiponectin is important in the fibrogenic response concerns the putative role of adipose tissue in modulating susceptibility to liver fibrosis. In one study, fibroblast growth factor-treated stromal cells derived from adipose tissue had the potential to reverse fibrotic liver injury.34 Although this is an exciting finding, the biology underlying this phenomenon is not completely understood. One possible mechanistic link between adipose tissue and stellate cells is their exclusive ability to store retinoids, which appear to play a pivotal role in maintaining stellate cell quiescence and modulation of fibrosis.35 Many studies, including work in our own laboratory, have shown that the metabolic syndrome is associated with extensive inflammation (and fibrosis) of adipose tissue.36–38 Although stellate cells in the liver and pancreas and in related cells in other tissues clearly regulate fibrosis in these organs, the cellular drivers of fibrosis in adipose tissue are unknown, even though adipocytes may contribute to the phenomenon.39 A particularly intriguing possibility is that a stellate cell-like cell type may exist in adipose tissue. A final important question related to adipose tissue and adiponectin is whether healthy adipose tissue that expresses and releases high levels of adiponectin may have antifibrotic therapeutic potential.
Footnotes
Supported by the NIH (R01-DK50574 to D.C.R.; and R01-DK55758, R01-CA112023, RC1-DK086629, and P01-DK088761 to P.E.S.).
M.S.S. and S.S. contributed equally to this manuscript.
References
- 1.Kamada Y., Takehara T., Hayashi N. Adipocytokines and liver disease. J Gastroenterol. 2008;43:811–822. doi: 10.1007/s00535-008-2213-6. [DOI] [PubMed] [Google Scholar]
- 2.Rombouts K., Marra F. Molecular mechanisms of hepatic fibrosis in non-alcoholic steatohepatitis. Dig Dis. 2010;28:229–235. doi: 10.1159/000282094. [DOI] [PubMed] [Google Scholar]
- 3.Farrell G.C., Larter C.Z. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 4.Friedman S.L., Rockey D.C., Bissell D.M. Hepatic fibrosis 2006: report of the Third AASLD Single Topic Conference. Hepatology. 2007;45:242–249. doi: 10.1002/hep.21459. [DOI] [PubMed] [Google Scholar]
- 5.Tsukamoto H., She H., Hazra S., Cheng J., Miyahara T. Anti-adipogenic regulation underlies hepatic stellate cell transdifferentiation. J Gastroenterol Hepatol. 2006;21(Suppl 3):S102–S105. doi: 10.1111/j.1440-1746.2006.04573.x. [DOI] [PubMed] [Google Scholar]
- 6.She H., Xiong S., Hazra S., Tsukamoto H. Adipogenic transcriptional regulation of hepatic stellate cells. J Biol Chem. 2005;280:4959–4967. doi: 10.1074/jbc.M410078200. [DOI] [PubMed] [Google Scholar]
- 7.Yoneda M., Iwasaki T., Fujita K., Kirikoshi H., Inamori M., Nozaki Y., Maeyama S., Wada K., Saito S., Terauchi Y., Nakajima A. Hypoadiponectinemia plays a crucial role in the development of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus independent of visceral adipose tissue. Alcohol Clin Exp Res. 2007;31(1 Suppl):S15–S21. doi: 10.1111/j.1530-0277.2006.00281.x. [DOI] [PubMed] [Google Scholar]
- 8.Wang J., Brymora J., George J. Roles of adipokines in liver injury and fibrosis. Expert Rev Gastroenterol Hepatol. 2008;2:47–57. doi: 10.1586/17474124.2.1.47. [DOI] [PubMed] [Google Scholar]
- 9.Shetty S., Kusminski C.M., Scherer P.E. Adiponectin in health and disease: evaluation of adiponectin-targeted drug development strategies. Trends Pharmacol Sci. 2009;30:234–239. doi: 10.1016/j.tips.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Asano T., Watanabe K., Kubota N., Gunji T., Omata M., Kadowaki T., Ohnishi S. Adiponectin knockout mice on high fat diet develop fibrosing steatohepatitis. J Gastroenterol Hepatol. 2009;24:1669–1676. doi: 10.1111/j.1440-1746.2009.06039.x. [DOI] [PubMed] [Google Scholar]
- 11.Fukushima J., Kamada Y., Matsumoto H., Yoshida Y., Ezaki H., Takemura T., Saji Y., Igura T., Tsutsui S., Kihara S., Funahashi T., Shimomura I., Tamura S., Kiso S., Hayashi N. Adiponectin prevents progression of steatohepatitis in mice by regulating oxidative stress and Kupffer cell phenotype polarization. Hepatol Res. 2009;39:724–738. doi: 10.1111/j.1872-034X.2009.00509.x. [DOI] [PubMed] [Google Scholar]
- 12.Arvaniti V.A., Thomopoulos K.C., Tsamandas A., Makri M., Psyrogiannis A., Vafiadis G., Assimakopoulos S.F., Labropoulou-Karatza C. Serum adiponectin levels in different types of non alcoholic liver disease: Correlation with steatosis, necroinflammation and fibrosis. Acta Gastroenterol Belg. 2008;71:355–360. [PubMed] [Google Scholar]
- 13.Nakayama H., Otabe S., Yuan X., Ueno T., Hirota N., Fukutani T., Wada N., Hashinaga T., Yamada K. Effects of adiponectin transgenic expression in liver of nonalcoholic steatohepatitis model mice. Metabolism. 2009;58:901–908. doi: 10.1016/j.metabol.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Ma H., Gomez V., Lu L., Yang X., Wu X., Xiao S.Y. Expression of adiponectin and its receptors in livers of morbidly obese patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2009;24:233–237. doi: 10.1111/j.1440-1746.2008.05548.x. [DOI] [PubMed] [Google Scholar]
- 15.Nannipieri M., Cecchetti F., Anselmino M., Mancini E., Marchetti G., Bonotti A., Baldi S., Solito B., Giannetti M., Pinchera A., Santini F., Ferrannini E. Pattern of expression of adiponectin receptors in human liver and its relation to nonalcoholic steatohepatitis. Obes Surg. 2009;19:467–474. doi: 10.1007/s11695-008-9701-x. [DOI] [PubMed] [Google Scholar]
- 16.Ding X., Saxena N.K., Lin S., Xu A., Srinivasan S., Anania F.A. The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol. 2005;166:1655–1669. doi: 10.1016/S0002-9440(10)62476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hazra S., Xiong S., Wang J., Rippe R.A., Krishna V., Chatterjee K., Tsukamoto H. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J Biol Chem. 2004;279:11392–11401. doi: 10.1074/jbc.M310284200. [DOI] [PubMed] [Google Scholar]
- 18.Nawrocki A.R., Rajala M.W., Tomas E., Pajvani U.B., Saha A.K., Trumbauer M.E., Pang Z., Chen A.S., Ruderman N.B., Chen H., Rossetti L., Scherer P.E. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281:2654–2660. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- 19.Combs T.P., Pajvani U.B., Berg A.H., Lin Y., Jelicks L.A., Laplante M., Nawrocki A.R., Rajala M.W., Parlow A.F., Cheeseboro L., Ding Y.Y., Russell R.G., Lindemann D., Hartley A., Baker G.R., Obici S., Deshaies Y., Ludgate M., Rossetti L., Scherer P.E. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology. 2004;145:367–383. doi: 10.1210/en.2003-1068. [DOI] [PubMed] [Google Scholar]
- 20.Wu J.B., Chuang H.R., Yang L.C., Lin W.C. A standardized aqueous extract of Anoectochilus formosanus ameliorated thioacetamide-induced liver fibrosis in mice: the role of Kupffer cells. Biosci Biotechnol Biochem. 2010;74:781–787. doi: 10.1271/bbb.90824. [DOI] [PubMed] [Google Scholar]
- 21.He W., Barak Y., Hevener A., Olson P., Liao D., Le J., Nelson M., Ong E., Olefsky J.M., Evans R.M. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci USA. 2003;100:15712–15717. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rockey D.C., Chung J.J. Endothelin antagonism in experimental hepatic fibrosis: Implications for endothelin in the pathogenesis of wound healing. J Clin Invest. 1996;98:1381–1388. doi: 10.1172/JCI118925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shafiei M.S., Rockey D.C. The role of integrin-linked kinase in liver wound healing. J Biol Chem. 2006;281:24863–24872. doi: 10.1074/jbc.M513544200. [DOI] [PubMed] [Google Scholar]
- 24.Knodell R.G., Ishak K.G., Black W.C., Chen T.S., Craig R., Kaplowitz N., Kiernan T.W., Wollman J. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1:431–435. doi: 10.1002/hep.1840010511. [DOI] [PubMed] [Google Scholar]
- 25.Kamada Y., Tamura S., Kiso S., Matsumoto H., Saji Y., Yoshida Y., Fukui K., Maeda N., Nishizawa H., Nagaretani H., Okamoto Y., Kihara S., Miyagawa J., Shinomura Y., Funahashi T., Matsuzawa Y. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125:1796–1807. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 26.Hellemans K., Michalik L., Dittie A., Knorr A., Rombouts K., De Jong J., Heirman C., Quartier E., Schuit F., Wahli W., Geerts A. Peroxisome proliferator-activated receptor-beta signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology. 2003;124:184–201. doi: 10.1053/gast.2003.50015. [DOI] [PubMed] [Google Scholar]
- 27.Yang L., Chan C.C., Kwon O.S., Liu S., McGhee J., Stimpson S.A., Chen L.Z., Harrington W.W., Symonds W.T., Rockey D.C. Regulation of peroxisome proliferator-activated receptor-gamma in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G902–G911. doi: 10.1152/ajpgi.00124.2006. [DOI] [PubMed] [Google Scholar]
- 28.Da Silva Morais A., Abarca-Quinones J., Horsmans Y., Stärkel P., Leclercq I.A. Peroxisome proliferated-activated receptor gamma ligand, pioglitazone, does not prevent hepatic fibrosis in mice. Int J Mol Med. 2007;19:105–112. [PubMed] [Google Scholar]
- 29.Adachi M., Brenner D.A. High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology. 2008;47:677–685. doi: 10.1002/hep.21991. [DOI] [PubMed] [Google Scholar]
- 30.Asterholm I.W., Scherer P.E. Enhanced metabolic flexibility associated with elevated adiponectin levels. Am J Pathol. 2010;176:1364–1376. doi: 10.2353/ajpath.2010.090647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou M., Xu A., Tam P.K., Lam K.S., Chan L., Hoo R.L., Liu J., Chow K.H., Wang Y. Mitochondrial dysfunction contributes to the increased vulnerabilities of adiponectin knockout mice to liver injury. Hepatology. 2008;48:1087–1096. doi: 10.1002/hep.22444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saxena N.K., Ikeda K., Rockey D.C., Friedman S.L., Anania F.A. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762–771. doi: 10.1053/jhep.2002.32029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J.Y., van de Wall E., Laplante M., Azzara A., Trujillo M.E., Hofmann S.M., Schraw T., Durand J.L., Li H., Li G., Jelicks L.A., Mehler M.F., Hui D.Y., Deshaies Y., Shulman G.I., Schwartz G.J., Scherer P.E. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamada Y., Yoshida Y., Saji Y., Fukushima J., Tamura S., Kiso S., Hayashi N. Transplantation of basic fibroblast growth factor-pretreated adipose tissue-derived stromal cells enhances regression of liver fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G157–G167. doi: 10.1152/ajpgi.90463.2008. [DOI] [PubMed] [Google Scholar]
- 35.Hautekeete M.L., Dodeman I., Azais-Braesco V., Van den Berg K., Seynaeve C., Geerts A. Hepatic stellate cells and liver retinoid content in alcoholic liver disease in humans. Alcohol Clin Exp Res. 1998;22:494–500. [PubMed] [Google Scholar]
- 36.Halberg N., Schraw T.D., Wang Z.V., Kim J.Y., Yi J., Hamilton M.P., Luby-Phelps K., Scherer P.E. Systemic fate of the adipocyte-derived factor adiponectin. Diabetes. 2009;58:1961–1970. doi: 10.2337/db08-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan T., Muise E.S., Iyengar P., Wang Z.V., Chandalia M., Abate N., Zhang B.B., Bonaldo P., Chua S., Scherer P.E. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29:1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keophiphath M., Achard V., Henegar C., Rouault C., Clément K., Lacasa D. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol Endocrinol. 2009;23:11–24. doi: 10.1210/me.2008-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halberg N., Khan T., Trujillo M.E., Wernstedt-Asterholm I., Attie A.D., Sherwani S., Wang Z.V., Landskroner-Eiger S., Dineen S., Magalang U.J., Brekken R.A., Scherer P.E. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29:4467–4483. doi: 10.1128/MCB.00192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]