

Abstract
Recent findings have focused attention on the molecular consequences of the microenvironment in tumor progression, but events occurring in cancer cells themselves in response to their ambient conditions remain obscure. Here, we identify receptor activator of nuclear factor κB ligand (RANKL) as a microenvironment-specific factor essential for tumorigenesis in vivo, using head and neck squamous cell carcinoma (HNSCC) as a model. In human HNSCC tissues, RANKL is abundantly expressed, and its expression level correlates with the histological grade of differentiation. RANKL levels are significantly higher in poorly differentiated SCCs than in well or moderately differentiated SCCs. In contrast, all HNSCC cell lines tested displayed extremely low RANKL expression; however, RANKL is efficiently up-regulated when these cell lines are inoculated in the head and neck region of mice. RANKL expression is restored in a microenvironment-specific manner, and cannot be observed when the cells are inoculated in the hindlimbs. Forced expression of RANKL compensates for tumor growth in the hindlimb milieu, promotes epithelial mesenchymal transition, and induces tumor angiogenesis, in a manner independent of vascular endothelial growth factor (VEGF). These results implicate RANKL expression causatively in tumor growth and progression in HNSCC in vivo. RANKL may provide a novel functional marker for biological malignancy and a therapeutic target based on the specific nature of the microenvironment.
The microenvironment (ie, the sanctuary in which a tumor originates) plays a critical role in tumor initiation and progression; it is created by a complex relationship between tumor cells and their surrounding tissues consisting of extracellular matrices, extracellular molecules, and host cells.1–7 Knowledge of the complicated interplay within these niches is still limited, however. In particular, the exact mechanism of how the host cells that comprise normal stroma are altered during tumor progression and how they reciprocally influence tumor cells during tumorigenesis is poorly understood. In addressing these fundamental issues, we ultimately envisage development of therapeutic strategies targeted at specific interactions between the tumor and its microenvironment.
Given that the head and neck region is an environment challenged by a large variety of insults, including pathogens, foods, and chemicals, the relationship between cancer cells and inflammatory stroma might be of particular importance for malignancies arising there. Head and neck cancers, >90% of which are squamous cell carcinomas (SCCs),8 represent approximately 6% of all new cancers in the United States,9 and consistently rank as one of the top 10 cancers worldwide.10 Of more concern is that the incidence of head and neck cancer appears to be increasing in many parts of the world.11–14 Head and neck squamous cell carcinoma (HNSCC) is characterized by a high degree of local invasiveness and a high rate of metastasis to the cervical lymph nodes.15 Survival of patients with HNSCC has not improved in the last 40 years, despite recent advances in surgical procedures and the availability of new chemotherapeutic agents. In addition, surgical treatment results in significant functional and cosmetic deficits. It is important, therefore, to develop conservative therapeutics, and identification of markers for HNSCC aggressiveness would be useful in deciding upon the most suitable treatment for each patient from among therapeutic options.16
As with other cancers, most of the deaths from head and neck cancer are accounted for by local invasion and distant metastasis. The landmark of carcinoma progression during the invasive and metastatic phase is epithelial cell plasticity and dedifferentiation, which is similar to the epithelial-mesenchymal transition (EMT) that occurs during embryonic development. In the EMT process, cells undergo a switch from a polarized, epithelial phenotype to a motile mesenchymal phenotype.17–20 Loss of epithelial cell polarity and acquisition of motility results from the disappearance of cell junction adherence molecules, reorganization of cytoskeleton, and redistribution of organelles.21,22 Uncovering the mechanism for and identifying a marker or markers of EMT could lead to strategies for predicting tumor progression and possibly for therapeutic intervention. This goal is complicated, however, by the diversity of molecular mechanisms contributing to the plasticity of epithelial cells in different tissues,21 especially in cancer tissues in vivo.
We have previously reported that parathyroid hormone-related protein (PTHrP) promotes malignant conversion of head and neck cancers in a paracrine or autocrine manner.23 This finding raises the possibility that PTHrP induces the expression of receptor activator of nuclear factor-κB ligand (RANKL), a member of the tumor necrosis factor (TNF) family, in a manner analogous to that of osteoblasts.24–26 Here, we report that RANKL is preferentially expressed in poorly differentiated HNSCC and plays a critical role in tumor progression in vivo in a microenvironment-specific fashion. With the use of RANKL-expressing cancer cells, RANKL expression is revealed to induce poorly differentiated histology, EMT, and tumor angiogenesis, presumably in a vascular endothelial growth factor (VEGF)-independent manner. In view of the fact that tumors cannot exist outside of their respective microenvironments, these findings highlight RANKL and its downstream signaling as an in vivo specific marker of tumor progression and an attractive therapeutic target in HNSCC.
Materials and Methods
Cell Culture
Human head and neck squamous cell carcinoma (HNSCC) cell lines HSC-2 (JCRB0622), HSC-3 (JCRB0623), HSC-4 (JCRB0624), SAS (JCRB0260), and Ca9-22 (JCRB0625) were obtained from the Japanese Collection of Research Bioresources cell bank (JCRB, Osaka, Japan). Human gingival fibroblasts (CRL-1740) and murine leukemic monocytes/macrophage cell line RAW 264.7 cells (TIB-71) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Except for RAW 264.7 cells, all cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS; complete DMEM) at 37°C under a humidified atmosphere containing 5% CO2. RAW 264.7 cells were maintained in RPMI 1640 medium supplemented with 10% FBS. Human umbilical vein endothelial cells (HUVECs) were obtained from Lonza (Walkersville, MD) and were maintained in complete endothelial basal medium (EBM-2; Lonza).
Antibodies and Reagents
Antibodies to RANKL, TCF8 (encoded by the ZEB1 gene, also known as ZEB1 or EF1), and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Flag (M2) was obtained from Stratagene (La Jolla, CA) and anti-E-cadherin or anti-N-cadherin antibodies were obtained from BD Biosciences Pharmingen (San Diego, CA). In immunohistochemical (IHC) analysis, additional monoclonal antibodies against E-cadherin (Zymed/Invitrogen, Carlsbad, CA) and N-cadherin (Takara Bio, Otsu, Japan) were used. Anti-CD31 was obtained from Abcam (Cambridge, UK) and Slug antibodies were from Cell Signaling Technology (Danvers, MA). Human recombinant osteoprotegerin (OPG; TNFRSF11B)/Fc chimera, an anti-VEGF antibody, and a human VEGF enzyme-linked immunosorbent assay kit were from R&D Systems (Minneapolis, MN). Human recombinant RANKL and VEGF were from PeproTech (Rocky Hill, NJ).
Ethics
Tumor tissues for this study were from patients who had signed a written informed consent document. We also obtained approval from the Institutional Review Board of Hokkaido University Hospital.
RNA Isolation and PCR
Total RNA isolation, first-strand cDNA synthesis, and RT-PCR were performed as described previously.27 The sequences for primers used are given in Table 1. The RT-PCR was performed using a thermal cycler, as follows: denaturation at 94°C for 30 seconds, annealing at 58°C (for RANKL) or 60°C (for GAPDH) for 30 seconds, extension at 72°C for 30 seconds, and a final incubation at 72°C for 10 minutes. RT-PCR products were subjected to 1% agarose gel electrophoresis, stained with ethidium bromide, and detected using an image analyzer hardware (Atto, Tokyo, Japan). Quantitative real-time PCR (qPCR) was performed as described previously,28 using a StepOne real-time PCR system (Applied Biosystems, Foster City, CA) and the primers used are given in Table 1. Data were normalized by the expression level of GAPDH and are expressed as fold increase relative to control. Of note, all primers except those for mouse VEGF were designed to amplify human mRNAs.
Table 1.
Primers Used for PCR
| Target transcripts | Direction | Sequence |
|---|---|---|
| RANKL⁎ | Forward | 5′-TGGCACTCACTGCATTTATAGAATT-3′ |
| Reverse | 5′-AGTTGAAGATACTCTGTAGCTAGGT-3′ | |
| RANKL† | Forward | 5′-CGTTGGATCACAGCACATCAG-3′ |
| Reverse | 5′-GCTCCTCTTGGCCAGATCTAAC-3′ | |
| E-cadherin† | Forward | 5′-TCCATTTCTTGGTCTACGCC-3′ |
| Reverse | 5′-CACCTTCAGCCATCCTGTTT-3′ | |
| N-cadherin† | Forward | 5′-GTGCCATTAGCCAAGGGAATTCAGC-3′ |
| Reverse | 5′-GCGTTCCTGTTCCACTCATAGGAGG-3′ | |
| TCF8† | Forward | 5′-GCTAAGAACTGCTGGGAGGAT-3′ |
| Reverse | 5′-TCCTGCTTCATCTGCCTGA-3′ | |
| Slug† | Forward | 5′-TGGTTGCTTCAAGGACACAT-3′ |
| Reverse | 5′-GTTGCAGTGAGGGCAAGAA-3′ | |
| Snail† | Forward | 5′-GCTGCAGGACTCTAATCCAGA-3′ |
| Reverse | 5′-ATCTCCGGAGGTGGGATG-3′ | |
| Twist† | Forward | 5′-GGCATCACTATGGACTTTCTCTATT-3′ |
| Reverse | 5′-GGCCAGTTTGATCCCAGTATT-3′ | |
| Human VEGF⁎ | Forward | 5′-GGCTCTAGATCGGGCCTCCGAAACCAT-3′ |
| Reverse | 5′-GGCTCTAGAGCGCAGAGTCTCCTCTTC-3′ | |
| Mouse VEGF⁎ | Forward | 5′-ACATCTTCAAGCCGTCCTGTGTGC-3′ |
| Reverse | 5′-AAATGGCGAATCCAGTCCCACGAG-3′ | |
| GAPDH⁎† | Forward | 5′-GAAATCCCATCACCATCTTCCAGG-3′ |
| Reverse | 5′-CATGTGGGCCATGAGGTCCACCAC-3′ |
Except for mouse VEGF, all primers are designed to amplify human mRNAs specifically.
RT-PCR.
qPCR.
Pathological Examination and IHC
Formalin-fixed, paraffin-embedded tissue sections (4-μm thick) of head and neck cancer samples were deparaffinized and rehydrated. These deparaffinized sections were stained with H&E by the conventional method. Histological classifications were performed by two pathologists (M.S. and Y.O.) independently, according to two common sets of criteria for SCC. The first set is according to grades of differentiation, in which histological differentiation is divided into well differentiated (stratified squamous cell nest ≥50%), poorly differentiated (<5%), and moderately differentiated SCCs (the remainder). The second is the Yamamoto-Kohama classification, a histological grading of mode of invasion, in which tumor tissues are categorized into four groups: 1 = well-defined borderline; 2 = cords, less marked borderline; 3 = groups of cells, no distinct border line; and 4 = diffuse invasion.29
The sections were also immersed in 10 mmol/L citrate buffer (pH 6.0) and heated in a pressure cooker for antigen retrieval, followed by incubation in 3% H2O2 peroxidase blocking solution. For RANKL staining, the specimens were treated with 100 mmol/L glycine solution (pH 3.0) for 20 minutes before the blocking step. After incubation in 1% bovine serum albumin blocking solution for 30 minutes, the sections were incubated with a primary antibody for RANKL (FL-317), E-cadherin (4A2 C7), N-cadherin (M142), or CD31 (ab28364) for 1 hour, followed by further incubation with the labelled polymer (EnVision+ System-HRP, Dako, Carpinteria, CA). Microscopic observation was performed after counterstaining with hematoxylin.
To semiquantify RANKL expression, staining intensity, evaluated by two pathologists (M.S. and Y.O.) independently, was categorized into five groups: 1 = no detectable immunoreactivity; 2 = weak staining; 3 = moderate staining; 4 = moderate to intense staining; 5 = intense staining. The staining intensity of the surrounding tissue was used as a basal-level reference.
Immunoblotting
Cells were lysed in a solution containing 10 mmol/L Tris-HCl (pH 7.4), 5 mmol/L EDTA, 150 mmol/L NaCl, 10% glycerol, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 50 mmol/L NaF, 1 mmol/L Na3VO4, and complete (EDTA-free) protease inhibitor (Roche, Indianapolis, IN) for 20 minutes on ice and clarified by microcentrifugation at 18,000 × g for 10 minutes at 4°C. Supernatants were subjected to SDS-PAGE, and separated proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA). The membranes were incubated with primary antibodies, followed by horseradish peroxidase-labeled secondary antibodies. Signals were developed using enzymatic chemiluminescence Western blotting detection reagent (GE Healthcare, Chalfont St. Giles, UK; Piscataway, NJ) and detected using an LAS-1000UVmini image analyzer (Fujifilm, Tokyo, Japan).
Establishment of RANKL-Expressing Cancer Cells
Full-length cDNA for human RANKL was kindly provided by Dr. Hiroshi Takayanagi (Tokyo Medical and Dental University, Tokyo, Japan) and was subcloned into the XhoI and NotI site of a pCXN2-Flag expression vector.30
HSC-3 cells were then transfected with pCXN2-Flag-RANKL or its control vector without RANKL expression, with the use of Fugene HD reagent (Roche). Starting at 2 days after transfection, the cells were cultured in complete DMEM containing 0.5 mg/mL G418 (Sigma-Aldrich). After 10 days, the resistant cells were together harvested and cultured for 7 days more. Stably transfected cells, in which RANKL expression was examined by RT-PCR and immunoblotting analyses, were maintained in culture media supplemented with 0.2 mg/mL G418.
In Vivo Tumor Formation in Nude Mice
Mouse breeding and experiments were approved by the institutional animal care and experiment committee of Hokkaido University. Nude mice (BALB/cA Jc1 nu/nu) were injected with HNSCC cells and control or RANKL-expressing HSC-3 cells in their muscles in masseter or hindlimb regions. Of note, the masseter region is one of the established sites for an oral cancer orthotopic model,31–33 and the hindlimb region was chosen as a muscle existing far from the oral tissue. Total RNA from developed tumors was isolated using the RNeasy mini kit (Qiagen, Valencia, CA) and was analyzed as described above (under RNA Isolation and PCR). Proteins were extracted through lysis as described above (under Immunoblotting). Formalin-fixed paraffin sections were also prepared and stained with H&E by conventional methods.
Osteoclastogenesis
RAW 264.7 cells and RANKL-expressing or control HSC-3 cells were cocultured for 6 days. The cells were fixed with 8% glutaraldehyde and subjected to tartrate-resistant acid phosphatase (TRAP) staining as described previously.33 As a positive control for TRAP staining, RAW 264.7 cells were also cultured in the presence of 100 ng/mL RANKL for 4 days. RAW 264.7 cells were cultured in a 96-well plate. After 24 hours, cancer cells stained with Hoechst 33342 fluorescent dye (Molecular Probes, Eugene, OR) were added on a RAW 264.7 cell monolayer and allowed to adhere for 30 minutes. The medium was then removed, and the adherent cells were quantified by measuring the fluorescence at 480 nm with an excitation wavelength of 375 nm.
Cell Proliferation and Wound-Healing Assays
The cell proliferation was measured by counting every other day for 8 days after 5 × 104 cell plating. A wound-healing assay was performed as described previously.23 Briefly, confluent cells were wounded by scraping with a P200 pipette tip. Cell movements were then observed by phase-contrast microscopy with a 10× objective lens.
Three Dimensional Culture and Colony Formation in Gels
Collagen gel and Matrigel (BD Biosciences–Discovery Labware, Bedford, MA) cultures were performed essentially as described previously,34 with some modifications and according to the manufacturer's protocol. Briefly, cells (2 × 104) were resuspended in 0.5 mL complete DMEM containing 0.3% collagen (type I-A; Nitta Gelatin, Osaka, Japan), and plated on a 12-well dish. After the collagen solution had gelled, 1 mL complete DMEM was added to each well and changed every 7 days. Alternatively, the cells were resuspended in 0.8 mL Matrigel, and plated on a 24-well dish. After the Matrigel had gelled, 0.5 mL complete DMEM was added to each well and changed every 7 days. After 21 days, colonies were photographed.
Endothelial Cell Migration Assay
The chemotaxis assay was performed using 24-well cell culture inserts with 8.0-μm pores (Nunc, Kamstrupvej, Denmark), as described previously.28 Conditioned media obtained were added as a chemoattractant into the lower chamber, and HUVECs (1.5 × 104) were seeded in the upper chamber. In some experiments, osteoprotegerin (100 ng/mL) or an anti-VEGF antibody (1 μg/mL) was added to the medium. Recombinant human VEGF (10 ng/mL) was also used as a control. After 24 hours incubation at 37°C, transferred HUVECs to the lower surface of filters were fixed and stained by 0.2% crystal violet, and the cell number in randomly selected fields was counted. Levels of secreted VEGF in conditioned media were analyzed by enzyme-linked immunosorbent assay according to the manufacturer's recommendations.
Statistical Analysis
All data, unless otherwise specified, are expressed as means ± SD; data were subjected to one-way analysis of variance, followed by comparison using a Student's t-test, a Mann-Whitney U-test, or a Spearman's test to evaluate the difference between the samples. A P value of <0.05 was considered significant in each test.
Results
RANKL Expression in Human Head and Neck Cancer Tissues and Cell Lines
To explore the possible involvement of RANKL in HNSCC progression, we first examined the expression of RANKL mRNA by qPCR in 20 human HNSCC samples, including those in the tongue and the gingiva. In all cases, high expression (from six- to 123-fold) was observed, relative to human gingival fibroblasts (Figure 1A). Next, to clarify the relationship between RANKL expression and HNSCC progression, HNSCC samples were categorized into various groups, based on difference in clinical staging or histological differentiation, and RANKL expression levels were compared across the groups using nonparametric analyses. The expression level of RANKL was positively correlated with the histological grade of differentiation (Figure 1B), but not with tumor-node-metastasis TNM staging (ρ = 0.0264, P = 0.497). Because factors related to TNM status can be affected by the phase at which patients are diagnosed, this result does not rule out the possibility that RANKL is implicated in progression and biological malignancies of HNSCC. Indeed, when we used the Yamamoto-Kohama classification, which is based on histological architecture and mode of invasion,29 strikingly high expression was observed in a YK-4 group, a diffuse invasive type (Figure 1C).
Figure 1.
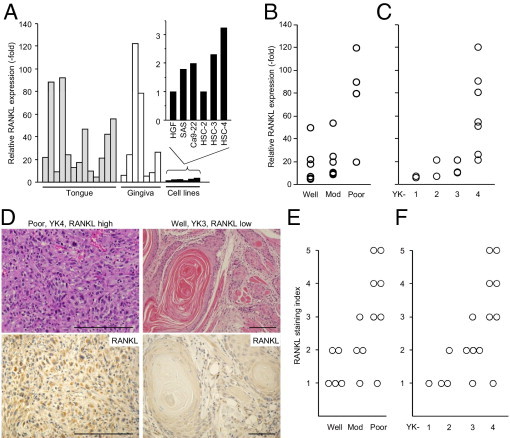
Expression of RANKL in human head and neck cancers. A: The mRNA levels of RANKL in surgical specimens of human head and neck cancers and HNSCC cell lines were analyzed by qPCR. Human gingival fibroblasts (HGF) were used as a control. B: The expression level of RANKL mRNA in well, moderately, and poorly differentiated tumors in each tumor was plotted. Spearman's test was used to evaluate the difference between samples (ρ = 0.629, P < 0.05). C: The expression level of RANKL mRNA was plotted according to invasion mode, categorized into four groups according to the Yamamoto-Kohama (YK) classification (ρ = 0.876, P < 0.01). D: Typical histology is shown for tumors with poorly differentiated SCC (left) and well-differentiated SCC (right); expression levels of RANKL were also analyzed by IHC staining using anti-RANKL antibody. Scale bars = 100 μm. E and F: Staining intensity for RANKL was categorized into five groups and plotted against grades of differentiation (E) (ρ = 0.705, P < 0.01) and invasion mode (F) (ρ = 0.756, P < 0.01). Criteria for differentiation, invasion mode, and staining intensity are described under Materials and Methods.
When samples were available for histological examination (n = 16), IHC analysis was also performed to further evaluate the role of the RANKL protein in HNSCC malignancy. Poorly differentiated SCC (YK-4), in which atypical cancer cells diffusely invaded surrounding tissues, expressed abundant RANKL proteins (Figure 1D), whereas the level was low in well-differentiated SCC (YK-3), in which tumor cells showed expanded growth with obvious keratinization (Figure 1D). Statistical analyses also revealed that RANKL expression was correlated with the histological grade of differentiation and mode of invasion (Figure 1, E and F). Thus, the expression level of RANKL is intimately associated with the grade of histological differentiation of HNSCC. Nevertheless, and to our surprise, none of the tested HNSCC cell lines (namely, HSC-2, HSC-3, HSC-4, SAS, and Ca9-22) displayed such abundant RANKL expression as that observed in vivo, albeit the expression in these cell lines, except for HSC-2, is higher than that in human gingival fibroblasts (∼3.2-fold) (Figure 1A). In particular, although the HSC-2, HSC-3, and SAS cell lines are established from moderately to poorly differentiated SCC with aggressive invasiveness, and in fact displayed similar characteristics when the cells were inoculated into murine oral tissue (see next section), the RANKL expression level in these cell lines might be repressed under culture conditions.
Environment-Dependent Expression of RANKL
We therefore hypothesized that repressed RANKL expression under culture conditions is due to a lack of environmental cues that are required for the maintenance of its expression. To test this possibility, HSC-3 cells were inoculated into the masseter muscles of mice (one of the established sites for a head and neck cancer orthotopic model),31–33 and the amount of RANKL mRNA was analyzed by RT-PCR and qPCR using a primer set specific for human RANKL. As expected, human RANKL expression in all tumor tissues tested (n = 3) was dramatically augmented, compared with cultured control cells (Figure 2, A and B). RANKL protein was also up-regulated in vivo, compared with culture conditions (Figure 2A). Moreover, when all other available HNSCC cell lines (ie, HSC-2, HSC-4, SAS, and Ca9-22) were inoculated into mouse masseter regions, HSC-2 and SAS could form obvious tumors, and RANKL up-regulation was observed in these tumors (Figure 2B). Histological examination revealed that HSC-3 and SAS displayed poorly differentiated SCC, whereas HSC-2 showed moderate differentiation (Figure 2C), in accordance with our clinicopathological findings that RANKL expression correlated with histological grade and invasion mode (Figure 1). In addition, consistent with the established role for RANKL in bone resorption,35–37 we could also observe bone destructive lesions accompanied by the accumulation of TRAP-positive, mature osteoclasts (Figure 2D), confirming that inoculated HNSCCs expressed functional RANKL protein.
Figure 2.
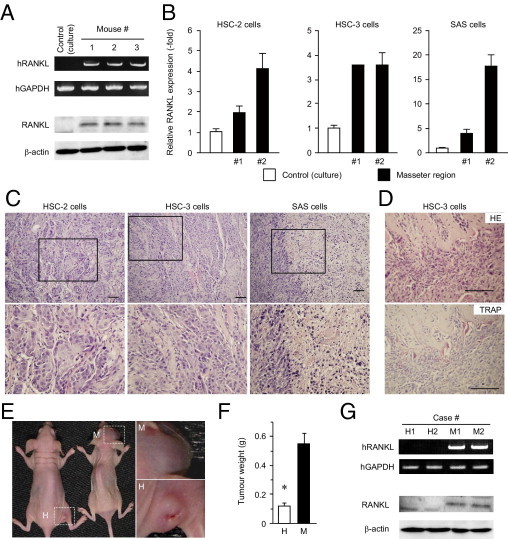
Induction of RANKL expression in the head and neck environment. A: Human head and neck cancer cells (HSC-3) were inoculated into nude mice and allowed to form tumors. Expression levels of RANKL mRNA (upper rows) and protein (lower rows) were analyzed by RT-PCR and immunoblotting, respectively. Cells under culture condition before inoculation to mice were used as a control. B: Cells of the HNSCC lines were inoculated into nude mice and allowed to form tumors. RANKL mRNA levels were evaluated by qPCR. Of the cell lines tested, HSC-2, HSC-3, and SAS cells formed tumors. Cells under culture conditions before inoculation were used as a control (open column). C and D: The tissues of tumors formed by the three cell lines were subjected to histopathological examination under H&E staining (HE) (C and D, top) and tartrate-resistant acid phosphatase staining (TRAP) (D, bottom) are shown. Scale bars = 100 μm. E–G: HSC-3 cells were injected into the masseter (M) or hindlimb (H) region of the mice. After 28 days, the tumors that formed were photographed (E) and weighed (F) (*P < 0.05). Expression levels of RANKL mRNA and protein in the masseter region (M1 and M2) or the hind leg region (H1 and H2) of nude mice were analyzed by RT-PCR (G, upper rows) and immunoblotting (G, lower rows).
To assess the contribution of the oral environment to RANKL expression and subsequent tumor formation, HSC-3 cells were also injected into the muscle of hindlimbs, a location selected in analogy to the orthotopic site (intramuscular) and for its distance from the head and neck region. Tumors formed in the hindlimbs were significantly smaller than those in the masseter region (Figure 2, E and F). In parallel with tumor weight, RANKL was detected in the masseter region tumors, but not in the hindlimb tumors, at both mRNA and protein levels (Figure 2G). Essentially similar results were obtained when HSC-2 and SAS cells were used (data not shown). Thus, RANKL expression requires the orthotopic environment and correlates with tumor formation ability.
RANKL Expression Accelerates Tumor Formation
Because none of the HNSCC cell lines expressed RANKL plentifully, compared with the in vivo condition, we established HNSCC cell lines that stably express RANKL, to further confirm the role for RANKL in HNSCC tumor formation. Of the cell lines that were able to form tumors in the masseter region (Figure 2), we used HSC-3 cells, which display poorly differentiated, invasive SCC uniformly without apparent necrosis (Figure 2C). Among several established cell lines, two control vector-transfected cell lines (C1 and C2), and two RANKL-expressing cell lines (R1 and R2) were used in the following experiments. These R1 and R2 cells appeared to express sufficient amounts of RANKL mRNA and RANKL protein (Figure 3A). Moreover, in view of induction of mature osteoclasts from RAW 264.7 cells (Figure 3B) and adhesiveness to a RAW 264.7 cell monolayer (Figure 3, C and D), the expressed RANKL was apparently functional. Under these conditions, C1 and R2 cells were injected into the mouse hindlimbs in which both RANKL expression and tumor formation were abolished in parental HNSCC lines (Figure 2, E–G). To our surprise, RANKL-expressing cells achieved efficient tumor formation in the hindlimbs (Figure 4, A and B), whereas C1 cells, in which RANKL expression was not observed even after the inoculation (Figure 4, C and D), failed to form sizable tumors (Figure 4, A and B), similar to parental cells (Figure 2E). Together, these results demonstrate that RANKL expression, which ordinarily depends on the head and neck environment, has the potential to induce HNSCC formation.
Figure 3.
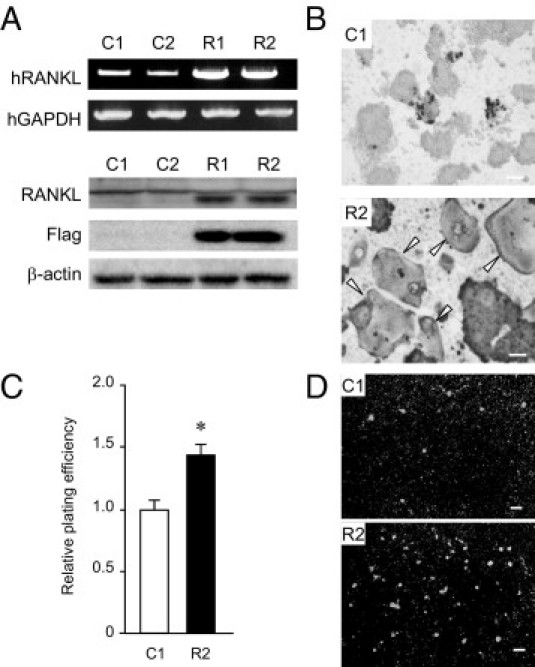
Establishment of RANKL-overexpressing cells. A: Expression of RANKL mRNA and protein were determined by RT-PCR at 36 cycles (upper rows) and immunoblotting using anti-RANKL and anti-Flag antibodies (lower rows), respectively. B: RAW 264.7 cells were cocultured with C1 (vector-transfected cells) or R2 (RANKL-expressing cells), and stained by TRAP. Arrowheads indicate mature osteoclasts. Scale bars = 300 μm. C and D: RAW 264.7 cells were precultured in 96-well plates. After 24 hours, C1 or R2 cells stained with Hoechst 33342 were added on the RAW cell monolayer and incubated for 30 minutes. After medium removal, the bound cells were quantified by measuring the fluorescence at an excitation wavelength of 357 nm. *P < 0.05 Representative photographs are shown. Scale bars = 30 μm.
Figure 4.
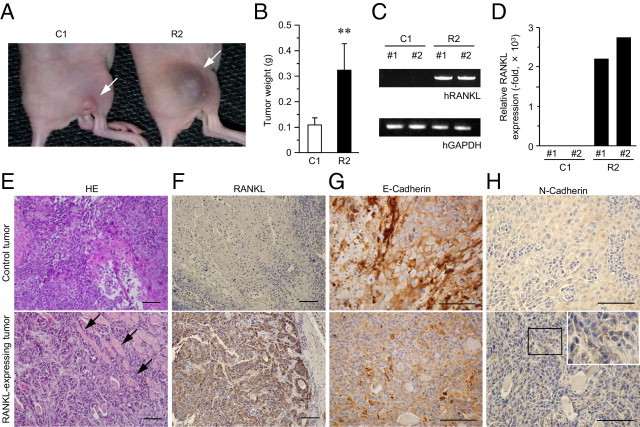
Tumor formation in hindlimbs rescued by RANKL expression. A–D: Control (C1) and RANKL-expressing (R2) cells were injected into hindlimb muscles and, after 28 days, the tumors were photographed (A) and weighed (B). **P < 0.01. RANKL mRNA in tumors formed by C1 or R2 cells was examined by RT-PCR (C) and qPCR (D). E–H: Histopathology of HNSCC in the presence or absence of RANKL expression. Pathological section from tumors formed by C1 and R2 cells were stained by H&E and photographed (E). The sections were also subjected to IHC using antibodies against RANKL (F), E-cadherin (G), and N-cadherin (H). Arrows indicate striated muscle. H, inset: Higher-magnification view of boxed area. Scale bars = 100 μm.
Given that RANKL expression positively correlated with histological grading of differentiation in human HNSCC samples (Figure 1, B–F), we examined the histology of the tumor formed by control and by RANKL-expressing cells. With H&E staining, it was revealed that RANKL-expressing, hindlimb-injected tumors exhibited more poorly differentiated and invasive characters than the control tumors (Figure 4E), consistent with results observed in human specimens (Figure 1, B–F). In these tumors, RANKL expression was detected at the protein level by IHC (Figure 4F). We further performed IHC analysis to visualize the localization of the cell-to-cell adhesion molecule E-cadherin, one of the most important hallmarks of epithelial cells. Loss of E-cadherin localization from the cell-to-cell contact sites was specifically observed in tumors formed by RANKL-expressing cells, whereas control tumors displayed typical E-cadherin localization at the cell-to-cell borders (Figure 4G). Moreover, in response to E-cadherin disappearance from the plasma membrane, intense staining for N-cadherin was observed in the tumor cell cytoplasm in RANKL-expressing tumors (Figure 4H). These findings raise the possibility that RANKL promotes loss of epithelial character; that is, it promotes EMT, a fundamental process in tumor development and progression.38
RANKL Induces Malignant Phenotypes in an in Vivo-Specific Manner
To test whether RANKL-expressing tumors in fact underwent EMT, we evaluated the expression levels of E-cadherin, N-cadherin, and several transcription factors implicated in EMT.21 However, we observed no differences in their expression in vitro (data not shown). Moreover, notwithstanding the dramatic increment in tumor formation of RANKL-expressing cells in the hindlimbs, in vitro proliferation of R2 cells was substantially slower than that of C1 cells (Figure 5A). In addition, there was no significant difference in in vitro motility (Figure 5B) or invasiveness (data not shown) between these two cell lines.
Figure 5.
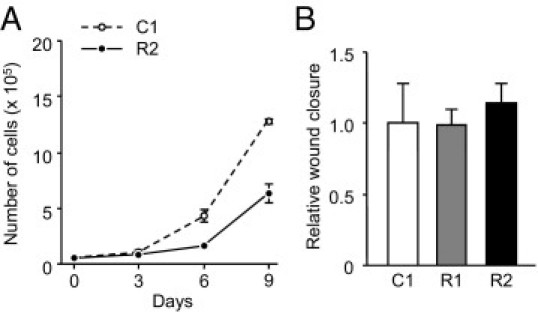
The effect of RANKL expression on cell proliferation and motility in vitro. A: The numbers of C1 and R2 cells were counted every 3 days and were plotted. B: C1, R1, and R2 cells were cultured on type I collagen-coated plates, allowed to form a cell monolayer, and subjected to a wound-healing assay. Wound closure is shown relative to C1 cells.
We therefore assumed that RANKL function differs between in vivo and in vitro conditions and, in particular, that RANKL can promote tumor growth and EMT in an in vivo-specific manner. Indeed, expression of E-cadherin was significantly decreased in RANKL-expressing tumor cells at both protein and mRNA levels, as measured by Western blotting (Figure 6A) and qPCR (Figure 6B), respectively. Accordingly, N-cadherin expression was dramatically up-regulated in tumor tissues expressing RANKL (Figure 6, A and B). An increase in N-cadherin expression was also observed in tumors arising from HNSCCs inoculated to the mouse masseter region (Figure 6C). These results thus confirmed that cadherin switching from E-cadherin to N-cadherin accrued in tumors expressing RANKL.
Figure 6.

HNSCC epithelial mesenchymal transition (EMT) promoted by RANKL. A and B: Alteration in expression levels of E-cadherin and N-cadherin. Protein and mRNA expression levels of these molecules in tumors formed by C1 and R2 cells were determined by immunoblotting (A) and qPCR (B), respectively. *P < 0.01; **P < 0.005. C: Expression levels of E-cadherin and N-cadherin mRNA in tumors formed by the indicated HNSCCs (as in Figure 2, B and C) were analyzed by qPCR. D: Expression of four transcription factors implicated in EMT was analyzed by qPCR. *P < 0.05. C1 cells were used as a control (B and D). E: Expression of Slug and TCF8 was analyzed by immunoblotting. F: C1 and R2 cells were seeded on 12-well plates at a density of 2 × 104 cells per well in 0.3 mg/mL type I collagen-containing DMEM or on 24-well plates in Matrigel, and cultured for 21 days. Representative photographs are shown. Scale bars = 100 μm.
We further examined the levels of several mRNA transcripts for transcription factors implicated in inducing EMT, including Slug, Snail, Twist, and TCF8. Of these, Slug and TCF8, but not Snail and Twist, were up-regulated at both mRNA and protein levels in RANKL-expressing tumors (Figure 6, D and E). Furthermore, when we inoculated cells in gels and observed the morphology of formed colonies, RANKL-expressing cells efficiently formed colonies with a higher invasive character (epithelial branching, an EMT-dependent event)39 than control cells in both a collagen gel and Matrigel (Figure 6F), thus indicating that RANKL promotes EMT and enhances invasiveness in a three-dimensional environment.
Tumor Angiogenesis Is Induced by RANKL
It is well established that tumor angiogenesis is critical not only for tumor growth but also for malignant properties, including invasiveness. Because RANKL-expressing tumors were grossly rich in blood vessels (R2 cells in Figure 7A), we investigated promotion of angiogenesis as another function of RANKL. IHC analysis using an antibody against CD31, a well-established marker for endothelial cells, revealed that RANKL-expressing tumors harbor significantly more abundant tumor microvessels than do control tumors (Figure 7, B and C).
Figure 7.
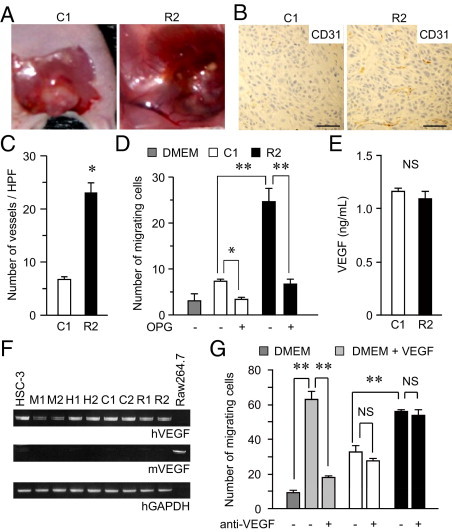
Tumor angiogenesis induced by RANKL. A: Photographs of gross examination of the same tumors as in Figure 4A. B: Sections (obtained as described for Figure 4, F–H) were stained with an anti-CD31 antibody. Representative photomicrographs are shown. Scale bars = 0.5 mm. C: The number of blood vessels visualized by CD31 was counted over three randomly selected high-power fields (HPFs); data are reported as means ± SD. *P < 0.001. D: The conditioned medium of C1 or R2 cells was used as a chemoattractant and HUVEC chemotaxis was examined in the presence or absence of osteoprotegerin (OPG), an inhibitor of RANKL. DMEM was used as a negative control. The numbers of HUVECs migrating to the lower surface of the chambers were counted and are reported as means ± SD. *P < 0.005; **P < 0.001. E: Concentrations of VEGF in conditioned media of C1 and R2 cells were determined by enzyme-linked immunosorbent assay (ELISA). F: Levels of human and mouse VEGF mRNA were determined by RT-PCR, using primers specific for each species. RAW 264.7 cells were used as a positive control for mouse VEGF. M1, M2, H1, and H2 represent samples of the tumors shown in Figure 2G. G: HUVEC migration toward the indicated conditioned media was examined in the presence (1 μg/mL) or absence of anti-VEGF antibody. DMEM and DMEM containing 10 ng/mL VEGF were used as negative and positive controls, respectively. **P < 0.01; NS, not significant.
A report indicating that endothelial cells are activated in response to RANKL via its cognate receptor RANK40 encouraged us to examine whether RANKL-expressing cells are capable of mobilizing endothelial cells. To do this, we used conditioned media from C1 and R2 cells as chemoattractants for HUVECs and evaluated their migration ability. The number of migrating HUVECs toward culture medium of C1 cells was marginally up-regulated, compared with that toward control DMEM. In contrast, conditioned medium of R2 cells substantially facilitated HUVEC migration, which was reverted in the presence of osteoprotegerin, a RANKL decoy receptor that inhibits RANK-RANKL signaling (Figure 7D). It is noteworthy that an equivalent level of VEGF was secreted in media of both C1 and R2 cells (Figure 7E). In addition, expression levels of human and murine VEGF were not altered, regardless of RANKL expression and regardless of whether cell growth conditions were in vivo or in vitro, as measured by RT-PCR using primers specific for respective species (Figure 7F). Moreover, the HNSCC- or RANKL-dependent HUVEC migration could not be hampered by an anti-VEGF antibody, which at a same concentration almost completely inhibited the migration induced by VEGF (Figure 7G). Together, these findings demonstrate that expressed RANKL promotes tumor angiogenesis in a manner independent of VEGF.
Discussion
In the present study, we established that RANKL expression is specifically detected in the intravital environment, which in turn promotes EMT, tumorigenesis, and angiogenesis of HNSCCs in vivo. These observations highlight RANKL as a biofunctional marker molecule that could be useful for both diagnosis and therapy of this disease.
One of the most serious clinical concerns accompanying HNSCC is a high potential for local invasion, frequently targeting the adjacent bone and thus requiring radical surgical procedures that put much strain on patients due to the deprivation of fundamental functions, including mastication and vocalization. In the future, after appropriate preclinical and clinical tests, the recognition of RANKL and its relevant signaling as potential targets for conservative therapy may enable us to hamper tumorigenesis and invasion by cutting the connection between the HNSCC and the tumor microenvironment. In addition to the conventional molecular targeted therapy (ie, small compounds and humanized antibodies), RANKL may constitute a better candidate for cancer immunotherapy. Several tumor antigens such as cancer-testis antigens provide specific targets for cancer cells because of their restricted expression patterns.41–43 However, in the case that these molecules are not essential for cancer cell survival, the cells can escape the challenge of the immune system by reducing the expression of the antigens. Because the expression of RANKL in response to the microenvironment is critical for HNSCC progression (Figures 2 and 4), there is a strong possibility that RANKL-RANK signaling is central to the conservative, multimodal treatment for this disease.
The epithelial-mesenchymal transition, characterized by the loss of epithelial characteristics and the acquisition of mesenchymal phenotypes, is an important event in the progression toward more invasive and metastatic cancerous cells. During EMT, tumor cells up-regulate mesenchymal markers, such as vimentin and Snail, and down-regulate epithelial markers, such as E-cadherin.22 There is a hierarchy of EMT-regulating transcription factors in SCC cells, in which Snail locates on the vertex, especially in cells with mesenchymal phenotypes.44 Several reports also suggest that, as in other malignancies, Snail is a highly potent E-cadherin suppressor.20,45–49 Most of these studies, however, used cultured cell lines and overexpression experiments in their analyses. Moreover, few common carcinoma cell types with a well-defined epithelial phenotype can complete EMT in vitro, probably because EMT is very sensitive to culture conditions, including substrates and the presence of serum.22 Indeed, Onoue et al15 reported that an SDF-1/CXCR4-dependent morphological change to a fibroblast phenotype is dependent on culture conditions, including serum starvation and low confluence. On the other hand, our present results clearly indicate that RANKL-induced EMT observed in vivo is accounted for by up-regulation of Slug and TCF8, but not Snail and Twist (Figure 6, D and E). In support of this view, in cells obtained from recurrent tumors, TCF8 and SIP1 (ZEB2) overexpression, but not Snail up-regulation, were specifically detected, relative to cells from primary tumors.48 Thus, expression of ZEB family proteins is sufficient to induce EMT in vivo. Furthermore, exogenous expression of Snail can further promote loss of E-cadherin even in these ZEB1/2-overexpressing cells. This is consistent with our observation that RANKL-expressing tumors preferentially displayed the up-regulation of N-cadherin; E-cadherin repression was not so dramatic (Figure 6B).
Potent tumorigenicity of RANKL-expressing cells is reminiscent of the properties of cancer stem cells. The invasive feature of these cells, however, appears to be dissimilar to the dormant nature of stem cells. On the other hand, recent accumulating evidence indicates that the EMT, a phase at which carcinoma cells ephemerally obtain a highly invasive mesenchymal phenotype, generates cells with stem cell attributes.50–53 Unfortunately, RANKL-expressing cells exhibit no stem cell phenotype (spheroid formation and marker expression) in vitro (data not shown). Because the effect of RANKL expression on the promotion of tumor formation and EMT is specific for in vivo conditions, and the proliferation and motility of these cells were slower than those of control cells (Figure 5), these results cannot rule out the possibility that RANKL-expressing cells might indeed behave like stem cells. In fact, our preliminary experiments revealed that the expression of the hyaluronic acid receptor CD44, recently implicated as a cell surface marker of tumor initiating cells of gastrointestinal tract and breast malignancies,54,55 is specifically observed in in vivo conditions, and that RANKL can facilitate the shedding of this molecule (T.Y., M.T., and Y.O., unpublished data). Further in-depth studies to clarify the significance of RANKL expression in vivo will be required for ablating the tumor-initiating cells, to prevent the occurrence and recurrence of cancers, which is a current goal among cancer scientists worldwide.
The mechanism by which RANKL is expressed in a manner specific to the microenvironment remains to be addressed. Given the specific character of the head and neck as a gatekeeper of organisms that is always challenged by every pathogen, inflammatory responses play an indispensable role in HNSCC tumor initiation and progression.56–60 In fact, genome-wide microarray analyses demonstrate the up-regulation of inflammation-associated molecules in this cancer.61,62 The head and neck region including the oral cavity is also known as a site abundant in a range of growth factors that are known to contribute to malignant conversion of HNSCC through activating diverse cancer-related signaling pathways.23,54,55,63 We therefore extended the investigation of RANKL-inducing agents from factors directly implicated in HNSCC malignancy (EGF or PTHrP) to those implicated in EMT (such as TGF-β), as well as inflammatory cytokines (data not shown), but failed to identify the RANKL-inducing agent. Because the morphological differences between control and RANKL-expressing cells were observed in colonies formed in a collagen gel and in Matrigel (Figure 6F), we thought that the engagement with extracellular matrices is likely attributable to RANKL induction. Unfortunately, however, no extracellular matrices per se could evoke RANKL expression in vitro (data not shown). We therefore infer that the efficient RANKL expression and the subsequent EMT promotion and tumor progression are orchestrated by the combination of intravital conditions, including growth factors, tumor-associated fibroblasts, extracellular matrices, infiltration of inflammatory cells with the production of cytokines, and cells composing vasculatures.
There are other possibilities that account for the specific observation of RANKL expression in vivo. For example, RANKL-expressing cells, which constitute a minor population under culture conditions, may preferentially survive under in vivo conditions. Given that the RANKL-expressing cells formed larger colonies than control cells, it is possible that the interaction between the cells and the specific extracellular matrices is consolidated to escape anoikis. Indeed, intimate association has been described between cell adhesion and NF-κB, a downstream effector of RANKL and its cognate receptor RANK,64–67 and recently integrin has been shown to play a role in escaping anoikis.68 Research addressing this issue should be continued. In either case, we can state that RANKL is an in vivo-specific functional marker for HNSCC malignancy.
In summary, RANKL expression was specifically observed in vivo, and expressed RANKL plays a hitherto unidentified role in tumor progression, namely inducing EMT and angiogenesis. It may be important that aggressiveness of angiogenesis in HNSCC correlated with RANKL expression level, but not the VEGF level, suggesting that VEGF-independent strategies should be taken into consideration for hampering angiogenesis in HNSCC. The function of RANKL as an EMT inducer was also specific for in vivo conditions. The present observations and the unveiling of remaining associated issues may contribute to the establishment of more rational, potent anti-cancer therapies that take into consideration the communication between cancer cells and their respective microenvironment.
Acknowledgments
We thank Jun-ichi Hamada and Masahiko Takahata for the cells, Hiroshi Takayanagi for RANKL cDNA, Jun-ichi Miyazaki for the pCXN2 vector, Noriko Toyoda for technical assistance, Stephanie Darmanin for critical reading of the manuscript, and members of our laboratories for helpful discussion.
Footnotes
Supported in part by grants-in-aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and the Japan Society for Promotion of Science, and grants from the Japan Science and Technology Agency, Akiyama Foundation, and the Mochida Memorial Foundation for Medical and Pharmaceutical Research.
References
- 1.Albini A., Sporn M.B. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 2.Joyce J.A. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513–520. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 3.Joyce J.A., Pollard J.W. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2008;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liotta L.A., Kohn E.C. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 5.Massagué J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orimo A., Weinberg R.A. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–1601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 7.Polyak K., Haviv I., Campbell I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–38. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Chen A.Y., Myers J.N. Cancer of the oral cavity. Dis Mon. 2001;47:275–361. doi: 10.1067/mcd.2001.109374. [DOI] [PubMed] [Google Scholar]
- 9.Whiteside S.T., Israël A. I kappa B proteins: structure, function and regulation. Semin Cancer Biol. 1997;8:75–82. doi: 10.1006/scbi.1997.0058. [DOI] [PubMed] [Google Scholar]
- 10.Reichart P.A. Identification of risk groups for oral precancer and cancer and preventive measures. Clin Oral Investig. 2001;5:207–213. doi: 10.1007/s00784-001-0132-5. [DOI] [PubMed] [Google Scholar]
- 11.Conway D.I., Stockton D.L., Warnakulasuriya K.A., Ogden G., Macpherson L.M. Incidence of oral and oropharyngeal cancer in United Kingdom (1990–1999)–Recent trends and regional variation. Oral Oncol. 2006;42:586–592. doi: 10.1016/j.oraloncology.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 12.Llewellyn C.D., Linklater K., Bell J., Johnson N.W., Warnakulasuriya S. An analysis of risk factors for oral cancer in young people: a case-control study. Oral Oncol. 2004;40:304–313. doi: 10.1016/j.oraloncology.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Popovtzer A., Shpitzer T., Bahar G., Marshak G., Ulanovski D., Feinmesser R. Squamous cell carcinoma of the oral tongue in young patients. Laryngoscope. 2004;114:915–917. doi: 10.1097/00005537-200405000-00025. [DOI] [PubMed] [Google Scholar]
- 14.Shiboski C.H., Schmidt B.L., Jordan R.C. Tongue and tonsil carcinoma: increasing trends in the U.S. population ages 20–44 years. Cancer. 2005;103:1843–1849. doi: 10.1002/cncr.20998. [DOI] [PubMed] [Google Scholar]
- 15.Onoue T., Uchida D., Begum N.M., Tomizuka Y., Yoshida H., Sato M. Epithelial-mesenchymal transition induced by the stromal cell-derived factor-1/CXCR4 system in oral squamous cell carcinoma cells. Int J Oncol. 2006;29:1133–1138. [PubMed] [Google Scholar]
- 16.Chang J.Y., Wright J.M., Svoboda K.K. Signal transduction pathways involved in epithelial-mesenchymal transition in oral cancer compared with other cancers. Cells Tissues Organs. 2007;185:40–47. doi: 10.1159/000101301. [DOI] [PubMed] [Google Scholar]
- 17.Hay E.D. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 18.Kalluri R., Weinberg R.A. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [Erratum appeared in J Clin Invest 2010;120:1786] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thiery J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Thiery J.P. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 21.Gotzmann J., Mikula M., Eger A., Schulte-Hermann R., Foisner R., Beug H., Mikulits W. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res. 2004;566:9–20. doi: 10.1016/s1383-5742(03)00033-4. [DOI] [PubMed] [Google Scholar]
- 22.Thiery J.P., Sleeman J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 23.Yamada T., Tsuda M., Ohba Y., Kawaguchi H., Totsuka Y., Shindoh M. PTHrP promotes malignancy of human oral cancer cell downstream of the EGFR signaling. Biochem Biophys Res Commun. 2008;368:575–581. doi: 10.1016/j.bbrc.2008.01.121. [DOI] [PubMed] [Google Scholar]
- 24.Leibbrandt A., Penninger J.M. RANK/RANKL: regulators of immune responses and bone physiology. Ann N Y Acad Sci. 2008;1143:123–150. doi: 10.1196/annals.1443.016. [DOI] [PubMed] [Google Scholar]
- 25.Mundy G.R. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 26.Roodman G.D. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 27.Inuzuka T., Tsuda M., Kawaguchi H., Ohba Y. Transcription factor 8 activates R-Ras to regulate angiogenesis. Biochem Biophys Res Commun. 2009;379:510–513. doi: 10.1016/j.bbrc.2008.12.101. [DOI] [PubMed] [Google Scholar]
- 28.Inuzuka T., Tsuda M., Tanaka S., Kawaguchi H., Higashi Y., Ohba Y. Integral role of transcription factor 8 in the negative regulation of tumor angiogenesis. Cancer Res. 2009;69:1678–1684. doi: 10.1158/0008-5472.CAN-08-3620. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto E., Kohama G., Sunakawa H., Iwai M., Hiratsuka H. Mode of invasion, bleomycin sensitivity, and clinical course in squamous cell carcinoma of the oral cavity. Cancer. 1983;51:2175–2180. doi: 10.1002/1097-0142(19830615)51:12<2175::aid-cncr2820511205>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 30.Niwa H., Yamamura K., Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 31.Cui N., Nomura T., Noma H., Yokoo K., Takagi R., Hashimoto S., Okamoto M., Sato M., Yu G., Guo C., Shibahala T. Effect of YM529 on a model of mandibular invasion by oral squamous cell carcinoma in mice. Clin Cancer Res. 2005;11:2713–2719. doi: 10.1158/1078-0432.CCR-04-1767. [DOI] [PubMed] [Google Scholar]
- 32.Nomura T., Shibahara T., Katakura A., Matsubara S., Takano N. Establishment of a murine model of bone invasion by oral squamous cell carcinoma. Oral Oncol. 2007;43:257–262. doi: 10.1016/j.oraloncology.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 33.Suda T., Jimi E., Nakamura I., Takahashi N. Role of 1 alpha,25-dihydroxyvitamin D3 in osteoclast differentiation and function. Methods Enzymol. 1997;282:223–235. doi: 10.1016/s0076-6879(97)82110-6. [DOI] [PubMed] [Google Scholar]
- 34.Montesano R., Schaller G., Orci L. Induction of epithelial tubular morphogenesis in vitro by fibroblast-derived soluble factors. Cell. 1991;66:697–711. doi: 10.1016/0092-8674(91)90115-f. [DOI] [PubMed] [Google Scholar]
- 35.Lacey D.L., Timms E., Tan H.L., Kelley M.J., Dunstan C.R., Burgess T., Elliott R., Colombero A., Elliott G., Scully S., Hsu H., Sullivan J., Hawkins N., Davy E., Capparelli C., Eli A., Qian Y.X., Kaufman S., Sarosi I., Shalhoub V., Senaldi G., Guo J., Delaney J., Boyle W.J. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi N., Udagawa N., Suda T. A new member of tumor necrosis factor ligand family, ODF/OPGL/TRANCE/RANKL, regulates osteoclast differentiation and function. Biochem Biophys Res Commun. 1999;256:449–455. doi: 10.1006/bbrc.1999.0252. [DOI] [PubMed] [Google Scholar]
- 37.Yasuda H., Shima N., Nakagawa N., Yamaguchi K., Kinosaki M., Mochizuki S., Tomoyasu A., Yano K., Goto M., Murakami A., Tsuda E., Morinaga T., Higashio K., Udagawa N., Takahashi N., Suda T. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J., Weinberg R.A. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Shintani Y., Wheelock M.J., Johnson K.R. Phosphoinositide-3 kinase-Rac1-c-Jun NH2-terminal kinase signaling mediates collagen I-induced cell scattering and up-regulation of N-cadherin expression in mouse mammary epithelial cells. Mol Biol Cell. 2006;17:2963–2975. doi: 10.1091/mbc.E05-12-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim Y.M., Lee Y.M., Kim H.S., Kim J.D., Choi Y., Kim K.W., Lee S.Y., Kwon Y.G. TNF-related activation-induced cytokine (TRANCE) induces angiogenesis through the activation of Src and phospholipase C (PLC) in human endothelial cells. J Biol Chem. 2002;277:6799–6805. doi: 10.1074/jbc.M109434200. [DOI] [PubMed] [Google Scholar]
- 41.Maio M., Coral S., Fratta E., Altomonte M., Sigalotti L. Epigenetic targets for immune intervention in human malignancies. Oncogene. 2003;22:6484–6488. doi: 10.1038/sj.onc.1206956. [DOI] [PubMed] [Google Scholar]
- 42.Nicholaou T., Ebert L., Davis I.D., Robson N., Klein O., Maraskovsky E., Chen W., Cebon J. Directions in the immune targeting of cancer: lessons learned from the cancer-testis Ag NY-ESO-1. Immunol Cell Biol. 2006;84:303–317. doi: 10.1111/j.1440-1711.2006.01446.x. [DOI] [PubMed] [Google Scholar]
- 43.Suri A. Cancer testis antigens–their importance in immunotherapy and in the early detection of cancer. Expert Opin Biol Ther. 2006;6:379–389. doi: 10.1517/14712598.6.4.379. [DOI] [PubMed] [Google Scholar]
- 44.Taki M., Verschueren K., Yokoyama K., Nagayama M., Kamata N. Involvement of Ets-1 transcription factor in inducing matrix metalloproteinase-2 expression by epithelial-mesenchymal transition in human squamous carcinoma cells. Int J Oncol. 2006;28:487–496. [PubMed] [Google Scholar]
- 45.Batlle E., Sancho E., Francí C., Domínguez D., Monfar M., Baulida J., García De Herreros A. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 46.Cano A., Pérez-Moreno M.A., Rodrigo I., Locascio A., Blanco M.J., del Barrio M.G., Portillo F., Nieto M.A. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 47.De Craene B., Gilbert B., Stove C., Bruyneel E., van Roy F., Berx G. The transcription factor Snail induces tumor cell invasion through modulation of the epithelial cell differentiation program. Cancer Res. 2005;65:6237–6244. doi: 10.1158/0008-5472.CAN-04-3545. [DOI] [PubMed] [Google Scholar]
- 48.Takkunen M., Grenman R., Hukkanen M., Korhonen M., García de Herreros A., Virtanen I. Snail-dependent and -independent epithelial-mesenchymal transition in oral squamous carcinoma cells. J Histochem Cytochem. 2006;54:1263–1275. doi: 10.1369/jhc.6A6958.2006. [DOI] [PubMed] [Google Scholar]
- 49.Yokoyama K., Kamata N., Hayashi E., Hoteiya T., Ueda N., Fujimoto R., Nagayama M. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 2001;37:65–71. doi: 10.1016/s1368-8375(00)00059-2. [DOI] [PubMed] [Google Scholar]
- 50.Hollier B.G., Evans K., Mani S.A. The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J Mammary Gland Biol Neoplasia. 2009;14:29–43. doi: 10.1007/s10911-009-9110-3. [DOI] [PubMed] [Google Scholar]
- 51.Klarmann G.J., Hurt E.M., Mathews L.A., Zhang X., Duhagon M.A., Mistree T., Thomas S.B., Farrar W.L. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin Exp Metastasis. 2009;26:433–446. doi: 10.1007/s10585-009-9242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mani S.A., Guo W., Liao M.J., Eaton E.N., Ayyanan A., Zhou A.Y., Brooks M., Reinhard F., Zhang C.C., Shipitsin M., Campbell L.L., Polyak K., Brisken C., Yang J., Weinberg R.A. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morel A.P., Lièvre M., Thomas C., Hinkal G., Ansieau S., Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagano O., Saya H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. 2004;95:930–935. doi: 10.1111/j.1349-7006.2004.tb03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ponta H., Sherman L., Herrlich P.A. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 56.Allen C.T., Ricker J.L., Chen Z., Van Waes C. Role of activated nuclear factor-kappaB in the pathogenesis and therapy of squamous cell carcinoma of the head and neck. Head Neck. 2007;29:959–971. doi: 10.1002/hed.20615. [DOI] [PubMed] [Google Scholar]
- 57.Choi S., Myers J.N. Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dent Res. 2008;87:14–32. doi: 10.1177/154405910808700104. [DOI] [PubMed] [Google Scholar]
- 58.Ferris R.L., Grandis J.R. NF-kappaB gene signatures and p53 mutations in head and neck squamous cell carcinoma. Clin Cancer Res. 2007;13:5663–5664. doi: 10.1158/1078-0432.CCR-07-1544. [DOI] [PubMed] [Google Scholar]
- 59.Lin D.T., Subbaramaiah K., Shah J.P., Dannenberg A.J., Boyle J.O. Cyclooxygenase-2: a novel molecular target for the prevention and treatment of head and neck cancer. Head Neck. 2002;24:792–799. doi: 10.1002/hed.10108. [DOI] [PubMed] [Google Scholar]
- 60.Pries R., Nitsch S., Wollenberg B. Role of cytokines in head and neck squamous cell carcinoma. Expert Rev Anticancer Ther. 2006;6:1195–1203. doi: 10.1586/14737140.6.9.1195. [DOI] [PubMed] [Google Scholar]
- 61.Ginos M.A., Page G.P., Michalowicz B.S., Patel K.J., Volker S.E., Pambuccian S.E., Ondrey F.G., Adams G.L., Gaffney P.M. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004;64:55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- 62.Ziober A.F., Patel K.R., Alawi F., Gimotty P., Weber R.S., Feldman M.M., Chalian A.A., Weinstein G.S., Hunt J., Ziober B.L. Identification of a gene signature for rapid screening of oral squamous cell carcinoma. Clin Cancer Res. 2006;12:5960–5971. doi: 10.1158/1078-0432.CCR-06-0535. [DOI] [PubMed] [Google Scholar]
- 63.Todd R., Wong D.T. Epidermal growth factor receptor (EGFR) biology and human oral cancer. Histol Histopathol. 1999;14:491–500. doi: 10.14670/HH-14.491. [DOI] [PubMed] [Google Scholar]
- 64.Lin T.H., Rosales C., Mondal K., Bolen J.B., Haskill S., Juliano R.L. Integrin-mediated tyrosine phosphorylation and cytokine message induction in monocytic cells: A possible signaling role for the Syk tyrosine kinase. J Biol Chem. 1995;270:16189–16197. doi: 10.1074/jbc.270.27.16189. [DOI] [PubMed] [Google Scholar]
- 65.Narayanan R., Higgins K.A., Perez J.R., Coleman T.A., Rosen C.A. Evidence for differential functions of the p50 and p65 subunits of NF-kappa B with a cell adhesion model. Mol Cell Biol. 1993;13:3802–3810. doi: 10.1128/mcb.13.6.3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qwarnström E.E., Ostberg C.O., Turk G.L., Richardson C.A., Bomsztyk K. Fibronectin attachment activates the NF-kappa B p50/p65 heterodimer in fibroblasts and smooth muscle cells. J Biol Chem. 1994;269:30765–30768. [PubMed] [Google Scholar]
- 67.Sokoloski J.A., Sartorelli A.C., Rosen C.A., Narayanan R. Antisense oligonucleotides to the p65 subunit of NF-kappa B block CD11b expression and alter adhesion properties of differentiated HL-60 granulocytes. Blood. 1993;82:625–632. [PubMed] [Google Scholar]
- 68.Desgrosellier J.S., Barnes L.A., Shields D.J., Huang M., Lau S.K., Prévost N., Tarin D., Shattil S.J., Cheresh D.A. An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat Med. 2009;15:1163–1169. doi: 10.1038/nm.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]