

Abstract
Detection of reagent-free specific biomolecular interactions through sensing of nanoscopic magnetic labels provides one of the promising routes to biosensing with solid-state devices. In particular, Hall sensors based on semiconductor heterostructures have shown exceptional magnetic moment sensitivity over a large dynamic field range suitable for magnetic biosensing using superparamagnetic labels. Here we demonstrate the capability of such micro-Hall sensors to detect specific molecular binding using biotin-streptavidin as a model system. We apply dip-pen nanolithography to selectively biotinylate the active areas of InAs micro-Hall devices with nanoscale precision. Specific binding of complementarily functionalized streptavidin-coated superparamagnetic beads to the Hall crosses occur via molecular recognition, and magnetic detection of the assembled beads is achieved at room temperature using phase-sensitive micro-Hall magnetometry. The experiment constitutes the first unambiguous demonstration of magnetic detection of specific biomolecular interactions with semiconductor micro-Hall sensors, and the selective molecular functionalization and resulting localized bead assembly demonstrate the possibility of multiplexed sensing of multiple target molecules using a single device with an array of micro-Hall sensors.
Keywords: biological sensor, Hall sensor, dip-pen nanolithography, molecular recognition
1. Introduction
Biomolecular sensing platforms based on solid-state devices and electrical/magnetic transduction offer many potential advantages over existing assays that employ optical signatures, such as fluorescence. Most significantly, they promise unprecedented speed and portability and may be valuable for applications including rapid, in-home medical diagnosis and battlefield detection of biological/chemical agents. Detection of specific biomolecular interactions through sensing of nanoscopic magnetic labels provides one of the most promising routes towards biosensing with solid state devices [1–12]. Among these, Hall sensors based on semiconductor heterostructures have shown exceptional magnetic moment sensitivity over a large dynamic field range suitable for magnetic biosensing using superparamagnetic labels [9–12]. Important general attributes of semiconductor microelectronics, such as high performance/sensitivity, high-density integration, and mass manufacturability, make it technologically and economically attractive to explore their use as ultrasensitive, real-time, portable and inexpensive biological sensors. A necessary step in realizing biosensing is the integration of chemical and biological (soft) molecules with solid state devices. Typically, the active area of a device should be decorated with linker molecules, and the binding of complementary target molecules then lead to an electrical or magnetic signal. So far, the functionalization and assembly processes in magnetic sensors involve blanket assembly of linker molecules on the device without any specificity to the active region of the device. This scheme may be usable in the detection of a single type of target molecules; however, simultaneous detection of multiple target molecules using a single device with a sensor array requires selective functionalization of each sensor with respective linker molecules. Figure 1 shows the schematic diagrams depicting the magnetic sensing schemes for labeled and label-free biomolecules. In both cases, linker molecules (e.g., DNA strands, antibodies, etc.) are first deposited on the active area of a device. In the case of detection of labeled molecules, the target molecules (complementary DNA strands, antigens, etc.) are attached to magnetic particles. Selective assembly of the magnetic particles onto the device through specific molecular binding, detected magnetically, verifies the presence of the complementary target molecules as shown in figure 1(a). Figure 1(b) shows a scheme for detection of label-free biomolecules by functionalizing the device and magnetic particles with two different, non-interacting molecules, both of which are complementary to different parts of the target molecule. A magnetic signal can be obtained only when the label-free target molecules are present in the solution.
Figure 1.

Schema for detection of magnetically labeled (a) or label-free (b) biomolecules (not to scale). Linker molecules are deposited on the functionalized, active regions of SiO2-insulated micro-Hall devices (two are shown in each panel); other areas are passivated to minimize nonspecific binding elsewhere. Complementary target molecules of interest bind from solution under analysis, yielding a magnetic signal when superparamagnetic beads (orange spheres in both panels) bind at the active areas of Hall devices, i.e., when either target molecules coat the magnetic beads (a) or in a sandwich assay where the magnetic beads are coated with a third molecule that also binds to secondary sites on the unlabeled target molecules (b).
Constant miniaturization and nanoscale engineering of solid-state devices have resulted in remarkable improvement in their sensitivity. Of the magnetic sensors, magnetoresistive and Hall devices offer the best room-temperature performance and are most adaptable for operation in an aqueous environment. Miniaturized magnetoresistive and Hall devices both offer high room-temperature magnetic moment sensitivities adequate for the detection of individual micron and even sub-micron magnetic particles [8, 12–14]. Comparing the two, magnetoresistive devices typically have better magnetic field sensitivity but their response saturates at low applied magnetic field; Hall sensors, especially those based on semiconductor heterostructures, offer exceptional magnetic moment sensitivity over a large dynamic field range which is particularly useful in enhancing the detectable magnetic moment of superparamagnetic beads [9–12]. So far, magnetic detection of micron or sub-micron sized beads with excellent sensitivity has been demonstrated with both types of sensors, and prediction of detection of much smaller magnetic particles has been made. Until now, however, the magnetic beads were either physically placed on a device’s active region utilizing a micromanipulator [11, 12] or assembled via non-selective blanket self-assembly [10]. The non-selective assembly presents a significant drawback if these devices are to be used in practical multiple target sensing applications. Assembling biomolecules onto selective areas of micro- and nano-scale devices has been one of the major challenges in developing solid-state devices for biological sensing involving finite numbers of magnetic nanoparticles or beads. For magnetic sensors, this difficulty restricted investigations to statistical counting of an ensemble of particles on the device’s active region [15]. The development of various tools for parallel or serial deposition of organic molecules - micro-contact printing (μCP) [16–18], and dip-pen nanolithography (DPN) [19–21] in particular – has made highly specific assembly of biological molecules onto the active area of micro-Hall devices with nanoscale precision and registry possible.
Herein, we detect superparamagnetic beads tethered to the active regions of a micro-Hall device through biotin-streptavidin linkage. The biotin-streptavidin system is used as a model system due to specificity of the interaction, its implications on other ligand-receptor pairs, and its widespread use in linking other biomolecules. We apply DPN to selectively biotinylate the active areas of InAs micro-Hall devices with nanoscale precision to create a bio-molecular template for the attachment of streptavidin-coated superparamagnetic beads. Specific binding of complementarily functionalized, streptavidin-coated superparamagnetic beads onto the biotinylated micro-Hall crosses occur via biomolecular recognition. Magnetic detection of the assembled beads using ac phase sensitive Hall magnetometry [12], much smaller in size in comparison to those reported in literature, is achieved at room temperature. The experiment constitutes an unambiguous demonstration of magnetic detection of specific biomolecular interactions with semiconductor micro-Hall sensors; in particular, the selective functionalization, specific assembly, and successful magnetic detection have demonstrated the efficacy of the micro-Hall devices as a sensing platform for simultaneous detection of multiple molecular targets.
2. Experimental Details
Commercial magnetic beads (Bangs Laboratories, Inc.) were first characterized using scanning electron microscope (SEM), superconducting quantum interference device (SQUID) magnetometry and x-ray diffraction (XRD) to determine the size of the beads, size and type of the constituent nanoparticles, magnetic moment of the nanoparticles, etc. The quality of streptavidin coating on the beads was verified via biotinylation of a plain Au surface, attachment of the streptavidin-coated beads and further biotinylation with fluorescently labeled biotin. Fluorescence was observed only in the presence of streptavidin-coated beads, confirming biotin-streptavidin binding. The beads, which consist of superparamagnetic nanoparticles embedded in a polymer matrix, are widely used for tagging biological molecules. SEM experiments showed that the beads used in our experiments had diameters ranging from ~150 – 250 nm, and consisted of Fe3O4 nanoparticles as confirmed from XRD. The mean diameter of the latter was determined to be 15 nm by fitting the measured magnetization of the beads to the Langevin function. The nanoparticles are distributed in a sphere of non-magnetic matrix covered with a shell of dextran, and functionalized with streptavidin which can be used for assembly via biotinylated ligands. For the assembly and detection, we first established the specificity of the surface chemistry and the yield of the bead self-assembly on macroscale substrates, then worked out the precise procedure for selective functionalization of micro-Hall devices by DPN on patterned mimic structures, and finally demonstrated directed self-assembly and magnetic detection of biologically tagged superparamagnetic beads on actual InAs QW micro-Hall devices.
2.1. Large scale directed assembly of biologically functionalized beads
To identify the appropriate surface chemistry and optimal concentration of streptavidin-coated beads in the solution, and also to obtain bead assembly with macroscale regularity while controlling the average number of beads in each pattern and minimizing nonspecific binding, we first performed millimeter-scale assembly of streptavidin-coated magnetic beads. We used two different methods - μCP and DPN - for generating organic molecular templates with self-assembled monolayer (SAM) of 16-mercaptohexadecanoic acid (MHA) on Au substrate. μCP is a suitable method for generating microscale molecular templates over macroscale area, limited only by size of the mold used for the fabrication of elastomeric stamp [22]. The elastomeric stamp was fabricated from polydimethylsiloxane (PDMS) from Dow Corning Corp (Sylgard 184) [23]. To make a patterned stamp, PDMS was thoroughly mixed with the curing agent at a ratio of 10:1 by weight and the mixture was poured onto the photoresist master mold generated via photolithography on Si substrate. It was then cured at 60°C for 12 hours. The stamp was sonicated in acetone after having been peeled off from the master and rinsed in ethanol. It was then coated evenly with MHA molecules by applying 1 mM MHA solution (in ethanol) using a cotton swab. To remove excess molecules on the surface, the stamp was lightly rinsed with ethanol and blown-dry with dry N2. The substrate used for the molecular template was Si/SiO2 with thermally deposited film of 5 nm Ti/20 nm Au. MHA templates were created on Au by bringing the MHA-coated elastomeric stamp in contact with the Au substrate for 8 seconds. Finally, the sample was immersed in 1 mM 1-octadecanethiol (ODT) solution (in ethanol) for 2 min to passivate the areas without MHA molecules.
Chemisorption of thiolated molecules such as MHA and ODT when they come in contact with a Au surface results in a very stable molecular platform for hierarchical directed assembly of biological molecules [24]. Due to the high yield of molecular template patterning on macroscale, μCP offers a clear advantage for studying surface chemistry because it readily enables the examination of reproducibility and faithfulness of the molecular pattern generation, and the extent of nonspecific binding in the assembly process. Figure 2(a) shows the lateral force microscopy (LFM) image of the resulting molecular pattern. The carboxylic group of MHA was activated by reacting with 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) in the presence of N-hydroxysuccinimide (NHS) to form semi-stable amine reactive NHS ester as shown in figure 2(b)[25]. In the reaction pathway, the o-acylisourea ester formed during the reaction of carboxylic group and EDC is very unstable and may alternatively undergo hydrolysis in aqueous solution to regenerate the carboxylic group [25]. To prevent this unwanted hydrolysis reaction, the sample with MHA/ODT molecular patterns was immersed in a vial with 300 μL of 200 mM NHS solution prepared in 100 mM morpholinoethanesulfonic acid monohydrate solution (MES buffer) adjusted to pH 6.5. 300 μL of freshly prepared 100 mM EDC solution (prepared in 100 mM MES buffer) was added to the vial for EDC to react with the hydroxyl group of MHA and form a zero length linker, o-acylisourea which immediately reacts with NHS to form semi-stable NHS ester as shown in figure 2(b). After 30 minutes of the reaction, the sample was rinsed with MES buffer, sonicated in ethanol, and then rinsed in ethanol and deionized water. Finally the sample was dipped in a biotin hydrazide solution, prepared by making 50 mM solution in dimethylformamide (DMF) and diluting in MES buffer to 5 mM, for 12 hours to form stable amide bond between biotin hydrazide and NHS. After biotin molecules are immobilized on the MHA patterns, the sample was rinsed in MES buffer and then in phosphate buffer saline (PBS) solution (pH 7.4). The presence of ODT passivation layers outside the MHA patterns confines the assembly of biotin hydrazide exclusively to MHA regions, effectively eliminates nonspecific interactions, and results in a template for hierarchical directed assembly of streptavidin-coated superparamagnetic beads which is performed immediately after biotinylation of MHA patterns.
Figure 2.

(a) Lateral force microscopy (LFM) image of MHA/ODT patterns. Dark regions are μCP patterned MHA on Au. Bright surround results from backfilling with ODT. (b) Reaction pathway for biotinlyation of MHA’s carboxylic group, and possible side reactions for the intermediate following activation with EDC/NHS. (c) SEM image of the assembly of 110 nm diameter streptavidin-coated superparamagnetic beads on biotinylated MHA patterns. Biotin-functionalized MHA binds streptavidin with high affinity, thus immobilizing streptavidin-coated superparamagnetic beads over the MHA patterns. The surrounding ODT monolayer effectively passivates the rest of the surface by minimizing nonspecific binding of beads outside of the MHA patterns.
The magnetic beads were cleaned before being used for assembly through repetitive magnetic segregation and resuspension in PBS solution at 7.4 pH. The beads were segregated using a permanent magnet and the supernatant liquid was exchanged with PBS solution. The beads were resuspended in PBS by vortexing. This process was repeated at least 5 times. At the end, PBS solution was added to obtain the desired concentration. Then 20 μL of bead solution was pipetted onto the sample to cover the entire surface and was left on the sample for 3 hours, with the sample constantly agitated to enhance the biotin-streptavidin interaction. The sample was then rinsed in deionized water to remove the unattached beads and blown dry with N2. The results of the bead assembly were examined with non-contact AFM and SEM. The stable biotin-streptavidin linkage between assembled biotin and streptavidin on the beads immobilize the beads onto the organic molecular templates. Figure 2(c) shows an SEM image of such an assembly over a large area. Evidently, a high degree of specificity and excellent yield were achieved: over macroscopic areas there was very little nonspecific binding of the beads on the ODT regions, while on the biotinylated MHA patterns there was relatively uniform, high density coverage. Physisorption of some beads during the assembly process could occur due to surface roughness. These nonspecifically attached beads can be easily removed by rinsing vigorously with deionized water or slight ultrasonication, and the chemisorbed beads through biotin-streptavidin binding were unaffected by such treatment. The density of the bead assemblies can be effectively controlled by the bead concentration in the solution and is demonstrated in a separate experiment. Figures 3(a) and 3(b) show results of bead assembly on two identical templates from bead solutions of 50 mg/mL and 20 mg/mL concentrations respectively; the SEM images reveal a clear correlation between the average number of assembled beads in a pattern and the bead solution concentration.
Figure 3.

Concentration dependence of the directed assembly of streptavidin-coated superparamagnetic beads onto biotin-MHA patterned surfaces. Biotin-MHA patterns were generated via μCP as illustrated in figure 2. The sample in (a) was treated with a bead solution (50 mg/mL) that was 2.5 times more concentrated than the sample in (b).
To establish the specificity of the bead assembly as originating from biotin-streptavidin binding and rule out nonspecific interactions, we carried out a control experiment in which two substrates with biotinylated-MHA/ODT and unbiotinylated-MHA/ODT patterns respectively were exposed to a solution of fluorescently-labeled (Alexa Fluor 488 from Molecular Probes Inc) streptavidin. After the assembly, fluorescence microscopy revealed strong fluorescence on the biotinylated sample from the well-defined MHA regions, while the unbiotinylated sample showed negligible fluorescence across the entire substrate. These results provided evidence that the highly selective assembly of magnetic beads on the biotinylated template is indeed due to the binding between biotin and streptavidin.
2.2. Directed assembly of biologically functionalized beads with high spatial registry
While the method of μCP is an efficient way of exploring surface chemistries for the assembly, it cannot be easily used for generating molecular patterns on a prefabricated device with sufficient spatial resolution and registry. Once the appropriate surface chemistry is identified, DPN can be used for molecular patterning with the help of nanoscale imaging and registering capabilities of an atomic force microscopy (AFM). The alignment procedure and DPN deposition parameters were first worked out on mimic micro-Hall devices created on Si substrates. The device structures were defined by standard electron-beam lithography, which provides the flexibility of generating any number of crosses of defined size. To perform DPN on a prefabricated Hall device, an AFM tip coated with MHA was used. An AFM tip was coated with MHA molecules by holding it in 1 mM MHA solution (in ethanol) for 30 sec and then blowing dry with N2. If necessary, the coating process was repeated to get sufficient molecular coating on the tip. When the tip and the substrate are brought in contact, the organic molecules on the tip is deposited to the substrate due to concentration gradient between substrate and tip. The diffusion on the substrate is governed by the relative strength of intermolecular interaction and molecule-substrate interaction [20]. Circular or square MHA patterns were created on a Hall cross by keeping the tip stationary on the substrate for a desired duration or scanning it with an appropriate speed, respectively. The size of the pattern can be calculated by using isotropic diffusion in case of molecules with strong interaction with the substrate (e.g., MHA on Au). The area of the pattern is directly proportional to the time of contact of the tip to the substrate [20],
where, r is the radius of the circular molecular pattern and t is the time of contact. To confine deposition of MHA molecules exclusively to the active area of a micro-Hall cross, the coordinates of the cross were obtained by pre-scanning (with the MHA-coated tip) two other crosses at the ends of the same device, and then bringing the tip directly into contact with the center of the cross of interest, as shown in figure 4(a). Finally, ODT passivation of the rest of the device surface, biotinylation of the MHA, and directed assembly of streptavidin-coated magnetic beads were performed as described earlier. Figure 4(b) shows an SEM image of two Hall crosses at the center of the mimic device, with seven beads assembled on one cross and a single bead on the other; the inset to figure 4(b) shows a close-up view of the cross with a single bead. Most importantly, ODT passivation resulted in minimization of nonspecific binding; in the areas around the device, no beads were found in the ODT passivated region. In a separate control experiment, we fabricated an array of 28 mimic Hall crosses and completely coated with ODT by dipping the sample in 1 mM ODT solution, without any MHA patterns. No beads were observed in any of the micro-Hall crosses. Combined with the fluorescence microscopy results described earlier, this confirms that the presence of the streptavidin-coated magnetic beads are the direct result of the biotin-streptavidin linkage and not due to any nonspecific binding or simple physisorption onto the crosses.
Figure 4.

(a) Schematic depiction of molecular functionalization of micro-Hall devices using DPN. To confine the MHA deposition exclusively to the active regions of Hall crosses, the two crosses at the edges were first scanned with an MHA-coated AFM tip to register coordinates of the micro-Hall device. Without losing contact with the substrate the tip was brought to the center of the other Hall crosses to generate MHA patterns. For this sample, molecular patterns were produced by diffusion of MHA from a stationary AFM tip. (b) SEM image of two mimic Hall crosses with a single (right) or seven (left) superparamagnetic beads assembled at the centers of the crosses. The inset shows the single bead assembly at higher magnification.
2.3. Directed assembly of biologically functionalized beads on micro-Hall devices
The hierarchical directed assembly and detection experiment on an InAs quantum well (QW) micro-Hall device was performed after the alignment procedure and the deposition parameters were worked out. The micro-Hall device with 1 μm x 1 μm crosses was fabricated from an MBE-grown InAs QW wafer using photolithography and wet etching. Details of the fabrication and initial characterization of the devices have been described elsewhere [26]. The patterned device was first sputter-coated with 60 nm of SiO2 to provide both electrical insulation for the device from the Ti (5 nm)/Au (20 nm) layer which was subsequently deposited on the SiO2, and also for protection of the device from the solutions used during assembly of the beads on the micro-Hall crosses. The Au surface served as a platform for the molecular assembly as described before. An MHA SAM was formed selectively on the cross region by scanning the MHA-coated AFM tip over the center area of each cross as illustrated in figure 4a. The active area of each Hall cross was located before DPN by prescanning the two crosses at the ends of the pattern as done in the mimic device with the MHA-coated tip. After DPN of MHA on the active area of the micro-Hall crosses, the rest of the Au surface was then passivated using ODT, and biotinylation of MHA and directed assembly of streptavidin-coated beads were performed as described before in Section 2.1 and shown in figure 2(b). Figure 5(a) shows an SEM image of two micro-Hall crosses at the center of the device after bead assembly. The biotinylated MHA regions are visible at the center of the crosses. It is evident that the assembly had a high degree of registry and specificity and had resulted from the specific biotin-streptavidin binding. On one cross, three beads assembled while none were present on the other; the difference is well within the fluctuations in the assembly density as revealed in figure 3(b), since a 10 mg/mL bead solution, half as concentrated as the one for figure 3(b), was used here for the assembly.
Figure 5.
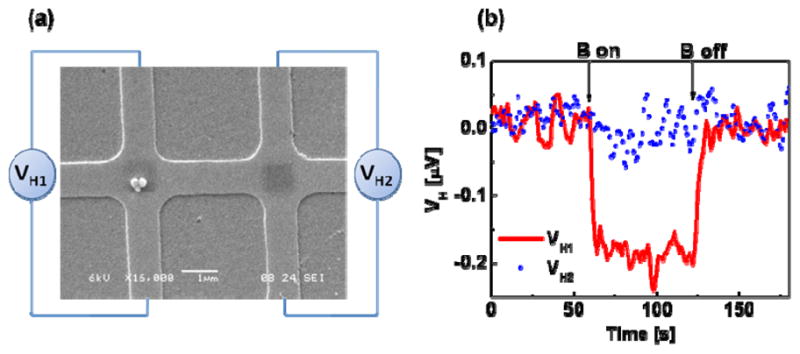
(a) SEM image of a micro-Hall device that was patterned from an InAs QW wafer via photolithography and wet etching, then coated with an insulating layer of SiO2, followed by a Au layer for molecular interfacing. MHA was patterned by DPN (figure 4), then biotinylated (figure 2), and is evident as dark squares in the central, active regions of the Hall crosses; the surrounding Au surface was passivated with ODT (figure 2). Three streptavidin-coated magnetic beads assembled on the biotinylated region of the left cross. (b) ac Hall voltage as a function of time for the two crosses shown in (a). Due to the presence of magnetic beads on the left cross, the ac Hall voltage VH1 (red) decreased by ~0.19 μV when a dc magnetic field B = 70.6 mT was applied. In contrast, no change in Hall voltage VH2 (blue) was observed when B was applied to the empty cross.
3. Results of magnetic detection of superparamagnetic beads
As demonstrated in figure 5(a), three superparamagnetic beads assembled on the micro-Hall cross were detected using ac phase sensitive Hall magnetometry. Details of the setup and the detection method have been reported earlier [12, 26]. Here the Hall device was biased with a dc current of 30 μA and an ac magnetic field (B0, 3.76 mT at 96.3 Hz) was applied perpendicular to the sensor plane. The signal was measured by a lock-in amplifier with a time constant of 1 sec and a 12 dB roll-off giving equivalent bandwidth, Δf, of 0.125 Hz. In this initial state, ac Hall voltage arising from a direct detection of B0 was nulled by using the zero-offset option of the lock-in amplifier. An NdFeB magnet was then placed over the setup to generate a dc magnetic field of 70.6 mT at the Hall crosses. In the presence of the superparamagnetic bead(s), the dc field induces a drop in the measured ac Hall voltage, a consequence of monotonically decreasing susceptibility of the bead with increasing magnetic field as given by [12]
where, ΔM̃ is the difference in the induced ac magnetizations of the bead before and after the static field has been applied. Figure 5(b) shows the result of the detection experiment performed on the micro-Hall cross with 3 assembled beads. The application of the dc field resulted in a drop in the ac Hall voltage of 0.19 μV (red curve), which corresponds to an average stray magnetic field of 33.05 μT. In contrast, the Hall cross without magnetic beads did not exhibit any change in the ac Hall voltage when the dc magnetic field was applied (blue curve). The rms voltage noise, determined from standard deviation of the ac Hall voltage, was 16.1 nV, which corresponds to a minimum detectable stray magnetic field of 2.86 μT. Hence with the presence of 3 beads on the cross, the signal-to-noise ratio was 11.6 (21.3 dB). The magnetic detection results provided a definitive indication of the presence of superparamagnetic bead(s) on the functionalized Hall cross and hence provided confirmation of the specific biotin-streptavidin binding.
The beads shown in figure 5(a) are 224 nm, 240 nm, and 146 nm in diameter; their contributions to the average stray magnetic field can be calculated by integrating the magnetic flux of the dipole field from each bead using the following equation [9, 11]
where x, y, z are the position of a bead from the center of the cross, Ms and V are the saturation magnetization and the volume of the bead. In our experiment where the beads are situated relatively in the center region of the cross, the average field due to each bead depends more strongly upon the size of the bead whereas the location of the bead has less impact on the value of the average field as suggested by the above equation. In the present setup, our calculation shows that the Hall voltage noise of 16.1 nV is equivalent to the signal from a similar bead of a diameter of 130 nm placed at the center of the cross; and a bead of 164 nm diameter would give a detectable signal with a signal-to-noise ratio of 2. On the other hand, since the low-frequency noise of the InAs micro-Hall devices is dominated by 1/f noise, at a higher frequency (~90 kHz), the noise level can be reduced to the thermal noise limit (~ 4.2 nV and a noise-equivalent magnetic field sensitivity of 0.19 μT), which would improve the magnetic moment sensitivity of the Hall devices to the equivalent of a single Fe3O4 nanoparticle of less than 20 nm in diameter. The Hall detection of the binding of a single superparamagnetic nanoparticle would then be a definitive indicator of specific interactions between only a small number of biomolecules—just two or three in the limit—during the assembly process.
4. Conclusions
To summarize, we have demonstrated highly specific directed self-assembly of streptavidin-coated superparamagnetic beads on Au films with organic molecular templates, as well as effective biofunctionalization of InAs QW micro-Hall devices with nanoscale spatial registry using DPN, which enabled selective assembly of the magnetic beads onto the active areas of a micro-Hall device through specific biotin-streptavidin binding. With the successful demonstration of magnetic detection of biotin-streptavidin as a model system for complementary pairs of ligand-receptor proteins, this work can be readily extended to the detection of other biomedically significant complementary biomolecular pairs (e.g., complementary strands of DNA, antibody-antigen, etc.). The selective molecular functionalization of the micro-Hall device led to highly localized binding of the magnetic labels and minimal nonspecific binding. This result reveals the possibility of functionalizing different micro-Hall crosses with different linker molecules and hence simultaneously detecting multiple target molecules in a solution with a single device. Furthermore, the signal-to-noise ratio exhibited in our measurements indicated the feasibility of detecting a single superparamagnetic nanoparticle down to ~ 20 nm, which could pave the way for single molecule detection and single-molecule dynamics study using magnetic tweezers.
Acknowledgments
We acknowledge financial support by NIH (NIGMS grant GM079592) and NSF (NIRT grant ECS-0210332).
Footnotes
Publisher's Disclaimer: This is an author-created, un-copyedited version of an article accepted for publication in the journal Nanotechnology. IOP Publishing Ltd is not responsible for any errors or omissions in this version of the manuscript or any version derived from it. The definitive publisher authenticated version is available online at DOI: 10.1088/0957-4484/20/35/355501.
References
- 1.Ferreira HA, Graham DL, Freitas PP, Cabral JMS. J Appl Phys. 2003;93:7281–7286. [Google Scholar]
- 2.Wang SX, Li G. IEEE Trans Magn. 2008;44:1687–1702. doi: 10.1109/TMAG.2008.2002795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baselt DR, Lee GU, Natesan M, Metzger SW, Sheehan PE, Colton RJ. Biosens Bioelectron. 1998;13:731–739. doi: 10.1016/s0956-5663(98)00037-2. [DOI] [PubMed] [Google Scholar]
- 4.Edelstein RL, Tamanaha CR, Sheehan PE, Miller MM, Baselt DR, Whitman LJ, Colton RJ. Biosens Bioelectron. 2000;14:805–813. doi: 10.1016/s0956-5663(99)00054-8. [DOI] [PubMed] [Google Scholar]
- 5.Miller MM, Sheehan PE, Edelstein RL, Tamanaha CR, Zhong L, Bounnak S, Whitman LJ, Colton RJ. J Magn Magn Mater. 2001;225:138–144. [Google Scholar]
- 6.Graham DL, Ferreira HA, Freitas PP. Trends Biotechnol. 2004;22:455–462. doi: 10.1016/j.tibtech.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Millen RL, Kawaguchi T, Granger MC, Porter MD. Anal Chem. 2005;77:6581–6587. doi: 10.1021/ac0509049. [DOI] [PubMed] [Google Scholar]
- 8.Shen W, Liu X, Mazumdar D, Xiao G. Appl Phys Lett. 2005;86(253901):1–3. [Google Scholar]
- 9.Besse P-A, Boero G, Demierre M, Pott V, Popovic R. Appl Phys Lett. 2002;80:4199–4201. [Google Scholar]
- 10.Sandhu A, Kumagai Y, Lapicki A, Sakamoto S, Abe M, Handa H. Biosens Bioelectron. 2007;22:2115–2120. doi: 10.1016/j.bios.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 11.Landry G, Miller MM, Bennett BR, Johnson M, Smolyaninova V. Appl Phys Lett. 2004;85:4693–4695. [Google Scholar]
- 12.Mihajlović G, Xiong P, von Molnár S, Ohtani K, Ohno H, Field M, Sullivan GJ. Appl Phys Lett. 2005;87(112502):1–3. [Google Scholar]
- 13.Cardoso FA, Germano J, Ferreira R, Cardoso S, Martins VC, Freitas PP, Piedade MS, Sousa L. J Appl Phys. 2008;103(07A301):1–3. [Google Scholar]
- 14.Li G, Sun S, Wilson RJ, White RL, Pourmand N, Wang SX. Sens Actuators A. 2006;126:98–106. doi: 10.1016/j.sna.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li G, Wang SX, Sun S. IEEE Trans Magn. 2004;40:3000–3002. [Google Scholar]
- 16.Kumar A, Biebuyck HA, Whitesides GM. Langmuir. 1994;10:1498–1511. [Google Scholar]
- 17.Kumar A, Whitesides GM. Appl Phys Lett. 1993;63:2002–2004. [Google Scholar]
- 18.Xia Y, Tien J, Qin D, Whitesides GM. Langmuir. 1996;12:4033–4038. [Google Scholar]
- 19.Basnar B, Willner I. Small. 2009;5:28–44. doi: 10.1002/smll.200800583. [DOI] [PubMed] [Google Scholar]
- 20.Manandhar P, Jang J, Schatz GC, Ratner MA, Hong S. Phys Rev Lett. 2003;90(115505):1–4. doi: 10.1103/PhysRevLett.90.115505. [DOI] [PubMed] [Google Scholar]
- 21.Piner RD, Zhu J, Xu F, Hong S, Mirkin CA. Science. 1999;283:661–663. doi: 10.1126/science.283.5402.661. [DOI] [PubMed] [Google Scholar]
- 22.Quist AP, Pavlovic E, Oscarsson S. Anal Bioanal Chem. 2005;381:591–600. doi: 10.1007/s00216-004-2847-z. [DOI] [PubMed] [Google Scholar]
- 23.Choi KM, Rogers JA. J Am Chem Soc. 2003;125:4060–4061. doi: 10.1021/ja029973k. [DOI] [PubMed] [Google Scholar]
- 24.Manandhar P, Huang L, Grubich JR, Hutchinson JW, Chase PB, Hong S. Langmuir. 2005;21:3213–3216. doi: 10.1021/la047227i. [DOI] [PubMed] [Google Scholar]
- 25.Pierce Biotechnology. http://www.piercenet.com.
- 26.Mihajlović G, Xiong P, von Molnár S, Field M, Sullivan GJ. J Appl Phys. 2007;102(034056):1–9. [Google Scholar]