

Abstract
Accumulation of various lipid-rich extracellular matrix (ECM) deposits under the retinal pigment epithelium (RPE) has been observed in eyes with age-related macular degeneration (AMD). RPE-derived matrix metalloproteinase (MMP)-2, MMP-14, and basigin (BSG) are major enzymes involved in the maintenance of ECM turnover. Hypertension (HTN) is a systemic risk factor for AMD. It has previously been reported that angiotensin II (Ang II), one of the most important hormones associated with HTN, increases MMP-2 activity and its key regulator, MMP-14, in RPE, inducing breakdown of the RPE basement membrane, which may lead to progression of sub-RPE deposits. Ang II exerts most of its actions by activating the mitogen-activated protein kinase (MAPK) signaling pathway. Herein is explored the MAPK signaling pathway as a potential key intracellular modulator of Ang II–induced increase in MMP-2 activity and MMP-14 and BSG protein expression. It was observed that Ang II stimulates phosphorylation of extracellular signal-regulated kinase (ERK) and p38 MAPK in RPE cells and ERK/p38 and Jun N-terminal kinase (JNK) in mice. These effects were mediated by Ang II type 1 receptors. Blockade of ERK or p38 MAPK abrogated the increase in MMP-2 activity and MMP-14 and BSG proteins in ARPE-19 cells. A better understanding of the molecular events by which Ang II induces ECM dysregulation is of critical importance to further define its contribution to the progression of sub-RPE deposits in AMD patients with HTN.
Age-related macular degeneration (AMD) is the leading cause of vision loss in the elderly, and in Western societies, its incidence increases as the population ages.1 The retinal pigment epithelium (RPE) supports photoreceptor cell function and is recognized as the initial pathogenic target in AMD.2 Other than age, epidemiologic risk factors most consistently associated with AMD are systemic or environmental.3–5 Among them, hypertension (HTN) is of special interest as a potential link between cardiovascular diseases and AMD.6–9
Angiotensin II (Ang II) is the most important HTN-associated hormone that has a pivotal role in pathologic complications.10,11 Expression of Ang II and other components of the renin-angiotensin system has been demonstrated in the RPE and other eye tissues.12–15 In addition to evidence in support of the existence of a local renin-angiotensin system in the adult eye, expression of Ang II receptor types 1 and 2 (AT1 and AT2, respectively) has been demonstrated in the retina and the RPE15–18, which provides the molecular basis for a biological function of Ang II in the eye.
Numerous biochemical and anatomical changes occur in Bruch's membrane (BrM) as part of aging retina in the absence of apparent disease including collagenous thickening, calcification, and lipid infiltration.2 However, the presence of large numbers and extensive areas of additional deposits (drusen) beneath the RPE and within the inner collagenous layer of BrM is a defining histopathologic landmark associated with dry AMD,2,19–21 the most common type of the disease, accounting for approximately 90% of all AMD cases. To date, the underlying pathogenic mechanism by which these sub-RPE deposits traverse the RPE basement membrane to form drusen remains unknown. It was previously hypothesized that progression of sub-RPE deposits into BrM requires degradation of extracellular matrix (ECM) proteins, which are critical components of the RPE basement membrane and adjacent BrM,22,23 and that ECM turnover up-regulation is necessary for breakdown of these physical barriers. In this regard, matrix metalloproteinase (MMP)−2, the most abundant and important MMP enzyme synthesized by the RPE, is crucial for degradation of ECM proteins in the RPE basement membrane and BrM.18,22–26
It has been well established that action of Ang II regulates expression of ECM proteins in various tissues.27–30 It has been recently demonstrated that Ang II induces AT1-mediated breakdown of the RPE basement membrane by increasing MMP-2 activity in RPE.18,26 A concomitant increase in MMP-14 expression has also been demonstrated, which might account, at least in part, for the Ang II–induced increase in MMP-2 activity.18 Another potential regulator of MMPs is basigin (BSG), also known as extracellular MMP inducer or CD147, which is a protein that induces MMP-2 and MMP-14 in various cell types.31–34 BSG is expressed in human RPE and ARPE-19 cells,35 although its function in the retina and regulation by Ang II in the RPE remain unknown. Altogether, the signaling events involved in Ang II-stimulated MMP-2 activity in RPE remain to be elucidated.
The majority of intracellular actions of Ang II in most tissues is mediated by activation of mitogen-activated protein kinases (MAPKs). However, nothing is known about Ang II signaling in the RPE. MAPKs are a group of serine/threonine kinases36–38 that can be divided into three major groups, that is, extracellular signal-regulated protein kinase (ERK), p38, and Jun N-terminus kinase (JNK), and participate in a wide array of cellular responses including proliferation, differentiation, migration, and stress responses, among others.39–41 In the present study, involvement of MAPKs signal transduction pathways in Ang II–induced effects on MMP-2 activity and MMP-14 and BSG protein expression was examined in RPE. A greater understanding of the underlying cellular and molecular mechanisms involved in the cause of early AMD is mandatory to develop novel therapeutic approaches for management of this disease that causes a great deal of discomfort and disability.
Materials and Methods
Compounds and Drugs
PBS, Dulbecco's modified Eagle's medium–Ham's nutrient mixture F-12, fetal bovine serum (FBS), HEPES, L-glutamine, penicillin/streptomycin, Tris-buffered saline solution (TBS), Na2HCO3, DAPI stain, 4% to 20% Tris-glycine gels, 10% gelatin zymograms, and ProLong Antifade reagent with DAPI were purchased from Invitrogen Corp. (Carlsbad, CA). Angiotensin II, NaCl, KH2PO4, CaCl2, MgSO4, l-glucose, bovine serum albumin, EDTA, l-glycine, SDS, Tris, Brilliant Blue R solution, dispase, and inhibitors PD98059, PD123319, and SB203580 were purchased from Sigma-Aldrich Corp. (St. Louis, MO). Mouse monoclonal anti-endothelial cells (CD146) and mouse monoclonal anti–MMP-14 antibodies were purchased from Chemicon International Inc. (Temecula, CA). Rabbit anti-ERK antibody was purchased from Promega Corp. (Madison, WI). Mouse phospho-ERK, mouse monoclonal anti–cytokeratin 18, and horseradish peroxidase–linked donkey anti-rabbit or anti-mouse antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit polyclonal anti-SAPK/JNK, mouse monoclonal anti–phospho-SAPK/JNK, rabbit polyclonal anti-p38, mouse monoclonal anti–phospho-p38, rabbit monoclonal anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies, control small-interfering RNA (siRNA), SignalSilence p38 MAPK siRNA, and SignalSilence p44/42 MAPK (ERK) siRNA were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Mouse polyclonal anti-BSG was purchased from Novus Biologicals LLC (Littleton, CO). A detergent-compatible protein quantification kit was purchased from Bio-Rad Laboratories, Inc. (Hercules, CA). Protein lysis buffer M-PER and ECL Western blotting substrate were purchased from Pierce Chemical Co. (Rockford, IL). Candesartan was generously provided by Dr. Raij from the Veterans Administration Medical Center (Miami, FL). The plasma renin activity GammaCoat radioimmunoassay kit was purchased from Diasorin Inc. (Stillwater, MN).
Cell Culture
The human retinal pigment epithelium ARPE-19 cell line was purchased from the American Type Culture Collection (Manassas, VA). ARPE-19 cells at passages 20 to 22 were plated at subconfluent density on T-75 (75 cm2) flasks and grown to confluence in maintenance medium [Dulbecco's modified Eagle's medium–Ham's nutrient mixture F-12 (1:1 v/v) supplemented with 10% FBS, 100 μg/mL penicillin/streptomycin, and 0.348% Na2HCO3]. Cells were maintained at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% O2 air. For the experiments, confluent cells were split and plated at subconfluent density (2 × 105 cells) and grown to confluence unless otherwise indicated. Cells were prepared for the experiment by changing the maintenance medium to assay medium (ie, maintenance medium without phenol red) for 2 days. This medium was then replaced with assay medium supplemented with 1% FBS for 1 day. Subsequently, the medium was replaced with assay medium supplemented with 0.1% FBS. Cells were then treated with one of the following protocols: i) dose-response stimulating cells with various Ang II concentrations (10−11 to 10−7 mol/L) for 5 minutes; ii) time-course analysis exposing cells to 10−7 mol/L Ang II for various times (1, 2, 5, 10, 20, or 60 minutes); iii) treatment of cells with 10−7 mol/L Ang II alone or in combination with 10−6 mol/L candesartan, an AT1 blocker, PD123319 (10−6 mol/L), an AT2 blocker, and/or a combination of both inhibitors for 1 hour before Ang II (10−7 mol/L) exposure for 5 minutes; iv) same as protocol 3 but exposure of cells to Ang II for 24 hours rather than 5 minutes; and v) same as protocol 3 but with pretreatment of cells for 1 hour with 20 μmol/L SB203580, a pyridinyl imidazole highly selective inhibitor of the kinase activity of p38α and p38βMAPK isoforms, 40 μmol/L of the ERK inhibitor PD98059, and/or a combination of both inhibitors rather than Ang II. The culture medium was removed, and cells were either washed three times with PBS containing 1 mmol/L sodium orthovanadate before they were harvested (protocols 1 to 3) or washed twice with PBS and incubated in fresh medium supplemented with 0.1% FBS for 24 hours (protocols 4 and 5). Cell homogenates and conditioned media were collected for protein determination using a detergent-compatible kit based on the Bradford method.
Mice
Mice used in the study were handled in accordance with the Association for Research in Vision and Ophthalmology Guidelines for the Use of Animals in Ophthalmic and Vision Research. All experimental protocols were approved by the University of Miami Care and Use Committee. Experiments were performed in 9-month-old C57BL6 female mice (National Institute of Aging, Bethesda, MD). Animals were fed a normal sodium diet (UAR A.40, Na+ 104 mmol/kg). Food and water intake was monitored daily.
Subsequently, mice were randomly divided into four groups of five mice each to investigate the effect of exogenous Ang II administration. Animals were anesthetized using 2% isoflurane in oxygen, and an osmotic minipump (Azet model 2001; Alza Corp., Palo Alto, NC) was implanted subcutaneously between the scapulae. Mice were infused for different durations with saline solution; Ang II (3.0 μg/kg/min) in saline solution; Ang II in combination with candesartan (10 mg/kg/d), an AT1 antagonist administered in drinking water; and Ang II in combination with PD123319 (50 mg/kg/d), an AT2 antagonist administered in drinking water. The dosages of candesartan and PD123319 effectively block AT1 and AT2, respectively.26,42–45 Systolic blood pressure was recorded using the tail-cuff plethysmography method. Animals were sacrificed at 1, 7, 14, or 30 days after treatment, and eyes were removed for recovery of RPE sheets.
To evaluate the effect of Ang II signaling, experiments were performed in one-kidney, one-clip (1K1C; n = 5) mice with renovascular HTN. Animals were anesthetized using ketamine 42.8 mg/kg in combination with xylazine 8.5 mg/kg, acepromazine 1.4 mg/kg, and buprenorphine 0.05 to 0.1 mg/kg. A silver clip was placed around the left renal artery, and the contralateral kidney was removed. Control 1K1C mice [simulated 1K1C (s1K1C); n = 5] underwent sham clipping and right nephrectomy. Blood pressure was measured as described above. Mice were sacrificed 4 weeks after sham clipping and nephrectomy, blood was collected to measure plasma renin activity, and eyes were removed for recovery of RPE sheets.
Plasma Renin Activity
Blood was collected in a 75-μL hematocrit tube containing 1 μL EDTA (125 mmol/L) in its tip. Red blood cells and plasma were separated by centrifugation and frozen until used for renin determination. With the use of a fivefold dilution of 2 μL plasma, renin concentration was measured using a radioimmunoassay as the generation of Ang I after addition of excess rat substrate with final plasma dilutions varying between 1:500 and 1:1000. Ang I generation was determined for a 1-hour incubation period at 37°C, and was expressed as an hourly mean. In each assay, background Ang I formation was determined by incubating substrate without plasma for the same time, and was subtracted from the plasma-containing samples if necessary. In addition, background Ang I concentrations were determined in a plasma aliquot kept frozen without addition of substrate until assay. All plasma samples were stored at −20°C until assay.
RPE Isolation
RPE sheets were isolated as previously described.46 In brief, the eyes were enucleated, rinsed with 10% gentamicin for sterilization and twice with 1X PBS, and placed in a dish containing 1X PBS. Using a dissecting microscope, eyes were opened via a circumferential incision at the ora serrata. The anterior segment was removed, and the vitreous-retina was separated from the RPE and choroid eyecup using a round-tipped disposable blade (K20-1504) and Tennant forceps (K5-5230) (both from Katena Products, Inc., Denville, NJ). The remaining eyecup was incubated with dispase in 1X PBS at 37°C for 30 minutes. With use of a Barraquer spatula (K3-2310; Katena Products, Inc.), the RPE monolayer was dissected from BrM and the choroid. The RPE sheets were transferred into individual tubes (Eppendorf, Fremont, CA) and homogenized on ice with a pestle. Subsequently, proteins were extracted, quantified, and stored at −80°C.
Aliquots from the isolated RPE sheets were stained using mouse anti–cytokeratin-18 antibody as a positive control for epithelial cells and with mouse anti–endothelial cell (CD146) monoclonal antibody to ensure that there was no contamination with endothelial cells.
Western Blot Analysis
Lysates from ARPE-19 cells and mice RPE sheets were harvested, and protein concentration was determined using a detergent-compatible protein assay. Protein extracts (20 to 40 μg) were denatured in Laemmli's sample buffer, followed by boiling for 5 minutes, and then resolved on a 4% to 20% Tris-glycine gel. After electrophoresis (120 V for 2 hours), proteins were transferred in 1X transfer buffer [25 mmol/L Tris, 192 mmol/L glycine, 0.1% SDS, and 20% methanol (pH 8.4)] to a Hybond-ECL nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ) using constant current (350 mA for 45 minutes). Membranes were blocked in 5% nonfat dry milk–TBS solution for 1 hour at room temperature. Blots were incubated overnight at 4°C using one of the following primary antibodies: rabbit polyclonal anti-ERK, mouse monoclonal anti–phospho-ERK, rabbit polyclonal anti-SAPK/JNK, mouse monoclonal anti–phospho-SAPK/JNK, rabbit polyclonal anti-p38, mouse monoclonal anti–phospho-p38, rabbit monoclonal anti-GAPDH, mouse polyclonal anti-BSG, and mouse monoclonal anti–MMP-14. Membranes were washed three times with TBS-Tween 20 then incubated with horseradish peroxidase–linked donkey anti-mouse or anti-rabbit antibody for 2 hours at room temperature, and then washed three times in TBS-Tween 20. Detection was performed using ECL Western blotting substrate.
Zymography
MMP-2 activity was determined as previously described.47 In brief, conditioned culture medium was collected after cell treatment and clarified at centrifugation at 15,000 × g for 30 minutes at 4°C. Protein concentration was determined and MMP-2 activity assessed using 10% gelatin zymography gels. Protein extracts, 10 μg, were subjected to electrophoresis (120 V for 2 hours) and subsequently incubated in 2.5% Triton X-100 for 1½ hours. Gels were next incubated in 50 mmol/L Tris buffer for 18 hours, enabling determination of total proteolytic MMP-2 activity with no interference from their associated tissue inhibitors. Gels were stained using Brilliant Blue R solution (Sigma-Aldrich Corp.) for 2 hours. Densitometry was performed using ImageJ 1.17 software (National Institutes of Health, Bethesda, MD).
Immunocytochemistry
Subconfluent ARPE-19 cells were grown on chamber slides and treated with or without Ang II (10−7 mol/L) for 5 minutes in the absence or presence of a 30-minute pretreatment with either AT1 blocker candesartan (10−6 mol/L) or AT2 blocker PD123319 (10−6 mol/L). Cells were fixed in 4% paraformaldehyde for 10 minutes, washed with PBS, and permeabilized in 0.1% Triton X-100 in PBS for 20 minutes at room temperature. Cells were next washed with PBS, blocked in 5% bovine serum albumin for 1 hour, and incubated with mouse monoclonal anti–phospho-ERK or anti–phospho-p38 antibodies (dilution 1:200) for 3 hours at 4°C. Thereafter, cells were washed with PBS, incubated with anti-mouse antibody conjugated to Alexa-488 (dilution 1:200) for 1 hour, washed again with PBS, and mounted with ProLong Antifade with DAPI. Omission of the primary antibody was used as negative control.
Gene Silencing
ARPE-19 cells grown in six-well plates were allowed to reach approximately 90% confluence before transfection. Then cells were transiently transfected with 100 nmol/L p38 MAPK siRNA, ERK siRNA, or scrambled control siRNA according to the manufacturer's protocol. Twenty-four hours after transfection, cells were prepared for the experiment by replacing the medium with phenol red–free 1% FBS medium for 24 hours. Subsequently, cells were treated with Ang II (10−7 mol/L) for 24 hours in phenol red–free medium supplemented with 0.1% FBS. The p38 MAPK siRNA, ERK siRNA, and control siRNA consisted of single proprietary sequences. The efficiency of siRNA in reducing endogenous p38 MAPK and ERK protein was confirmed using Western blot analysis. Cell lysates and conditioned media were collected and processed for either Western blot analysis or zymography as described above.
Statistical Analysis
All experiments were performed three or four times, with reproducible results. Data are expressed as mean ± SEM. Statistical comparisons were performed using one-way analysis of variance and the Dunnett multiple comparisons as post hoc test when differences were significant. P < 0.05 was considered statistically significant.
Results
Ang II Activates ERK and p38 MAPKs in RPE Cells
First, it was confirmed that the three major members of the MAPKs family are expressed in ARPE-19 cells (Figure 1). Second, a dose-dependent evaluation of ERK, p38, and JNK MAPK phosphorylation on cell lysates from ARPE-19 cells exposed to various concentrations of Ang II for 5 minutes demonstrated that stimulation with 10−7 mol/L Ang II resulted in an approximately 150% increase in ERK phosphorylation (values give as mean ± SEM; 255.5% ± 27.7% versus 100.0% ± 10.2%; P < 0.05) (Figure 1A). In addition, strong induction of p38 phosphorylation was observed after treatment with 10−7 mol/L (311.1% ± 33.3% versus 100.0% ± 9.1%; P < 0.01) and 10−8 mol/L Ang II (288.8% ± 2.6% versus 100.0% ± 9.1%; P < 0.01). In contrast, Ang II failed to induce phosphorylation of JNK at the concentrations tested (Figure 1C). Next, it was demonstrated that Ang II at 10−7 mol/L time-dependently induced rapid and robust phosphorylation of both ERK (Figure 2A) and p38 (Figure 2B) MAPKs, beginning within 2 minutes, reaching a peak level at 5 minutes, remaining elevated at 10 minutes, and gradually declining thereafter. JNK was not activated in response to Ang II in ARPE-19 cells (Figure 2C). These findings demonstrate for the first time, to our knowledge, that Ang II at physiologically relevant concentrations activates ERK and p38 MAPKs signaling pathways in RPE cells.
Figure 1.
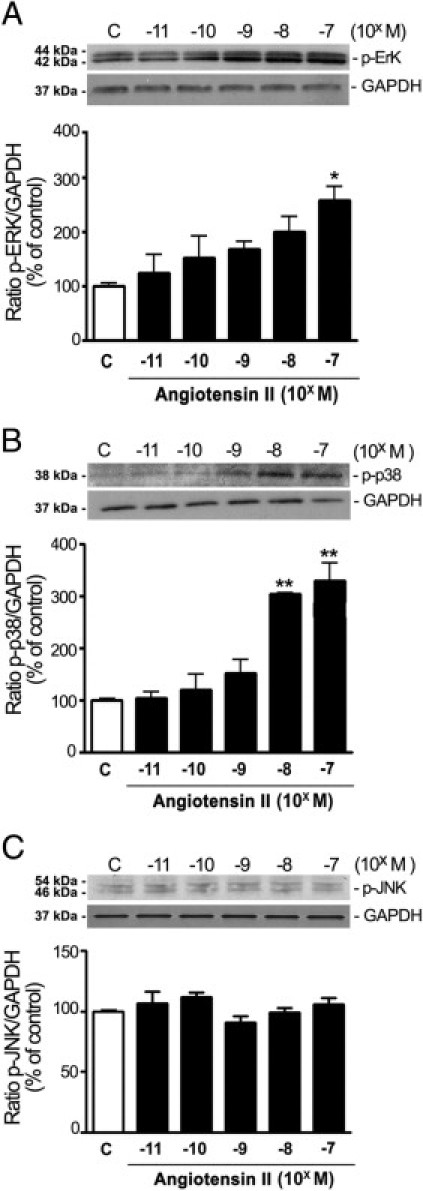
Ang II activates MAPKs in ARPE-19 cells incubated with increasing concentrations of Ang II (10−11 to 10−7 mol/L) for 5 minutes. Phosphorylation dose-response of ERK (A), p38 MAPK (B), and JNK (C). Top panels: Representative Western blots for evaluation of phosphorylated protein in cell lysates. Numbers on the left represent protein molecular weight in kilodaltons. GAPDH protein is shown as the loading control. Bottom panels: Bar graphs corresponding to mean results of three independent experiments normalized to their corresponding loading controls. Results are expressed as mean ± SEM. *P < 0.05 and **P < 0.01 compared with untreated control cells.
Figure 2.
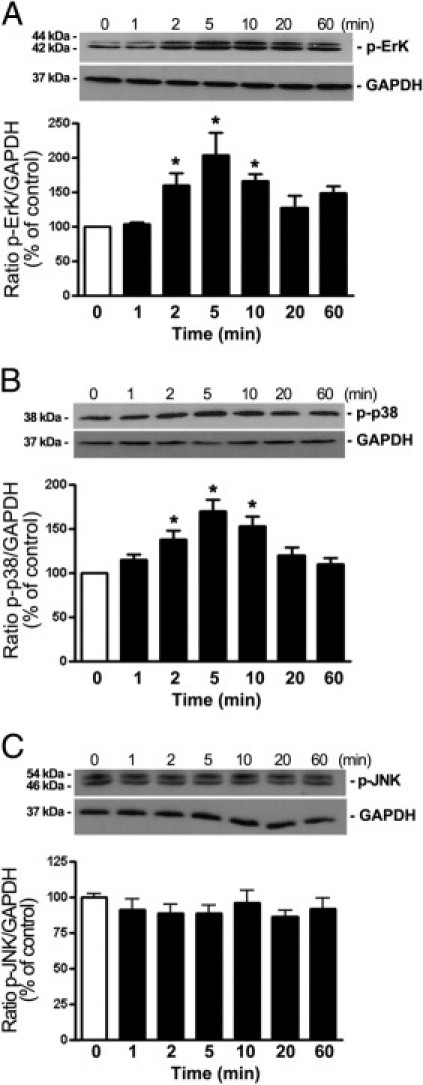
Phosphorylation time course of MAPKs ERK, p38, and JNK in ARPE-19 cells stimulated with Ang II. A–C: Time-dependent ERK phosphorylation (A), p38 phosphorylation (B), and JNK phosphorylation (C) assessed using Western blot analysis on cell lysates from ARPE-19 cells treated with Ang II (10−7 mmol/L) for various periods. Top panels: Western blots from a representative experiment. Numbers on the left represent protein molecular weight in kilodaltons. GAPDH protein is shown as the loading control. Bottom panels: Bar graphs corresponding to the mean results of three independent experiments normalized to their corresponding loading controls. Results are expressed as percentage of control, and are the mean ± SEM of three independent experiments run in duplicate. *P < 0.05 compared with untreated control cells.
AT1 Mediates Ang II–Induced MAPKs Activation in RPE Cells
Next evaluated was the role of the two subtypes of Ang II receptors in Ang II–induced activation of ERK and p38 MAPK cascades in ARPE-19 cells using specific antagonists for AT1 and AT2. Exposure of ARPE-19 cells to 10−7 mol/L Ang II for 5 minutes elicited an approximately 102% increase in ERK phosphorylation relative to control (202.2% ± 42.3% versus 100.0% ± 5.2%; P < 0.05), which was abrogated in the presence of the AT1 blocker candesartan (112.7% ± 12.3% versus 202.2% ± 42.3%; P < 0.05). In contrast, AT2 antagonist PD123319 did not have any significant effect on Ang II–induced increase in phosphorylated ERK (181.4% ± 30.0% versus 202.2% ± 42.3%; P < 0.05) (Figure 3A). As expected, the combination of both AT1 and AT2 was as effective as the use of AT1 blocker alone in blunting ARPE-19 cell response to Ang II (Figure 3A). Similarly, the AT1 antagonist candesartan but not PD123319 abolished Ang II–induced p38 MAPK phosphorylation (116.1% ± 9.4% versus 183.0 ± 27.6%; P < 0.05) (Figure 3B). AT1 and AT2 blockers used in combination resulted in the same effect as AT1 alone.
Figure 3.
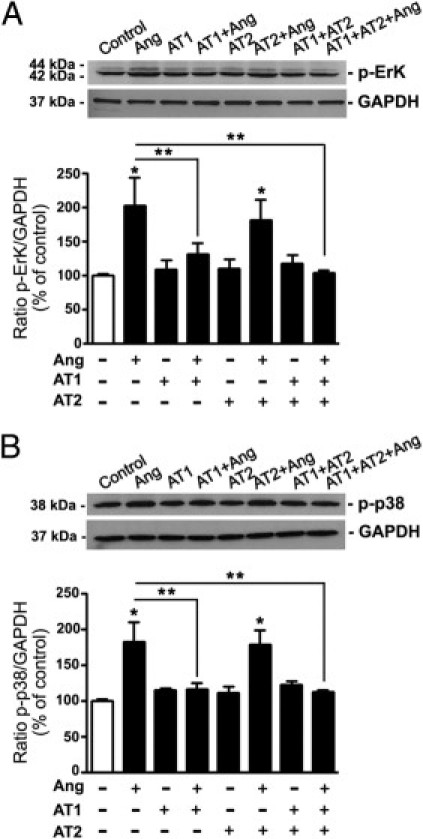
AT1 blocks Ang II-induced ERK and p38 phosphorylation in ARPE-19 cells. ARPE-19 cells were stimulated with Ang II in the presence of Ang II. p-ERK protein (A) and p-p38 protein (B) assessed using Western blot analysis. Top panels: Representative Western blots for p-ERK and p-p38. Numbers on the left represent protein molecular mass in kilodaltons. p-ERK and p-p38 protein expression was normalized to GAPDH. Bottom panels: Ratios of p-ERK/GAPDH or p-p38/GAPDH. Data are expressed as percentage of control, and are the mean ± SEM of three independent experiments run in duplicate. *P < 0.05 compared with untreated control cells; **P < 0.05 versus Ang II alone.
Results of Western blot analysis were subsequently confirmed by immunocytochemistry. In brief, stimulation with 10−7 mol/L Ang II for 5 minutes led to phosphorylation of ERK (Figure 4G) and p38 (Figure 4Q) MAPKs. This effect was abolished by candesartan (Figure 4H and 4R) but not by PD123319 (Figure 4I and 4S). As described above, the effect of candesartan and PD123319 in combination was similar to that with AT1 antagonist alone. Together, these results demonstrate that Ang II activates ERK and p38 MAPK signaling pathways via AT1 in ARPE-19 cells.
Figure 4.
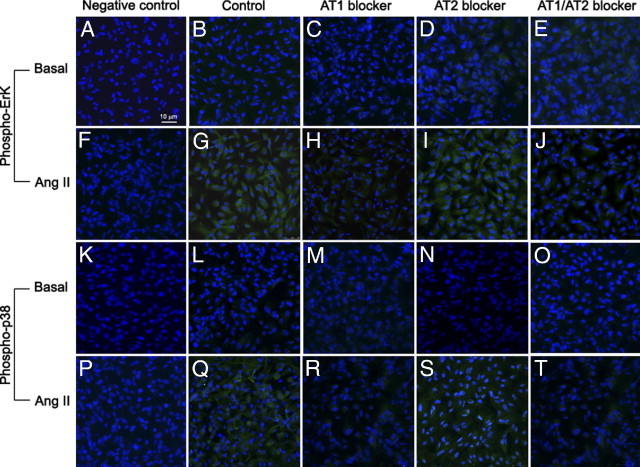
Representative immunofluorescent double staining of phosphorylated ERK and p38 MAPK (green) and nuclei (blue) in Ang II–stimulated ARPE-19 cells in the presence of Ang II in the presence or absence of Ang II receptor blockers AT1 and AT2. Figure is a composite image demonstrating the immunostaining of nontreated (basal) (A–E and K–O) and treated (Ang II) (F–J and P–T) cells in the absence (control) (B, G, L, and Q) or presence of candesartan (AT1 blocker) (C, H, M, and R) or PD123319 (AT2 blocker) (D, I, N, and S) or both (AT1/2 blockers) (E, J, O, and T). Negative controls were generated by omission of the primary antibody. Sections were analyzed using confocal microscopy (×40 magnification).
Ang II Up-Regulates BSG Expression via AT1 in RPE Cells
BSG, a key regulator of MMP-2 activity, is expressed in human RPE.35 Herein, it was investigated whether Ang II modulates BSG expression in cultured RPE cells. To address this, ARPE-19 cells were exposed to various concentrations of Ang II for 24 hours, and BSG protein expression was assessed using Western blot analysis. Ang II up-regulated BSG expression at all concentrations tested (Figure 5A). The maximal stimulatory effect resulting in an approximately 80% increase in BSG levels relative to control was achieved at 10−10 mol/L (183.12% ± 3.03% versus 100.0% ± 3.12%; P < 0.01) and 10−9 mol/L (180% ± 6.86% versus 100.0% ± 3.12%; P < 0.01) Ang II. Ang II 10−8 mol/L and 10−7 mol/L increased BSG protein expression by approximately 60% (164.37% ± 15.6% and 159.12% ± 18.7%, respectively, versus 100.0% ± 3.12%; P < 0.05). At the lowest concentrations tested (10−11 mol/L), Ang II induced only small changes in BSG expression compared with control (131.78% ± 6.25% versus 100.0% ± 3.12%; P < 0.05) (Figure 5A).
Figure 5.
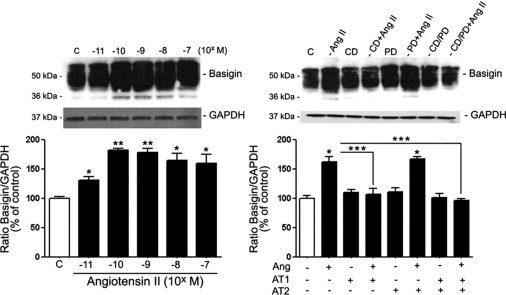
Ang II up-regulated BSG protein expression through AT1 activation in RPE-19 cells. A: RPE-derived BSG protein expression in the presence of various concentrations of Ang II for 24 hours. B: BSG protein expression in the presence of candesartan (CD) alone, PD123319 (PD) alone, or a combination of both Ang II receptor blockers before treatment with Ang II. Top panels: Representative Western blots for BSG. Numbers on the left represent protein molecular weight in kilodaltons. Bottom panels: Ratios of BSG/GAPDH. Data are expressed as percentage of control, and are the mean ± SEM of three independent experiments run in duplicate. *P < 0.05 and **P < 0.01 versus control. ***P < 0.05 versus Ang II alone.
Next evaluated was the contribution of AT1 and AT2 in Ang II–induced up-regulation of BSG expression. Treatment with the AT1 blocker candesartan suppressed Ang II–induced increase in BSG protein expression (105.8% ± 8.8% versus 163.7% ± 5.7%; P < 0.05) (Figure 5B). The AT2 blocker PD123319 had no effect (Figure 5B). Treatment with candersartan and PD123319 in combination was as effective as AT1 antagonist alone in preventing the increase in BSG expression (Figure 5B). These observations establish that the stimulatory effect of Ang II on BSG protein expression is mediated by AT1, which may have a key role in regulation of MMP-2 activity in ARPE-19 cells.
Ang II-Induced Increase in MMP-2 Activity Is ERK- and p38 MAPK–Mediated in RPE Cells
Next explored was the potential role of ERK and p38 MAPK in Ang II–induced increase in MMP-2 activity in cultured RPE cells. ARPE-19 cells were stimulated with Ang II (10−7 mol/L) for 24 hours in the presence or absence of selective inhibitors of p38 and ERK MAPKs. MMP-2 activity was increased by approximately 58% (157.7% ± 17.7% versus 100.0% ± 3.2%; P < 0.05) in response to Ang II compared with control (Figure 6A). Treatment with either PD98059 or SB203580, alone or in combination, suppressed Ang II–induced increase in MMP-2 activity, which suggests that this effect is mediated by ERK and p38 MAPKs in ARPE-19 cells.
Figure 6.
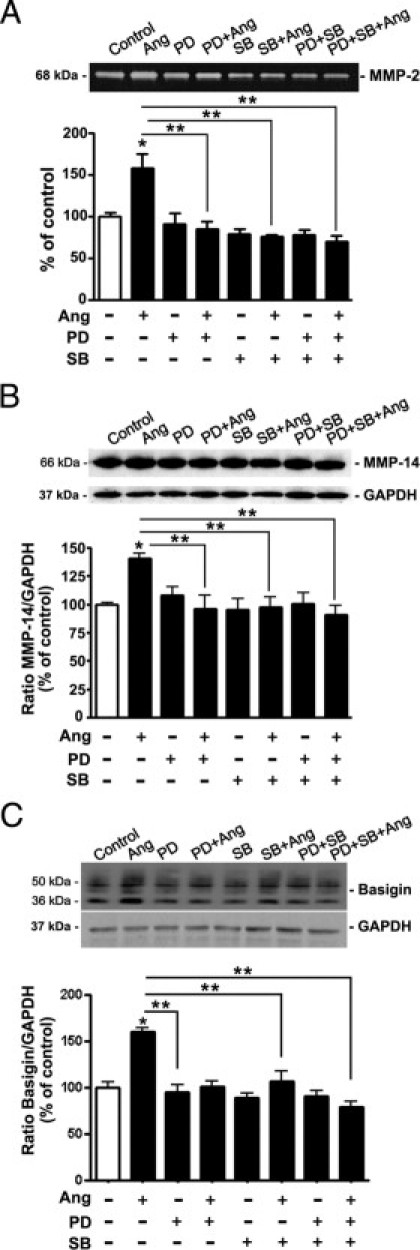
Inhibition of ERK and p38 MAPK blocks Ang II–induced MMP-2 activity and MMP-14 and BSG protein expression in ARPE-19 cells. A: Inhibition of Ang II–induced MMP-2 activity by the selective inhibitor of ERK PD98059 (PD), and p38α and p38β MAPK isoforms, SB203580 (SB). Top: Representative gelatin zymogram. Numbers on the left represent protein molecular weight in kilodaltons. Bottom: Bar graph corresponding to the mean results of three independent experiments. B: Inhibition of Ang II–induced MMP-14 protein expression by PD and SB. Top: Representative Western blot for MMP-14. Numbers on the left represent protein molecular weight in kilodaltons. Bottom: Ratio of MMP-14/GAPDH. C: Ang II effect on expression of BSG in ARPE-19 cells in the presence or absence of MAPKs inhibitors. Top: Representative Western blot for BSG. Numbers on the left represent protein molecular weight in kilodaltons. Bottom: Ratio of BSG/GAPDH. Data are expressed as percentage of control, and are the mean ± SEM of three independent experiments run in duplicate. *P < 0.05 versus control; **P < 0.05 versus Ang II alone.
Ang II–Induced Increase in MMP-14 and BSG Expression Is ERK- and p38 MAPK–Mediated in RPE Cells
BSG and MMP-14 induce MMP-2 activity 48–50. Herein, it was investigated whether the effect of Ang II on MMP-14 and BSG expression might be ERK and/or p38 MAPK–dependent in RPE cells. It was demonstrated that MMP-14 protein levels were increased by approximately 35% compared with control (134.6% ± 3.9% versus 100.01% ± 3.0%; P < 0.05) (Figure 6B), and BSG protein expression was up-regulated by approximately 60% (163.4% ± 3.9% versus 100.01% ± 6.4%; P < 0.05) in response to stimulation with Ang II (10−7 mol/L) for 24 hours (Figure 6C). Pretreatment of cells with inhibitors PD98059 or SB203580 to block ERK and p38 MAPK signaling pathways, respectively, completely blocked Ang II–induced expression of MMP-14 (Figure 6B) and BSG (Figure 6C), which suggests that this effect is mediated by both ERK and p38 MAPK in ARPE-19 cells.
Gene Silencing of ERK and p38 MAPK Abolishes the Effect of Ang II on MMP-2 Activity and MMP-14 and BSG Protein Expression in ARPE-19 Cells
The siRNA-based RNA interference method was used as a genetic approach to validate the above observations generated using pharmacologic tools. Compared with cells transfected with scrambled siRNA, constitutive expression of ERK was reduced by approximately 57% in ARPE-19 cells transfected with ERK siRNA (43.7% ± 6.2% versus 93.8% ± 3.1%; P < 0.05) (Figure 7A), and p38 protein levels were decreased by approximately 49% (55.5% ± 4.6% versus 102.7% ± 2.6%; P < 0.05) in cells transfected with p38 siRNA (Figure 7B). After exposure to Ang II (10−7 mol/L) for 24 hours, MMP-2 activity was increased by approximately 64% (164.6% ± 4.2% versus 100.0% ± 2.1%; P < 0.05) in nontransfected cells, and approximately 39% (138.9% ± 5.0% versus 100.0% ± 2.1%; P < 0.05) in cells transfected with scrambled siRNA compared with control (Figure 8A). Ang II–induced increase in MMP-2 activity was completely abolished in cells transfected with either ERK or p38 siRNA alone or in combination (Figure 7C).
Figure 7.
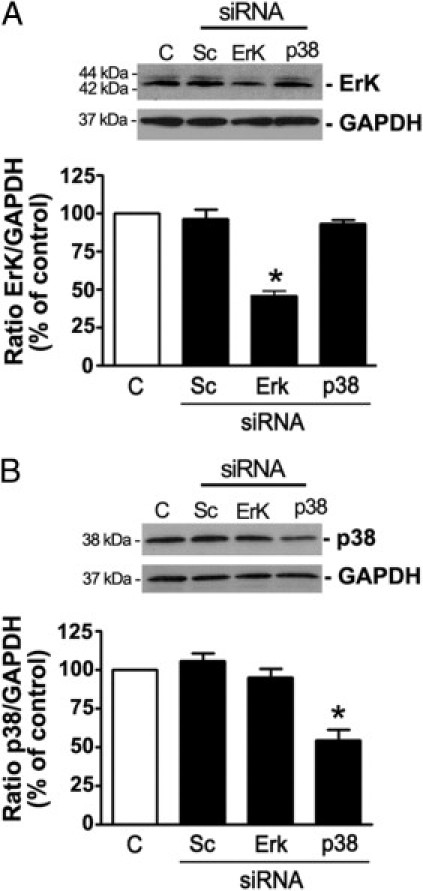
Effect of ERK and p38 MAPK gene silencing on ERK and p38 protein expression. ARPE-19 cells were transiently transfected with either a scrambled sequence (Sc) or a specific siRNA against ERK or p38 MAPK (p38). A: Reduction in endogenous ERK1/2 protein expression by siRNA against ERK MAPK. B: Reduction in endogenous p38 MAPK protein expression by siRNA against p38 MAPK. Efficiency of siRNA against ERK or p38 MAPK in reducing endogenous ERK or P38 protein was confirmed using Western blot analysis. Top panels: Representative Western blots for ERK (A) or p38 MAPK (B). Numbers on the left represent protein molecular mass in kilodaltons. ERK and p38 MAPK protein expression was normalized to GAPDH. Bottom panels: Mean results of four independent experiments run in duplicate on ARPE-19 cells. Data are expressed as percentage of control. *P < 0.05 compared with nontransfected control cells.
Figure 8.
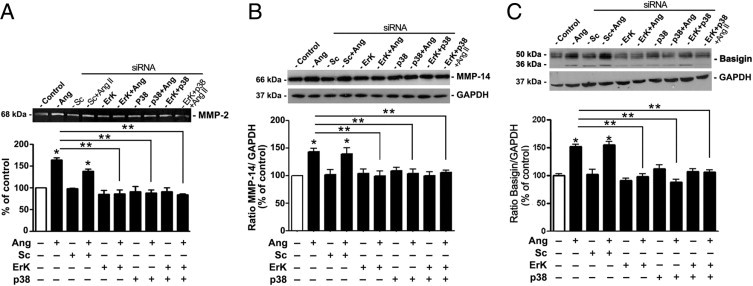
Effect of ERK and p38 MAPK gene silencing on Ang II–induced MMP-2 activity and MMP-14 and BSG protein expression in ARPE-19 cells. A: Effect of ERK and p38 MAPK siRNA on Ang II–induced MMP-2 activity. Top: Gelatin zymogram from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Bar graph corresponding to mean results of three independent experiments. B: Effect of ERK and p38 MAPK siRNA on Ang II–induced MMP-14 protein expression. C: Effect of ERK and p38 MAPK siRNA on Ang II–induced BSG protein expression. Top: Representative Western blot for MMP-14 (B) and BSG (C). Numbers on the left represent protein molecular weight in kilodaltons. MMP-14 and BSG protein expression was normalized to GAPDH. Bottom: Ratio of MMP-14/GAPDH (B) or BSG/GAPDH (C). Results are expressed as percentage of control, and are the mean ± SEM of four independent experiments. *P < 0.05 compared with nontransfected control cells; **P < 0.05 versus nontransfected Ang II–treated cells.
MMP-14 protein expression was increased by approximately 50% in nontransfected cells (148.7% ± 5.3% versus 100.0% ± 1.3%; P < 0.05), and approximately 44% (143.9% ± 11.0% versus 100.0 ± 1.3%; P < 0.05) in cells transfected with scrambled siRNA compared with control in response to 10−7 mol/L Ang II for 24 hours (Figure 8B). Following the pattern of MMP-2 activity, transfection with either ERK or p38 siRNA alone or in combination completely abrogated Ang II–induced increase in MMP-14 protein expression (Figure 8B).
In addition, BSG protein expression was increased by approximately 58% in nontransfected cells (156.12% ± 9.3% versus 100.0% ± 4.15%; P < 0.05), and approximately 60% (162.9% ± 11.0% versus 100.0% ± 4.15%; P < 0.05) in cells transfected with scrambled siRNA compared with control in response to 10−7 mol/L Ang II for 24 hours (Figure 8C). Transfection with either ERK or p38 siRNA alone or in combination abrogated Ang II–induced increase in BSG protein expression (Figure 8C). These data demonstrate that ERK and p38 MAPKs have an essential role in regulation of MMP-2 activity through MMP-14 and BSG in response to Ang II in ARPE-19 cells.
Exogenous Ang II Increases Systolic Blood Pressure in Mice
Ang II was administered long term in mice to induce HTN. After 24 hours of treatment, no difference was observed in systolic blood pressure between mice infused with Ang II and control mice given saline solution (112.8 ± 5.3 mmHg versus 109.9 ± 2.7 mmHg) (Figure 9A). After Ang II infusion for 7 days, blood pressure increased significantly in treated mice compared with control mice (152.6 ± 10.5 mmHg versus 115.8 ± 6.8 mmHg; P < 0.05), and after 14 days, it increased to 172.4 ± 5.2 mmHg versus 115.7 ± 4.6 mmHg in control mice (P < 0.01) (Figure 9A). This increase persisted after 30 days of treatment with Ang II (179.2 ± 7.4 mmHg versus 116.8 ± 5.2 mmHg in control mice; P < 0.01) (Figure 9A).
Figure 9.
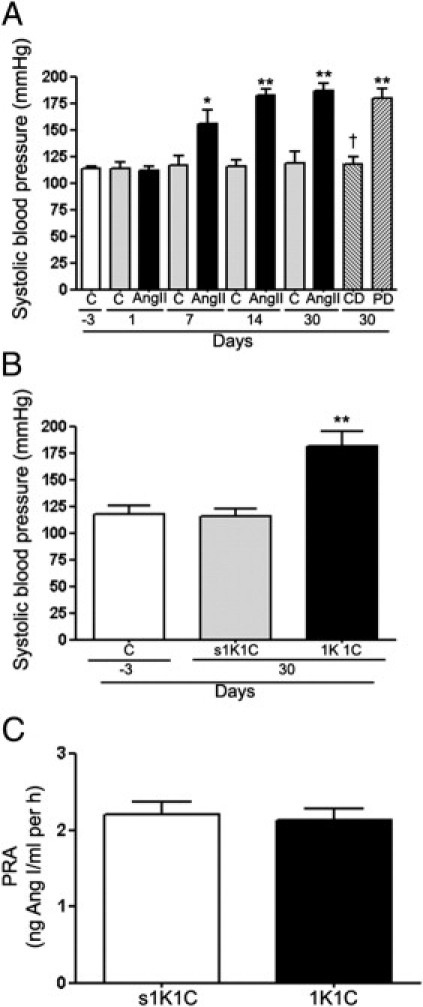
A: Systolic blood pressure in response to administration of Ang II alone for 1, 7, 14, and 30 days and Ang II in combination with the AT1 antagonist candesartan (CD) or the AT2 antagonist PD123319 (PD) for 30 days in C57BL/6 mice. *P < 0.05 and **P < 0.01 versus their respective basal blood pressure; †P < 0.01 versus Ang II administration for 30 days. B: Systolic blood pressure and (C) plasma renin activity (PRA) in 1k1C mice and in the corresponding sham-operated control group (s1K1C). Data are expressed as mean ± SEM. **P < 0.01 compared with corresponding control and sham-operated control groups.
Pharmacologic studies using AT1 and AT2 blockers performed during the 30-day treatment with Ang II revealed that the AT1 antagonist candesartan abolished Ang II–induced increase in blood pressure, whereas AT2 antagonist PD123319 did not have any effect (Figure 9A). These observations demonstrate that in mice, the Ang II–mediated effect on blood pressure is AT1-dependent.
In parallel experiments, the 1K1C experimental model of renovascular Ang II–independent (normal to low renin) HTN was used. Blood pressure was significantly higher in 1K1C mice compared with sham-operated animals (173.7 ± 17.4 mmHg versus 118.6 ± 6.2 mmHg; P < 0.01) (Figure 8B). No significant difference was reported in blood pressure between control and sham-operated animals (Figure 9B).
Plasma Renin Activity Remains Unchanged in 1K1C Mice with HTN
Plasma renin activity was measured in 1K1C mice with HTN to assess development of normal to low renin HTN. Plasma renin activity in 1K1C animals with HTN was similar to that in the sham-operated group, which confirms that HTN is independent of Ang II in this model (Figure 9C).
Ang II Induces ERK, p38, and JNK MAPKs Phosphorylation Through AT1 Activation in Mice RPE Sheets
In agreement with in vitro results, it was demonstrated for the first time, to our knowledge, that ERK (Figure 10A) and p38 (Figure 11A) phosphorylation was increased in RPE sheets from mice treated with Ang II. No activation was observed of either ERK (Figure 10C) or p38 (Figure 11C) MAPK signaling pathways in RPE from 1K1C hypertensive mice.
Figure 10.
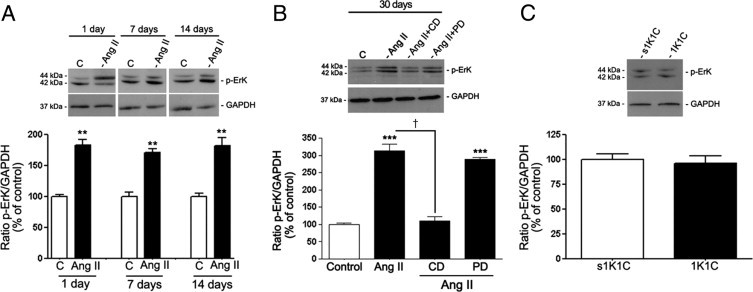
Regulation of phosphorylated ERK MAPK in dissected RPE sheets from mice treated with Ang II infused subcutaneously via osmotic minipump. Proteins were extracted from RPE sheets (n = 6 eyes per group). A: Phosphorylated p-ERK protein expression evaluated using Western blot analysis in RPE from mice treated with Ang II for 1, 7, and 14 days. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Ratio of p-ERK/GAPDH. Data are the mean ± SEM results from six eyes expressed as percentage of control. **P < 0.01 versus control. B: RPE-derived p-ERK protein expression evaluated using Western blot analysis in RPE sheets from mice treated with saline solution, Ang II, or Ang II in combination with candesartan (CD) or PD123319 (PD) for 30 days. Protein was extracted from RPE sheets (n = 6 eyes per group). Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Ratio of p-ERK/GAPDH. Data are expressed as percentage of control. Shown are mean ± SEM values. ***P < 0.001 versus control mice. †P < 0.001 versus mice treated with Ang I. C: RPE-derived p-ERK protein expression evaluated using Western blot analysis in RPE sheets from 1K1C mice with renovascular HTN and control 1K1C mice (s1K1C) underwent sham clipping and right nephrectomy. Top: p-ERK protein expression evaluated using Western blot analysis from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Ratio of p-ERK/GAPDH. Data are expressed as percentage of control. Shown are mean ± SEM values.
Figure 11.
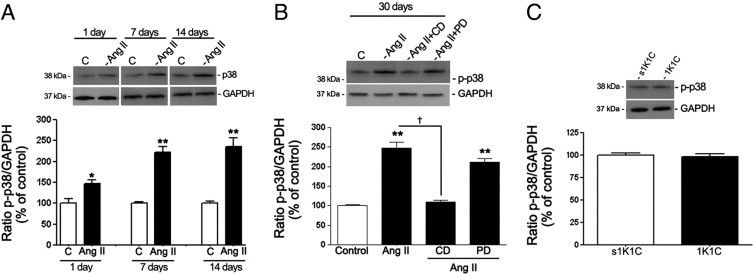
Regulation of phosphorylated p38 (p-p38) MAPK in dissected RPE sheets from mice treated with Ang II. A: Expression of p-p38 protein evaluated using Western blot analysis in RPE sheets from mice treated with Ang II for 1, 7, and 14 days. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Ratio of p-p38/GAPDH protein. Data are the mean results from six eyes and are expressed as percentage of control. Shown are mean ± SEM values. *P < 0.05 and **P < 0.01 versus control. B: RPE-derived p-p38 protein expression evaluated using Western blot analysis in RPE sheets from mice treated with saline solution, Ang II alone, or Ang II in combination with candesartan (CD) or PD123319 (PD) for 30 days. Protein was extracted from RPE sheets (n = 6 eyes per group). Top: Expression of p-p38 protein evaluated using Western blot analysis from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Ratio of p-p38/GAPDH protein expression. Data are expressed as percentage of control. Shown are mean ± SEM values. **P < 0.01 versus control. †P < 0.01 versus mice treated with Ang II. C: RPE-derived p-p38 protein expression evaluated using Western blot analysis in RPE sheets from 1K1C mice with renovascular HTN and control 1K1C mice (s1K1C) that underwent sham clipping and right nephrectomy. Top: Ratio of p-p38/GAPDH protein expression evaluated using Western blot analysis from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Data are expressed as percentage of control. Shown are mean ± SEM values.
A rapid, robust, sustained increase was observed (approximately 80%; 183.7% ± 11.6% versus 100.1% ± 3.6%, P < 0.01 at day 1; 169.7% ± 8.1% versus 100.04% ± 7.8%, P < 0.01 at day 7; and 181.2% ± 16.5% versus 100.0% ± 4.1%, P < 0.01 at day 14) in ERK phosphorylation in RPE sheets from mice throughout treatment with Ang II (Figure 9A). Maximal activation of approximately 2.8-fold was achieved after administration of Ang II for 30 days (285% ± 14.2% versus 100.0% ± 4.2%; P < 0.001) (Figure 10B).
It was also observed that p38 phosphorylation was increased approximately 40% relative to controls (145.5% ± 9.09% versus 100.0% ± 10.2%; P < 0.05) after 1 day of treatment with Ang II. After 7, 14, and 30 days, p38 was strongly activated by approximately 2.3-fold compared with controls (229.3% ± 10.01% versus 100.0% ± 2.8%, P < 0.01; 236.6% ± 18.5% versus 100.0% ± 5.2%, P < 0.01; and 238% ± 9.5% versus 100.0% ± 1.1%, P < 0.01, respectively) (Figure 11, A and B). In addition, the effect of Ang II on ERK and p38 phosphorylation was blocked by the AT1 antagonist candesartan, whereas the AT2 antagonist PD123319 had no effect (Figures 10B and 11B).
In contrast with in vitro observations, it was demonstrated that long-term administration of Ang II in mice resulted in rapid, robust, sustained activation of the JNK signaling pathway in RPE sheets. No activation of JNK was detected in RPE from 1K1C mice with HTN (Figure 12C). JNK phosphorylation was increased approximately 80% after treatment with Ang II for 1 day (185.2% ± 18.1% versus 100.0% ± 3.1%; P < 0.05) and 7 days (180.9% ± 13.5% versus 100.01% ± 14.2%; P < 0.05) (Figure 12A). After 14 and 30 days, RPE sheets from mice exhibited an even stronger approximately 2.4-fold increase in JNK stimulation (245.5% ± 18% versus 100.2% ± 15.1%, P < 0.01; and 233.07% ± 18% versus 100.02% ± 3.1%, P < 0.01, respectively) (Figure 12, A and B). Furthermore, the effect of Ang II on JNK phosphorylation was abolished by the AT1 antagonist candesartan, whereas AT2 blocker PD123319 had no effect (Figure 12B). Together, these data demonstrate that prolonged exposure to Ang II induces AT1-mediated activation of ERK, p38, and JNK MAPKs signaling pathways in mice RPE sheets. In addition, observations in 1K1C mice with HTN firmly established that MAPKs phosphorylation in mice treated with Ang II is Ang II–dependent.
Figure 12.
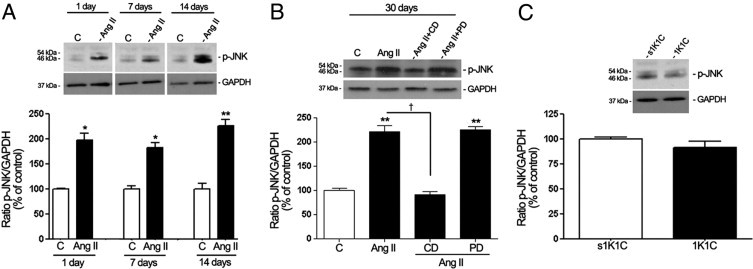
Regulation of phosphorylated JNK MAPK in dissected RPE sheets from mice treated with Ang II infused subcutaneously using an osmotic minipump. Proteins were extracted from RPE sheets (n = 6 eyes per group). A: Phosphorylated JNK (p-pJNK) protein expression evaluated using Western blot analysis in RPE from mice treated with Ang II for 1, 7, and 14 days. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Ratio of p-JNK/GAPDH. Data are the mean results from six eyes expressed as percentage of control. Shown are mean ± SEM values. *P < 0.05 and **P < 0.01 versus control. B: RPE-derived p-JNK protein expression evaluated using Western blot analysis in RPE sheets from mice treated with saline solution, Ang II, or Ang II in combination with candesartan (CD) or PD123319 (PD). Protein was extracted from RPE sheets (n = 6 eyes per group). Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Ratio of p-JNK/GAPDH. Data are expressed as percentage of control. Shown are mean ± SEM values. **P < 0.01 versus control; †P < 0.01 versus mice treated with Ang II. C: RPE-derived p-JNK protein expression evaluated using Western blot analysis in RPE sheets from 1K1C mice with renovascular HTN and control 1K1C mice (s1K1C) that underwent sham clipping and right nephrectomy. Top: Ratio of p-JNK/GAPDH protein expression evaluated using Western blot analysis from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons Bottom: Data are expressed as percentage of control. Shown are mean ± SEM values.
MMP-14 and BSG Protein Expression Increases in Mouse RPE Sheets after Exposure to Ang II
MMP-14 (Figure 13, A and B) and BSG (Figure 14, A and B) protein expression was increased in RPE sheets from mice exposed to Ang II, whereas 1K1C mice with renovascular HTN exhibited no effect (Figures 13C and 14C). MMP-14 protein was increased by approximately 66% relative to controls in RPE from mice exposed to Ang II for 1, 7, or 14 days (159.6% ± 11.2% versus 100.1% ± 10.5%, P < 0.05; 171.2% ± 5.6% versus 100.0% ± 3.4%, P < 0.05; and 166.3% ± 3.4% versus 100.0% ± 2.2%, P < 0.05, respectively) (Figure 12A). After Ang II infusion for 30 days, MMP-14 protein levels were up-regulated by approximately 34% (134.4% ± 12.0% versus 100.2% ± 6.2%; P < 0.05) (Figure 13B).
Figure 13.
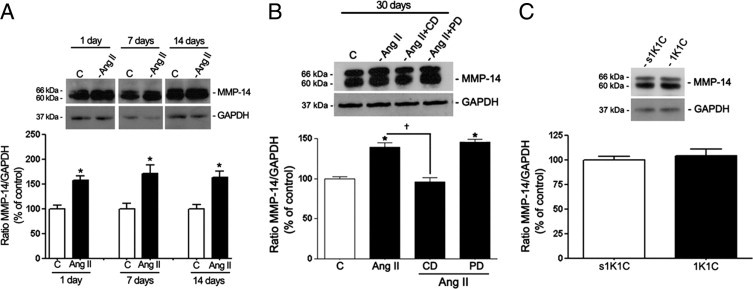
MMP-14 protein expression in isolated RPE sheets from control and treated mice. A: Effect on MMP-14 protein expression after 1, 7, and 14 days of treatment with saline solution or Ang II. B: RPE-derived MMP-14 protein expression in mice treated with saline solution, Ang II, or Ang II in combination with candesartan (CD) or PD123319 (PD) for 30 days. C: MMP-14 protein expression in RPE sheets from 1K1C mice and from the corresponding sham-operated control group (s1K1C). Mice were sacrificed at 30 days after initiation of renovascular HTN. Top panels: Ratio of MMP-14/GAPDH protein expression evaluated using Western blot analysis from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom panels: Mean densitometry results. Data are expressed as percentage of control. Shown are mean ± SEM values. *P < 0.05 versus control; †P < 0.05 versus Ang II.
Figure 14.
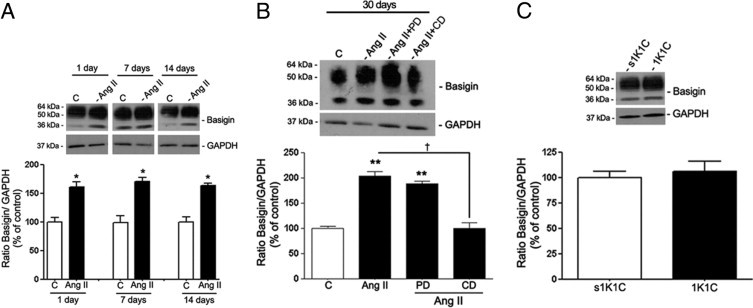
BSG protein expression in isolated RPE sheets from control and treated mice. A: RPE BSG protein expression in mice treated with saline solution or Ang II for 1, 7, and 14 days. B: BSG protein expression in mice treated with saline solution, Ang II, or Ang II in combination with candesartan (CD) or PD123319 (PD) for 30 days. C: BSG protein expression in RPE sheets from 1K1C mice and from the corresponding sham-operated control group (s1K1C). Mice were sacrificed at 30 days after initiation of renovascular HTN. Top panels: Ratio of BSG/GAPDH protein expression evaluated using Western blot analysis from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom panels: Mean densitometry results. Data are expressed as percentage of control. Shown are mean ± SEM values. *P < 0.05; **P < 0.01 versus control; †P < 0.01 versus Ang II.
In addition, RPE sheets from mice exposed to Ang II for 1, 7, and 14 days exhibited an approximately 60% increase in BSG protein expression (158.6% ± 11.2% versus 100.0% ± 9.8%, 169.2% ± 5.6% versus 100.0% ± 8.8%, and 163.3% ± 3.4% versus 100.0% ± 6.8%, respectively; P < 0.05) compared with control mice (Figure 14A). Maximal up-regulation was achieved after 30 days of exposure to Ang II (210.6% ± 4.9% versus 100.0% ± 1.1%; P < 0.01) (Figure 14B). In agreement with in vitro results, it was confirmed that Ang II–induced increase in MMP-14 and BSG protein expression was suppressed by the AT1 blocker candesartan (Figures 13B and 14B). Together, these data demonstrate that prolonged exposure to Ang II induces an AT1-mediated activation of MMP-14 and BSG protein expression in mouse RPE sheets, which may have a key role in regulating MMP-2 activity.
Discussion
The present study shows for the first time, to our knowledge, that Ang II induces an AT1-mediated increase in MMP-2 activity and in MMP-14 and BSG protein expression through ERK- and p38 MAPK-dependent signaling pathways in cultured human ARPE-19 cells. In vivo, it was demonstrated that exogenous Ang II activates ERK, p38, and JNK MAPK cascades and up-regulates MMP-14 and BSG protein levels in mouse RPE sheets. These effects were AT1-mediated and independent of blood pressure.
Dry AMD is characterized by accumulation of specific deposits under the RPE and within BrM.51 However, the mechanisms by which these sub-RPE deposits traverse the RPE basement membrane remain unexplored. To date, there is little consensus on the exact pathophysiologic mechanisms underlying AMD, although HTN is a significant risk factor.7–9 A study by Nagai et al 52 suggested that Ang II is involved in development of choroidal neovascularization in wet AMD through AT1 activation . However, there is complete lack of information about the contribution of Ang II signaling in the pathogenesis of dry AMD.
Dysregulation of ECM turnover also seems to be important in the pathogenesis of AMD.1,18,23,26 In this regard, MMP-2 is the most abundant and important MMP enzyme synthesized by the RPE relevant to retinal disorders such as AMD, in which its activity is dysregulated.53,54 Moreover, MMP-2 is crucial in degradation of ECM proteins in the RPE basement membrane and BrM 4,19,55. The mechanism by which sub-RPE deposits traverse the RPE basement membrane and invade BrM remains unexplored. It has been hypothesized that progression of sub-RPE deposits into BrM requires degradation of ECM proteins, which are critical components of RPE and basement membrane adjacent to BrM (19,4), and that ECM turnover up-regulation via MMP-2 activation is necessary for breakdown of these physical barriers. Ang II signaling might contribute to this process, thereby providing a pathogenic mechanism for its potential role in patients with HTN with dry AMD. In this regard, it has been previously reported that Ang II induces breakdown of the RPE basement membrane by increasing MMP-2 activity in RPE.18,26 However, the intracellular signaling events that link Ang II to the subsequent increase in MMP-2 activity have not been explored.
Many Ang II–induced effects on intracellular signal transduction pathways that regulate gene expression are mediated by activation and nuclear translocation of MAPKs. Multiple lines of evidence indicate that Ang II activates ERK, p38, and JNK MAPKs in various tissues.56–59 However, no study has explored Ang II–induced MAPK signaling in the RPE. Several reports have demonstrated an increase in phosphorylated ERK, p38, and JNK MAPKs in response to Ang II in vitro and in vivo.60–63 In agreement with these previous studies in various cellular systems,60–63 an increase in both ERK and p38 phosphorylation was observed in ARPE-19 cells stimulated with Ang II. However, unlike other cell types,64,65 JNK MAPK was not regulated by Ang II in RPE cells. Cell type–specific differences in Ang II signaling may account for this discrepancy. In addition, the possibility of JNK regulation after prolonged exposure to Ang II cannot be ruled out.
In an effort to translate in vitro observations into more physiologic in vivo settings, the Ang II–induced HTN experimental model26 and the 1K1C model of renovascular HTN without activation of the renin-angiotensin system were used. In agreement with results in ARPE-19 cells, it was observed that phosphorylated ERK and p38 MAPKs expression was increased in RPE from mice exposed to Ang II. However, in contrast to observations in ARPE-19 cells, levels of phosphorylated JNK were dramatically increased in mice infused with Ang II. The possible reasons for the disparity between in vitro and in vivo observations regarding as JNK regulation in response to Ang II are not clear, but may be explained in that human RPE cells were transiently exposed to Ang II for short periods, whereas mice were treated for several days, which might result in a differential regulation of the JNK signaling pathway. Moreover, the mammalian organism milieu and a fully formed RPE monolayer constitute an exponentially more complex system than isolated RPE cells in culture. Species difference between human and mouse RPE cells may also account for the discrepant results. After 1 day, Ang II infusion did not induce HTN while regulating MAPKs signaling pathways, and elevation of blood pressure in 1K1C mice with HTN had no effect on MAPKs activation. Thus, the data clearly demonstrate that in vivo activation of MAPKs in RPE sheets is a direct HTN-independent effect of Ang II. The results provide evidence that phosphorylated ERK, p38, and JNK MAPKs may have a major role as key intracellular mediators of Ang II–induced effects on MMP-2 activity in RPE.
Ang II exerts its intracellular actions through binding to one of two well-characterized plasma membrane receptor types, AT1 or AT2.66,67 These receptor types are coupled to different signal transduction mechanisms. Although there is consensus about the existence of a regular pattern of intracellular pathways regulated by each receptor type, evidence suggests that this pattern may vary depending on a particular tissue or cell type68,69 Expression of both receptor types (ie, AT1 and AT2) in ARPE-19 cells, mouse RPE, and human retina has been demonstrated.16–18,26 Ang II receptor expression has also been confirmed in the RPE-choroid complex in human and mouse choroidal neovascularization tissues 52. Using selective antagonists of either AT1 or AT2, it has been demonstrated that Ang II–induced activation of ERK and p38 MAPKs was AT1-mediated in ARPE-19 cells. These findings are in accordance with those of studies performed in other tissues and cell types in which Ang II–induced activation of ERK and p38 MAPKs was mediated by its interaction with the AT1 receptor.70 In agreement with in vitro observations, it was observed that Ang II induced activation of ERK and p38 cascade signals via AT1 in mice RPE sheets. Moreover, Ang II induced activation of JNK via AT1 in vivo.
It has been suggested that dysregulation of MMP-2 is implicated in the pathophysiology of AMD.71,72 MMP-14 and BSG, major regulators of MMP-2 activity,73–75 are considered candidate downstream effectors of Ang II. Several reports have demonstrated that Ang II positively regulates MMP-2 activity18,76,77 and that MMP-14 in combination with BSG is involved in MMP-2 activation in ARPE-19 cells.38 Using a pharmacologic approach combined with siRNA technology, it was demonstrated that Ang II stimulates MMP-2 activity, and MMP-14 and BSG protein expression via AT1 by signaling through ERK and p38 MAPKs in cultured RPE cells. These observations confirm and extend the recently published finding that demonstrates that Ang II induces MMP-14 protein expression through AT1 activation in vitro.18 In addition, these data are consistent with those of a previous study by Chen et al78 that found that BSG expression up-regulated by Ang II was suppressed by AT1 blocker in human smooth muscle cells. ERK and p38 MAPKs inhibition did not work in an additive manner to prevent Ang II–induced increase in MMP-2 activity and MMP-14 and BSG expression. Intracellular signaling networks consist of both distinct and overlapping signaling pathways. A substantial body of evidence suggests cross-talk between p38 and ERK MAPK cascades in a variety of cells79–82 including human RPE cells, as recently demonstrated.83 Evidence was reported that p38 MAPK exerts a tonic inhibition on the ERK pathway that positively modulates p38 signaling under basal conditions in ARPE-19 cells.83 The signaling events responsible for such cross-talk between p38 and ERK signaling cascades are poorly understood. Cross-talk between parallel pathways of the MAPK cascades depends on a complex interplay involving kinases and phosphatases.84 The dynamic balance between phosphorylation and dephosphorylation of kinase reactions determines the extent of feedback control between two parallel signaling pathways and, therefore, potential interactions between p38 and ERK. Another possibility to be considered is a direct physical interaction between phosphorylated p38 and ERK.85 The complexity of these interdependent signaling pathways may explain why no additive effect was observed when ERK and p38 were blocked simultaneously.
Another major new finding in the present study is that Ang II–induced up-regulation of MMP-14 and BSG protein expression is AT1-mediated in mouse RPE sheets. These new observations reinforce the validity and significance of the in vitro observations. It has been previously demonstrated that infusion of Ang II for 4 weeks up-regulated MMP-2 activity via AT1 activation in mouse RPE, which may explain, at least in part, the breakdown of the RPE basement membrane.18,26 Ang II–mediated MAPK activation in mouse RPE, the data indicate that Ang II is solely responsible for the in vivo effects on BSG and MMP-14 expression. MMP-14 and BSG are major inducers of MMP-2, the results lead to speculation that MMP-14 and BSG might regulate Ang II–induced MMP-2 activity through MAPKs- and AT1-dependent signaling pathways in the RPE in vitro and in vivo. To test this hypothesis, further studies with ERK and p38 MAPK inhibitors are needed in mice infused with Ang II.
In summary, the present data demonstrate that AT1 mediates Ang II–induced increase in MMP-2 activity through MMP-14 and BSG and a signaling cascade dependent on MAPKs in the RPE in vitro and in vivo. These original observations highlight the potential importance of this signaling pathway as a potential mediator of RPE response to Ang II–induced ECM dysregulation and breakdown of the RPE basement membrane, believed to be involved in progression of sub-RPE deposits in the pathogenesis of AMD. On the basis of these observations, MAPKs inhibitors and AT1 blocker may prevent these ECM changes essential in early AMD development and provide a potential future clinical tool for prevention of AMD.
Acknowledgment
We thank Dr. Gabriel Gaidosh, Ph.D. (Bascom Palmer Eye Institute, University of Miami), for technical assistance with confocal microscopy.
Footnotes
Supported by grants R01 EY015249-01A1 and EY015249-01A1S1 from the National Eye Institute (M.E.M.-C.) and unrestricted grant P30-EY14801 from Research to Prevent Blindness (M.E M.-C., O.A., and M.P.).
References
- 1.Fine S.L., Berger J.W., Maguire M.G., Ho A.C. Age-related macular degeneration. N Engl J Med. 2000;342:483–492. doi: 10.1056/NEJM200002173420707. [DOI] [PubMed] [Google Scholar]
- 2.Green W.R. Histopathology of age-related macular degeneration. Mol Vis. 1999;5:27. [PubMed] [Google Scholar]
- 3.Clemons T.E., Milton R.C., Klein R., Seddon J.M., Ferris F.L., III Risk factors for the incidence of advanced age-related macular degeneration in the Age-Related Eye Disease Study (AREDS): AREDS report No. 19. Ophthalmology. 2005;112:533–539. doi: 10.1016/j.ophtha.2004.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans J.R. Risk factors for age-related macular degeneration. Prog Retin Eye Res. 2001;20:227–253. doi: 10.1016/s1350-9462(00)00023-9. [DOI] [PubMed] [Google Scholar]
- 5.McConnell V., Silvestri G. Age-related macular degeneration. Ulster Med J. 2005;74:82–92. [PMC free article] [PubMed] [Google Scholar]
- 6.Hayreh S.S., Servais G.E., Virdi P.S. Macular lesions in malignant arterial hypertension. Ophthalmologica. 1989;198:230–246. doi: 10.1159/000310001. [DOI] [PubMed] [Google Scholar]
- 7.Hyman L., Schachat A.P., He Q., Leske M.C. Age-Related Macular Degeneration Risk Factors Study Group: Hypertension, cardiovascular disease, and age-related macular degeneration. Arch Ophthalmol. 2000;118:351–358. doi: 10.1001/archopht.118.3.351. [DOI] [PubMed] [Google Scholar]
- 8.Jonas J.B., Hayreh S.S., Martus P. Influence of arterial hypertension and diet-induced atherosclerosis on macular drusen. Graefes Arch Clin Exp Ophthalmol. 2003;241:125–134. doi: 10.1007/s00417-002-0615-3. [DOI] [PubMed] [Google Scholar]
- 9.Klein R., Klein B.E., Tomany S.C., Cruickshanks K.J. The association of cardiovascular disease with the long-term incidence of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology. 2003;110:636–643. doi: 10.1016/S0161-6420(02)01448-3. [DOI] [PubMed] [Google Scholar]
- 10.Lee M.A., Bohm M., Paul M., Ganten D. Tissue renin-angiotensin systems: their role in cardiovascular disease. Circulation. 1993;87:IV7–IV13. [PubMed] [Google Scholar]
- 11.Taylor W.R. Hypertensive vascular disease and inflammation: mechanical and humoral mechanisms. Curr Hypertens Rep. 1999;1:96–101. doi: 10.1007/s11906-999-0079-5. [DOI] [PubMed] [Google Scholar]
- 12.Berka J.L., Stubbs A.J., Wang D.Z., DiNicolantonio R., Alcorn D., Campbell D.J., Skinner S.L. Renin-containing Muller cells of the retina display endocrine features. Invest Ophthalmol Vis Sci. 1995;36:1450–1458. [PubMed] [Google Scholar]
- 13.Kohler K., Wheeler-Schilling T., Jurklies B., Guenther E., Zrenner Angiotensin II in the rabbit retina. Vis Neurosci. 1997;14:63–71. doi: 10.1017/s0952523800008762. [DOI] [PubMed] [Google Scholar]
- 14.Wagner J., Jan Danser A.H., Derkx F.H., de Jong T.V., Paul M., Mullins J.J., Schalekamp M.A., Ganten D. Demonstration of renin mRNA, angiotensinogen mRNA, and angiotensin converting enzyme mRNA expression in the human eye: evidence for an intraocular renin-angiotensin system. Br J Ophthalmol. 1996;80:159–163. doi: 10.1136/bjo.80.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wheeler-Schilling T.H., Kohler K., Sautter M., Guenther E. Angiotensin II receptor subtype gene expression and cellular localization in the retina and non-neuronal ocular tissues of the rat. Eur J Neurosci. 1999;11:3387–3394. doi: 10.1046/j.1460-9568.1999.00787.x. [DOI] [PubMed] [Google Scholar]
- 16.Savaskan E., Loffler K.U., Meier F., Muller-Spahn F., Flammer J., Meyer P. Immunohistochemical localization of angiotensin-converting enzyme, angiotensin II and AT1 receptor in human ocular tissues. Ophthalmic Res. 2004;36:312–320. doi: 10.1159/000081633. [DOI] [PubMed] [Google Scholar]
- 17.Senanayake P., Drazba J., Shadrach K., Milsted A., Rungger-Brandle E., Nishiyama K., Miura S., Karnik S., Sears J.E., Hollyfield J.G. Angiotensin II and its receptor subtypes in the human retina. Invest Ophthalmol Vis Sci. 2007;48:3301–3311. doi: 10.1167/iovs.06-1024. [DOI] [PubMed] [Google Scholar]
- 18.Striker G.E., Praddaude F., Alcazar O., Cousins S.W., Marin-Castano M.E. Regulation of angiotensin II receptors and extracellular matrix turnover in human retinal pigment epithelium: role of angiotensin II. Am J Physiol Cell Physiol. 2008;295:C1633–C1646. doi: 10.1152/ajpcell.00092.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burns R.P., Feeney-Burns L. Clinico-morphologic correlations of drusen of Bruch's membrane. Trans Am Ophthalmol Soc. 1980;78:206–225. [PMC free article] [PubMed] [Google Scholar]
- 20.Ishibashi T., Patterson R., Ohnishi Y., Inomata H., Ryan S.J. Formation of drusen in the human eye. Am J Ophthalmol. 1986;15:342–353. doi: 10.1016/0002-9394(86)90830-5. 101. [DOI] [PubMed] [Google Scholar]
- 21.Zhu Z.R., Goodnight R., Nishimura T., Sorgente N., Ogden T.E., Ryan S.J. Experimental changes resembling the pathology of drusen in Bruch's membrane in the rabbit. Curr Eye Res. 1998;7:581–592. doi: 10.3109/02713688809031814. [DOI] [PubMed] [Google Scholar]
- 22.Atkison S.J., Petterson M.L., Butler M.J., Murphy G. Membrane type 1 matrix metalloproteinase and gelatinase A synergistically degrade type I collagen in a cell model. FEBS Lett. 2001;491:222–226. doi: 10.1016/s0014-5793(01)02204-9. [DOI] [PubMed] [Google Scholar]
- 23.Deryugina E.I., Bourdon M.A., Reisfeld R.A., Strongin A. Remodeling of collagen matrix by human tumor cells requires activation and cell surface association of matrix metalloproteinase-2. Cancer Res. 1989;58:3743–3750. [PubMed] [Google Scholar]
- 24.Zhuge Y., Xu J. Rac1 mediates type I collagen-dependent MMP-2 activation: role in cell invasion across collagen barrier. J Biol Chem. 2001;276:16248–16256. doi: 10.1074/jbc.m010190200. [DOI] [PubMed] [Google Scholar]
- 25.Ebihara I., Nakamura T., Tomino Y., Koide H. Effect of a specific endothelin receptor A antagonist and an angiotensin-converting enzyme inhibitor on glomerular mRNA levels for extracellular matrix components, metalloproteinases (MMP) and a tissue inhibitor of MMP in aminonucleoside nephrosis. Nephrol Dial Transplant. 1997;12:1001–1006. doi: 10.1093/ndt/12.5.1001. [DOI] [PubMed] [Google Scholar]
- 26.Praddaude F., Cousins S.W., Pecher C., Marin-Castano M.E. Angiotensin II–induced hypertension regulates AT1 receptor subtypes and extracellular matrix turnover in mouse retinal pigment epithelium. Exp Eye Res. 2009;89:109–118. doi: 10.1016/j.exer.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crabos M., Roth M., Hahn A.W., Erne P. Characterization of angiotensin II receptors in cultured adult rat cardiac fibroblasts: coupling to signaling systems and gene expression. J Clin Invest. 1994;93:2372–2378. doi: 10.1172/JCI117243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwami K., Ashizawa N., Do Y.S., Graf K., Hsueh W.A. Comparison of ANG II with other growth factors on Egr-1 and matrix gene expression in cardiac fibroblasts. Am J Physiol. 1996;270:H2100–H2107. doi: 10.1152/ajpheart.1996.270.6.H2100. [DOI] [PubMed] [Google Scholar]
- 29.Kato H., Suzuki H., Tajima S., Ogata Y., Tominaga T., Sato A., Saruta T. Angiotensin II stimulates collagen synthesis in cultured vascular smooth muscle cells. J Hypertens. 1991;9:17–22. [PubMed] [Google Scholar]
- 30.Kim S., Iwao H. Molecular and cellular mechanisms of angiotensin II–mediated cardiovascular and renal diseases. Pharmacol Rev. 2000;52:11–34. [PubMed] [Google Scholar]
- 31.Guo H., Zucker S., Gordon M.K., Toole B.P., Biswas C. Stimulation of matrix metalloproteinase production by recombinant extracellular matrix metalloproteinase inducer from transfected Chinese hamster ovary cells. J Biol Chem. 1997;272:24–27. [PubMed] [Google Scholar]
- 32.Kanekura T., Chen X., Kanzaki T. Basigin (CD147) is expressed on melanoma cells and induces tumor cell invasion by stimulating production of matrix metalloproteinases by fibroblasts. Int J Cancer. 2002;99:520–528. doi: 10.1002/ijc.10390. [DOI] [PubMed] [Google Scholar]
- 33.Kataoka H., DeCastro R., Zucker S., Biswas C. Tumor cell–derived collagenase-stimulatory factor increases expression of interstitial collagenase, stromelysin, and 72-kDa gelatinase. Cancer Res. 1993;53:3154–3158. [PubMed] [Google Scholar]
- 34.Sameshima T., Nabeshima K., Toole B.P., Yokogami K., Okada Y., Goya T., Koono M., Wakisaka S. Glioma cell extracellular matrix metalloproteinase inducer (EMMPRIN) (CD147) stimulates production of membrane-type matrix metalloproteinases and activated gelatinase A in co-cultures with brain-derived fibroblasts. Cancer Lett. 2000;157:177–184. doi: 10.1016/s0304-3835(00)00485-7. [DOI] [PubMed] [Google Scholar]
- 35.Alcazar O., Hawkridge A.M., Collier T.S., Cousins S.W., Bhattacharya S.K., Muddiman D.C., Marin-Castano M.E. Proteomics characterization of cell membrane blebs in human retinal pigment epithelium cells. Mol Cell Proteomics. 2009;8:2201–2211. doi: 10.1074/mcp.M900203-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ichijo H., Nishida E., Irie K., ten Dijke P., Saitoh M., Moriguchi T., Takagi M., Matsumoto K., Miyazono G., Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 37.Sugden P.H., Clerk A. Regulation of the ERK subgroup of MAP kinase cascades through G protein–coupled receptors. Cell Signal. 1997;9:337–351. doi: 10.1016/s0898-6568(96)00191-x. [DOI] [PubMed] [Google Scholar]
- 38.Taniyama Y., Ushio-Fukai M., Hitomi H., Rocic P., Kingsley M.J., Pfahnl C., Weber D.S., Alexander R.W., Griendling K.K. Role of p38 MAPK and MAPKAPK-2 in angiotensin II–induced Akt activation in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2004;287:C494–C499. doi: 10.1152/ajpcell.00439.2003. [DOI] [PubMed] [Google Scholar]
- 39.Chen Z., Gibson T.B., Robinson F., Silvestro L., Pearson G., Xu B., Wright A., Vanderbilt C., Cobb M.H. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 40.Lewis T.S., Shapiro P.S., Ahn N.G. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 41.Pearson G., Robinson F., Beers Gibson T., Xu B.E., Karandikar M., Berman K., Cobb M.H. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 42.Makino N., Sugano M., Otsuka S., Hata T. Molecular mechanism of angiotensin II type 1 and type 2 receptors in cardiac hypertrophy of spontaneously hypertensive rats. Hypertension. 1997;30:796–802. doi: 10.1161/01.hyp.30.4.796. [DOI] [PubMed] [Google Scholar]
- 43.Gigante B., Piras O., de Paolis P., Porcellini A., Natale A., Volpe M. Role of angiotensin II AT2-subtype receptors in the blood pressure–lowering effect of losartan in salt-restricted rats. J Hypertens. 1998;16:2039–2043. doi: 10.1097/00004872-199816121-00027. [DOI] [PubMed] [Google Scholar]
- 44.Thai H., Wollmuth J., Goldman S., Gaballa M. Angiotensin subtype 1 receptor (TA1) blockade improves vasorelaxation in heart failure by up-regulation of endothelial nitric-oxide synthase via activation of the AT2 receptor. J Pharmacol Exp Ther. 2003;370:1171–1178. doi: 10.1124/jpet.103.054916. [DOI] [PubMed] [Google Scholar]
- 45.Banes-Berceli A.K., Ketsawatsomkron P., Ogbi S., Patel B., Pollock C.M., Marrero G.A. Angiotensin II and endothelin-1 augment the vascular complications of diabetes via JAK2 activation. Am J Physiol Heart Circ Physiol. 2007;293:H1291–H1299. doi: 10.1152/ajpheart.00181.2007. [DOI] [PubMed] [Google Scholar]
- 46.Cousins S.W., Marin-Castaño M.E., Espinosa-Heidmann D.G., Alexandridou A., Striker L., Elliot S. Female gender, estrogen loss, and sub-RPE deposit formation in aged mice. Invest Ophthalmol Vis Sci. 2003;44:50–59. doi: 10.1167/iovs.02-0285. [DOI] [PubMed] [Google Scholar]
- 47.Marin-Castaño M.E., Elliot S.J., Potier M., Karl M., Striker L.J., Striker G.E., Csaky K.G., Cousins S.W. Regulation of estrogen receptors and MMP-2 expression by estrogens in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2003;44:50–59. doi: 10.1167/iovs.01-1276. [DOI] [PubMed] [Google Scholar]
- 48.Wang M., Takagi G., Asai K., Resuello R.G., Natividad F.F., Vatner D.E., Vatner S.F., Lakatta E.G. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension. 2002;41:1308–1316. doi: 10.1161/01.HYP.0000073843.56046.45. [DOI] [PubMed] [Google Scholar]
- 49.Strongin A.Y., Collier I., Bannikov G., Marmer B.L., Grant G.A., Goldberg G.I. Mechanism of cell surface activation of 72-kDa type IV collagenase: isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- 50.Itoh Y., Takamura A., Ito N., Maru Y., Sato H., Suenaga N., Aoki T., Seiki M. Homophilic complex formation of MT1-MMP facilitates proMMP-2 activation on the cell surface and promotes tumor cell invasion. EMBO J. 2002;20:4782–4793. doi: 10.1093/emboj/20.17.4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abdelsalam A., Del Priore L., Zarbin M.A. Drusen in age-related macular degeneration: pathogenesis, natural course, and laser photocoagulation–induced regression. Surv Ophthalmol. 1999;44:1–29. doi: 10.1016/s0039-6257(99)00072-7. (review) [DOI] [PubMed] [Google Scholar]
- 52.Nagai N., Oike Y., Izumi-Nagai K., Urano T., Kubota Y., Noda K., Ozawa Y., Inoue M., Tsubota K., Suda T., Ishida S. Angiotensin II type 1 receptor–mediated inflammation is required for choroidal neovascularization. Arterioscler Thromb Vasc Biol. 2006;26:2252–2259. doi: 10.1161/01.ATV.0000240050.15321.fe. [DOI] [PubMed] [Google Scholar]
- 53.Lambert V., Wielockx B., Munaut C., Galopin C., Jost M., Itoh T., Werb Z., Baker A., Libert C., Krell H.W., Foidart J.M., Noel A., Rakic J.M. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. FASEB J. 2003;17:2290–2292. doi: 10.1096/fj.03-0113fje. [DOI] [PubMed] [Google Scholar]
- 54.Plantner J.J., Jiang C., Smine A. Increase in interphotoreceptor matrix gelatinase A (MMP-2) associated with age-related macular degeneration. Exp Eye Res. 1998;67:637–645. doi: 10.1006/exer.1998.0552. [DOI] [PubMed] [Google Scholar]
- 55.Ryder J.W., Fahlman R., Wallberg-Henriksson H., Alessi D.R., Krook A., Zierath J.R. Effect of contraction on mitogen-activated protein kinase signal transduction in skeletal muscle: involvement of the mitogen- and stress-activated protein kinase 1. J Biol Chem. 2000;275:1457–1462. doi: 10.1074/jbc.275.2.1457. [DOI] [PubMed] [Google Scholar]
- 56.Eguchi S., Matsumoto T., Motley E.D., Utsunomiya H., Inagami T. Identification of an essential signaling cascade for mitogen-activated protein kinase activation by angiotensin II in cultured rat vascular smooth muscle cells: possible requirement of Gq-mediated p21ras activation coupled to a Ca2+/calmodulin-sensitive tyrosine kinase. J Biol Chem. 1986;271:14169–14175. doi: 10.1074/jbc.271.24.14169. [DOI] [PubMed] [Google Scholar]
- 57.Fischer T.A., Singh K., O'Hara D.S., Kaye D.M., Kelly R.A. Role of AT1 and AT2 receptors in regulation of MAPKs and MKP-1 by ANG II in adult cardiac myocytes. Am J Physiol. 1998;275(3 Pt 2):H906–H916. doi: 10.1152/ajpheart.1998.275.3.H906. [DOI] [PubMed] [Google Scholar]
- 58.Terada Y., Tomita K., Homma M.K., Nonoguchi H., Yang T., Yamada T., Yuasa Y., Krebs E.G., Marumo F. Sequential activation of MAP kinase cascade by angiotensin II in opossum kidney cells. Kidney Int. 1995;48:1801–1809. doi: 10.1038/ki.1995.478. [DOI] [PubMed] [Google Scholar]
- 59.Xu Q., Liu Y., Gorospe M., Udelsman R., Holbrook N.J. Acute hypertension activates mitogen-activated protein kinases in arterial wall. J Clin Invest. 1996;97:508–514. doi: 10.1172/JCI118442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pellieux C., Sauthier T., Aubert J.F., Brunner H.R., Pedrazzini T. Angiotensin II–induced cardiac hypertrophy is associated with different mitogen-activated protein kinase activation in normotensive and hypertensive mice. J Hypertens. 2000;18:1307–1317. doi: 10.1097/00004872-200018090-00017. [DOI] [PubMed] [Google Scholar]
- 61.Kudoh S., Komuro I., Mizuno T., Yamazaki T., Zou Y., Shiojima I., Takekoshi N., Yazaki Y. Angiotensin II stimulates c-Jun NH2-terminal kinase in cultured cardiac myocytes of neonatal rats. Circ Res. 1997;80:139–146. doi: 10.1161/01.res.80.1.139. [DOI] [PubMed] [Google Scholar]
- 62.Force T., Bonventre J.V. Growth factors and mitogen-activated protein kinases. Hypertension. 1998;31:152–161. doi: 10.1161/01.hyp.31.1.152. [DOI] [PubMed] [Google Scholar]
- 63.Zou Y., Komuro I., Yamazaki T., Kudoh S., Aikawa R., Zhu W., Shiojima I., Hiroi Y., Tobe K., Kadowaki T., Yazaki Y. Cell type–specific angiotensin II–evoked signal transduction pathways: critical roles of Gbetagamma subunit, Src family, and Ras in cardiac fibroblasts (review) Circ Res. 1998;82:337–345. doi: 10.1161/01.res.82.3.337. [DOI] [PubMed] [Google Scholar]
- 64.Schmitz U., Ishida T., Ishida M., Surapisitchat J., Hasham M.I., Pelech S., Berk B.C. Angiotensin II stimulates p21-activated kinase in vascular smooth muscle cells: role in activation of JNK. Circ Res. 1998;82:1272–1278. doi: 10.1161/01.res.82.12.1272. [DOI] [PubMed] [Google Scholar]
- 65.Zohn I.E., Yu H., Li X., Cox A.D., Earp H.S. Angiotensin II stimulates calcium-dependent activation of c-Jun N-terminal kinase. Mol Cell Biol. 1995;15:6160–6168. doi: 10.1128/mcb.15.11.6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mukoyama M., Nakajima M., Horiuchi M., Sasamura H., Pratt R.E., Dzau V.J. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J Biol Chem. 1993;268:24539–24542. [PubMed] [Google Scholar]
- 67.Murphy T.J., Alexander R.V., Griendling K.K., Runge M.S., Bernstein K.E. Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature. 1991;351:233–236. doi: 10.1038/351233a0. [DOI] [PubMed] [Google Scholar]
- 68.D'Amore A., Black M.J., Thomas W.G. The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor–mediated hypertrophy. Hypertension. 2005;46:1347–1354. doi: 10.1161/01.HYP.0000193504.51489.cf. [DOI] [PubMed] [Google Scholar]
- 69.Landon E.J., Inagami T. Beyond the G protein: the saga of the type 2 angiotensin II receptor. Arterioscler Thromb Vasc Biol. 2005;25:15–16. doi: 10.1161/01.ATV.0000153047.93274.5c. [DOI] [PubMed] [Google Scholar]
- 70.Mehta P.K., Griendling K.K. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 71.Lambert V., Wielockx B., Munaut C., Galopin C., Jost M., Itoh T., Werb Z., Baker A., Libert C., Krell H.W., Foidart J.M., Noel A., Rakic J.M. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. FASEB J. 2003;17:2290–2292. doi: 10.1096/fj.03-0113fje. [DOI] [PubMed] [Google Scholar]
- 72.Plantner J.J., Jiang C., Smine A. Increase in interphotoreceptor matrix gelatinase A (MMP-2) associated with age-related macular degeneration. Exp Eye Res. 1998;67:637–645. doi: 10.1006/exer.1998.0552. [DOI] [PubMed] [Google Scholar]
- 73.Strongin A.Y., Collier I., Bannikov G., Marmer B.L., Grant G.A., Goldberg G.I. Mechanism of cell surface activation of 72-kDa type IV collagenase: isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- 74.Itoh Y., Takamura A., Ito N., Maru Y., Sato H., Suenaga N., Aoki T., Seiki M. Homophilic complex formation of MT1-MMP facilitates proMMP-2 activation on the cell surface and promotes tumor cell invasion. EMBO J. 2002;20:4782–4793. doi: 10.1093/emboj/20.17.4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caudroy S., Polette M., Tournier J.M., Burlet H., Toole B., Zucker S., Birembaut P. Expression of the extracellular matrix metalloproteinase inducer (EMMPRIN) and the matrix metalloproteinase-2 in bronchopulmonary and breast lesions. J Histochem Cytochem. 1999;47:1575–1580. doi: 10.1177/002215549904701209. [DOI] [PubMed] [Google Scholar]
- 76.Jimenez E., Perez de la Blanca E., Urso L., Gonzalez I., Salas J., Montiel M. Angiotensin II induces MMP 2 activity via FAK/JNK pathway in human endothelial cells. Biochem Biophys Res Commun. 2009;380:769–774. doi: 10.1016/j.bbrc.2009.01.142. [DOI] [PubMed] [Google Scholar]
- 77.Saygili E., Rana O.R., Meyer C., Gemein C., Andrzejewski M.G., Ludwig A., Weber C., Schotten U., Kruttgen A., Weis J., Schwinger R.H., Mischke K., Rassaf T., Kelm M., Schauerte P. The angiotensin-calcineurin-NFAT pathway mediates stretch-induced up-regulation of matrix metalloproteinases-2/-9 in atrial myocytes. Basic Res Cardiol. 2009;104:435–438. doi: 10.1007/s00395-008-0772-6. [Epub ahead of press January 15, 2009] [DOI] [PubMed] [Google Scholar]
- 78.Chen X.F., Wang J.A., Hou J., Gui C., Tang L.J., Chen Q.X., Xie X.J., Jiang J., Cai F.J., Chen H.S., Lu H.S., Chen H. Extracellular matrix metalloproteinase inducer (EMMPRIN) is present in smooth muscle cells of human aneurysmal aorta and is induced by angiotensin II in vitro. Clin Sci (Lond) 2009;116:819–826. doi: 10.1042/CS20080235. [DOI] [PubMed] [Google Scholar]
- 79.Chen G., Hitomi M., Han J., Stacey D.W. The p38 pathway provides negative feedback for Ras proliferative signaling. J Biol Chem. 2000;275:38973–38980. doi: 10.1074/jbc.M002856200. [DOI] [PubMed] [Google Scholar]
- 80.Wang Z., Yang H., Tachado S.D., Capo-Aponte J.E., Bildin V.N., Koziel H., Reinach P.S. Phosphatase-mediated crosstalk control of ErK and p38 MAPK signaling in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47:5267–5275. doi: 10.1167/iovs.06-0642. [DOI] [PubMed] [Google Scholar]
- 81.Kogkopoulou O., Tzakos E., Mavrothalassitis G., Baldari C.T., Paliogianni F., Young H.A., Thyphronitis G. Conditional up-regulation of IL-2 production by p38 MAPK inactivation is mediated by increased Erk1/2 activity. J Leukoc Biol. 2006;79:1052–1060. doi: 10.1189/jlb.0705418. [DOI] [PubMed] [Google Scholar]
- 82.Liu Q., Hofmann P.A. Protein phosphatase 2A-mediated cross-talk between p38 MAPK and ErK in apoptosis of cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H2204–H2212. doi: 10.1152/ajpheart.01050.2003. [DOI] [PubMed] [Google Scholar]
- 83.Pons M., Cousins S.W., Csaky K.G., Striker G., Marin-Castaño M.E. Cigarette smoke–related hydroquinone induces filamentous actin reorganization and heat shock protein 27 phosphorylation through p38 and extracellular signal-regulated kinase 1/2 in retinal pigment epithelium: implications for age-related macular degeneration. Am J Pathol. 2010;177:1198–1213. doi: 10.2353/ajpath.2010.091108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Keyse S.M. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 85.Zhang H., Shi X., Hampong M., Blanis L., Pelech S. Stress-induced inhibition of ErK1 and ErK2 by direct interaction with p38 MAP kinase. J Biol Chem. 2001;276:6905–6908. doi: 10.1074/jbc.C000917200. [DOI] [PubMed] [Google Scholar]