

Abstract
Obesity and hypertension, major risk factors for the metabolic syndrome, render individuals susceptible to an increased risk of cardiovascular complications, such as adverse cardiac remodeling and heart failure. There has been much investigation into the role that an increase in the renin-angiotensin-aldosterone system (RAAS) plays in the pathogenesis of metabolic syndrome and in particular, how aldosterone mediates left ventricular hypertrophy and increased cardiac fibrosis via its interaction with the mineralocorticoid receptor (MR). Here, we review the pertinent findings that link obesity with elevated aldosterone and the development of cardiac hypertrophy and fibrosis associated with the metabolic syndrome. These studies illustrate a complex cross-talk between adipose tissue, the heart, and the adrenal cortex. Furthermore, we discuss findings from our laboratory that suggest that cardiac hypertrophy and fibrosis in the metabolic syndrome may involve cross-talk between aldosterone and adipokines (such as adiponectin).
1. Introduction
The metabolic syndrome is characterized by a collection of cardiovascular risk factors that include obesity, dyslipidemia, hypertension, and glucose intolerance/insulin resistance and together predispose individuals to an increased risk of heart disease, stroke and diabetes [1, 2]. The increased prevalence of obesity and the metabolic syndrome portends a greater risk of cardiovascular disease such as heart failure and premature death [3–6]. Nearly 70 million adults in the USA are obese (defined as a body mass index (BMI) ≥30 kg/m2) [7, 8]. Although the pathogenesis of the metabolic syndrome has yet to be fully elucidated, increasing evidence has linked the renin-angiotensin-aldosterone system (RAAS) with the associated risk factors, including obesity and hypertension [9–11]. Elevated plasma aldosterone levels are found in patients with the metabolic syndrome [12], in resistant hypertension [13, 14], and are associated with the development of left ventricular (LV) hypertrophy (LVH) and increased cardiac fibrosis [13, 15, 16]. In addition, elevated RAAS activity is evident in patients with congestive heart failure [17]. Importantly, patients with obesity, hypertension, or diabetes mellitus are prone to diastolic heart failure which may be characterized by LVH. Profibrotic and proinflammatory effects observed in heart failure patients have been attributed to elevated angiotensin II (Ang-II) levels [18, 19]; however, it is now believed that an elevation in plasma aldosterone may also be in part responsible for these events [17]. This paper focuses on the link between obesity, aldosterone, and cardiac remodeling associated with the metabolic syndrome.
2. Aldosterone Production
Aldosterone is primarily synthesized in the zona glomerulosa of the adrenal cortex and requires the coordinated activity of several enzymes including that of aldosterone synthase (CYP11B2), which mediates deoxycorticosterone catalysis into aldosterone [20]. Angiotensin remains a primary stimulus for aldosterone secretion in response to blood volume depletion [21], but the production of aldosterone is also increased in response to adrenocorticotropin, potassium (K+), and other lipid soluble factors [9]. Normally, aldosterone can be extracted from the circulation and concentrated in the heart in both normal patients and those with chronic heart failure [22, 23]. Although controversial, there is conflicting evidence as to whether aldosterone may be synthesized in the heart under certain pathologic conditions and may be related to species, strain, and pathological conditions.
For example, rats subjected to myocardial infarction showed an increase in both cardiac aldosterone synthase (CYP11B2) and aldosterone levels [24], whereas aldosterone synthase mRNA was not found in Sprague Dawley rat hearts unless the rats were subjected to chronic Ang-II infusions [25]. Others have found an all together complete lack of aldosterone synthase expression in rats [26]. Aldosterone synthase expression has also been found to be elevated in humans with heart failure [27], and aldosterone levels in the coronary sinus were reported to be significantly lower than in the aortic root, in humans with congestive heart failure and dilated cardiomyopathy, suggesting that plasma aldosterone was extracted through the heart in these patients [28]. However, it should be noted that mRNA levels of aldosterone synthase are about 100 to 10,000 times lower in the human heart than in the human adrenal gland [29]. Similarly, others have reported only aldosterone synthase mRNA expression in the fetal heart [30]. Thus, the healthy adult human heart does not appear to express aldosterone synthase mRNA, whereas human fetal and failing adult hearts do [29].
2.1. Adipose Tissue Can Regulate Aldosterone Secretion
In addition to its role as a primary energy source, adipose tissue serves as a highly active endocrine organ, capable of secreting various factors (i.e., leptin, IL-6, TNF-α, and adiponectin) into the circulation. These factors can affect adrenal functions by influencing steroidogenesis [31, 32], which may ultimately affect the cardiovascular system [33]. There has been a reported association between obesity and hypertension via stimulation of aldosterone production and the subsequent renal sodium retention [34]. Likewise, adipocytes can mediate adrenal secretion of aldosterone via the release of the so-called “aldosterone-stimulating factors” [11, 13]. These secreted factors from visceral or subcutaneous adipose tissues reach the adrenal gland via the circulation to stimulate aldosterone release and thus function in an endocrine manner [35, 36]. Adipose tissue is also located on the adrenal gland and is therefore in the proximity of adrenocortical cells, indicating that adipocyte-derived factors may also act in a paracrine manner [37]. Early work by Goodfriend et al. showed in patients with visceral obesity that aldosterone levels increased independently of renin or K+ concentrations, suggesting the possibility that an adipocyte-derived factor was stimulating aldosterone secretion [38].
Obesity is associated with an increase in plasma fatty acid and enhanced oxidative stress [39, 40] and has led to the hypothesis that oxidized free fatty acids might lead to aldosterone secretion [41]. 12,13-epoxy-9-keto-10(trans)-octadecanoic acid (EKODE), a highly potent oxidized derivative of linoleic acid, was found to stimulate aldosteroneogenesis in rat adrenal glomerulosa cells [41, 42], and EKODE directly correlated with aldosterone levels in human subjects with an elevated BMI and in African Americans with hypertension [41]. These findings suggest a link between oxidative stress, aldosterone, and hypertension in obesity.
Ehrhart-Bornstein et al. also showed that human adipocytes secrete potent mineralocorticoid-releasing factor which can act directly on the adrenal gland to increase mineralocorticoid secretion [37]. Here, conditioned media from human derived adipocytes was added to human derived NCI-H295R adrenocortical cells, and these cells increased the secretion of mineralocorticoids including cortisol, dehydroepiandrosterone (DHEA), and most prominently, that of aldosterone. Although it was previously shown that Ang-II led to an increased aldosterone secretion through interaction with the Ang-II type-1 (AT1) receptor in NCI-H295R cells [43], AT1 antagonism with valsartan had no effect on aldosterone secretion in the fat cell-conditioned media, eliminating the effect of Ang-II. In addition to Ang-II, plasma K+ is reported to induce aldosterone secretion [44], but because K+ concentration in the cell media was not different among treatment groups [43], K+ was ruled out as a mediator of aldosterone release in this study [43]. Although the investigators were unable to identify or further characterize the identity of these adipose-derived mineralocorticoid-releasing factors, they were able to determine that these effects were not mediated by leptin, adiponectin, IL-6, or TNF-α. Interestingly, aldosterone may in turn promote adipogenesis through its interaction with the mineralocorticoid receptor (MR) [45], and although adipocytes are thought not to synthesize aldosterone, they express the MR [46]. However, recent data from the Touyz lab suggests that adipocytes may produce aldosterone [47]. Thus, it is plausible that aldosterone secretion mediated by adipose-derived factors or “aldosterone-stimulating factors” [11, 13] may promote further adipogenesis resulting in a vicious cycle linking hyperaldosteronism and obesity, compounding the adverse effects of the metabolic syndrome [9] (Figure 1).
Figure 1.
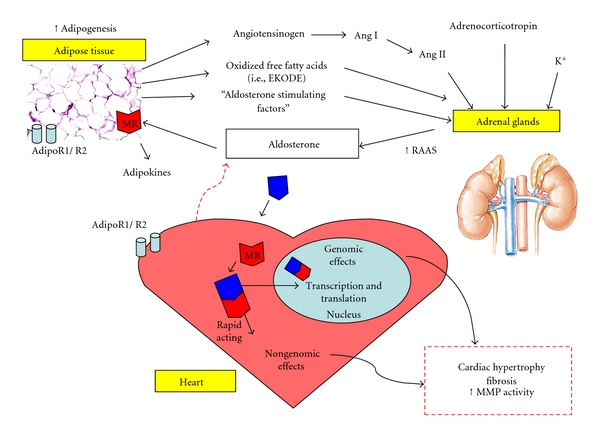
Proposed schematic representation of the interplay between adipose tissue, adrenal aldosterone production, and cardiac remodeling in the metabolic syndrome. Aldosterone production is increased in response to molecular factors including angiotensin, K+, and adrenocorticotropin, as well as a number of adipocyte-derived “aldosterone-stimulating factors.” Aldosterone mediates both genomic and nongenomic effects on the heart through its interaction with the mineralocorticoid receptor (MR), which may ultimately result in adverse cardiac remodeling. In addition, aldosterone can target adipocyte-specific MRs which may enhance adipogenesis. Furthermore, elevated aldosterone binding to adipocyte MRs may modulate adiponectin production. The presence of adiponectin receptors (AdipoR1 and AdipoR2) on the cardiomyocyte and that both aldosterone and adiponectin are synthesized in the failing human heart suggest a potential for crosstalk between adiponectin and aldosterone in pathological conditions.
2.2. Adiponectin and Aldosterone (Figure 1)
In obesity and hypertension, aldosterone levels are increased [48], whereas adiponectin levels are decreased [49, 50]. Evidence for adiponectin and aldosterone crosstalk exists. In diabetic db/db mice, inhibition of aldosterone binding to the MRs in adipose tissue modulated obesity-related changes in cardiac adiponectin expression [51]. Adipocytes express adiponectin receptors [52], MRs [46], Ang-II type 1 receptors, and angiotensinogen [53], suggesting an interaction between the RAAS and adiponectin in adipose tissue. Interestingly, whilst the MR is also present on cardiomyocytes [54], the adiponectin receptors, AdipoR1 and AdipoR2 [55], are also present. It is unknown whether crosstalk between adiponectin and aldosterone occurs at the receptor level in the heart and has not been explored.
The proinflammatory milieu induced by aldosterone contributes to the progression of hypertension to diastolic HF [56, 57]. We have shown that lack of adiponectin exacerbates the progression from hypertension to diastolic HF [57]. It is likely that the effect of adiponectin is not purely salutary but represents an important interaction in the pathophysiology of hypertension-related cardiac disease. Although it has been demonstrated that the heart synthesizes aldosterone [27], it is unclear whether the heart synthesizes physiological significant amounts of aldosterone [58]; however the MRs present on cardiomyocytes may be accessed by both aldosterone and cortisol [59, 60] (Figure 2). Although recent data (in abstract form) suggests that adipocytes may produce aldosterone in both 3T3-L1 adipocytes and obese db/db mice via activation of AT1-R [47], the majority of findings strongly suggest that adipocytes do not synthesize aldosterone. Although adipocytes [61] and cardiomyocytes, in pathological situations, synthesize adiponectin [51, 62], only cardiomyocytes [27] appear to synthesize aldosterone. On the other hand, it is also worth noting that adipose tissue secretes angiotensinogen [63–65] and accounts for increased plasma angiotensinogen levels found in obese humans [66]. Angiotensinogen has also been suggested as a link between obesity and the development of the metabolic syndrome [67].
Figure 2.
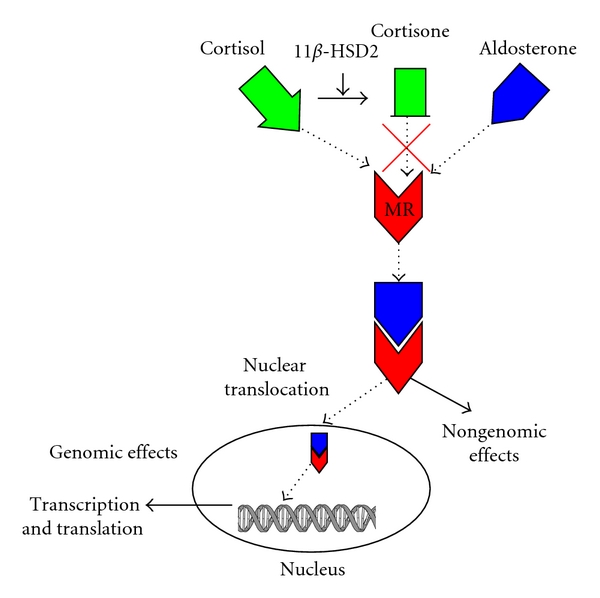
Cortisol and aldosterone bind the MR with equal affinity, but cortisol levels are significantly higher relative to aldosterone and, thus, occupy these receptors. The enzyme 11β-HSD2 acts to convert cortisol to cortisone, which is unable to bind the MR, thus allowing aldosterone to bind the available receptors. Once bound, the MR can translocate to the nucleus and engage DNA promoter sites to affect transcription and translation (genomic effects). Aldosterone can also mediate rapid nongenomic effects via the MR, which does not involve gene expression and protein synthesis.
Further evidence of an interaction between aldosterone and adiponectin is provided by the following: AdipoR1 and AdipoR2 have been found in the histologically normal human adrenal cortex and in aldosterone-producing adenomas [68]. In normal subjects, a high-salt diet suppresses RAAS and increases adiponectin levels. The decreased Ang-II levels are in proportion the decreased renin and aldosterone levels [69]. Whilst adiponectin levels are depressed in the metabolic syndrome where hypertension is prevalent, a high incidence of metabolic syndrome occurs in primary aldosteronism where hypertension is prevalent. Chronic infusion of aldosterone in mice decreased adiponectin production from fat in wild-type mice and was undetectable in adiponectin-deficient mice [57]. Similarly in vitro experiments in aldosterone-treated adipocytes decreased adiponectin transcript [51]. This may be due to direct (aldosterone excess) or indirect mechanisms [70] but the interaction with the receptors for aldosterone and adiponectin has not been elucidated in the metabolic syndrome.
3. Aldosterone Mediates Its Effects via the Mineralocorticoid Receptor (MR)
Aldosterone binds to the MR, which is a ligand-specific transcription factor belonging to the steroid super-family member of receptors [71]. Evolutionarily, the MR appeared earlier than the CYP11B2 enzyme [21, 72], suggesting that other possible ligands existed for the MR. For example, cortisol also serves as a potent ligand for the MR that competes with aldosterone with equal affinity [73]. However, because cortisol concentrations are at the very least tenfold and up to 1000-fold higher than that of aldosterone in the cardiomyocyte [17, 74] and at least 100-fold higher in the circulating plasma [75], it primarily will occupy the MR site. Once cortisol is converted to its inactive form, cortisone, by the actions of 11β hydroxysteroid dehydrogenase 2 (11β-HSD2), aldosterone is then permitted to occupy the MR [73] (Figure 2). Many tissues including the kidney, colon, endothelium, and vascular smooth muscle cells express 11β-HSD2, but there has been controversy about its expression in cardiac tissue [73]. While many argue that the heart expresses little if any of this specific enzyme [21, 59, 73, 76, 77], Lombes et al. has shown that the human heart does expressed 11β-HSD2 [54]. Additionally, aldosterone has been shown to be produced in the failing human heart by increased expression of CYP11B2 (aldosterone synthase), the enzyme catalyzing the terminal step in aldosterone synthesis [78].
Aldosterone may lead to the development of the metabolic syndrome via both genomic and nongenomic effects of the activated MR [79]. Increased expression of proinflammatory cytokines (TNF-α, MCP-1) and prothrombotic factor PAI-1 is inhibited by the selective antagonist (eplerenone) indicating a role for MR activation and increased inflammation in obese diabetic db/db mice [51]. Furthermore, MR activation can lead to the generation of increased cardiovascular oxidative stress [17, 80], which can contribute to increased cardiac hypertrophy and fibrosis associated with the metabolic syndrome [81]. In the clinical setting, there has been a link between MR activation, LVH and the metabolic syndrome in humans with primary aldosteronism [15].
Upon ligation, the MR will translocate to the nucleus where it regulates gene expression by binding to the hormone/steroid response element (HRE/SRE) [82] or negative response element (nSRE) [83] DNA sequences. In addition to its expression in kidney, colon, and brain [84, 85], the MR is found throughout the cells in the vasculature (i.e., smooth muscle cells and endothelial cells) [86], on cardiac myocytes [54, 85], and on cardiac fibroblasts, where aldosterone induces fibroblast growth through a Ras-Raf-MEK-ERK signaling cascade activated by the MR [87]. Because aldosterone can mediate detrimental effects in the heart by interactions with the MR, MR inhibition/antagonism is an attractive therapeutic strategy, a topic discussed in a later section.
Aldosterone can also mediate rapid (<5 min) nongenomic effects that do not require transcription or protein synthesis in order to mediate these effects [22, 88]. It was initially believed that these nongenomic effects occurred through a MR-independent mechanisms [21]. However, it is now generally accepted that these nongenomic effects may also be mediated in part through the MR [89]. Eplerenone, a selective MR antagonist, has been shown to inhibit nongenomic effects of aldosterone [90]. Aldosterone mediates nongenomic effects in many cells including vascular smooth muscle cells and arteriole endothelial cells, as well as in cardiomyocytes [91–93]. Effects on the cardiomyocyte include a rapid increase in Na+/K+/2Cl− cotransporter as well as a concomitant decreased Na+/K+ pump via a nongenomic protein kinase C-ε- (PKC-ε-) dependent mechanism [73, 93]. A reduced Na+/K+ pump activity would subsequently raise intracellular Ca2+ concentration, which is known to promote prohypertrophic signaling pathways in the cardiomyocyte [94]. In addition to a nongenomic rise in intracellular Ca2+, aldosterone rapidly increases intracellular Na+ concentration and cell volume in adult rat ventricular myocytes (ARVM) [95]. Our laboratory showed that, in isolated rat ventricular myocytes, aldosterone stimulated rapid extracellular-regulated kinase (ERK) phosphorylation that was not inhibited by RNA and protein synthesis inhibitors. In addition, this rapid increase in ERK1/2 phosphorylation was inhibited by spironolactone indicating that these nongenomic effects in cardiomyocytes were mediated by the MR [96]. Others have shown rapid activation by aldosterone of the prohypertrophic ERK/mitogen-activated protein kinase (MAPK) pathway, phosphorylation of Src, Jun N-terminal kinases (JNK), and the nuclear factor kappa B (NF-κB) signaling molecules [73, 89]. In addition, aldosterone induces a rapid decrease in PKC activity in neonatal rat ventricular myocytes [97]. Aldosterone also mediates rapid nongenomic effects on the electrophysiological properties of the heart (i.e., increased monophasic action potential) in patients with ventricular arrhythmias [98]. Interestingly, recent evidence suggests an interaction between nongenomic and genomic effects of aldosterone, specifically with the genomic effects being dependent upon the nongenomic [88, 89]. Non-genomic rapid ERK activation by aldosterone enhances aldosterone's genomic responses in vitro, likely due to the induction of the MR nuclear-cytoplasmic translocation [99]. Inhibition of early aldosterone-mediated PKCα activation also subsequently reduced MR transactivation [100]. Thus, the nongenomic-mediated activation of the prolonged genomic effects, coupled with the rapid nongenomic effects themselves, suggests a synergistic response to aldosterone [89].
4. Adverse Effects of Aldosterone on Cardiac Remodeling
RAAS activation in hypertension is associated with LVH and cardiac remodeling [101]. Initially LVH serves as an adaptive or compensatory response to a pathologic stimuli such as myocardial infarction [102], hypertension [103], and other causes of cardiovascular oxidative stress, for example, ischemia-reperfusion injury [104]. However, excessive LVH can result in cardiac dysfunction [105]. Early investigations focused on the actions of Ang-II and its receptors (AT-1 and AT-2) in the development of LVH [106, 107], whilst the potential for aldosterone to directly affect cardiac hypertrophy through the action of the MR was unclear. However experimental and clinical studies began to present data suggesting that aldosterone induced LVH and cardiac fibrosis [108–110]. These effects could be attenuated by MR antagonism with spironolactone or eplerenone indicating the involvement of the MR [108, 111–113]. However it was not determined whether these effects were directly due to aldosterone or to the secondary effects of aldosterone. Okoshi et al. was able to show that aldosterone directly induced cardiac hypertrophy and atrial natriuretic peptide (ANP) mRNA expression (a molecular marker of cardiac hypertrophy) in cultured neonatal rat ventricular myocytes (NRVMs) independently of Na+/K+ balance. However, MR antagonism with spironolactone revealed that this was dependent on binding to the MR [114]. Additionally, they found that this involved rapid activation (≈5 min) of the ERK1/2 and JNK MAPK cascade and the PKC pathway, suggesting nongenomic effects of aldosterone/MR association. In another study, transgenic mice overexpressing 11β-HSD2 in cardiomyocytes, which allows for enhanced occupation of the MR by aldosterone (and not cortisol), exhibited spontaneous LVH, fibrosis, and subsequent heart failure and premature death [59]. These effects were independent of blood pressure and were ameliorated by eplerenone, further confirming the role of the MR in direct aldosterone-mediated LVH.
More recent studies have further identified aldosterone's direct effects on the potential mechanism involved in the development of cardiac hypertrophy. NRVMs stimulated with aldosterone led to the interaction of the MR with p300, a GATA4 transcriptional coactivator involved in cardiac hypertrophy. The resultant ANP gene expression and increase in myocyte size were attenuated by spironolactone [115]. Others have shown that aldosterone-mediated hypertrophy involves a MR-p38 MAPK-dependent pathway, which leads to increased protein expression of cardiotropin-1, a prohypertrophic cytokine [116], as well as increased protein expression of IL-18 and subsequent hypertrophy, acting through a Rho/Rho-kinase and PPAR/NF-κB pathway [117]. Finally, aldosterone activation of the MR can increase cardiovascular reactive oxygen species (ROS) generation via NAD(P)H oxidase activity [96], which are known to induce cardiac hypertrophy and remodeling [118, 119].
Cardiac fibroblasts are also affected by aldosterone and further contribute to adverse hypertrophy and remodeling by excessive proliferation, matrix deposition, and increased matrix metalloproteinase (MMP) activity both in vitro and in vivo [96, 120–122]. Aldosterone-infused rats on a high-salt diet exhibited increased inflammation and fibrosis, which were prevented by MR inhibition [123, 124]. Increased expression of proinflammatory molecules by aldosterone acting via the MR may help to explain the potential mechanisms involved. For example, aldosterone increases expression of TGF-β via MR activation, which promotes fibrosis, tissue remodeling and production of matrix proteins [17]. Interestingly, aldosterone enhanced a TGF-β-mediated downregulation of inducible nitric oxide synthase (iNOS) and nitric oxide (NO) in a dose-dependent manner via the MR [125]. This appears contradictory given that iNOS is proinflammatory; however, long-term inhibition of iNOS may result in cardiac fibrosis suggesting that NO may play a role in preventing fibrosis [126]. In additional experiments, it was found that NO acts to quench ROS through peroxnitrite formation, which may protect against fibrosis [127]. Similarly, iNOS inhibition decreased the NO/ROS ratio, favoring the development of fibrosis. Furthermore, decreased endothelial NOS-derived NO has been shown previously to be central in the development of cardiovascular disorders [128] and LVH [129]. Aldosterone also increases the expression of plasminogen activator inhibitor (PAI)-1, a serine protease inhibitor which is important in fibrinolysis. PAI-1 is also involved in angiogenesis and atherogenesis [130]. PAI-1 is secreted by endothelial cells, vascular smooth muscle cells, hepatocytes, platelets, and adipocytes [131]. In obesity, the majority of the circulating PAI-1 is from adipose tissue [132]. Thus, not surprisingly, in addition to elevated aldosterone levels, obese subjects have higher levels of PAI-1 [133], as well as increased expression of endothelin-1 [134], that may contribute to enhanced cardiac fibrosis and collagen secretion.
In many cases, the MAPK intracellular signaling cascade plays an essential role in mediating these responses. Stockand and Meszaros found that aldosterone acting through the MR stimulates proliferation of adult rat cardiac fibroblasts in vitro through a Kirsten-(Ki-) RasA-ERK1/2-dependent pathway [87]. Additionally, p38 MAPK cascade is central in aldosterone-MR-mediated expression of connective tissue growth factor (CTGF) in embryonic rat cardiomyocytes [135].
The contribution of RAAS activation to cardiac fibrosis in the metabolic syndrome is evident not only in the conditions that constitute the metabolic syndrome, such as hypertension or insulin resistance, but also has been demonstrated in an elegant study by Matsui et al. [136]. They demonstrate that MR activation leads to cardiac fibrosis in an experimental model of metabolic syndrome. Metabolic syndrome was induced by a derivative of the spontaneously hypertensive rat (SHR) with leptin receptor deficiency. In this study, MR activation with salt caused LV diastolic dysfunction and cardiac fibrosis. Increased oxidative stress and enhanced MR activation accelerated the pathogenesis of salt-induced diastolic dysfunction in this model of metabolic syndrome [136].
5. Treatments/MR Antagonists
Several therapeutic strategies are being applied to alleviate the adverse effects of aldosterone in cardiovascular disorders associated with cardiac hypertrophy and fibrosis. While the use of angiotensin converting enzyme (ACE) inhibitors in heart failure patients reduces plasma aldosterone levels, this is only a temporary effect (referred to as “aldosterone escape”), as even maximal doses of ACE inhibitors are associated with increased Ang-II and aldosterone levels, and were accompanied by impaired exercise capacity [137, 138]. Two important clinical trials, the Randomized Aldosterone Evaluation Study (RALES) [139] and the Eplerenone Heart Failure and Survival Study (EPHESUS) [140], demonstrated that addition of MR antagonists to ACE inhibitors reduced mortality in patients with LV systolic dysfunction and postmyocardial infarction with heart failure, respectively. In addition to these findings, further experimental investigation into MR antagonism by such pharmacological agents as spironolactone and eplerenone that may prevent cardiac complications in patients has proved promising [15, 16] with ongoing trials being explored in patients with diastolic HF. In vitro studies demonstrated that spironolactone inhibited aldosterone-induced [14C]-phenylalanine incorporation in NRVMs [114] and aldosterone-induced [3H]-thymidine incorporation in adult rat cardiac fibroblasts [87]. Likewise, in vivo experimental studies showed that spironolactone prevented ventricular fibrosis [141]. Eplerenone also attenuated LVH in clinical trials and may be a more attractive therapy, because of its fewer side effects compared to spironolactone [137, 142]. Our laboratory has previously reported that eplerenone treatment in mice subjected to ascending aortic constriction (a model for chronic pressure overload) improved LVH, indirectly linking the involvement of aldosterone with chronic pressure overload [80]. It was noted that, because aldosterone-mediated LVH and fibrosis and was in part due to increased oxidative stress and inflammation, it is possible that the beneficial effects of eplerenone may be due to a reduction in oxidative stress [143, 144]. Additionally, aldosterone antagonists can work to downregulate MMP activity [96]. In the RALES study, spironolactone-treated patients had reduced matrix turnover and fibrosis, which has appeared to be the most important effect of spironolactone in the heart [137, 139].
RAAS blockade with angiotensin receptor blockers (ARBs) and ACE inhibition have all been shown to mitigate the adverse hemodynamic and remodeling effects in several animal models of obesity [145–148]. For example, losartan, an AT1 receptor antagonist, improved cardiac function via a PKB/Akt-dependent mechanism in a rat model of diet-induced obesity [145]. Similarly, candesartan, another ARB, when used in combination with pioglitazone, a PPAR-γ agonist, decreased inflammation, oxidative stress, and fibrosis in obese, type 2 diabetic mice [148]. ACE inhibition with captopril restored insulin signaling, improved fatty acid oxidation and glycolysis regulation, and improved energy status by reducing AMPK activity in hearts from obese ob/ob mice [146]. Enalapril, another ACE inhibitor, reduced blood pressure and improved autonomic dysfunction in obese ob/ob mice [147]. The effects seen with enalapril were similar to those seen after leptin replacement in these mice [147].
Other therapeutic strategies against the development of aldosterone-induced cardiac remodeling that target other potential pathways have been explored. Past work from our laboratory has looked at fenofibrate, a PPAR-α agonist, which has been reported to suppress NF-κB activity, macrophage recruitment, and the development of cardiac dysfunction [149–151]. Furthermore, fenofibrate inhibits ERK1/2 kinase phosphorylation and MMP activity in aldosterone-stimulated isolated ARVMs [152]. Further in vivo work has shown that fenofibrate decreases LVH, matrix turnover, and fibrosis, as well as improved LV chamber size and function [153].
Currently, there are several clinical trials underway exploring aldosterone inhibition in obesity. These include modulation of RAAS activity and lipid/carbohydrate metabolism in adipose and skeletal muscle tissue in obese patients with hypertension (http://www.clinicaltrials.gov/, NCT00498433). Also, investigators are exploring aldosterone inhibition in obesity specifically addressing endothelial and fibrinolytic dysfunction and measuring PAI-1 levels to determine if there is a reduction in the risk of stroke in obesity (http://www.clinicaltrials.gov/, NCT00608465).
6. Summary
Obesity and its association with other factors of the metabolic syndrome (specifically hypertension and insulin resistance) remain a significant and growing problem among the population. Adverse cardiac remodeling can further contribute to these cardiovascular complications, resulting in a vicious cycle that may ultimately lead to heart failure. Furthermore, an association between adipose tissue and aldosterone is evident, as adipocytes release factors (such as “aldosterone-stimulating factors” and adipokines) that directly or indirectly stimulate aldosterone secretion. Much work has explored the role that hypertension and the RAAS (in particular of Ang-II) has on adverse cardiac remodeling, as new insights have emerged about the direct effect of aldosterone on the MR (nongenomic and/or genomic). Whilst MR antagonists have been important therapeutic tools against cardiac hypertrophy and fibrosis, further understanding of the intricate relationship between obesity, aldosterone, and adverse cardiac remodeling may yield more effective therapeutic treatments.
Acknowledgments
E. E. Essick is supported by funding from the National Institutes of Health (NIH) T32HL007224, and F. Sam is supported by funding from the NIH HL079099, HL095891, and HL102631.
References
- 1.Kaplan NM. The deadly quartet. Upper-body obesity, glucose intolerance, hypertriglyceridemia, and hypertension. Archives of Internal Medicine. 1989;149(7):1514–1520. doi: 10.1001/archinte.149.7.1514. [DOI] [PubMed] [Google Scholar]
- 2.Mulè G, Cerasola G. The metabolic syndrome and its relationship to hypertensive target organ damage. Journal of Clinical Hypertension. 2006;8(3):195–201. doi: 10.1111/j.1524-6175.2006.04716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24(2):e13–e18. doi: 10.1161/01.ATV.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 4.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. The Lancet. 2005;365(9468):1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 5.Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care. 2005;28(7):1769–1778. doi: 10.2337/diacare.28.7.1769. [DOI] [PubMed] [Google Scholar]
- 6.Isomaa B, Almgren P, Tuomi T, et al. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24(4):683–689. doi: 10.2337/diacare.24.4.683. [DOI] [PubMed] [Google Scholar]
- 7.Ginsberg HN, Maccallum PR. The obesity, metabolic syndrome, and type 2 diabetes mellitus pandemic: part I. Increased cardiovascular disease risk and the importance of atherogenic dyslipidemia in persons with the metabolic syndrome and type 2 diabetes mellitus. Journal of the CardioMetabolic Syndrome. 2009;4(2):113–119. doi: 10.1111/j.1559-4572.2008.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park YW, Zhu S, Palaniappan L, Heshka S, Carnethon MR, Heymsfield SB. The metabolic syndrome: prevalence and associated risk factor findings in the US population from the Third National Health and Nutrition Examination Survey, 1988–1994. Archives of Internal Medicine. 2003;163(4):427–436. doi: 10.1001/archinte.163.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whaley-Connell A, Johnson MS, Sowers JR. Aldosterone: role in the cardiometabolic syndrome and resistant hypertension. Progress in Cardiovascular Diseases. 2010;52(5):401–409. doi: 10.1016/j.pcad.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bomback AS, Klemmer PJ. Interaction of aldosterone and extracellular volume in the pathogenesis of obesity-associated kidney disease: a narrative review. American Journal of Nephrology. 2009;30(2):140–146. doi: 10.1159/000209744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. Journal of the American Society of Nephrology. 2006;17(12):3438–3446. doi: 10.1681/ASN.2006080944. [DOI] [PubMed] [Google Scholar]
- 12.Bochud M, Nussberger J, Bovet P, et al. Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension. 2006;48(2):239–245. doi: 10.1161/01.HYP.0000231338.41548.fc. [DOI] [PubMed] [Google Scholar]
- 13.Calhoun DA, Sharma K. The role of aldosteronism in causing obesity-related cardiovascular risk. Cardiology Clinics. 2010;28(3):517–527. doi: 10.1016/j.ccl.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall JE. The renin-angiotensin system: renal actions and blood pressure regulation. Comprehensive Therapy. 1991;17(5):8–17. [PubMed] [Google Scholar]
- 15.Giacchetti G, Turchi F, Boscaro M, Ronconi V. Management of primary aldosteronism: its complications and their outcomes after treatment. Current Vascular Pharmacology. 2009;7(2):244–249. doi: 10.2174/157016109787455716. [DOI] [PubMed] [Google Scholar]
- 16.Marcy TR, Ripley TL. Aldosterone antagonists in the treatment of heart failure. American Journal of Health-System Pharmacy. 2006;63(1):49–58. doi: 10.2146/ajhp050041. [DOI] [PubMed] [Google Scholar]
- 17.Marney AM, Brown NJ. Aldosterone and end-organ damage. Clinical Science. 2007;113(5-6):267–278. doi: 10.1042/CS20070123. [DOI] [PubMed] [Google Scholar]
- 18.Xu J, Carretero OA, Liao T-D, et al. Local angiotensin II aggravates cardiac remodeling in hypertension. American Journal of Physiology. 2010;299(5):H1328–H1338. doi: 10.1152/ajpheart.00538.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paul M, Mehr AP, Kreutz R. Physiology of local renin-angiotensin systems. Physiological Reviews. 2006;86(3):747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 20.Mornet E, Dupont J, Vitek A, White PC. Characterization of two genes encoding human steroid 11β-hydroxylase (P-450(11β)) Journal of Biological Chemistry. 1989;264(35):20961–20967. [PubMed] [Google Scholar]
- 21.Funder JW. Minireview: aldosterone and mineralocorticoid receptors: past, present, and future. Endocrinology. 2010;151(11):5098–5102. doi: 10.1210/en.2010-0465. [DOI] [PubMed] [Google Scholar]
- 22.Shieh FK, Kotlyar E, Sam F. Aldosterone and cardiovascular remodelling: focus on myocardial failure. Journal of the Renin-Angiotensin-Aldosterone System. 2004;5(1):3–13. doi: 10.3317/jraas.2004.005. [DOI] [PubMed] [Google Scholar]
- 23.Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. Journal of the American College of Cardiology. 2000;36(3):838–844. doi: 10.1016/s0735-1097(00)00796-8. [DOI] [PubMed] [Google Scholar]
- 24.Xiu JC, Wu P, Xu JP, et al. Effects of long-term enalapril and losartan therapy of heart failure on cardiovascular aldosterone. Journal of Endocrinological Investigation. 2002;25(5):463–468. doi: 10.1007/BF03344039. [DOI] [PubMed] [Google Scholar]
- 25.Rudolph AE, Blasi ER, Delyani JA. Tissue-specific corticosteroidogenesis in the rat. Molecular and Cellular Endocrinology. 2000;165(1-2):221–224. doi: 10.1016/s0303-7207(00)00258-6. [DOI] [PubMed] [Google Scholar]
- 26.Ye P, Kenyon CJ, MacKenzie SM, et al. The aldosterone synthase (CYP11B2) and 11β-hydroxylase (CYP11B1) genes are not expressed in the rat heart. Endocrinology. 2005;146(12):5287–5293. doi: 10.1210/en.2005-0370. [DOI] [PubMed] [Google Scholar]
- 27.Young MJ, Clyne CD, Cole TJ, Funder JW. Cardiac steroidogenesis in the normal and failing heart. Journal of Clinical Endocrinology and Metabolism. 2001;86(11):5121–5126. doi: 10.1210/jcem.86.11.7925. [DOI] [PubMed] [Google Scholar]
- 28.Tsutamoto T, Wada A, Maeda K, et al. Transcardiac gradient of aldosterone before and after spironolactone in patients with congestive heart failure. Journal of Cardiovascular Pharmacology. 2003;41(supplement 1):S19–S22. [PubMed] [Google Scholar]
- 29.Gomez-Sanchez EP, Ahmad N, Romero DG, Gomez-Sanchez CE. Origin of aldosterone in the rat heart. Endocrinology. 2004;145(11):4796–4802. doi: 10.1210/en.2004-0295. [DOI] [PubMed] [Google Scholar]
- 30.Kayes-Wandover KM, White PC. Steroidogenic enzyme gene expression in the human heart. Journal of Clinical Endocrinology and Metabolism. 2000;85(7):2519–2525. doi: 10.1210/jcem.85.7.6663. [DOI] [PubMed] [Google Scholar]
- 31.Ehrhart-Bornstein M, Hinson JP, Bornstein SR, Scherbaum WA, Vinson GP. Intraadrenal interactions in the regulation of adrenocortical steroidogenesis. Endocrine Reviews. 1998;19(2):101–143. doi: 10.1210/edrv.19.2.0326. [DOI] [PubMed] [Google Scholar]
- 32.Marx C, Ehrhart-Bornstein M, Scherbaum WA, Bornstein SR. Regulation of adrenocortical function by cytokines—relevance for immune-endocrine interaction. Hormone and Metabolic Research. 1998;30(6-7):416–420. doi: 10.1055/s-2007-978907. [DOI] [PubMed] [Google Scholar]
- 33.Poulos SP, Hausman DB, Hausman GJ. The development and endocrine functions of adipose tissue. Molecular and Cellular Endocrinology. 2010;323(1):20–34. doi: 10.1016/j.mce.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Bogaert YE, Linas S. The role of obesity in the pathogenesis of hypertension. Nature Clinical Practice Nephrology. 2009;5(2):101–111. doi: 10.1038/ncpneph1022. [DOI] [PubMed] [Google Scholar]
- 35.Kim S, Moustaid-Moussa N. Secretory, endocrine and autocrine/paracrine function of the adipocyte. Journal of Nutrition. 2000;130(12):3110S–3115S. doi: 10.1093/jn/130.12.3110S. [DOI] [PubMed] [Google Scholar]
- 36.Trayhurn P, Beattie JH. Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proceedings of the Nutrition Society. 2001;60(3):329–339. doi: 10.1079/pns200194. [DOI] [PubMed] [Google Scholar]
- 37.Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A, et al. Human adipocytes secrete mineralocorticoid-releasing factors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(2):14211–14216. doi: 10.1073/pnas.2336140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodfriend TL, Egan BM, Kelley DE. Aldosterone in obesity. Endocrine Research. 1998;24(3-4):789–796. doi: 10.3109/07435809809032689. [DOI] [PubMed] [Google Scholar]
- 39.Keaney JF, Larson MG, Vasan RS, et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham study. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(3):434–439. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- 40.Surya Prabha P, Das UN, Koratkar R, Sagar PS, Ramesh G. Free radical generation, lipid peroxidation and essential fatty acids in uncontrolled essential hypertension. Prostaglandins Leukotrienes and Essential Fatty Acids. 1990;41(1):27–33. doi: 10.1016/0952-3278(90)90127-7. [DOI] [PubMed] [Google Scholar]
- 41.Goodfriend TL, Ball DL, Egan BM, Campbell WB, Nithipatikom K. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension. 2004;43(2):358–363. doi: 10.1161/01.HYP.0000113294.06704.64. [DOI] [PubMed] [Google Scholar]
- 42.Gardner HW, Crawford CG. Degradation of linoleic acid hydroperoxides by a cysteine. FeCl3 catalyst as a model for similar biochemical reactions. III. A novel product, trans-12,13-epoxy-11-oxo-trans-9-octadecenoic acid, from 13-L(S)-hydroperoxy-cis-9,trans-11-octadecadienoic acid. Biochimica et Biophysica Acta. 1981;665(1):126–133. doi: 10.1016/0005-2760(81)90240-x. [DOI] [PubMed] [Google Scholar]
- 43.Hilbers U, Peters J, Bornstein SR, et al. Local renin-angiotensin system is involved in K+-induced aldosterone secretion from human adrenocortical NCI-H295 cells. Hypertension. 1999;33(4):1025–1030. doi: 10.1161/01.hyp.33.4.1025. [DOI] [PubMed] [Google Scholar]
- 44.Quinn SJ, Williams GH. Regulation of aldosterone secretion. Annual Review of Physiology. 1988;50:409–426. doi: 10.1146/annurev.ph.50.030188.002205. [DOI] [PubMed] [Google Scholar]
- 45.Caprio M, Fève B, Claës A, Viengchareun S, Lombès M, Zennaro MC. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB Journal. 2007;21(9):2185–2194. doi: 10.1096/fj.06-7970com. [DOI] [PubMed] [Google Scholar]
- 46.Pascual-Le TL, Lombès M. The mineralocorticoid receptor: a journey exploring its diversity and specificity of action. Molecular Endocrinology. 2005;19(9):2211–2221. doi: 10.1210/me.2005-0089. [DOI] [PubMed] [Google Scholar]
- 47.Briones AM, Dinh Cat AN, Callera GE, et al. Production of aldosterone by adipocytes: implications for obesity and vascular function. Hypertension. 2010;56, article e73 [Google Scholar]
- 48.Calhoun DA. Aldosteronism and hypertension. Clinical Journal of the American Society of Nephrology. 2006;1(5):1039–1045. doi: 10.2215/CJN.01060306. [DOI] [PubMed] [Google Scholar]
- 49.Arita Y, Kihara S, Ouchi N, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochemical and Biophysical Research Communications. 1999;257(1):79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 50.Iwashima Y, Katsuya T, Ishikawa K, et al. Hypoadiponectinemia is an independent risk factor for hypertension. Hypertension. 2004;43(6):1318–1323. doi: 10.1161/01.HYP.0000129281.03801.4b. [DOI] [PubMed] [Google Scholar]
- 51.Guo C, Ricchiuti V, Lian BQ, et al. Mineralocorticoid receptor blockade reverses obesity-related changes in expression of adiponectin, peroxisome proliferator-activated receptor-γ, and proinflammatory adipokines. Circulation. 2008;117(17):2253–2261. doi: 10.1161/CIRCULATIONAHA.107.748640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beylot M, Pinteur C, Peroni O. Expression of the adiponectin receptors AdipoR1 and AdipoR2 in lean rats and in obese Zucker rats. Metabolism. 2006;55(3):396–401. doi: 10.1016/j.metabol.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 53.Saiki A, Ohira M, Endo K, et al. Circulating angiotensin II is associated with body fat accumulation and insulin resistance in obese subjects with type 2 diabetes mellitus. Metabolism. 2009;58(5):708–713. doi: 10.1016/j.metabol.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 54.Lombes M, Alfaidy N, Eugene E, Lessana A, Farman N, Bonvalet JP. Prerequisite for cardiac aldosterone action: mineralocorticoid receptor and 11β-hydroxysteroid dehydrogenase in the human heart. Circulation. 1995;92(2):175–182. doi: 10.1161/01.cir.92.2.175. [DOI] [PubMed] [Google Scholar]
- 55.Ding G, Qin Q, He N, et al. Adiponectin and its receptors are expressed in adult ventricular cardiomyocytes and upregulated by activation of peroxisome proliferator-activated receptor γ. Journal of Molecular and Cellular Cardiology. 2007;43(1):73–84. doi: 10.1016/j.yjmcc.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilson RM, De Silva DS, Sato K, Izumiya Y, Sam F. Effects of fixed-dose isosorbide dinitrate/hydralazine on diastolic function and exercise capacity in hypertension-induced diastolic heart failure. Hypertension. 2009;54(3):583–590. doi: 10.1161/HYPERTENSIONAHA.109.134932. [DOI] [PubMed] [Google Scholar]
- 57.Sam F, Duhaney TAS, Sato K, et al. Adiponectin deficiency, diastolic dysfunction, and diastolic heart failure. Endocrinology. 2010;151(1):322–331. doi: 10.1210/en.2009-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gomez-Sanchez CE, Foecking MF, Gomez-Sanchez EP. Aldosterone esters and the heart. American Journal of Hypertension. 2001;14(6) doi: 10.1016/s0895-7061(01)02089-1. [DOI] [PubMed] [Google Scholar]
- 59.Qin W, Rudolph AE, Bond BR, et al. Transgenic model of aldosterone-driven cardiac hypertrophy and heart failure. Circulation Research. 2003;93(1):69–76. doi: 10.1161/01.RES.0000080521.15238.E5. [DOI] [PubMed] [Google Scholar]
- 60.Mano A, Tatsumi T, Shiraishi J, et al. Aldosterone directly induces myocyte apoptosis through calcineurin-dependent pathways. Circulation. 2004;110(3):317–323. doi: 10.1161/01.CIR.0000135599.33787.CA. [DOI] [PubMed] [Google Scholar]
- 61.Ouchi N, Kihara S, Arita Y, et al. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 1999;100(25):2473–2476. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 62.Piñeiro R, Iglesias MJ, Gallego R, et al. Adiponectin is synthesized and secreted by human and murine cardiomyocytes. FEBS Letters. 2005;579(23):5163–5169. doi: 10.1016/j.febslet.2005.07.098. [DOI] [PubMed] [Google Scholar]
- 63.DeClercq V, Taylor C, Zahradka P. Adipose tissue: the link between obesity and cardiovascular disease. Cardiovascular and Hematological Disorders—Drug Targets. 2008;8(3):228–237. doi: 10.2174/187152908785849080. [DOI] [PubMed] [Google Scholar]
- 64.Gilardini L, Zulian A, Girola A, Redaelli G, Conti A, Invitti C. Predictors of the early impairment of renal disease in human obesity. International Journal of Obesity. 2010;34(2):287–294. doi: 10.1038/ijo.2009.227. [DOI] [PubMed] [Google Scholar]
- 65.Engeli S, Schling P, Gorzelniak K, et al. The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? International Journal of Biochemistry and Cell Biology. 2003;35(6):807–825. doi: 10.1016/s1357-2725(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 66.Yasue S, Masuzaki H, Okada S, et al. Adipose tissue-specific regulation of angiotensinogen in obese humans and mice: impact of nutritional status and adipocyte hypertrophy. American Journal of Hypertension. 2010;23(4):425–431. doi: 10.1038/ajh.2009.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zulet MA, Puchau B, Navarro C, Martí A, Martínez Hernández JA. Inflammatory biomarkers: the link between obesity and associated pathologies. Nutricion Hospitalaria. 2007;22(5):511–527. [PubMed] [Google Scholar]
- 68.Rossi GP, Sticchi D, Giuliani L, et al. Adiponectin receptor expression in the human adrenal cortex and aldosterone-producing adenomas. International Journal of Molecular Medicine. 2006;17(6):975–980. [PubMed] [Google Scholar]
- 69.Lely AT, Krikken JA, Bakker SJL, et al. Low dietary sodium and exogenous angiotensin II infusion decrease plasma adiponectin concentrations in healthy men. Journal of Clinical Endocrinology and Metabolism. 2007;92(5):1821–1826. doi: 10.1210/jc.2006-2092. [DOI] [PubMed] [Google Scholar]
- 70.Fallo F, Della Mea P, Sonino N, et al. Adiponectin and insulin sensitivity in primary aldosteronism. American Journal of Hypertension. 2007;20(8):855–861. doi: 10.1016/j.amjhyper.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 71.Funder JW. Aldosterone and mineralocorticoid receptors in the cardiovascular system. Progress in Cardiovascular Diseases. 2010;52(5):393–400. doi: 10.1016/j.pcad.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 72.Hu X, Funder JW. The evolution of mineralocorticoid receptors. Molecular Endocrinology. 2006;20(7):1471–1478. doi: 10.1210/me.2005-0247. [DOI] [PubMed] [Google Scholar]
- 73.Tirosh A, Garg R, Adler GK. Mineralocorticoid receptor antagonists and the metabolic syndrome. Current Hypertension Reports. 2010;12:252–257. doi: 10.1007/s11906-010-0126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mihailidou AS, Funder JW. Nongenomic effects of mineralocorticoid receptor activation in the cardiovascular system. Steroids. 2005;70(5–7):347–351. doi: 10.1016/j.steroids.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 75.Muller O, Pradervand S, Berger S, et al. Identification of corticosteroid-regulated genes in cardiomyocytes by serial analysis of gene expression. Genomics. 2007;89(3):370–377. doi: 10.1016/j.ygeno.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 76.Chai W, Danser AHJ. Why are mineralocorticoid receptor antagonists cardioprotective? Naunyn-Schmiedeberg’s Archives of Pharmacology. 2006;374(3):153–162. doi: 10.1007/s00210-006-0107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nagata K, Obata K, Xu J, et al. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and failure in low-aldosterone hypertensive rats. Hypertension. 2006;47(4):656–664. doi: 10.1161/01.HYP.0000203772.78696.67. [DOI] [PubMed] [Google Scholar]
- 78.Yoshimura M, Nakamura S, Ito T, et al. Expression of aldosterone synthase gene in failing human heart: quantitative analysis using modified real-time polymerase chain reaction. Journal of Clinical Endocrinology and Metabolism. 2002;87(8):3936–3940. doi: 10.1210/jcem.87.8.8731. [DOI] [PubMed] [Google Scholar]
- 79.Briet M, Schiffrin EL. The role of aldosterone in the metabolic syndrome. Current Hypertension Reports. 2011;13(2):163–172. doi: 10.1007/s11906-011-0182-2. [DOI] [PubMed] [Google Scholar]
- 80.Kuster GM, Kotlyar E, Rude MK, et al. Mineralocorticoid receptor inhibition ameliorates the transition to myocardial failure and decreases oxidative stress and inflammation in mice with chronic pressure overload. Circulation. 2005;111(4):420–427. doi: 10.1161/01.CIR.0000153800.09920.40. [DOI] [PubMed] [Google Scholar]
- 81.Delbosc S, Paizanis E, Magous R, et al. Involvement of oxidative stress and NADPH oxidase activation in the development of cardiovascular complications in a model of insulin resistance, the fructose-fed rat. Atherosclerosis. 2005;179(1):43–49. doi: 10.1016/j.atherosclerosis.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 82.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor super-family: the second decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Booth RE, Johnson JP, Stockand JD. Aldosterone. American Journal of Physiology. 2002;26(1–4):8–20. doi: 10.1152/advan.00051.2001. [DOI] [PubMed] [Google Scholar]
- 84.Bonvalet JP. Regulation of sodium transport by steroid hormones. Kidney International. 1998;53(65):S49–S56. [PubMed] [Google Scholar]
- 85.Pearce P, Funder JW. High affinity aldosterone binding sites (type I receptors) in rat heart. Clinical and Experimental Pharmacology and Physiology. 1987;14(11-12):859–866. doi: 10.1111/j.1440-1681.1987.tb02422.x. [DOI] [PubMed] [Google Scholar]
- 86.Takeda Y, Miyamori I, Inaba S, et al. Vascular aldosterone in genetically hypertensive rats. Hypertension. 1997;29(1):45–48. doi: 10.1161/01.hyp.29.1.45. [DOI] [PubMed] [Google Scholar]
- 87.Stockand JD, Meszaros JG. Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling. American Journal of Physiology. 2003;284(1):H176–H184. doi: 10.1152/ajpheart.00421.2002. [DOI] [PubMed] [Google Scholar]
- 88.Chun TY, Pratt JH. Non-genomic effects of aldosterone: new actions and questions. Trends in Endocrinology and Metabolism. 2004;15(8):353–354. doi: 10.1016/j.tem.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 89.Grossmann C, Gekle M. New aspects of rapid aldosterone signaling. Molecular and Cellular Endocrinology. 2009;308(1-2):53–62. doi: 10.1016/j.mce.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 90.Michea L, Delpiano AM, Hitschfeld C, Lobos L, Lavandero S, Marusic ET. Eplerenone blocks nongenomic effects of aldosterone on the Na+/H+ exchanger, intracellular Ca2+ levels, and vasoconstriction in mesenteric resistance vessels. Endocrinology. 2005;146(3):973–980. doi: 10.1210/en.2004-1130. [DOI] [PubMed] [Google Scholar]
- 91.Liu SL, Schmuck S, Chorazcyzewski JZ, Gros R, Feldman RD. Aldosterone regulates vascular reactivity: short-term effects mediated by phosphatidylinositol 3-kinase-dependent nitric oxide synthase activation. Circulation. 2003;108(19):2400–2406. doi: 10.1161/01.CIR.0000093188.53554.44. [DOI] [PubMed] [Google Scholar]
- 92.Uhrenholt TR, Schjerning J, Hansen PB, et al. Rapid inhibition of vasoconstriction in renal afferent arterioles by aldosterone. Circulation Research. 2003;93(12):1258–1266. doi: 10.1161/01.RES.0000106135.02935.E1. [DOI] [PubMed] [Google Scholar]
- 93.Mihailidou AS, Mardini M, Funder JW. Rapid, nongenomic effects of aldosterone in the heart mediated by ε protein kinase C. Endocrinology. 2004;145(2):773–780. doi: 10.1210/en.2003-1137. [DOI] [PubMed] [Google Scholar]
- 94.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nature Reviews Molecular Cell Biology. 2006;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 95.Matsui S, Satoh H, Kawashima H, et al. Non-genomic effects of aldosterone on intracellular ion regulation and cell volume in rat ventricular myocytes. Canadian Journal of Physiology and Pharmacology. 2007;85(2):264–273. doi: 10.1139/y07-017. [DOI] [PubMed] [Google Scholar]
- 96.Rude MK, Duhaney TAS, Kuster GM, et al. Aldosterone stimulates matrix metalloproteinases and reactive oxygen species in adult rat ventricular cardiomyocytes. Hypertension. 2005;46(3):555–561. doi: 10.1161/01.HYP.0000176236.55322.18. [DOI] [PubMed] [Google Scholar]
- 97.Sato A, Liu JP, Funder JW. Aldosterone rapidly represses protein kinase C activity in neonatal rat cardiomyocytes in vitro. Endocrinology. 1997;138(8):3410–3416. doi: 10.1210/endo.138.8.5352. [DOI] [PubMed] [Google Scholar]
- 98.Tillmann HC, Schumacher B, Yasenyev O, et al. Acute effects of aldosterone on intracardiac monophasic action potentials. International Journal of Cardiology. 2002;84(1):33–39. doi: 10.1016/s0167-5273(02)00115-8. [DOI] [PubMed] [Google Scholar]
- 99.Grossmann C, Benesic A, Krug AW, et al. Human mineralocorticoid receptor expression renders cells responsive for nongenotropic aldosterone actions. Molecular Endocrinology. 2005;19(7):1697–1710. doi: 10.1210/me.2004-0469. [DOI] [PubMed] [Google Scholar]
- 100.Le Moëllic C, Ouvrard-Pascaud A, Capurro C, et al. Early nongenomic events in aldosterone action in renal collecting duct cells: PKCα activation, mineralocorticoid receptor phosphorylation, and cross-talk with the genomic response. Journal of the American Society of Nephrology. 2004;15(5):1145–1160. [PubMed] [Google Scholar]
- 101.Ma TKW, Kam KKH, Yan BP, Lam YY. Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: current status. British Journal of Pharmacology. 2010;160(6):1273–1292. doi: 10.1111/j.1476-5381.2010.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Byrne JA, Grieve DJ, Cave AC, Shah AM. Oxidative stress and heart failure. Archives des Maladies du Coeur et des Vaisseaux. 2003;96(3):214–221. [PubMed] [Google Scholar]
- 103.Matavelli LC, Zhou X, Frohlich ED. Hypertensive renal vascular disease and cardiovascular endpoints. Current Opinion in Cardiology. 2006;21(4):305–309. doi: 10.1097/01.hco.0000231399.02481.de. [DOI] [PubMed] [Google Scholar]
- 104.Grieve DJ, Byrne JA, Cave AC, Shah AM. Role of oxidative stress in cardiac remodelling after myocardial infarction. Heart Lung and Circulation. 2004;13(2):132–138. doi: 10.1016/j.hlc.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 105.Dhalla NS, Saini-Chohan HK, Rodriguez-Leyva D, Elimban V, Dent MR, Tappia PS. Subcellular remodelling may induce cardiac dysfunction in congestive heart failure. Cardiovascular Research. 2009;81(3):429–438. doi: 10.1093/cvr/cvn281. [DOI] [PubMed] [Google Scholar]
- 106.Sadoshima JI, Izumo S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts: critical role of the AT1 receptor subtype. Circulation Research. 1993;73(3):413–423. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- 107.Lorell BH. Role of angiotensin AT1 and AT2 receptors in cardiac hypertrophy and disease. American Journal of Cardiology. 1999;83(12A):48H–52H. doi: 10.1016/s0002-9149(99)00258-1. [DOI] [PubMed] [Google Scholar]
- 108.Takeda Y, Yoneda T, Demura M, Miyamori I, Mabuchi H. Cardiac aldosterone production in genetically hypertensive rats. Hypertension. 2000;36(4):495–500. doi: 10.1161/01.hyp.36.4.495. [DOI] [PubMed] [Google Scholar]
- 109.Kagiyama S, Matsumura K, Fukuhara M, Sakagami K, Fujii K, Iida M. Aldosterone-and-salt-induced cardiac fibrosis is independent from angiotensin II type 1a receptor signaling in mice. Hypertension Research. 2007;30(10):979–989. doi: 10.1291/hypres.30.979. [DOI] [PubMed] [Google Scholar]
- 110.Matsumura K, Fujii K, Oniki H, Oka M, Iida M. Role of aldosterone in left ventricular hypertrophy in hypertension. American Journal of Hypertension. 2006;19(1):13–18. doi: 10.1016/j.amjhyper.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 111.Suzuki G, Morita H, Mishima T, et al. Effects of long-term monotherapy with eplerenone, a novel aldosterone blocker, on progression of left ventricular dysfunction and remodeling in dogs with heart failure. Circulation. 2002;106(23):2967–2972. doi: 10.1161/01.cir.0000039104.56479.42. [DOI] [PubMed] [Google Scholar]
- 112.Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents post-infarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation. 2003;107(20):2559–2565. doi: 10.1161/01.CIR.0000068340.96506.0F. [DOI] [PubMed] [Google Scholar]
- 113.Pitt B, Reichek N, Willenbrock R, et al. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: the 4E-left ventricular hypertrophy study. Circulation. 2003;108(15):1831–1838. doi: 10.1161/01.CIR.0000091405.00772.6E. [DOI] [PubMed] [Google Scholar]
- 114.Okoshi MP, Yan X, Okoshi K, et al. Aldosterone directly stimulates cardiac myocyte hypertrophy. Journal of Cardiac Failure. 2004;10(6):511–518. doi: 10.1016/j.cardfail.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 115.Yoshida Y, Morimoto T, Takaya T, et al. Aldosterone signaling associates with p300/GATA4 transcriptional pathway during the hypertrophic response of cardiomyocytes. Circulation Journal. 2010;74(1):156–162. doi: 10.1253/circj.cj-09-0050. [DOI] [PubMed] [Google Scholar]
- 116.López-Andrés N, Iñigo C, Gallego I, Díez J, Fortuño MA. Aldosterone induces cardiotrophin-1 expression in HL-1 adult cardiomyocytes. Endocrinology. 2008;149(10):4970–4978. doi: 10.1210/en.2008-0120. [DOI] [PubMed] [Google Scholar]
- 117.Doi T, Sakoda T, Akagami T, et al. Aldosterone induces interleukin-18 through endothelin-1, angiotensin II, Rho/Rho-kinase, and PPARs in cardiomyocytes. American Journal of Physiology. 2008;295(3):H1279–H1287. doi: 10.1152/ajpheart.00148.2008. [DOI] [PubMed] [Google Scholar]
- 118.Mohammed SF, Ohtani T, Korinek J, et al. Mineralocorticoid accelerates transition to heart failure with preserved ejection fraction via “Nongenomic Effects”. Circulation. 2010;122:370–378. doi: 10.1161/CIRCULATIONAHA.109.915215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stas S, Whaley-Connell A, Habibi J, et al. Mineralocorticoid receptor blockade attenuates chronic overexpression of the renin-angiotensin-aldosterone system stimulation of reduced nicotinamide adenine dinucleotide phosphate oxidase and cardiac remodeling. Endocrinology. 2007;148(8):3773–3780. doi: 10.1210/en.2006-1691. [DOI] [PubMed] [Google Scholar]
- 120.Brilla CG, Matsubara LS, Weber KT. Antifibrotic effects of spironolactone in preventing myocardial fibrosis in systemic arterial hypertension. American Journal of Cardiology. 1993;71(3) doi: 10.1016/0002-9149(93)90239-9. [DOI] [PubMed] [Google Scholar]
- 121.Brilla CG, Zhou G, Matsubara L, Weber KT. Collagen metabolism in cultured adult rat cardiac fibroblasts: response to angiotensin II and aldosterone. Journal of Molecular and Cellular Cardiology. 1994;26(7):809–820. doi: 10.1006/jmcc.1994.1098. [DOI] [PubMed] [Google Scholar]
- 122.Campbell SE, Janicki JS, Weber KT. Temporal differences in fibroblast proliferation and phenotype expression in response to chronic administration of angiotensin II or aldosterone. Journal of Molecular and Cellular Cardiology. 1995;27(8):1545–1560. doi: 10.1016/s0022-2828(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 123.Brilla CG, Weber KT. Mineralocorticoid excess, dietary sodium, and myocardial fibrosis. Journal of Laboratory and Clinical Medicine. 1992;120(6):893–901. [PubMed] [Google Scholar]
- 124.Rocha R, Martin-Berger CL, Yang P, Scherrer R, Delyani J, McMahon E. Selective aldosterone blockade prevents angiotensin II/salt-induced vascular inflammation in the rat heart. Endocrinology. 2002;143(12):4828–4836. doi: 10.1210/en.2002-220120. [DOI] [PubMed] [Google Scholar]
- 125.Chun TY, Bloem LJ, Pratt JH. Aldosterone inhibits inducible nitric oxide synthase in neonatal rat cardiomyocytes. Endocrinology. 2003;144(5):1712–1717. doi: 10.1210/en.2002-220956. [DOI] [PubMed] [Google Scholar]
- 126.Pecháňová O, Bernátová I, Pelouch V, Babál P. L-NAME-induced protein remodeling and fibrosis in the rat heart. Physiological Research. 1999;48(5):353–362. [PubMed] [Google Scholar]
- 127.Ferrini MG, Vernet D, Magee TR, et al. Antifibrotic role of inducible nitric oxide synthase. Nitric Oxide. 2002;6(3):283–294. doi: 10.1006/niox.2001.0421. [DOI] [PubMed] [Google Scholar]
- 128.Carlström M, Persson AEG, Larsson E, et al. Dietary nitrate attenuates oxidative stress, prevents cardiac and renal injuries, and reduces blood pressure in salt-induced hypertension. Cardiovascular Research. 2011;89(3):574–585. doi: 10.1093/cvr/cvq366. [DOI] [PubMed] [Google Scholar]
- 129.Hunter JC, Zeidan A, Javadov S, Kilić A, Rajapurohitam V, Karmazyn M. Nitric oxide inhibits endothelin-1-induced neonatal cardiomyocyte hypertrophy via a RhoA-ROCK-dependent pathway. Journal of Molecular and Cellular Cardiology. 2009;47(6):810–818. doi: 10.1016/j.yjmcc.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 130.Gils A, Declerck PJ. Plasminogen activator inhibitor-1. Current Medicinal Chemistry. 2004;11:2323–2334. doi: 10.2174/0929867043364595. [DOI] [PubMed] [Google Scholar]
- 131.Juhan-Vague I, Vague P. Hypofibrinolysis and insulin-resistance. Diabete et Metabolisme. 1991;17(1, part 2):96–100. [PubMed] [Google Scholar]
- 132.Mertens I, van Gaal LF. Obesity, haemostasis and the fibrinolytic system. Obesity Reviews. 2002;3(2):85–101. doi: 10.1046/j.1467-789x.2002.00056.x. [DOI] [PubMed] [Google Scholar]
- 133.Skurk T, Hauner H. Obesity and impaired fibrinolysis: role of adipose production of plasminogen activator inhibitor-1. International Journal of Obesity. 2004;28(11):1357–1364. doi: 10.1038/sj.ijo.0802778. [DOI] [PubMed] [Google Scholar]
- 134.Park JB, Schiffrin EL. Cardiac and vascular fibrosis and hypertrophy in aldosterone-infused rats: role of endothelin-1. American Journal of Hypertension. 2002;15(2):164–169. doi: 10.1016/s0895-7061(01)02291-9. [DOI] [PubMed] [Google Scholar]
- 135.Lee YS, Kim JA, Kim KL, et al. Aldosterone upregulates connective tissue growth factor gene expression via p38 MAPK pathway and mineralocorticoid receptor in ventricular myocytes. Journal of Korean Medical Science. 2004;19(6):805–811. doi: 10.3346/jkms.2004.19.6.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Matsui H, Ando K, Kawarazaki H, et al. Salt excess causes left ventricular diastolic dysfunction in rats with metabolic disorder. Hypertension. 2008;52(2):287–294. doi: 10.1161/HYPERTENSIONAHA.108.111815. [DOI] [PubMed] [Google Scholar]
- 137.Bauersachs J, Fraccarollo D. Aldosterone antagonism in addition to angiotensin-converting enzyme inhibitors in heart failure. Minerva Cardioangiologica. 2003;51(2):155–164. [PubMed] [Google Scholar]
- 138.Jewell CW, Watson LE, Mock J, Dostal DE. Aldosterone receptor antagonists and cardiovascular disease: do we need a change of the guard? Cardiovascular and Hematological Agents in Medicinal Chemistry. 2006;4(2):129–153. doi: 10.2174/187152506776369935. [DOI] [PubMed] [Google Scholar]
- 139.Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. The New England Journal of Medicine. 1999;341(10):709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 140.Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. The New England Journal of Medicine. 2003;348(14):1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 141.Brilla CG. Aldosterone and myocardial fibrosis in heart failure. Herz. 2000;25(3):299–306. doi: 10.1007/s000590050024. [DOI] [PubMed] [Google Scholar]
- 142.Abuannadi M, O'Keefe JH. Eplerenone: an underused medication? Journal of Cardiovascular Pharmacology and Therapeutics. 2010;15(4):318–325. doi: 10.1177/1074248410371946. [DOI] [PubMed] [Google Scholar]
- 143.Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart: role of oxidative stress. American Journal of Pathology. 2002;161(5):1773–1781. doi: 10.1016/S0002-9440(10)64454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. American Journal of Physiology. 2001;280(1):C53–C60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 145.Huisamen B, Pêrel SJC, Friedrich SO, Salie R, Strijdom H, Lochner A. ANG II type i receptor antagonism improved nitric oxide production and enhanced eNOS and PKB/Akt expression in hearts from a rat model of insulin resistance. Molecular and Cellular Biochemistry. 2011;349(1-2):21–31. doi: 10.1007/s11010-010-0656-6. [DOI] [PubMed] [Google Scholar]
- 146.Tabbi-Anneni I, Buchanan J, Cooksey RC, Abel ED. Captopril normalizes insulin signaling and insulin-regulated substrate metabolism in obese (ob/ob) mouse hearts. Endocrinology. 2008;149(8):4043–4050. doi: 10.1210/en.2007-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hilzendeger AM, Da Costa Goncalves AC, Plehm R, et al. Autonomic dysregulation in ob/ob mice is improved by inhibition of angiotensin-converting enzyme. Journal of Molecular Medicine. 2010;88(4):383–390. doi: 10.1007/s00109-009-0569-6. [DOI] [PubMed] [Google Scholar]
- 148.Fukuda M, Nakamura T, Kataoka K, et al. Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. Journal of Hypertension. 2010;28(2):340–352. doi: 10.1097/HJH.0b013e32833366cd. [DOI] [PubMed] [Google Scholar]
- 149.Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARα activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99(24):3125–3131. doi: 10.1161/01.cir.99.24.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kooistra T, Verschuren L, de Vries-van der Weij J, et al. Fenofibrate reduces atherogenesis in ApoE*3Leiden mice: evidence for multiple antiatherogenic effects besides lowering plasma cholesterol. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(10):2322–2330. doi: 10.1161/01.ATV.0000238348.05028.14. [DOI] [PubMed] [Google Scholar]
- 151.Ogata T, Miyauchi T, Sakai S, Takanashi M, Irukayama-Tomobe Y, Yamaguchi I. Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-B pathway. Journal of the American College of Cardiology. 2004;43(8):1481–1488. doi: 10.1016/j.jacc.2003.11.043. [DOI] [PubMed] [Google Scholar]
- 152.Duhaney T-AS, Cui L, Rude MK, et al. Peroxisome proliferator-activated receptor α-independent actions of fenofibrate exacerbates left ventricular dilation and fibrosis in chronic pressure overload. Hypertension. 2007;49(5):1084–1094. doi: 10.1161/HYPERTENSIONAHA.107.086926. [DOI] [PubMed] [Google Scholar]
- 153.LeBrasseur NK, Duhaney TAS, De Silva DS, et al. Effects of fenofibrate on cardiac remodeling in aldosterone-induced hypertension. Hypertension. 2007;50(3):489–496. doi: 10.1161/HYPERTENSIONAHA.107.092403. [DOI] [PubMed] [Google Scholar]