

Abstract
Clinical evidence accumulated from hemophilic patients during prophylaxis with recombinant activated factor VII (rFVIIa) suggests that the duration of the hemostatic action of rFVIIa exceeds its predicted plasma half-life. Mechanisms involved in this outcome have not been elucidated. We have investigated in vitro the redistribution of rFVIIa in platelets from healthy donors, patients with FVII deficiency, and one patient with Bernard-Soulier syndrome. Platelet-rich plasma was exposed to rFVIIa (3 to 60 μg/mL). Flow cytometry, immunocytochemistry, and coagulation tests were applied to detect and quantify rFVIIa. The hemostatic effect of rFVIIa associated to platelets was evaluated using perfusion models. Our studies revealed a dose-dependent association of rFVIIa to the platelet cytoplasm with redistribution into the open canalicular system, and α granules. Mechanisms implicated in the internalization are multiple, involve GPIb and GPIV, and require phospholipids and cytoskeletal assembly. After platelet activation with thrombin, platelets exposed rFVIIa on their membrane. Perfusion studies revealed that the presence of 30% of platelets containing FVIIa improved platelet aggregate formation and enhanced fibrin generation (P < 0.01 versus control). Our results indicate that, at therapeutic concentrations, rFVIIa can be internalized into platelets, where it is protected from physiological clearance mechanisms and can still promote hemostatic activity. Redistribution of rFVIIa into platelets may explain the prolonged prophylactic effectiveness of rFVIIa in hemophilia.
Hemophilic patients with inhibitors to coagulation factor VIII (FVIII) or factor IX (FIX) cannot benefit from prophylaxis with these coagulation factors. Recombinant activated coagulation factor VII (rFVIIa) was developed for the treatment of bleeding episodes in these patients, facilitating their clinical management.1 The rFVIIa, which has the same structure and activity as the human coagulation factor, restores hemostasis by favoring thrombin generation.2
Notably, rFVIIa has proven useful to control active bleeding episodes not only in hemophilia, but also in other hemostatic deficiencies, including platelet and coagulation disorders.1,3,4 The main mechanism by which rFVIIa exerts its hemostatic action in the control of active bleeding in congenital and acquired disorders of hemostasis could be explained by an enhanced thrombin generation at damaged vessels.5,6 Tissue factor (TF) exposed at sites of vascular damage would help to localize the hemostatic response, favoring fibrin generation and platelet recruitment in more stable thrombi.7–9
Pharmacokinetic studies performed on rFVIIa by different groups have established a half-life of 2.7 hours in adults and 1.3 hours in children.10–12 Clinical experience from exploratory phase II trials, however, suggests that the hemostatic action of rFVIIa exceeds its predicted plasma half-life in patients subjected to prophylaxis.13–15 Recent publications have highlighted the potential role of rFVIIa in prophylaxis of hemophilic patients with inhibitors.14,16,17
Although the mechanisms of action of rFVIIa in the correction of active bleeding have been widely studied, those involved in the apparent long-lasting effects of rFVIIa for prophylactic treatment remain to be clarified. It has been speculated that a portion of the rFVIIa infused into patients could diffuse to the extravascular space and, once there, become available at the site of injury.18
Several research groups have already suggested the presence of TF in platelets.19–21 Indeed, recent investigations from our own group have demonstrated that platelets possess mechanisms to internalize TF-rich microvesicles.22 Of note, one of the TF preparations used in these studies was known to contain traces of FVII.23 It was therefore hypothesized that platelets may be able to incorporate FVIIa or even TF-FVIIa complexes. Redistribution of rFVIIa into platelets could protect this factor from physiological clearance mechanisms and thus explain the prolonged hemostatic action of rFVIIa under some clinical conditions.
In the present study, we investigated the possible redistribution of rFVIIa into intravascular compartments, with specific focus on platelets. To detect the possible traffic of rFVIIa into platelets, and to evaluate its potential implications on its hemostatic capacity, we applied a combination of flow cytometry, electron microscopy, coagulometry, and perfusion techniques.
Materials and Methods
This study was approved by the Ethics Committee of the Hospital Clinic in Barcelona. (2008/4624).
Reagents and Antibodies
Whole blood was anticoagulated with citrate/phosphate/dextrose buffer (CPD) to a final concentration of citrate of 19 mmol/L, or with low molecular weight heparin (Fragmin, Pharmacia, Madrid, Spain) at a final concentration of 20 U/mL. rFVIIa was supplied as NovoSeven by Novo Nordisk (Bagsvaerd, Denmark). PBS was from Gibco BRL Life Technologies (Paisley, UK). Antibody against CD41a was from BD Biosciences (San Jose, CA). Antibody to CD62-P (clone CLBThromb/6) was from Immunotech (Marseille, France). The Alexa Fluor 488 microscale protein labeling kit, was from Invitrogen Molecular Probes (Eugene, OR). IntraPrep permeabilization kit was from Beckman Coulter (Fullerton, CA). For detection of unlabeled human rFVIIa, a polyclonal rabbit anti-human antibody was used (Agrisera, Vännäs, Sweden). The secondary antibody was a goat anti-rabbit IgG fluorescein isothiocyanate-conjugated (BD Biosciences San Jose, CA). For immunocytochemical techniques, the same antibody against FVIIa previously described was used (polyclonal rabbit anti-human antibody; Agrisera, Vännäs, Sweden), which in turn was detected with protein A coupled to colloidal gold particles 10 nm in diameter (from the Cell Microscopy Center, Department of Cell Biology, University Medical Center Utrecht, The Netherlands). Bovine serum albumin, Cohn V fraction, was from Sigma-Aldrich (Steinheim, Germany). Thrombin from human plasma used for platelet activation was from Sigma-Aldrich (Buchs SG, Switzerland). The Staclot VIIa-rTF kit was from Diagnostica Stago (Asnieres, France). The monoclonal mouse anti-human CD36 antibody (clone 185-1G2) was from LifeSpan BioSciences (Seattle, WA). The monoclonal mouse anti-human CD42b antibody (clone SZ2) was from Immunotech. For the inhibitory strategies, we used Fab fragments of the chimeric anti-GIIbIIIa MoAb (clone 7E3, ReoPro, Abciximab) from Lilly (Madrid, Spain). The recombinant human activated protein C (rhAPC, activated Drotrecogin α) was from Lilly Pharma (Giessen, Germany). Annexin V was prepared as described previously.24 Cytochalasin B, tyrphostin-47, and wortmannin were from Sigma-Aldrich (Steinheim, Germany). Fibrillar collagen type I was from Chrono-Log (Havertown, PA). The embedding kit JB-4 was from Polyscience (Warrington, PA).
Experimental Design
The present study was designed to evaluate the possible redistribution of rFVIIa into platelets. Platelet-rich plasma samples from normal donors or from four patients diagnosed with FVII deficiency (one with mild deficiency, 57% FVII, and three with severe deficiency, <2% FVII) and one patient with Bernard-Soulier syndrome were exposed for 2 hours to rFVIIa (0, 3, 6, 12, 30, and 60 μg/mL, final concentration). Presence of intraplatelet rFVIIa and membrane rFVIIa was evaluated using flow cytometry and immunolabeling techniques. The amount of FVIIa in platelets was determined with a coagulometry assay specifically designed to evaluate only the activated form of FVII (FVIIa). The ability of platelets to expose previously internalized rFVIIa was measured using flow cytometry after platelet activation with thrombin. Inhibitory strategies were introduced in several experiments, to determine the possible mechanisms through which FVIIa could be internalized into platelets. Some of these focused on the involvement of major glycoproteins, such as GPIIbIIIa and GPIb (the latter has been suggested as a possible receptor for FVIIa25). Moreover, the role of the scavenger receptor GPIV (CD36) was evaluated.
Other strategies were aimed at investigating the binding of FVIIa to anionic phospholipids or the involvement of the cytoskeleton arrangement. The involvement of a protein C receptor was also assessed, because it was suggested in endothelial cells.26–28 Finally, a combination of wortmannin and tyrphostin-47,29 was used to page the main intracellular signaling pathways involved in the internalization and traffic of particles independently of the mechanism of binding to the membrane.
The preservation of the hemostatic activity of intraplatelet FVIIa was tested using flow devices. The effects on thrombus formation and fibrin generation were assessed. Moreover, the effect of intraplatelet rFVIIa on viscoelastometric properties of the forming clot was evaluated using the ROTEM thromboelastometry analyzer (Pentapharm, Munich, Germany).
Labeling of FVIIa with Alexa Fluor 488
In some experimental procedures the use of a fluorescent-labeled rFVIIa was required. Labeling was performed with the Alexa Fluor 488 (AF488) microscale protein labeling kit according to the manufacturer's instructions. The final concentration was determined with a NanoDrop ND-1000 spectrophotometer (Isogen, IJsselstein, The Netherlands).
Isolation of Platelet-Rich Plasma and Incubation with rFVIIa
Blood samples were collected from healthy donors (n = 10) who had not taken any drug known to affect platelet function in the previous 10 days. Blood samples were anticoagulated with citrate/phosphate/dextrose (CPD) at a final citrate concentration of 19 mmol/L. Platelet-rich plasma was obtained by centrifugation at 120 × g for 15 minutes. Platelet-rich plasma aliquots were incubated with PBS, unlabeled rFVIIa (3, 6, 12, and 60 μg/mL, final concentration), or with the same concentrations of AF488-rFVIIa, for 2 hours at 37°C, in agreement with the calculated half-life of this drug. Afterward, platelet aliquots were fixed (0.3% paraformaldehyde) and washed three times with PBS and processed for flow cytometry.
Flow Cytometry Analysis
Fixed platelets were incubated with a CD41a-PerCP antibody to detect the platelet population through the glycoprotein GPIIbIIIa. Interaction of AF488-rFVIIa with platelets was evaluated directly by flow cytometry.
To assess whether unlabeled rFVIIa interacting with platelets was located at the platelet membrane or inside platelets, we applied a permeabilization procedure to arrive at the intraplatelet antigen. The method of permeabilization used has proved to be feasible and is widely used for this purpose.30,31 Labeling of FVIIa was performed in parallel for surface and intraplatelet rFVIIa, according to the instructions provided in the IntraPrep kit. To determine membrane rFVIIa, solution 2 (saponin) from the kit was replaced by PBS. Samples were incubated with 14 μg/mL of primary rabbit anti-human FVII/VIIa antibody for 30 minutes at room temperature. Because the anti-FVII/VIIa primary antibody was not fluorescent, a second step of incubation was performed with a secondary goat anti-rabbit IgG FITC-conjugated, in dark conditions (30 minutes at room temperature). This second incubation was performed with PBS for the membrane antigen or solution 2 (saponin), because saponin induces a reversible permeabilization, to reach the intracellular antigen. The final pellet was resuspended in paraformaldehyde (1%) and was kept at room temperature in darkness until analysis by flow cytometry.
Platelets were analyzed by flow cytometry using a FACScan flow cytometer (BD Biosciences, Mountain View, CA) at an excitation wavelength of 488 nm. Labeled platelets were analyzed using dual-color labeling as described previously.32 Platelets were differentiated by their characteristic forward versus side scatter. Histograms were generated with fluorescence data obtained in the logarithmic mode from 10,000 events analyzed in each sample. Data were expressed as the mean of fluorescence measured per platelet or as percentage of positive platelets. For the latter, an analytical marker was set in the corresponding fluorescence channel to define 2% of the resting platelet population with the highest membrane fluorescence at baseline level. This marker was used as a threshold to determine the proportion of platelets exhibiting immunofluorescence above this level in all subsequent samples.
Platelet Isolation
For other experimental techniques, such as immunolocalization, quantification and perfusion studies, after the incubation with PBS or rFVIIa for 2 hours at 37°C, platelet-rich plasma was washed three times with equal volumes of citrate-citric acid-dextrose (93 mmol/L sodium citrate, 7 mmol/L citric acid, and 140 mmol/L dextrose, pH 6.5, containing 5 mmol/L adenosine and 3 mmol/L theophylline) to eliminate the rFVIIa present in the solution and to obtain a platelet suspension containing only the rFVIIa internalized by platelets. The final platelet pellet was resuspended in Hanks balanced salt solution supplemented with dextrose and NaHCO3 (pH 7.2), and rested for 45 minutes at 37°C before use.
Immunolocalization on Platelet Cryosections
Platelet suspensions were chemically fixed at 4°C with a mixture of 2% paraformaldehyde and 0.1% glutaraldehyde in PBS buffer. After a washing with PBS containing 50 mmol/L glycine, cells were embedded in 12% gelatin and were infused in 2.3 mol/L sucrose. Mounted gelatin pages were frozen in liquid nitrogen. Thin sections were prepared in an ultracryomicrotome (Leica EM Ultracut UC6/FC6; Leica, Vienna, Austria). Ultrathin cryosections were collected with 2% methylcellulose in 2.3 mol/L sucrose. Cryosections were incubated at room temperature on drops of 2% gelatin in PBS for 20 minutes at 37°C, followed by 20 mmol/L glycine in PBS during 15 minutes, and 1% BSA in PBS during 15 minutes. Subsequently, the sections were incubated with anti-FVIIa (20 μg/mL) in 1% BSA in PBS for 60 minutes. After three washes with drops of 0.1% BSA in PBS for 10 minutes, sections were incubated for 20 minutes using protein A coupled to colloidal gold (10 nm in diameter), using a 1:40 dilution with 1% BSA. This was followed by three washes with drops of PBS for 10 minutes and two washes with distilled water. As a control for nonspecific binding of the colloidal gold-conjugated antibody, the primary polyclonal antibody was omitted.
Observations were performed using a transmission electron microscope (Tecnai Spirit; FEI, Eindhoven, The Netherlands) with a charge-coupled device camera SIS Megaview III.
Quantification of FVIIa in Platelet Lysates
Quantification of FVIIa was performed according to the instructions provided in the Staclot VIIa-rTF kit (Diagnostica Stago, Asnieres, France).
The Staclot is a clotting assay designed to quantify FVIIa. The test is based on a recombinant soluble tissue factor (rsTF) that possesses a cofactor function specific for FVIIa. The rsTF binds only to FVIIa, and does not activate FVII into FVIIa. We applied this assay using as sample not plasma aliquots but platelet lysates obtained through three rapid cycles of freeze/thaw of washed platelet suspensions (1.2 × 106 platelets/μL) containing or not containing rFVIIa. Washed platelet suspensions were obtained from control platelet-rich plasma aliquots incubated for 2 hours with different concentrations of rFVIIa (0, 3, 6, 12, 30, and 60 μg/mL).
Exposure of FVIIa on Platelet Membrane after Platelet Activation
To evaluate the ability of platelets previously incubated with rFVIIa to expose FVIIa on their membrane after activation with a physiological agonist: 225 μL of washed platelets previously incubated with rFVIIa were activated with 0.1 U/mL final concentration of thrombin at 37°C for 0, 1, 5, 10, and 15 minutes. Presence of FVIIa on activated platelets was evaluated using flow cytometry techniques, as described above. Concomitant exposure of CD62-P (P-Selectin), an activation-dependent marker of platelets, was also evaluated at the same time intervals.
Inhibitory Strategies
To determine the potential mechanisms through which rFVIIa is internalized, some inhibitory strategies were performed in the flow cytometry models. Some of them focused on the involvement of major glycoproteins, such as GPIb and GPIIbIIIa. For this purpose, washed platelets, at 0.3 × 106 platelets/μL, were incubated with antibodies against the glycoproteins [ie, the clone SZ2 (20 μg/mL), which binds to GPIb, or abciximab (20 μg/mL), which binds to GPIIbIIIa]. The involvement of the scavenger receptor GPIV (also known as CD36) was also evaluated, using an antibody (20 μg/mL) against this glycoprotein.
To investigate the possible binding of FVIIa to anionic phospholipids or the involvement of the cytoskeleton arrangement, platelets were incubated, respectively, with annexin V at 250 nmol/L in the presence of 2.5 mmol/L ionic calcium, or with cytochalasin B (25 μmol/L).
Moreover, because it has been described26–28 that FVIIa interacts with the endothelial cells through the endothelial protein C receptor (EPRC) and that platelets could interact with surfaces exposing activated protein C (APC), despite the lack of evidence of a receptor for APC in platelets, we performed a previous incubation using recombinant human APC (rhAPC) at 50 nmol/L to page this possible receptor before exposure to rFVIIa.
Finally, a combination of wortmannin and tyrphostin-4729 was used to page the main intracellular signaling pathways involved in the internalization and traffic of particles, independently of the mechanism of binding to the membrane. Tyrosine phosphorylation was prevented with tyrphostin-47 (100 μmol/L). Endo- and exocytosis processes were pageed using wortmannin (11.6 μmol/L). The combination of both inhibitors is known to preserve platelet stability during blood manipulation.29
In all these inhibitory strategies, platelets were incubated with the inhibitors for 30 minutes at 37°C. After incubation with cytochalasin B, platelets were washed. To determine the mechanisms involved in FVIIa internalization, inhibitory strategies were tested using two different approaches: in the presence of a supratherapeutic rFVIIa dose (30 μg/mL) and in the presence of a therapeutic dose equivalent to 6 μg/mL. The aim of this strategy was to evaluate the effect of different concentrations on the process of internalization. In the experiments with unlabeled FVIIa, platelet samples were processed after incubation for flow cytometry analysis as described above, including permeabilization to measure the intraplatelet antigen, whereas in the experiments with AF488-rFVIIa the samples were fixed in 0.3% paraformaldehyde, labeled with CD41a-PerCP, and directly analyzed. Results were expressed as mean fluorescence ± SEM (n = 3) or as percentage of inhibition.
Perfusion Studies
Adhesive and cohesive properties of platelets containing rFVIIa were assessed in perfusion models. Aliquots of blood samples anticoagulated with citrate (19 mmol/L) and with low molecular weight heparin (20 U/mL) were obtained from the same donor. Platelet-rich plasma from citrated blood was incubated with PBS or rFVIIa (6 μg/mL) for 2 hours at 37°C and washed as described above. Aliquots of washed platelets containing rFVIIa were added to blood anticoagulated with low molecular weight heparin to increase the initial platelet count by 30%.
Perfusion studies were performed with two different devices: a flat perfusion microchamber and an annular perfusion chamber.
Studies in Flat Perfusion Microchambers
Less than 1 mL of reconstituted blood was perfused through a flat microchamber using immobilized collagen as thrombogenic substrate.33 The collagen was sprayed on a Nunc Thermanox coverslip to achieve a coat concentration equivalent to 30.9 μg/cm2. Perfusions were performed at a shear rate of 600 s−1 for 5 minutes. After perfusion, surfaces were rinsed with PBS (0.15 mol/L), fixed with 0.5% glutaraldehyde (in 0.15 mol/L PBS) at 4°C for 24 hours, and stained with Toluidine Blue 0.05% for morphometric en face evaluation. Images from the perfused surfaces were obtained using a digital camera (Leica DC300F) adapted to a light microscope (Polyvar widefield photomicroscope; Reichert-Jung, Vienna, Austria). A software package (SigmaScan image analysis software, version 4.00.023; Jandel Scientific–Systat, Chicago, IL) provides a specialized program to determine the gray level histogram of each image.34 Through successive mathematical calculations, this computer program determines the percentage of surface covered by platelets (% P) versus the total area screened.
Studies in Annular Perfusion Devices
Twenty-two milliliters of reconstituted blood was perfused in an annular chamber using damaged vascular segments as thrombogenic substrate. Perfusions were performed at a shear rate of 600 s−1 for 10 minutes. After perfusion, vascular segments were rinsed with PBS (0.15 mol/L), fixed with 2.5% glutaraldehyde (in 0.15 mol/L PBS) at 4°C for 24 hours, and processed histologically for morphometric evaluation. Fibrin deposition and platelet interaction were evaluated by a light microscope connected to a computer, provided with software that automatically classifies and quantifies platelet and fibrin coverage.35 Platelet interaction was expressed as a percentage of the surface covered by platelets (% P) and as the mean area of platelet interaction (Area P; μm2). Furthermore, platelet interaction was classified as contact (C), adhesion (A), or thrombi (T). The presence of fibrin was also morphometrically quantified and expressed as a percentage of fibrin (% F) or as the mean area of the fibrin mesh (Area F; μm2).36
Thromboelastometry Studies
To evaluate the influence of intraplatelet rFVIIa on clot formation, we investigated the dynamic thromboelastography of whole-blood coagulation, using the ROTEM thromboelastometry analyzer (Pentapharm).37 This technique was performed according to the manufacturer's instructions. We used the EXTEM test, in which TF is used as activator and is sensitive to measure changes on the extrinsic pathway of coagulation. Although this technique provides several parameters, we have focused on clotting time (CT) and clot formation time (CFT), which indicate the dynamics of clot formation, as well as clot amplitude, which gives information about clot strength and stability, largely dependent on fibrinogen and platelets. The CT is defined as the time when the forming clot reaches 2 mm; the CFT is the time when this clot reaches 20 mm; and the clot amplitude after 10 minutes (A10) is a measure of clot firmness.
According to our experimental design, some modifications of the standard technique were included. Experiments were performed using blood anticoagulated with low molecular weight heparin (20 U/mL) from different healthy donors. Profiles of continuous whole-blood clot formation were recorded for 30 minutes.
Statistics
Data are reported as means ± SEM. The number of experiments for intraplatelet detection of rFVIIa by flow cytometry was at least n = 10 for healthy donors and n = 5 for patients (1 with mild FVII deficiency, 3 with severe FVII deficiency, and 1 with Bernard-Soulier syndrome). One-way analysis of variance test for independent experiments was applied when multiple comparisons were required, followed by Bonferroni's test if statistical significance was detected. Student's t-test for paired data was used for comparisons in perfusion experiments. The level of statistical significance was established at P < 0.05.
Results
Redistribution of rFVIIa into Platelets
Control Platelets
Flow cytometry analysis confirmed the interaction of rFVIIa with platelets from healthy donors and revealed a dose-dependent increase in the mean of fluorescence of samples incubated with AF488-rFVIIa (Figure 1A). A more accurate experimental procedure using permeabilization demonstrated that unlabeled rFVIIa was internalized by platelets in a dose-dependent manner (Figure 1B), whereas no changes were visible at the platelet surface (Figure 1C). All concentrations tested for rFVIIa resulted in a statistically significant dose-dependent increase relative to control samples (P < 0.05 for the lower doses and P < 0.001 for the higher doses).
Figure 1.
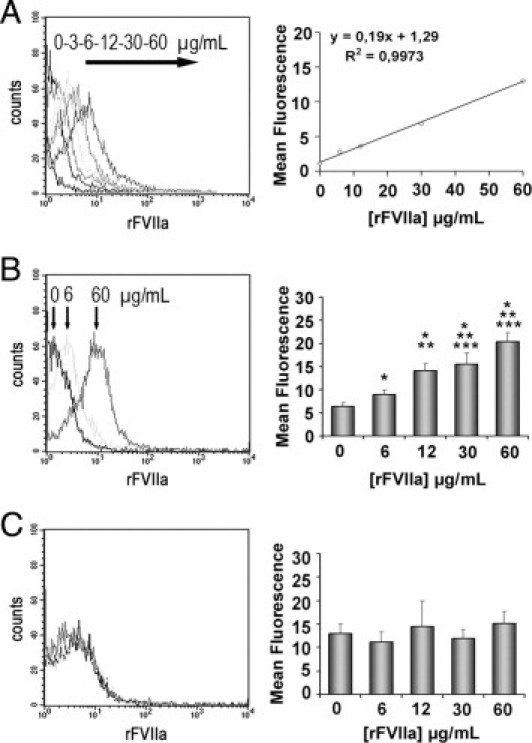
Flow cytometry detection of rFVIIa in platelets from healthy donors exposed to different doses of rFVIIa (0, 3, 6, 12, 30, and 60 μg/mL) for 2 hours. The leftpanels show the representative shift in the fluorescent profiles for the different concentrations of rFVIIa associated to the platelet population. A: Internalization of Alexa Fluor 488-labeled rFVIIa by platelets. The rightpanel shows the linear regression corresponding to the mean of fluorescence and the concentrations of AF488-rFVIIa used. B: Intraplatelet unlabeled rFVIIa detected through a two-step antibody method combined with a permeabilization procedure of the platelet membrane. The rightpanel shows a dose-dependent increase of intraplatelet mean fluorescence measured for each concentration of unlabeled rFVIIa tested. C: No statistical differences were observed in the mean of fluorescence corresponding to rFVIIa associated to the platelet membrane. *P < 0.01 versus PBS; **P < 0.05 versus rFVIIa 6 μg/mL; and ***P < 0.05 versus rFVIIa 12 μg/mL (n = 10).
FVII-Deficient Platelets
The same experiment performed with platelets from three patients diagnosed with severe FVII deficiency (<2% FVII) and one patient with moderate FVII deficiency (57% FVII versus healthy subjects), confirmed platelet rFVIIa internalization in a dose-dependent form, although in a smaller proportion (1.6-fold) than in control platelets. A significant increase in the percentage of positive events for the higher concentration of rFVIIa was also detected in these samples (Figure 2). No changes were visible after the analysis of platelet membrane-bound rFVIIa.
Figure 2.
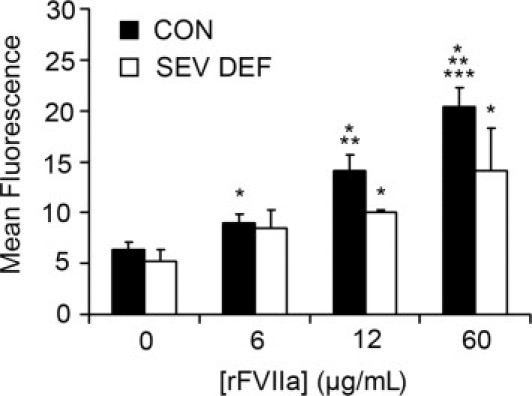
Internalization of FVIIa in platelets from patients** diagnosed with severe FVII deficiency (FVII < 2%). Data are reported as mean fluorescence associated to FVIIa by flow cytometry, in platelets from patients with severe FVII deficiency (SEV DEF, n = 3), compared with healthy donor control subjects (CON; n = 10). *P < 0.01 versus rFVIIa (0 μg/mL); **P < 0.05 versus rFVIIa (6 μg/mL); ***P < 0.05 versus rFVIIa (12 μg/mL).
Immunolocalization of Intraplatelet rFVIIa
The immunocytochemical techniques applied to platelet cryosections revealed positive immunogold labeling for rFVIIa mainly moving through the open canalicular system, in the platelet cytoplasm, and in the α granules. Labeling was also observed associated to the platelet surface (Figure 3). Quantification of positive labeling for rFVIIa in platelet cryosections resulted in a twofold increase in the presence of rFVIIa on their membranes, approximately a threefold increase related to locations of the open canalicular system, and up to fivefold increase on structures of the α granules. Despite the presence of traffic of FVIIa, platelets seem to preserve their original discoid shape, without signs of partial degranulation or activation.
Figure 3.
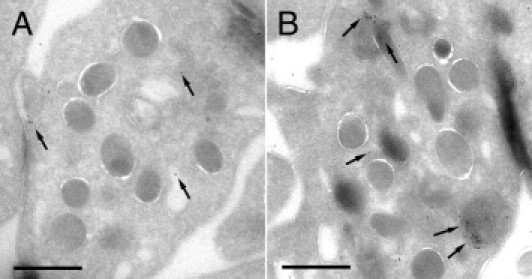
Immunostaining of FVIIa in platelets cryosections obtained from washed platelet suspensions previously incubated or not with rFVIIa 6 μg/mL for 2 hours. A: Immunoelectron micrograph showing background labeling for FVIIa (10-nm gold particles, arrows) on platelets that had not been incubated with rFVIIa. B: Micrograph corresponding to platelets incubated with rFVIIa. Positive labeling was detected for FVIIa in the membranes of the open canalicular system, in the platelet cytoplasm, and in the α granules. Labeling was also observed associated to the platelet surface after 2 hours of exposure to rFVIIa. Scale bars = 1000 nm.
Platelets not incubated with rFVIIa presented only background labeling for FVIIa, which was irregularly distributed and comparable to the labeling observed with the secondary antibody control.
Quantification of FVIIa Redistributed into Platelets
The coagulometric assessment of FVIIa in the platelet lysates revealed an increase in the activity of intraplatelet FVIIa along with the dose of rFVIIa used for incubations. Activity values of intraplatelet rFVIIa expressed as mU/mL were 0.20 ± 0.03, 1.19 ± 0.31, 2.58 ± 0.63, 8.47 ± 0.54, 21.09 ± 2.47, and 39.56 ± 5.67 for platelets incubated with rFVIIa at 0, 3, 6, 12, 30, and 60 μg/mL, respectively. The statistical significance among all of the samples was P < 0.001. Using the equivalences provided by the kit, results can be expressed as femtograms of FVIIa per platelet and therefore can be converted into nanograms of FVIIa per milliliter, considering a normal platelet count (200 × 103 platelets/μL) present in a healthy donor; as summarized in Figure 4. Of note, our results show that the amount FVIIa inside platelets after exposure to doses used in clinical practice exceeds by threefold (15.35 ng/mL) the quantity of FVIIa present in plasma (5 ng/mL) for a normal platelet count of 200 × 103 platelets/μL.
Figure 4.
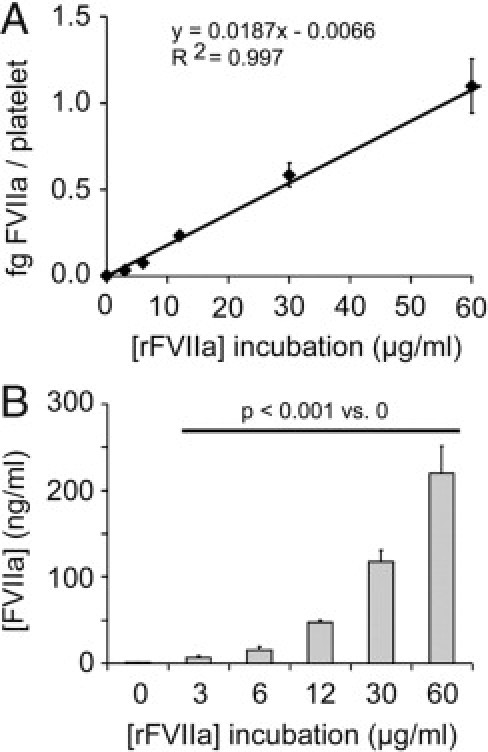
FVIIa activity associated to platelet lysates assessed by a coagulometric assay. A dose-associated increase was observed in the amount of intraplatelet FVIIa over a range of doses of rFVIIa used for incubations (0, 3, 6, 12, 30, and 60 μg/mL). Results can be expressed as femtograms of FVIIa per platelet (A), but also as nanograms of FVIIa per milliliter, considering a normal platelet count (200 × 103 platelets/μL) (B). The statistical significance among all of the samples was P < 0.001.
Exposure of FVIIa on Platelet Membrane after Platelet Activation
Platelet activation with thrombin showed that previously internalized rFVIIa was expressed on the platelet membranes after platelet activation induced by thrombin. Moreover, exposure of FVIIa on the platelet surface paralleled exposure of CD62-P, an activation marker stored also in α granules and exposed on platelet membrane after activation. In fact, we found a good correspondence between the exposure of both antigens. The decrease of levels of FVIIa after 15 minutes of activation could be also compatible with a release of the protein from the platelet membrane, as described for CD62-P. These results are summarized in Figure 5.
Figure 5.
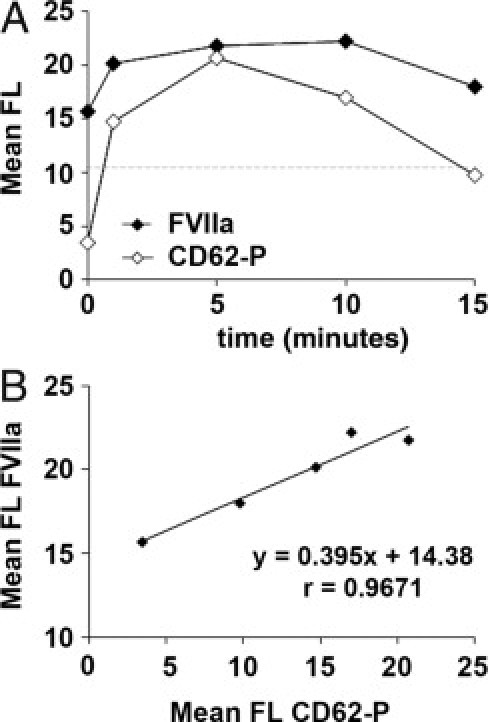
Exposure of FVIIa on platelet membrane after platelet activation with thrombin. Washed platelets previously incubated with 30 μg/mL of rFVIIa were activated with thrombin (0.1 U/mL) for 0, 1, 5, 10, and 15 minutes. A: Sequential flow cytometry analysis showing exposure of FVIIa (solid symbols) and CD62-P (open symbols) on the platelet membrane at the different time points. Results are expressed as mean fluorescence (FL). The dashed horizontal line indicates levels of mean fluorescence for FVIIa in platelets not exposed to rFVIIa. Platelet activation with thrombin resulted in the exposure of FVIIa on the platelet surface, which paralleled expression of CD62-P. B: Correlation studies between the mean of fluorescence measurements of CD62-P and FVIIa confirmed a good correspondence between the exposure of both antigens.
Evaluation of the Potential Mechanisms of Internalization
Experiments designed to investigate the mechanism through which platelets internalize FVIIa showed only partial inhibitions for the different strategies applied, except in the case of the Bernard-Soulier patient.
Experiments performed using blood samples from a patient diagnosed with Bernard-Soulier's syndrome to evaluate the role of the complex GPIb-IX-V showed fourfold reduction in the internalization of rFVIIa, compared with healthy donors (Table 1); however, the lack of GPIb did not totally page the internalization of rFVIIa into platelets. When high doses of AF488- rFVIIa (30 μg/mL) were used, incubation of the platelet suspension with an antibody against the α chain of GPIb reduced by approximately 15% the internalization of rFVIIa, reaching 20% inhibition at low doses of rFVIIa (6 μg/mL). However, the inhibition of the scavenger receptor GPIV by a specific antibody showed a 43% reduction (range, 35% to 50%) of rFVIIa associated to platelets for the lower doses of AF488-rFVIIa, whereas only a 15% inhibition was detected with the higher doses, suggesting a dose-dependent behavior.
Table 1.
Percentage of Positive Events for Intraplatelet rFVIIa in Healthy Control and Bernard Soulier Syndrome Donors
| rFVIIa (μg/mL) | Control (%) | Bernard-Soulier syndrome (%) |
|---|---|---|
| 0.0 | 2.0 | 2.0 |
| 12.0 | 20.8 | 4.7 |
| 60.0 | 82.0 | 19.6 |
Strategies directed to investigate the possible implication of platelet membrane phospholipids and cytoskeleton arrangement showed 15% reduction in the internalization of both doses of rFVIIa when anionic phospholipids were pageed. Inhibitions of approximately 25% were detected by pageing cytoskeletal arrangement with cytochalasin B. Additional studies in the presence of an excess of rhAPC revealed a partial inhibition (13% to 25%) of FVIIa internalization at high doses of rFVIIa and a 30% inhibition when low doses were used.
Of note, the maximum inhibition of internalization of rFVIIa (60%) was achieved when platelets were incubated with a combination of wortmannin and tyrphostin-47, used to page the main intracellular signaling pathways involved in the internalization and traffic of particles independently of the mechanism of binding to the membrane.
Hemostatic Activity Associated with Intraplatelet rFVIIa
Control Donors
Perfusion studies using collagen surfaces with flowing blood containing 30% of platelets previously incubated with PBS or with 6 μg/mL rFVIIa revealed a platelet-covered surface of 19.4 ± 3.4% and 28.8 ± 4.0%, respectively (P < 0.05) (Figure 6, A and B).
Figure 6.
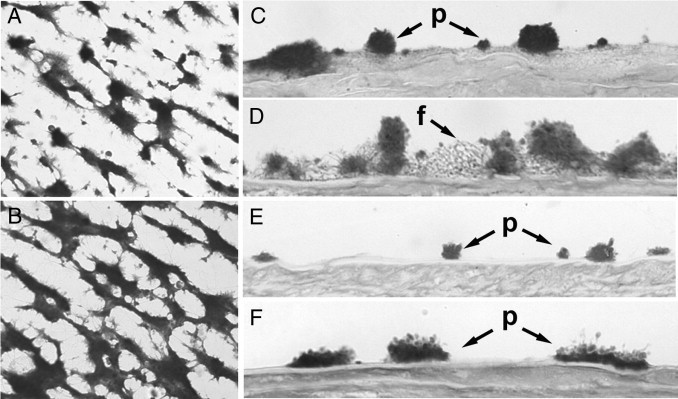
Hemostatic activity associated to intraplatelet rFVIIa. A and B: Experiments were performed by adding 30% of platelets previously incubated with PBS or with 6 μg/mL rFVIIa to blood samples. Platelets were washed to remove excess rFVIIa before being added to the perfusates. Images are representative fields of perfusion studies in microchambers using collagen surfaces with flowing blood from healthy donors containing platelets previously incubated with PBS (A) or with rFVIIa (B). C–F: Studies were performed with damaged vascular vessels adding platelets from healthy donors (C) or patients with severe FVII deficiency (E) incubated with PBS (D) or platelets incubated with rFVIIa (F). Perfused surfaces were washed, fixed and processed histologically. Micrographs were obtained in the bright field mode; original magnification, ×400. p, platelet interaction; f, fibrin mesh.
Studies performed with damaged vascular vessels resulted in a platelet coverage of 35.1 ± 6.0% and a 33.6 ± 8.8% of surface covered by fibrin for the control experiments. When platelets had been previously incubated with rFVIIa for 2 hours, the platelet coverage was 27.9 ± 7.0%. In these samples, an increase of fibrin formation was detected (46.0 ± 17.3; versus 33.6 ± 8.8%). A more detailed analysis of the fibrin net showed also a twofold increase of the mean area of the fibrin net (444.4 ± 182.6; versus 238.7 ± 78.7 μm2; P < 0.01). Micrographs representative of these results are shown in Figure 6, C and D.
FVII-Deficient Donor
Similar experiments were performed in triplicate, using blood and platelets from one patient diagnosed with severe deficiency of FVII (< 2% FVII) (Figure 6, E and F). In this patient, the additional 30% of platelets incubated with PBS resulted in 16.9% of surface covered by platelets, 1.3% corresponding to large platelet aggregates (>5 μm in height). No fibrin formation was observed. When platelet count was increased to 30% with platelets previously incubated with rFVIIa, we observed increased platelet deposition. The surface covered by platelets was 25.66%, with a marked increase of large platelet aggregates (15.78%), but no change in fibrin generation.
Thromboelastometry Studies
The presence of a proportion of platelets containing rFVIIa improved the viscoelastometry parameters during clot formation. The clotting time and the clot formation time were reduced, reaching levels of statistical significance in CT (173.8 ± 24.7 seconds versus 255.8 ± 27.7 seconds in control samples; P < 0.05), suggesting an accelerated clot formation. A moderate increase of clot firmness after 10 minutes was also observed in these experiments, although differences did not reach levels of statistical significance. These results are summarized in Figure 7.
Figure 7.
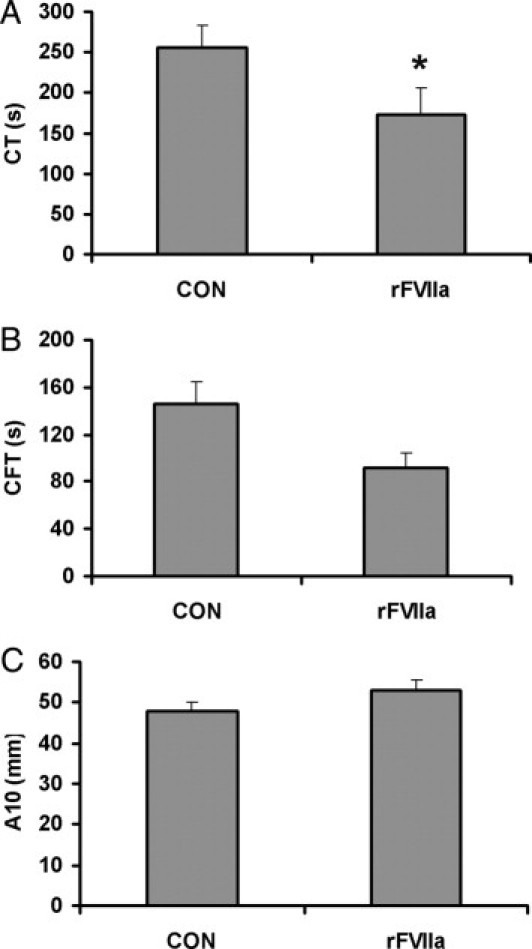
Effect of intraplatelet rFVIIa on thromboelastometric properties. Thromboelastometry experiments were performed by adding 30% of platelets previously incubated with PBS (control, CON) or with 6 μg/mL rFVIIa to blood samples. Results were obtained using the EXTEM test for clotting time (CT) (A), clotting formation time (CFT) (B), and clot amplitude (ie, firmness) measured after 10 minutes (A10) (C). Results are expressed as means ± SEM (n = 8). *P < 0.05.
Discussion
Results from the present studies demonstrate that rFVIIa can be internalized into platelets, where it will remain functional and capable of promoting hemostasis. Redistribution of rFVIIa into platelets may explain the prolonged hemostatic protection observed with this recombinant factor administered prophylactically to patients with hemophilia who have developed inhibitors.
It is well known that platelets can internalize different molecules and small particles through specific and nonspecific mechanisms.38–40 In a previous study, we demonstrated the internalization of TF-rich microparticles by platelets.22 Notably, one of the TF sources used was known to contain small amounts of FVII.23 The present data indicate that rFVIIa is internalized by platelets in a dose-dependent manner both in healthy subjects and in patients with severe FVII deficiency. To our knowledge, there have not been previous reports of FVIIa inside platelets. In 1997, Monroe and colleagues41,42 reported that rFVIIa could weakly bind to the membrane of activated platelets, where it could promote thrombin generation, but no reference was made to a possible presence of FVIIa inside platelets. Under physiological conditions, levels of FVIIa in plasma are very low (approximately 1% of the total circulating FVII). Minimal amounts of FVII become activated in vivo, and this activated form (FVIIa) has a short life in plasma.43 Once activated, FVIIa forms complexes with TF, becomes inactivated by the tissue factor pathway inhibitor (TFPI), and is rapidly cleared from the circulation. Of note, levels of rFVIIa achieved in patients subjected to therapeutic doses exceed by far those observed under physiological conditions. We believe that the elevated levels of rFVIIa attained in treated patients create a specific and physiologically unique situation. Such an unusual circumstance would explain why the presence FVIIa has not been previously reported in platelets.
Our present studies demonstrate that rFVIIa internalized by platelets is exposed after platelet activation with a physiological agonist and may promote hemostasis. Experiments with flowing blood showed that the presence of a relative proportion (30%) of platelets containing rFVIIa potentiated platelet aggregation and enhanced fibrin formation on damaged vascular areas. These observations suggest that platelets containing rFVIIa could locally promote the formation of a more effective hemostatic plug. Previous studies have demonstrated that the presence of exogenously added rFVIIa in blood from patients with different coagulopathies or thrombopathies improved fibrin generation at sites of vascular damage, but did not improve platelet aggregate formation.7,9,35,44 The thrombin generated in the previous situations influences the formation of a fibrin net and improves its structure and stability, which is not achieved at lower doses of rFVIIa.45 Notably, in our studies using blood samples from patients with FVII deficiency, addition of platelets containing rFVIIa promoted the formation of larger platelet aggregates, but did not result in an increased fibrin deposition. It is likely that the reduced plasma levels of FVIIa in these patients were insufficient to cause the generation of fibrin that occurs in healthy donors. Overall, our findings suggest that the existence of a reservoir of rFVIIa, latent inside platelets but that can be expressed on platelet activation, may locally enhance thrombin generation, promote the recruitment of platelets on the forming aggregate, and accelerate clot formation.
To investigate the mechanisms involved in the internalization of rFVIIa and to discern the contribution of passive versus metabolic mechanisms, we applied a series of inhibitory strategies. It is evident that internalization requires a previous binding of rFVIIa to the membrane. Our results suggest the contribution of several mechanisms to this initial binding. Although the most recognized mechanism of the hemostatic action of FVIIa is the binding to TF present in damaged vessels.9,35 It is also possible that platelets at the site of injury could incorporate some TF from the injured site and further bind FVIIa, improving hemostasis. Other authors have proposed that high doses of rFVIIa could also exert hemostatic action on the activated platelet membrane. The components of the membrane specifically involved have not been fully identified. Some authors have considered anionic phospholipids exposed on activated platelets as the main contributors.2,42,46 Our experiments with annexin V aimed at pageing anionic phospholipids showed a moderate effect (15% of inhibition). We cannot discard the binding of rFVIIa to anionic phospholipids as a possible mechanism, because our experiments were performed with resting washed platelets exposing a very limited proportion of anionic phospholipids. The contribution of this mechanism to the beneficial effect of rFVIIa in prophylaxis would be difficult to explain, because platelets from these patients should be expected to circulate in a resting state with very low proportion of exposed anionic phospholipids.
Other authors have considered the GPIb-IX-V complex as a potential binding site to localize FVIIa on the platelet membrane,25 and further suggested that this interaction would be involved in the enhanced thrombin generation mediated by rFVIIa on the activated platelet surface. We assessed the possible role of GPIb-IX-V complex in rFVIIa internalization using platelets from a patient with Bernard-Soulier syndrome or using a specific antibody directed toward the α chain of GPIb. Both strategies reduced, but never fully prevented, the internalization of rFVIIa. The pageage of GPIIb-IIIa, another major glycoprotein, did not affect internalization mechanisms.
Recent studies have confirmed that rFVIIa becomes redistributed into endothelial cells26–28 and still maintains its hemostatic activity after a vascular damage.47 This redistribution was mediated by endothelial cell protein C receptor (EPCR).26,27 Although platelets do not possess an equivalent for the EPCR, a recent report indicated that immobilized protein C (PC) or activated protein C (APC) support platelet adhesion and activation through mechanisms that could involve GPIbα.48 The latter finding indirectly suggests that platelets may at least express receptors that could mediate interactions with APC. In our studies, an excess of recombinant human APC in the incubation media inhibited up to 30%, but never completely pageed the rFVIIa association with platelets. It is thus evident that controversy already exists on the specificity of the mechanisms involved in the hemostatic action of rFVIIa. Interestingly, our data demonstrate that the scavenger receptor GPIV, known to be involved in the uptake of molecules or even microvesicles, has a role in the binding of rFVIIa to platelet membrane.49,50
Overall, our investigations also suggest that, although the binding of rFVIIa to the platelet membrane is contributed through different components of the platelet membrane, there is certainly a metabolic basis in the internalization process. The use of cytochalasin B resulted in a partial inhibition (25%), in accord with the results observed with a combination of wortmannin and tyrphostin-47 (60% inhibition), the most effective strategy tested. Interestingly, that last strategy blocks the main signaling pathways involved in the internalization and traffic of particles, and would act independently of the mechanism of rFVIIa binding to the platelet membrane.29 It is important to note that wortmannin also interferes with the cytoskeleton arrangement. A passive component to the binding cannot be discarded, because association of rFVIIa to platelets was not fully inhibited by the combined inhibition of the signaling pathways. Consequently, the inability of the multiple inhibitory strategies to page the binding of rFVIIa to membranes would be consistent with the fact that platelets can interact with molecules, surfaces, particles of different origins, and even with bacteria and viruses through both specific and nonspecific mechanisms.38–40
In conclusion, recent clinical reports have suggested the possibility of a prolonged hemostatic efficacy of rFVIIa in patients with hemophilia. Our present experimental studies demonstrate that rFVIIa can redistribute into platelets, where it would remain functionally active and promote hemostasis. Binding of rFVIIa to the platelet membrane is contributed by several mechanisms, but internalization of rFVIIa can be consistently reduced by strategies that prevent signaling mechanisms involved in endo/exocytosis. Redistribution of rFVIIa into other cellular compartments, such as endothelial cells or platelets, where it could be protected from plasma clearance, may explain the prolonged prophylactic action of rFVIIa in some clinical conditions. The existence of extravascular sources of FVIIa may provide new insights into the role of FVIIa in hemostasis. Our findings with FVIIa redistributing into platelets may also provide the basis for further investigations on the prolonged action of prophylactic FVIII infusions in patients with hemophilia.
Acknowledgments
We thank Montserrat Viñas, Fulgencio Navalon, Marc Pino, and Patricia Molina Moreno of the Service of Hemotherapy-Hemostasis of Hospital Clinic, and thank the staff of Servicios Cientifico-Técnicos, Universitat de Barcelona, for their technical assistance in electron microscopy.
Footnotes
Supported in part grants SAF 2009-10365, FIS CP04-00112, FIS PS09/00664 and Red HERACLES RD06/0009, from the Spanish government, and sponsored by an unrestricted grant from Novo Nordisk.
U.H. is an external advisor for Novo Nordisk. A.M.G. belongs to the Miguel Servet stabilization program from the “Direcció d'Estratègia i Coordinació del Departament de Salut de la Generalitat de Catalunya.”
References
- 1.Hedner U., Erhardtsen E. Potential role for rFVIIa in transfusion medicine. Transfusion. 2002;42:114–124. doi: 10.1046/j.1537-2995.2002.00017.x. [DOI] [PubMed] [Google Scholar]
- 2.Monroe D.M. Further understanding of recombinant activated factor VII mode of action. Semin Hematol. 2008;45(2 Suppl 1):S7–S11. doi: 10.1053/j.seminhematol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Hedner U., Ezban M. Tissue factor and factor VIIa as therapeutic targets in disorders of hemostasis. Annu Rev Med. 2008;59:29–41. doi: 10.1146/annurev.med.59.061606.095605. [DOI] [PubMed] [Google Scholar]
- 4.Hers I., Mumford A. Understanding the therapeutic action of recombinant factor VIIa in platelet disorders. Platelets. 2008;19:571–581. doi: 10.1080/09537100802406653. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman M., Monroe D.M., 3rd The action of high-dose factor VIIa (FVIIa) in a cell-based model of hemostasis. Semin Hematol. 2001;38:6–9. doi: 10.1016/s0037-1963(01)90140-4. [DOI] [PubMed] [Google Scholar]
- 6.Roberts H.R., Hoffman M., Monroe D.M. A cell-based model of thrombin generation. Semin Thromb Hemost. 2006;32(Suppl 1):32–38. doi: 10.1055/s-2006-939552. [DOI] [PubMed] [Google Scholar]
- 7.Galan A.M., Tonda R., Altisent C., Maragall S., Ordinas A., Escolar G. Recombinant factor VIIa (NovoSeven®) restores deficient coagulation: experience from an ex vivo model. Semin Hematol. 2001;38:10–14. doi: 10.1016/s0037-1963(01)90141-6. [DOI] [PubMed] [Google Scholar]
- 8.Lisman T., De Groot P.G. Mechanism of action of recombinant factor VIIa. J Thromb Haemost. 2003;1:1138–1139. doi: 10.1046/j.1538-7836.2003.00225.x. [DOI] [PubMed] [Google Scholar]
- 9.Galán A.M., Tonda R., Pino M., Reverter J.C., Ordinas A., Escolar G. Increased local procoagulant action: a mechanism contributing to the favorable hemostatic effect of recombinant FVIIa in PLT disorders. Transfusion. 2003;43:885–892. doi: 10.1046/j.1537-2995.2003.00427.x. [DOI] [PubMed] [Google Scholar]
- 10.Lindley C.M., Sawyer W.T., Macik B.G., Lusher J., Harrison J.F., Baird-Cox K., Birch K., Glazer S., Roberts H.R. Pharmacokinetics and pharmacodynamics of recombinant factor VIIa. Clin Pharmacol Ther. 1994;55:638–648. doi: 10.1038/clpt.1994.80. [DOI] [PubMed] [Google Scholar]
- 11.Girard P., Nony P., Erhardtsen E., Delair S., Ffrench P., Dechavanne M., Boissel J.P. Population pharmacokinetics of recombinant factor VIIa in volunteers anticoagulated with acenocoumarol. Thromb Haemost. 1998;80:109–113. [PubMed] [Google Scholar]
- 12.Erhardtsen E. Pharmacokinetics of recombinant activated factor VII (rFVIIa) Semin Thromb Hemost. 2000;26:385–391. doi: 10.1055/s-2000-8457. [DOI] [PubMed] [Google Scholar]
- 13.Ludlam C.A. The evidence behind inhibitor treatment with recombinant factor VIIa. Pathophysiol Haemost Thromb. 2002;32(Suppl 1):13–18. doi: 10.1159/000057294. [DOI] [PubMed] [Google Scholar]
- 14.Morfini M., Auerswald G., Kobelt R.A., Rivolta G.F., Rodriguez-Martorell J., Scaraggi F.A., Altisent C., Blatny J., Borel-Derlon A., Rossi V. Prophylactic treatment of haemophilia patients with inhibitors: clinical experience with recombinant factor VIIa in European Haemophilia Centres. Haemophilia. 2007;13:502–507. doi: 10.1111/j.1365-2516.2007.01455.x. [DOI] [PubMed] [Google Scholar]
- 15.Konkle B.A., Ebbesen L.S., Erhardtsen E., Bianco R.P., Lissitchkov T., Rusen L., Serban M.A. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost. 2007;5:1904–1913. doi: 10.1111/j.1538-7836.2007.02663.x. [DOI] [PubMed] [Google Scholar]
- 16.Yilmaz A.A., Yalcin S., Serdaroglu H., Sonmezer M., Uysalel A. Prophylaxis with recombinant-activated factor VII (rFVIIa) for minimally invasive surgery in a patient with congenital factor VII deficiency: a case report with a single-low dose of rFVIIa. Blood Coagul Fibrinolysis. 2008;19:693–695. doi: 10.1097/MBC.0b013e3282f544ff. [DOI] [PubMed] [Google Scholar]
- 17.Jiménez-Yuste V., Alvarez M.T., Martín-Salces M., Quintana M., Rodriguez-Merchan C., Lopez-Cabarcos C., Velasco F., Hernández-Navarro F. Prophylaxis in 10 patients with severe haemophilia A and inhibitor: different approaches for different clinical situations. Haemophilia. 2009;15:203–209. doi: 10.1111/j.1365-2516.2008.01915.x. [DOI] [PubMed] [Google Scholar]
- 18.Hedner U. Potential role of recombinant factor FVIIa in prophylaxis in severe hemophilia patients with inhibitors. J Thromb Haemost. 2006;4:2498–2500. doi: 10.1111/j.1538-7836.2006.02166.x. [DOI] [PubMed] [Google Scholar]
- 19.Müller I., Klocke A., Alex M., Kotzsch M., Luther T., Morgenstern E., Zieseniss S., Zahler S., Preissner K., Engelmann B. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J. 2003;17:476–478. doi: 10.1096/fj.02-0574fje. [DOI] [PubMed] [Google Scholar]
- 20.Mackman N., Tilley R.E., Key N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687–1693. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- 21.Schwertz H., Tolley N.D., Foulks J.M., Denis M.M., Risenmay B.W., Buerke M., Tilley R.E., Rondina M.T., Harris E.M., Kraiss L.W., Mackman N., Zimmerman G.A., Weyrich A.S. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J Exp Med. 2006;203:2433–2440. doi: 10.1084/jem.20061302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lopez-Vilchez I., Escolar G., Diaz-Ricart M., Fuste B., Galan A.M., White J.G. Tissue factor-enriched vesicles are taken up by platelets and induce platelet aggregation in the presence of factor VIIa. Thromb Haemost. 2007;97:202–211. [PubMed] [Google Scholar]
- 23.Orvim U., Roald H.E., Stephens R.W., Roos N., Sakariassen K.S. Tissue factor-induced coagulation triggers platelet thrombus formation as efficiently as fibrillar collagen at arterial blood flow conditions. Arterioscler Thromb. 1994;14:1976–1983. doi: 10.1161/01.atv.14.12.1976. [DOI] [PubMed] [Google Scholar]
- 24.Vanheerde W.L., Sakariassen K.S., Hemker H.C., Sixma J.J., Reutelingsperger C.P., de Groot P.G. Annexin v inhibits the procoagulant activity of matrices of TNF- stimulated endothelium under blood flow conditions. Arterioscler Thromb. 1994;14:824–830. doi: 10.1161/01.atv.14.5.824. [DOI] [PubMed] [Google Scholar]
- 25.Weeterings C., de Groot P.G., Adelmeijer J., Lisman T. The glycoprotein Ib-IX-V complex contributes to tissue factor-independent thrombin generation by recombinant factor VIIa on the activated platelet surface. Blood. 2008;112:3227–3233. doi: 10.1182/blood-2008-02-139113. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh S., Pendurthi U.R., Steinoe A., Esmon C.T., Rao L.V. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem. 2007;282:11849–11857. doi: 10.1074/jbc.M609283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.López-Sagaseta J., Montes R., Puy C., Diez N., Fukudome K., Hermida J. Binding of factor VIIa to the endothelial cell protein C receptor reduces its coagulant activity. J Thromb Haemost. 2007;5:1817–1824. doi: 10.1111/j.1538-7836.2007.02648.x. [DOI] [PubMed] [Google Scholar]
- 28.Gopalakrishnan R., Hedner U., Ghosh S., Nayak R.C., Allen T.C., Pendurthi U.R., Rao L.V. Bio-distribution of pharmacologically administered recombinant factor VIIa (rFVIIa) J Thromb Haemost. 2010;8:301–310. doi: 10.1111/j.1538-7836.2009.03696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diaz-Ricart M., Brunso L., Pino M., Navalon F., Jou J.M., Heras M., White J.G., Escolar G. Preanalytical treatment of EDTA-anticoagulated blood to ensure stabilization of the mean platelet volume and component measured with the ADVIA counters. Thromb Res. 2010;126:e30–e35. doi: 10.1016/j.thromres.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Sedlmayr P., Grosshaupt B., Muntean W. Flow cytometric detection of intracellular platelet antigens. Cytometry. 1996;23:284–289. doi: 10.1002/(SICI)1097-0320(19960401)23:4<284::AID-CYTO4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 31.Tenedini E., Fagioli M.E., Vianelli N., Tazzari P.L., Ricci F., Tagliafico E., Ricci P., Gugliotta L., Martinelli G., Tura S., Baccarani M., Ferrari S., Catani L. Gene expression profiling of normal and malignant CD34-derived megakaryocytic cells. Blood. 2004;104:3126–3135. doi: 10.1182/blood-2003-07-2597. [DOI] [PubMed] [Google Scholar]
- 32.Lozano M., Estebanell E., Cid J., Diaz-Ricart M., Mazzara R., Ordinas A., Escolar G. Platelet concentrates prepared and stored under currently optimal conditions: minor impact on platelet adhesive and cohesive functions after storage. Transfusion. 1999;39:951–959. doi: 10.1046/j.1537-2995.1999.39090951.x. [DOI] [PubMed] [Google Scholar]
- 33.Galán A.M., van Heerde W.L., Escolar G., Ordinas A., Sixma J., de Groot P.G. Antithrombotic action of annexin V proved as efficient as direct inhibition of tissue factor or thrombin. Eur J Clin Invest. 2006;36:633–639. doi: 10.1111/j.1365-2362.2006.01698.x. [DOI] [PubMed] [Google Scholar]
- 34.Diaz-Ricart M., Carretero M., Castillo R., Ordinas A., Escolar G. Digital image analysis of platelet-extracellular matrix interactions: studies in von Willebrand disease patients and aspirin-treated donors. Haemostasis. 1994;24:219–229. doi: 10.1159/000217105. [DOI] [PubMed] [Google Scholar]
- 35.Tonda R., Galán A.M., Pino M., Cirera I., Bosch J., Hernández M.R., Ordinas A., Escolar G. Hemostatic effect of activated recombinant factor VII (rFVIIa) in liver disease: studies in an in vivo model. J Hepatology. 2003;39:954–959. doi: 10.1016/s0168-8278(03)00454-9. [DOI] [PubMed] [Google Scholar]
- 36.Escolar G., Bastida E., Castillo R., Ordinas A. Development of a computer program to analyze the parameters of platelet-vessel wall interaction. Haemostasis. 1986;16:8–14. doi: 10.1159/000215263. [DOI] [PubMed] [Google Scholar]
- 37.Anderson L., Quasim I., Soutar R., Steven M., Macfie A., Korte W. An audit of red cell and blood product use after the institution of thromboelastometry in a cardiac intensive care unit. Transfus Med. 2006;16:31–39. doi: 10.1111/j.1365-3148.2006.00645.x. [DOI] [PubMed] [Google Scholar]
- 38.White J.G., Clawson C.C. Effects of small latex particle uptake on the surface connected canalicular system of blood platelets: a freeze-fracture and cytochemical study. Diagn Histopathol. 1982;5:3–10. [PubMed] [Google Scholar]
- 39.Morgenstern E., Ruf A., Patscheke H. Transport of anti-glycoprotein IIb/IIIa-antibodies into the alpha granules of unstimulated human blood platelets. Thromb Haemost. 1992;67:121–125. [Erratum appeared in Thromb Haemost 1992, 68:485] [PubMed] [Google Scholar]
- 40.White J.G. Platelets are covercytes, not phagocytes: uptake of bacteria involves channels of the open canalicular system. Platelets. 2005;16:121–131. doi: 10.1080/09537100400007390. [DOI] [PubMed] [Google Scholar]
- 41.Monroe D.M., Hoffman M., Oliver J.A., Roberts H.R. Platelet activity of high-dose factor VIIa is independent of tissue factor. Br J Haematol. 1997;99:542–547. doi: 10.1046/j.1365-2141.1997.4463256.x. [DOI] [PubMed] [Google Scholar]
- 42.Hoffman M., Monroe D.M. Platelet binding and activity of recombinant factor VIIa. Thromb Res. 2010;125(Suppl 1):S16–S18. doi: 10.1016/j.thromres.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 43.Morrissey J.H., Macik B.G., Neuenschwander P.F., Comp P.C. Quantitation of activated factor VII levels in plasma using a tissue factor mutant selectively deficient in promoting factor VII activation. Blood. 1993;81:734–744. [PubMed] [Google Scholar]
- 44.Tonda R., Galán A.M., Pino M., Lozano M., Ordinas A., Escolar G. Hemostatic effect of activated recombinant factor VIIa in Bernard-Soulier syndrome: studies in an in vitro model. Transfusion. 2004;44:1790–1791. doi: 10.1111/j.0041-1132.2004.00439.x. [DOI] [PubMed] [Google Scholar]
- 45.He S., Blombäck M., Jacobsson E.G., Hedner U. The role of recombinant factor VIIa (FVIIa) in fibrin structure in the absence of FVIII/FIX. J Thromb Haemost. 2003;1:1215–1219. doi: 10.1046/j.1538-7836.2003.00242.x. [DOI] [PubMed] [Google Scholar]
- 46.Hoffman M., Monroe D.M., 3rd, Roberts H.R. Activated factor VII activates factors IX and X on the surface of activated platelets: thoughts on the mechanism of action of high-dose activated factor VII. Blood Coagul Fibrinolysis. 1998;9(Suppl 1):S61–S65. [PubMed] [Google Scholar]
- 47.Lopez-Vilchez I., Tusell J., Hedner U., Altisent C., Escolar G., Galan A.M. Traffic of rFVIIa through endothelial cells and redistribution into subendothelium: implications for a prolonged hemostatic effect. J Coag Disorders. 2009;1:1–6. [Google Scholar]
- 48.White T.C., Berny M.A., Tucker E.I., Urbanus R.T., de Groot P.G., Fernandez J.A., Griffin J.H., Gruber A., McCarty O.J. Protein C supports platelet binding and activation under flow: role of glycoprotein Ib and apolipoprotein E receptor. J Thromb Haemost. 2008;6:995–1002. doi: 10.1111/j.1538-7836.2008.02979.x. [DOI] [PubMed] [Google Scholar]
- 49.Ghosh A., Li W., Febbraio M., Espinola R.G., McCrae K.R., Cockrell E., Silverstein R.L. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J Clin Invest. 2008;118:1934–1943. doi: 10.1172/JCI34904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valiyaveettil M., Podrez E.A. Platelet hyperreactivity, scavenger receptors and atherothrombosis. J Thromb Haemost. 2009;7(Suppl 1):218–221. doi: 10.1111/j.1538-7836.2009.03422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]