

Abstract
Mitogen activated protein kinases (MAPK), such as ERK1/2 and p38 MAPK and phosphatidylinositol-3 phosphate kinase (PI-3K) play a major role in the development of cardiac hypertrophy. Recently, we have shown their crucial role in the regulation of the myocardial function through their effects on crucial ion channels. It is the focus of this study to resolve the interaction between these pathways and its implication on the function of the normal and hypertrophied cardiomyocytes.
To that end, we created arteriovenous fistula in the adult rat that developed volume-overload eccentric cardiac hypertrophy over a 3-week period. We measured the relative activity of ERK1/2, p38 MAPKs and Akt through western blot analysis and assessed the functional density of the outward rectifier potassium current (IK) using the patch-damp technique.
The results showed a mutual negative auto-regulation between ERK1/2 and p38 in normal cardiomyocytes, which disappears during cardiac hypertrophy. In addition, PI-3K seems to assume a greater role in mediating IGF-1 effects on the MAPKs during cardiac hypertrophy. This was also relevant to IK functional density which was reduced by activation of both MAPKs and Akt by angiotensin II (ANGII) and insulin-like growth factor-1 (IGF-1), respectively; however, this reduction was reversed by inhibition of PI-3K alone in hypertrophied myocytes but not in normal ones. This raises an important implication relative to the role of IGF-1-dependent activation of PI-3K, which may translate into a differential prognostic for cardiac hypertrophy among ethnic groups. This is true in African Americans, having higher circulating IGF-1 levels, and especially true for the athletes among them.
Keywords: MAPK, PI-3K, IK, Cardiac Hypertrophy
Introduction
Cardiac hypertrophy is an adaptive response to the inciting stress produced by workload to the heart. Chronic volume-overload occurring during aortic insufficiency, mitral regurgitation or arteriovenous shunt leads to the development of eccentric cardiac hypertrophy. It is characterized by ventricular chamber enlargement disproportionate to ventricular wall thickening.1 This involves the action of specific growth factors which are activated during the development of cardiac hypertrophy.2 Insulin-like growth factor-1 (IGF-1) has been known for its major role in cellular proliferation and differentiation during heart development.3 Furthermore, in the volume-overload model, we have shown that cardiac myofibers have an increased receptor affinity to IGF-1, which corroborated with the induction of eccentric hypertrophy.4 This was confirmed by our recent findings on the major role that IGF-1-dependent intracellular signaling pathways, MAPK and PI-3 kinase, play in modulating myocardial electrophysiological function.5
Binding of IGF-1 to its receptor stimulates PI-3 kinase causing activation of Akt.6,7 Transgenic overexpression of Akt was sufficient to induce significant cardiac hypertrophy in mice without affecting systolic function.8 Furthermore, IGF-1 activated the mitogen-activated protein kinase (MAPK) cascade in addition to the PI-3K/Akt one.9 Lavandero et al,10 showed that short exposure of ventricular cardiocytes to IGF-1 leads to the development of cardiac hypertrophy through activation of downstream ERK-MAPK pathway. In addition, Shyu et. al11 found that the increased expression of myostatin in hypertrophied cells is mediated by IGF-1, partly through p38 MAP kinase. In the same line of evidence, we have recently shown that IGF-1 plays an important role in the development of eccentric cardiac hypertrophy, through the activation of MAPK and PI-3K pathways.5
However, there is still a lack of information regarding the interactions between the MAPKs and the PI-3K pathways in response to IGF-1 in vitro or in vivo. Therefore, the objective of this study was to determine the level of interaction between the MAPK (ERK1/2 and p38) as well as the PI-3K in mediating IGF-1 effects in the normal and hypertrophied hearts. We hypothesized that during the compensated phase of eccentric cardiac hypertrophy, IGF-1 activation of MAPKs and PI-3 kinase induces differential cross-talk between these kinases that can be a key element in modulating its effect through the progression of the disease.
Materials and Methods
All procedures used conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH) publication No. 85-23, revised 1996.
Eccentric Cardiac Hypertrophy
Induction of Volume-overload Cardiac Hypertrophy
Adult male Sprague-Dawley rats (200–250 g) were used. The abdominal aorta and vena cava were shunted at the union of me segment two-third caudal to the renal artery and the one-third cephalic to the aortic bifurcation with an 18G needle. The patency of the shunt was verified visually. The same procedure was performed on the age-matched sham rats, except for the insertion of the needle. Three weeks were allowed for the cardiac hypertrophy to develop.
Isolated Heart Retrograde Perfusion
Immediately after heart excision, it was hung on Langendorff apparatus for retrograde perfusion with normal Tyrode’s solution (mM): 140 NaCI, 5.4 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 20 NaHCO3, 10 D-glucose, 0.1 EGTA, 0.4 NaH2PO4, 20 taurine, 10 creatine. The protocol consisted of continuing the retrograde perfusion of the hearts for 15 minutes with Tyrode’s solutions containing either 1) regular Tyrode’s as control, 2) IGF-1, or 3) Tyrode’s containing specific kinase inhibitor followed by IGF-1. At the end of the experiment, hearts were quickly snap-frozen in liquid nitrogen for western blot processing.
Western Blotting
Activation of ERK1/2, p38 MAPK and Akt were assessed using western blot technique as previously described.5 Briefly, protein samples were prepared from perfused heart tissue using lysis buffer with protease inhibitors cocktail (Roche, Calif.), followed by incubation with phospho-specific antibodies for Akt, ERK1/2 or p38 MAPK (Cell Signaling Technology, Mass.). Membranes were then incubated with secondary antibody conjugated to horseradish peroxidase (Cell Signaling Technology, Mass.). Bands were visualized by Chemiluminescence. Films from at least three independent experiments were scanned and densities of the immunoreactive bands were evaluated using UnScanIT software. Kinase activities were evaluated as the ratio of phosphorylated kinase over total protein kinase per experiment.
Isolation of Adult Rat Cardiomyocytes
As previously described,5 retrograde perfusion of the coronary arteries was performed by Langendorff method. The heart was sequentially perfused with Tyrode’s solution, followed by Tyrode’s solution containing liberase Blenzyme 4 (Roche Diagnostics, Indiana) and protease type XIV (Sigma, Missouri). Then the heart was perfused with regular Tyrode’s solution. Finally, the ventricles were cut, minced and filtered. The cells were diluted and kept in Tyrode’s solution until experimentation.
Electrophysiological Studies
The whole-cell patch-damp technique was used in the voltage-clamp mode. Ventricular myocytes were perfused with a buffer containing (mM): 5 KCl, 1 MgCl2, 140 NaCl, 10 HEPES, 10 D-glucose, 1 CaCl2 and pH at 7.4. The pipette solution contained (mM): 130 K-giutamate, 20 KCl, 5 EGTA, 5 NaCl, 5 Mg-ATP, 1 MgCl2, 10 HEPES, and pH at 7.2. The cardiomyocytes were stimulated and analyzed using pClamp 9.0 software connected to an Axopatch-1B amplifier (Molecular Devices, Calif). The currents were divided by the cell capacitance to normalize for cell size changes between normal and hypertrophied cardiomyocytes.
Experimental Protocols for Potassium Currents
Step voltages (400 msec) in 10 mV increments between −40mV and +40mV were applied from a holding potential of −40 mV. Steady-state currents were plotted as a function of the command potential. The inhibitors for PI-3K (LY294002), ERK1/2 MAPK (PD98059) and p38 MAPK (SB203580) were purchased from Cell Signaling Technology (Mass.).
Statistical Analysis
All statistical analysis was performed using SigmaStat software. The paired Student’s f-test was used to compare data before and after drug treatment of the same animal group. The heteroscedastic two-sample unpaired Student’s t-test assuming unequal variances was used when comparing the drug effects between two different animal groups (sham vs shunt). In addition, we performed one-way ANOVA followed by ad hoc Dunnett’s test for comparing the differences between the effects of inhibitors to a control in each animal model (sham or shunt) and on each kinase. Using the null hypothesis, P≤ .05 was considered significant.
Results
Structural Parameters
The data on the structural parameters from sham and shunted adult rats confirmed the development of the eccentric cardiac hypertrophy within 3 weeks post-surgery, as seen in Table 1. The shunted rats did not show a significant difference in their body weight vs normal sham ones. However, the heart weights (P< .01), as well as the relative heart weight of shunted rats, were higher compared to the sham ones (P< .01). In addition, the cellular membrane capacitance was greater in the cardiomyocytes from hypertrophied hearts as compared to control ones (P<.01).
Table 1.
Differences in the structural parameters of sham and shunt hearts
| Body Weight (g) | Heart Weight (mg) | Relative Heart Weight (mg/100g body weight) | Membrane Capacitance (pF) | |
|---|---|---|---|---|
| Sham | 225 ± 12 | 1043 ± 22 | 474 ± 19 | 209 ± 14 |
| Shunt | 256 ± 21 | 1397 ± 77* | 674 ± 34* | 294 ± 24* |
n=12 for each group
P<.01 for sham versus shunt
Effects of IGF-1 on ERK1/2 and p38-MAPKs as well as Akt in Normal and Hypertrophied Hearts
The activation levels of ERK1/2, p38 MAPKS and Akt was higher in the hypertrophied cardiomyocytes compared to normal ones, as shown in Figure 1A. Cardiac hypertrophy activated ERK-1 by 56 ± 19% (P=0.05), ERK-2 by 227 ± 21% (P<.001), p38 MAPK by 75 ± 17% (P=.001) and Akt by 67 ± 19% (P=.02). Similarly (Fig. 2B), exposure of normal cardiomyocytes to 10 nM IGF-1 increased the activities of ERK1/2 (38 ± 7%; P<.001 and 93 ± 0.3%; P<.05), p38 MAPK (109 ± 16%; P<.001) and Akt (95 ± 13%; P<.001) as during eccentric hypertrophy. However, IGF-1 effect was blunted for ERK-1/2 and p38 MAPKs in the hypertrophied hearts; but not for Akt (158 ± 18%; P=.02 vs untreated shunt).
Fig 1.
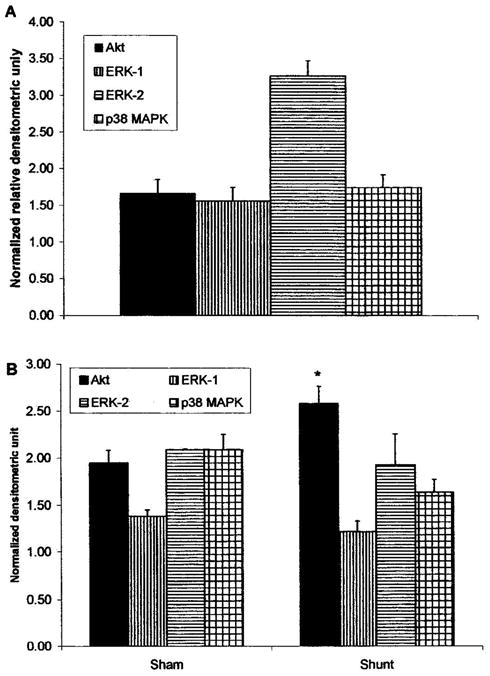
(A) Activation levek of Akt ERK1/2 and p38 MAPK in eccentrically hypertrophied hearts, relative to the levels found in normal hearts. (B) IGF-1-dependent activities of Akt, ERK1/2 and p38 MAPK in normal versus volume-overload-induced hypertrophied hearts. Data are expressed as the ratio of phosphorylated over total protein normalized to control untreated hearts. The data are presented as average ± SEM with n=3 (from different heart samples). *P<.01 as compared to normal hearts
Fig 2.

(A) Activation levels of ERK1/2 and p38 MAPK in untreated (control), 1uM LY294002-treated, and IGF-1 + LY294002 treated normal and hypertrophied hearts. (B) Activation levels of Akt and p38 MAPK in untreated (control), 1uM PD98059-treated, and IGF-1 + PD98059 treated normal and hypertrophied hearts. (Q Activation levels of Akt and ERK1/2 in untreated (control), 1uM SB203580, and IGF-1 + SB203580 treated normal and hypertrophied hearts. Data are expressed as the ratio of phosphorytated over total protein normalized to control untreated hearts. The data are presented as average ± SEM with n=3 (from different heart samples). *P<.05 as compared to normal hearts; # P<.05 as compared to untreated
Cross-talk Between ERK1/2 and p38 MAPKs Along with PI-3Kin Normal and Hypertrophied Hearts
Figure 2A shows that in the normal hearts, specific inhibition of PI-3K by LY294002 did not affect the activation level on any of the MAPKs. In the presence of the inhibitor, IGF-1 effect on ERK1/2 was abrogated but not on p38 MAPK This may imply that IGF-1 activated p38 MAPK through a parallel pathway to PI-3K/Akt, On the other hand, PI-3K inhibition in the hypertrophied hearts reduced the basal activity of ERK1/2. However, similar to the normal hearts, inhibition of PI-3K blocked the IGF-1 effect on ERK1/2 but not on p38 MAPKs.
Figure 2B shows mat in the normal hearts, specific inhibition of ERK1/2 by PD98059 did not affect the basal activation level of Akt but significantly increased that of p38 MAPK. In addition, IGF-1 effects on Akt and p38 MAPK were not altered by ERK1/2 inhibition. Conversely, in the hypertrophied hearts, ERK1/2 inhibition did not alter the basal activities of either kinase; thus the IGF-1 activation of Akt and p38 MAPK were not affected by such inhibition.
Figure 3B shows that specific inhibition of p38 MAPK by SB203580 in the normal hearts improved the basal activity of ERK1/2 but not that of Akt. Interestingly, p38 MAPK inhibition did not affect IGF-1 activation of Akt, but blunted that of ERK1/2. In comparison, p38 MAPK inhibition in the hypertrophied hearts only enhanced ERK-2 basal activity. Such inhibition did not affect IGF-1 on Akt but prevented ERK 1/2 activation.
Fig 3.

The combined effect of ANG II (10−8 M) and IGF-1 (10−8 M) on the current-voltage relationships of IK in sham (A) and shunt (B) cardiomyocytes, in the presence or absence of PI-3K inhibition (1uM LY294O02). Data are presented as average current density (n=5)
Functional Correlate for the Combined Effects of MAPKs and Akt on Normal and Hypertrophied Cardiomyocytes
Angiotensin II (ANG II) inotropic effects are known to be mainly mediated through MAPKs and not PI-3K. Therefore, the combined effects of ANG II and IGF-1 on the cardiomyocytes would reflect, to a certain extent, the translational outcome of the cross-talk found between both pathways. Figure 3 demonstrates that exposure of normal cardiomyocytes to ANG (10 nM) with IGF-1 (10 nM) reduces the functional density of the outward rectifier potassium channel (IK) to a greater level than in the hypertrophied ones. Interestingly, this combined effect of ANG II and IGF-1 was not altered by PI-3K inhibition in the normal cardiomyocytes, but was eliminated in the hypertrophied ones.
Discussion
Our study shows the existence of a cross-talk between intracellular MAPKs and PI-3 kinase pathways that can be translated into a functional significance, such as the activity of potassium channels, responsible for cardiac repolarization and relaxation. It has been shown that IGF-1 effects are mainly mediated by PI-3 kinase activation.12,13 We have previously confirmed this finding in addition to demonstrating a differential regulation of MAPKs and PI-3 kinase during the development of eccentric cardiac hypertrophy. Basically, the IGF-1 effect becomes transduced mainly via PI-3 kinase pathway. This corroborates with the reversal of the combined ANG II and IGF-1 effects by PI-3 kinase inhibition in the hypertrophied myocytes but not in the normal ones. Similar effects were observed using IGF-1 alone (data not shown).
Our data demonstrate that PI-3K has a positive influence on IGF-1-stimulated activation of ERK 1/2 but not p38 MAPK in both normal and hypertrophied hearts. In addition, IGF-1-dependent ERK1/2 activation is mediated by both PI-3K and p38 MAPKs in parallel. Therefore, both kinases seem to be upstream regulator for ERK1/2 activity. Furthermore, there is a mutual negative auto-regulatory cross-talk between basal p38 MAPK and ERK1/2 activities. Such mechanism seems to be dysfunctional during the development of cardiac hypertrophy. Thus, we can speculate that IGF-1 effects on ERK1/2 and p38 MAPK activities would be regulated to a greater extent by PI-3K during cardiac hypertrophy. This is in agreement with previous reports from our group and others that PI-3K plays a central role in mediating the hypertrophic and inotropic effects of IGF-1.3,5,13 Thus, the combined activation of MAPKs and PI-3K would be alleviated by PI-3K inhibition alone in the hypertrophied cardiomyocytes, but not in the normal ones. This is in accordance with our present findings on IK regulation by MAPKs and PI-3K in normal and hypertrophied cardiomyocytes.
These exciting cross-talk changes during cardiac hypertrophy are of great significance regarding the elucidation of the weaker prognostic in African Americans suffering from cardiac hypertrophy as compared to other ethnic groups.14,15 This is particularly important, since the circulating levels of free IGF-1 in African American has been reported to be more elevated than Caucasians, and more so in athletes among them.16,17
Acknowledgments
This work was supported in part by grants GM08016-38 NIGMS/NIH, 2G12 RR003048 RCMI, Division of Research Infrastructure, NCRR/NIH and SNRP to GEH.
References
- 1.Kidman RB, Houser SR. Outward currents in normal and hypertrophied feline ventricular myocytes. Am J Physiol. 1989;256(5 Pt 2):H1450–H1461. doi: 10.1152/ajpheart.1989.256.5.H1450. [DOI] [PubMed] [Google Scholar]
- 2.Modesri PA, Vanni S, Benolozzi I, et al. Early sequence of cardiac adaptations and growth factor formation in pressure- and volume-overload hypertrophy. Am J Physiol Heart Circ Physiol. 2000;279(3):H976–H985. doi: 10.1152/ajpheart.2000.279.3.H976. [DOI] [PubMed] [Google Scholar]
- 3.Huang CY, Hao LY, Buctow DE. Insulin-like growth factor-II induces hypertrophy of adult cardiomyocytes via two alternative pathways. Cell Biol Int. 2002;26(8):737–739. doi: 10.1006/cbir.2002.0919. [DOI] [PubMed] [Google Scholar]
- 4.Haddad GE, Blackwell K, Bikhazi A. Regulation of insulin-like growth factor-1 by the renin-angiotensin system during regression of cardiac eccentric hypertrophy through angiotensin-converting enzyme inhibitor and ATI antagonist. Con J Physiol Pharmacol. 2003;81(2):142–149. doi: 10.1139/y02-154. [DOI] [PubMed] [Google Scholar]
- 5.Teos LY, Zhao A, Alvin Z, Laurence GG, Li C, Haddad GE. Basal and IGF-I-dependent regulation of potassium channels by MAP kinases and P13-kinase during eccentric cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2008;295(5):H1834–H1845. doi: 10.1152/ajpheart.321.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeBosch B, Treskov I, Lupu TS, et al. Akt1 is required for physiological cardiac growth. Circulation. 2006;113(17):2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 7.McMullen JR, Shioi T, Huang WY, et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinosiride 3-kinase(pl lOalpha) pathway. J Biol Chem. 2004;279(6):4782–4793. doi: 10.1074/jbc.M310405200. [DOI] [PubMed] [Google Scholar]
- 8.Matsui T, Li L, Wu JC, et al. Phenotypic spectrum caused by transgenic over-expression of activated Akt in the heart. J Biol Chem. 2002;277(25):22896–22901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 9.Foncea R, Andersson M, Ketterman A, et al. Insulin-like growth factor-I rapidly activates multiple signal transduction pathways in cultured rat cardiac myocytes. J Biol Chem. 1997;272(31):19115–24. doi: 10.1074/jbc.272.31.19115. [DOI] [PubMed] [Google Scholar]
- 10.Lavandero S, Foncea R, Perez V, Sapag-Hagar M. Effect of inhibitors of signal transduction on IGF-1-induced protein synthesis associated with hypertrophy in cultured neonatal rat ventricular myocytes. FEBS Lett. 1998;422(2):193–196. doi: 10.1016/s0014-5793(98)00008-8. [DOI] [PubMed] [Google Scholar]
- 11.Shyu KG, Ko WH, Yang WS, Wang BW, Kuan P. Insulin-like growth (actor-1 mediates stretch-induced upregularion of myostatin expression in neonatal rat cardiomyocytes. Cardiovasc Res. 2005;68(3):405–414. doi: 10.1016/j.cardiores.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 12.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 13.Liu L, Zhao X, Pierre SV, Askari A. Association of PBK-Akt signaling pathway with digitalis-induced hypertrophy of cardiac myocytes. Am J Physiol Cell Physiol. 2007;293(5):C1489–C1497. doi: 10.1152/ajpcell.00158.2007. [DOI] [PubMed] [Google Scholar]
- 14.Havranek EP, Froshaug DB, Ernserman CD, et al. Left ventricular hypertrophy and cardiovascular mortality by race and ethnicity. Am J Med. 2008;121 (10):870–875. doi: 10.1016/j.amjmed.2008.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bahrami H, Kronmal R, Bluemke DA, et al. Differences in the incidence of congestive heart failure by ethnicity: the multi-ethnic study of atherosclerosis. Arch Intern Med. 2008;168(19):2138–2145. doi: 10.1001/archinte.168.19.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piatt EA, Pollak MN, Rimm EB, et al. Racial variation in insulin-like growth factor-1 and binding protein-3 concentrations in middle-aged men. Cancer Epidemiol Biomarkers Prev. 1999;8(12):1107–1110. [PubMed] [Google Scholar]
- 17.Tricoli JV, Winter DL, Hanlon AL, et al. Racial differences in insulin-like growth factor binding protein-3 in men at increased risk of prostate cancer. Urology. 1999;54(1):178–182. doi: 10.1016/s0090-4295(99)00129-6. [DOI] [PubMed] [Google Scholar]