

Abstract
Bronchopulmonary dysplasia is a chronic lung disease observed in premature infants requiring oxygen supplementation and ventilation. Although the use of exogenous surfactant and protective ventilation strategies has improved survival, the long-term pulmonary consequences of neonatal hyperoxia are unknown. Here, we investigate whether neonatal hyperoxia alters pulmonary function in aging mice. By 67 weeks of age, mice exposed to 100% oxygen between postnatal days 1 to 4 showed significantly a shortened life span (56.6% survival, n = 53) compared to siblings exposed to room air as neonates (100% survival, n = 47). Survivors had increased lung compliance and decreased elastance. There was also right ventricular hypertrophy and pathological evidence for pulmonary hypertension, defined by reduction of the distal microvasculature and the presence of numerous dilated arterioles expressing von Willebrand factor and α-smooth muscle actin. Consistent with recent literature implicating bone morphogenetic protein (BMP) signaling in pulmonary vascular disease, BMP receptors and downstream phospho-Smad1/5/8 were reduced in lungs of aging mice exposed to neonatal oxygen. BMP signaling alterations were not observed in 8-week-old mice. These data suggest that loss of BMP signaling in aged mice exposed to neonatal oxygen is associated with a shortened life span, pulmonary vascular disease, and associated cardiac failure. People exposed to hyperoxia as neonates may be at increased risk for pulmonary hypertension.
Premature infants are at significant risk for developing bronchopulmonary dysplasia (BPD), a chronic lung disease defined as a need for supplemental oxygen beyond 36 weeks postconceptional age.1 Infants born prematurely also have reduced antioxidant defenses and are sensitive to oxidant injury that can be exacerbated by therapeutic oxygen.2–5 Although improved clinical treatments have reduced the incidence of airway injury, inflammation, and fibrosis typically seen in the pre-surfactant era, infants dying from BPD today have simplified alveoli that are less vascularized.6 Analogous to the pathology seen in these infants, exposure of newborn rodents to ≥85% oxygen for 10 to 14 days causes alveolar simplification and abnormal vascular development.7,8 The oxygen-dependent loss of vascular endothelial cell growth factor (VEGF) likely contributes to these changes.9–11 Blocking VEGF signaling disrupts postnatal alveolar development,12–14 and VEGF gene therapy can partially protect the developing rat lung against hyperoxia.15,16 Besides VEGF, the number of circulating endothelial cell precursors found in blood and lung is reduced in neonatal mice exposed to hyperoxia17 and, when restored, can attenuate murine models of BPD.18,19 Collectively, a large body of data suggests that alveolar simplification and pulmonary hypertension seen in infants dying with BPD may be caused by oxidative stress disrupting microvascular development.
Fortunately, the use of exogenous surfactant, lung protective ventilation practices, and antenatal steroids has markedly reduced mortality attributed to BPD. However, children born prematurely have increased risk for asthma and are often rehospitalized when infected with respiratory syncytial virus.20,21 Abnormalities in airflow obstruction and air trapping persist into adolescence.22–26 Similarly, newborn animals exposed to ≥65% oxygen and recovered in room air develop long-term changes in airway responsiveness and increased lung volumes.27–31 We previously reported that 8-week-old adult mice exposed to ≥60% oxygen for the first 4 days of life have simplified alveoli attributed to changes in elastin expression and an imbalance in alveolar epithelial type I and II cells.27,32 Expression of vascular signaling genes was not significantly altered at this time, implying that longer exposures to hyperoxia might be needed to disrupt short-term vascular development and cause more severe disease. On the other hand, these mice were highly susceptible to influenza A virus infection, with greater inflammation, fibrosis, and mortality compared to siblings exposed to room air.33 Since children born prematurely also show changes in lung function and increased susceptibility to viral infections, this mouse model may provide an opportunity to investigate the long-term health effects attributable to neonatal oxygen exposure. Using this mouse model of “modern-era” BPD, we studied lung function and lung vascular structure in aging mice that were exposed to short-term hyperoxia as neonates.
Materials and Methods
Oxygen Exposures
Newborn C57Bl/6J mice from several litters were mixed on the morning of birth and randomly separated into two groups that were exposed to room air or 100% oxygen (hyperoxia). The gas was passed through deionized water–jacketed Nafiomembrane tubing (Perma Pure LLC, Toms River, NJ) and delivered into a sealed Lexan polycarbonate chamber holding the mice. Oxygen levels were monitored with an oxygen sensor (model #TED-60 from Teledyne Analytical Instruments, City of Industry, CA). Dams were rotated every 24 hours between pups exposed to room air or hyperoxia until morning of the fourth day when oxygen-exposed mice were returned to room air.27 Animals were then analyzed at 67 weeks of age, except for mRNA expression and heart weight measurements, which were done at 94 weeks. Data were predominantly collected from 47 mice exposed to room air and 53 mice exposed to oxygen, split between two separate exposures with similar numbers of mice in each group. Premature mortality was observed in a third group of mice, and a fourth group of mice was harvested at 8 weeks of age for studying bone morphogenetic protein (BMP) signaling. The University Committee on Animal Resources (UCAR) at the University of Rochester reviewed and approved these studies.
Physiology Measurements
Mice were anesthetized with 60 to 90 mg/kg of ketamine intraperitoneally (i.p.). The hair over their chests was clipped, and the mice were manually held in dorsal recumbency. The rectal probe (Reflectance R/E Sensor) from a SurgiVet V3402 pulse oximeter (Smiths Medical PM, Inc., Waukesha, WI) was applied to the chest over the heart. The probe was moved over the chest until the strongest signal was obtained, as indicated on the monitor. Heart rate and oxygen saturation were noted at this time. For pulmonary function measurements, mice were anesthetized with sodium pentobarbital (40 mL/kg) and their tracheas connected to a computer-controlled small animal mechanical ventilator (flexiVent; SCIREQ, Montreal, QC, Canada).34 Pancuronium (1 mg/kg) was injected i.p. to paralyze the diaphragm, and mice were ventilated with a tidal volume of 8 mL/kg at a rate of 150 breaths/minute with positive end-expiratory pressure of 2 cm H2O. Estimated tissue damping and tissue elastance were obtained from the flexiVent by fitting a model to each impedance spectrum using software provided by SCIREQ.35
Lung Fixation and Morphometry
Lungs were infused through the trachea with 10% neutral-buffered formalin at 25 cm gravity pressure and allowed to fix for 15 minutes. The trachea was tied; the lung was then removed and fixed further overnight at 4°C. The lung was dehydrated through graded alcohol, embedded in paraffin, and then cut into 5-μm sections. For morphometry, lung sections were stained with hematoxylin and eosin. Each slide contained tissue from several transverse slices of the left lobe. Ten randomly chosen areas from each section were photographed with the 10× objective of a Nikon Eclipse 80i microscope (Nikon Instruments Inc., Melville, NY). Mean airspace chord length was measured from each image using NIS-Elements AR (Nikon Instruments). The software allowed for manual identification and exclusion of large airways and vessels before mean chord length calculations.
Protein Expression and Immunohistochemistry
The left lobe was tied off and homogenized in ice-cold lysis buffer containing protease and phosphatase inhibitors.36 Soluble material was recovered by centrifugation, and protein was quantified using a modified Laemmli assay. Equivalent amounts of protein were resolved on Tris-HCl SDS–PAGE gel, transferred to a polyvinylidene difluoride (PVDF) membrane, and blocked in 5% nonfat dried milk. Membranes were incubated in rabbit anti-prosurfactant protein C (SP-C) (1:500; Millipore, Billerica, MA), hamster anti-T1α (also called podoplanin; Developmental Studies Hybridoma Bank at the University of Iowa, Iowa City, IA), rabbit anti-Clara Cell Secretory Protein (CCSP) (1:10,000; from Barry Stripp at Duke University, Durham, NC), goat anti-platelet endothelial cell adhesion molecule (PECAM, also called CD31) antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-vascular endothelial growth factor (VEGF) (1:200; Santa Cruz Biotechnology), rabbit anti–phospho-Smad1/5/8 (1:500; Cell Signaling Technology, Danvers, MA), rabbit anti-Smad1/5/8 (1:200; Santa Cruz Biotechnology), or anti–β-actin antibody (1:5000; Sigma, Saint Louis, MO) overnight at 4°C. Blots were extensively washed, and immune complexes were detected with horseradish peroxidase–conjugated appropriate secondary antibody (1:5000; from SouthernBiotech, Birmingham, AL) and visualized with enhanced chemiluminescence (ECL kit; GE Healthcare Life Sciences, Piscataway, NJ) using an α Innotech Fluorchem gel documentation system (Alpha Innotech, San Leandro, CA) or by exposure to blue sensitive film (Laboratory Products Sales, Rochester, NY). Band intensities were determined and normalized to room air treated samples arbitrarily set to a value of one, using ImageJ software (http://rsbweb.nih.gov/ij; last accessed October 24, 2008).
Paraffin-embedded sections (5 μm) were rehydrated and incubated with primary antibodies against α-smooth muscle actin (α-SMA) (1:100; DakoCytomation, Glostrup, Denmark) or von Willebrand factor (Millipore) overnight at 4°C. Immune complexes were captured with fluorescently labeled secondary antibodies (1:200; from Jackson ImmunoResearch Laboratories, West Grove, PA) before sections were counterstained with DAPI. Additional details for immunostaining and Hart's elastin staining have been previously described.32 Stained sections were visualized with a Nikon E800 fluorescence microscope (Nikon), and images were captured with a SPOT-RT digital camera (Diagnostic Instruments, Sterling Heights, MI).
To quantify the increase in α-SMA staining, a single slide with paraffin-embedded sections of room air–exposed (n = 3) or hyperoxia-exposed (n = 4) mouse lung was stained for SMA. Photographs (three to four from each lung) at 10× power were taken and analyzed by an observer blinded to the identity of the data. Acinar vessels (20 to 100 μm, not associated with a bronchiole) staining for SMA were counted and averaged over the number of photographs taken. Because this type of data is not obviously amenable to parametric analysis (continuous, normally distributed data), we took a conservative approach and analyzed the data with a nonparametric Mann-Whitney test.
RNA Isolation and Semiquantitative RT-PCR
Total RNA from each sample was isolated from unfixed lung tissue by Trizol (Invitrogen, Carslbad, CA) and purified by Turbo DNase (Ambion, Austin, TX). One microgram of total RNA was reverse transcribed in a 20-μL volume using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA). One microliter of this reaction was used as a template to amplify product using GoTaq DNA polymerase (Promega, Madison, WI) and 10 μmol/L isoform-specific primers synthesized by Integrated DNA Technologies, Coralville, IA. These primer sets for BMP/TGF-β receptor family members activin-like kinase 3 (Alk-3) and bone morphometric protein receptor (BMPR)1b (also called Alk-637), Alk-1 and BMPR-II,38 and actin,39 have been described previously. All reactions were run 35 cycles except actin, which was run for 30 cycles. Amplicons were separated on 1% agarose gels containing ethidium bromide and visualized under UV light. Failure to amplify product in the absence of reverse transcriptase confirmed that the original RNA sample was not contaminated with DNA. For quantitative real-time PCR of VEGF and PECAM, synthesis of cDNA was performed using an iScript cDNA synthesis kit and SYBR Green I dye on MyiQ 2 two-color real-time PCR detection system (Bio-Rad Laboratories). PCR products were amplified in triplicate with sequence-specific primers for mouse VEGF (forward, 5′-AGAGGCTTGGGGCAGCCGAG-3′; reverse, 5′-ACTCCCGGGCTGGTGAGTCC-3′) and PECAM (forward, 5′AGCCTCACCAAGAGAACGGAAGG-3′; reverse, 5′-ACGTGCACAGGACTCTCGCA-3′), with mouse 18S (forward, 5′-CGGCTACCACATCCAAGGAA-3′; reverse, 5′-GCTGGAATTACCGCGGCT-3′) used to confirm equal loading of template cDNA. Relative changes in expression were determined by normalizing threshold cycle (Ct) values of VEGF or PECAM with 18S.
Pulmonary Vascular Imaging Using X-Ray Microcomputed Tomography
The aorta was transected below the kidney, the right ventricle was cannulated, and heparinized saline was used to flush the lungs before infusion with a warm solution of 1% low-melting-point agarose and 30% barium (clinical radiology grade). The lungs were then inflation fixed with 10% paraformaldehyde and the pulmonary vasculature visualized on a Scanco vivaCT40 (Basserdorf, Switzerland).40 Computed tomography (CT) data were reconstructed as three-dimensional images and quantified using the Scanco software to produce a histogram of the number of microvessels. We integrated the counts of vessels smaller than 300 μm to generate average vessel counts within an individual animal.
Hematocrits and Arterial Wall Measurements
Retro-orbital bleeds were performed on anesthetized mice using a small capillary tube. The tubes were centrifuged and hematocrit determined as the percentage of packed red blood cell volume to total blood volume. Animals were then sacrificed, the hearts removed and dissected to isolate the free wall of the right ventricle from the left ventricle and septum. The ratios of right ventricular weight to final body weight, left ventricular + septal weight to final body weight, and right ventricle to left ventricle + septum were determined as a measure of cardiac disease.
Statistical Analyses
Statistical analysis of values obtained from room air– and oxygen-treated animals was performed by unpaired Student's t-test for single comparisons using Statview software (Abacus Concepts, Piscataway, NJ). For the micro-CT measurements, we did not assume the same variability for each group. The Wilcoxon two-sample test assuming nonparametric data was used to assess survival. Values are expressed as means ± SE with P < 0.05 being considered significant.
Results
Neonatal Oxygen Shortens Life Span in Aging Mice
Newborn mice were exposed to room air or 100% oxygen (hyperoxia) between postnatal days 1 and 4. Mice were then returned to room air and physically examined once per week for changes in general health. Although overt signs of disease were not apparent, four adult mice exposed to neonatal hyperoxia were reported dead by vivarium staff at around 20 weeks of age and were unavailable for analysis. Increasing mortality of oxygen-recovered mice was readily apparent in male and female mice after 40 weeks of age, with 43% mortality by 67 weeks of age (Figure 1). Close examination of mice used throughout this study failed to identify physical parameters indicative of morbidity or impending mortality. In contrast, sibling mice exposed to room air at birth had 100% survival during this time period.
Figure 1.
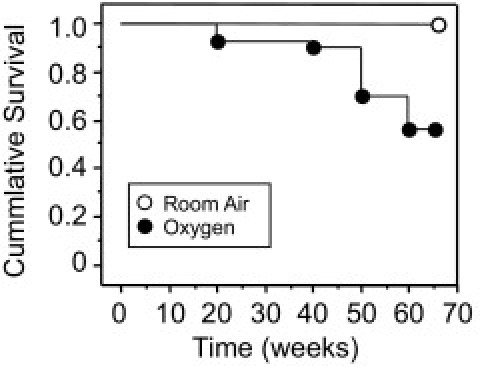
Neonatal oxygen shortens the life span in aging mice. Kaplan-Meier survival curve of aging mice exposed to room air (n = 43) or 100% oxygen (n = 53) between postnatal days 1 to 4. Neonatal oxygen significantly shortened the life span by 67 weeks of age, P < 0.0001. Statistical analysis was performed using Wilcoxon two-sample test assuming nonparametric data.
Changes in Lung Function and Development Persist in Aging Mice
To determine whether increased mortality might be attributed to progressive changes in lung function and structure previously seen at 8 weeks of age, pulmonary mechanics and expression of cell-specific proteins were investigated in surviving mice at 67 weeks of age. Consistent with findings seen at 8 weeks, lung compliance was increased whereas tissue and airway elastance, a measure of energy conservation, were decreased (Figure 2, A–E). In contrast, room air– and hyperoxia-exposed mice had identical tissue damping (a measure of energy dissipated into lung tissues) and airway resistance at 67 weeks; these measures had been significantly elevated in hyperoxia-exposed mice at 8 weeks.32 Similarly, as defined by quantifying mean alveolar chord length, the alveolar simplification seen at 8 weeks was no longer evident; in fact, mean chord length was slightly, but significantly, reduced in aged mice previously exposed to neonatal hyperoxia (Figure 2F).
Figure 2.
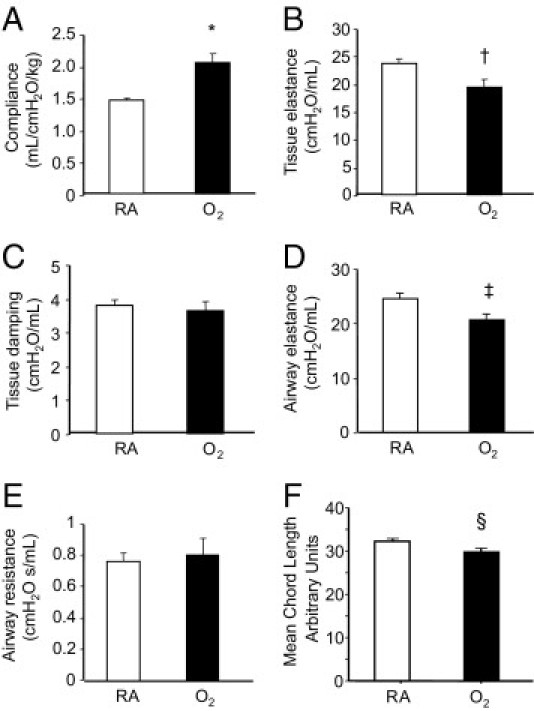
Neonatal oxygen disrupts normal pulmonary mechanics and alveolar structure. Lung compliance (A), tissue elastance (B), tissue damping (C), airway elastance (D), airway resistance (E), and mean cord length (F) were measured in 67-week-old mice exposed to room air or 100% oxygen between postnatal days 1–4. Oxygen significantly increased lung compliance (*P < 0.004), and reduced tissue elastance (†P < 0.05), airway elastance activity (‡P < 0.04), and mean alveolar cord length (§P < 0.05) based on an unpaired t-test. Values are means ± SE of five animals per group (A–E) or four animals per group (F). RA, room air.
Alveolar simplification and increased compliance observed at 8 weeks of age is associated with a loss of type II epithelial cells as defined by expression of proSP-C, an increase in type I cells as defined by expression of T1α, and minimal changes in vascular endothelial cells as defined by expression of PECAM.27,40 Expression of airway CCSP was also reduced. To determine whether these changes persist in aging mice, the expression of these proteins was investigated by Western blot analysis. Surprisingly, expression of proSP-C was increased, T1α was reduced, and CCSP was not different in aged mice exposed to neonatal oxygen (Figure 3, A and B). Moreover, expression of PECAM was markedly reduced, implying a loss in vascular endothelial cells that was not readily apparent at 8 weeks of age. Quantitative real-time PCR confirmed that PECAM mRNA in room air (1.00% ± 0.14%) was significantly reduced (0.53% ± 0.05%) by neonatal oxygen (n = 3, P < 0.04). Despite the loss of PECAM, levels of VEGF (Figure 3, A and B) were comparable between the mice. Quantitative real-time PCR confirmed VEGF mRNA in room air–treated (1.00% ± 0.17%) was not different from oxygen-treated (0.96% ± 0.13%) mice (n = 3, P < 0.9).
Figure 3.
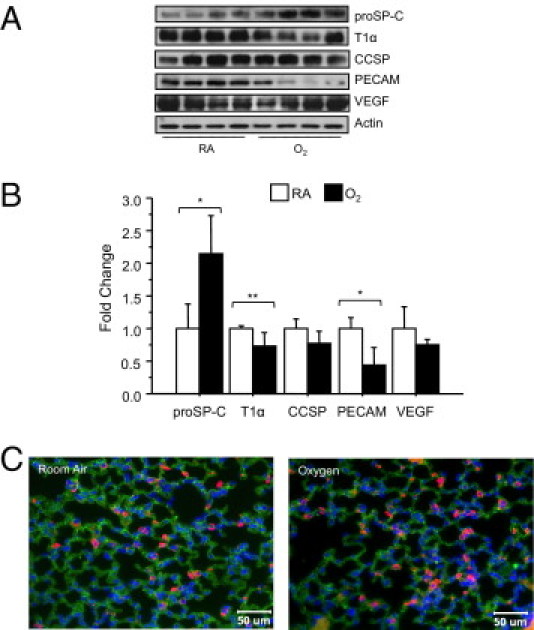
Neonatal oxygen alters expression of cell-specific markers in adult mice. Lungs of 67-week-old mice exposed to room air or 100% oxygen were immunoblotted with antibodies against proSP-C, T1α, CCSP, PECAM, VEGF, and actin as a loading control (A). Band intensities were quantified, normalized to expression of actin, and then graphed (B). Oxygen significantly increased expression of proSP-C, and decreased expression of T1α and PECAM based on an unpaired t-test (*P < 0.02, **P < 0.04). Values are means ± SE of three to four mice per group. Lungs of 67-week-old mice exposed to room air or oxygen at birth were immunostained for proSP-C (red) and T1α (green), and counterstained with DAPI (C). RA, room air.
Neonatal Hyperoxia Promotes Pathological Signs of Pulmonary Arterial Hypertension
Significant vascular remodeling became apparent in lungs of aging mice exposed to neonatal hyperoxia. The most prominent feature was the presence of large blood vessels of >50 μm that were not seen in aged mice exposed to room air as neonates (Figure 4, A and B). The vessel walls stained positive for von Willebrand factor, a protein expressed by endothelial cells, whereas the outer walls stained positive for α-smooth muscle actin (α-SMA), a protein expressed by smooth muscle cells and pericytes (Figure 4, C and D). Neonatal hyperoxia increased the number of α-SMA–positive arterioles (inter- and intra-acinar) from 4.8 ± 2.3 to 10.3 ± 3.6 (P < 0.0003 by Mann-Whitney test), but there was no associated increase in vessel wall thickness (Figure 4, E and F). However, thickened bundles of elastin lining alveolar epithelial walls observed at 8 weeks of age were still evident in the aged mice, implying that this might contribute to the increased alveolar compliance when alveolar simplification was not readily apparent.32
Figure 4.
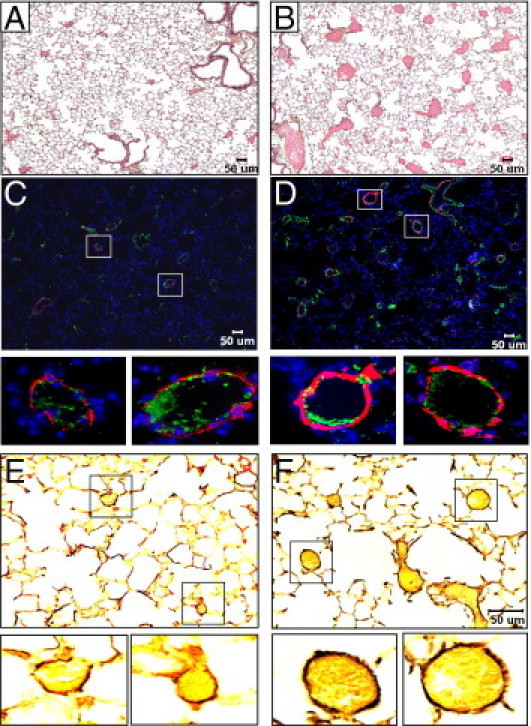
Neonatal oxygen promotes histological signs of pulmonary hypertension. H & E stains of 67-week-old mice exposed to room air (A) or 100% oxygen (B) as neonates. Von Willebrand factor (green) and α-smooth muscle actin (red) staining of 67-week-old mice exposed to room air (C) or 100% oxygen (D) as neonates. Boxed regions are enlarged below each image. Hart's elastin staining of 67-week-old mice exposed to room air (E) or 100% oxygen (F) as neonates. Boxed regions are enlarged below each image.
Pulmonary vascular imaging using micro-CT was used to quantify changes in small vessels in an unbiased, three-dimensional fashion. Lungs from room air mice displayed uniform perfusion of the distal arterial tree, whereas lungs exposed to neonatal oxygen showed significant vascular rarefaction (Figure 5, A and B). The numbers of vessels were automatically counted on the basis of vessel diameter. In room air littermates, a normal distribution of pulmonary vessels smaller than 300 μm was observed (Figure 5A, total vessel count = 1.1 × 108 ± 3.3 × 107, n = 3); in contrast, neonatal oxygen exposure dramatically reduced the number of microvessels (Figure 5B, total vessel count = 3.7 × 107 ± 1.7 × 107, n = 3; P < 0.01 in Student's t-test without assuming similar variances).
Figure 5.
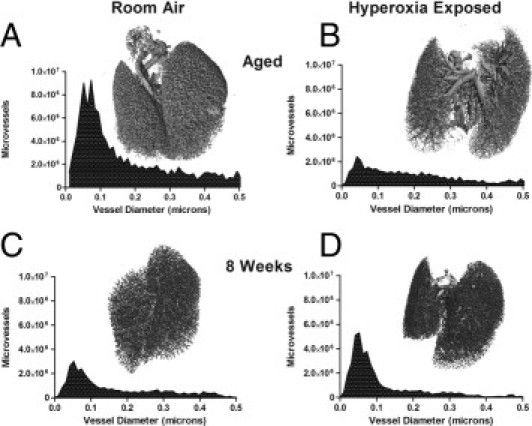
Distal microvessels are reduced in aged mice exposed to neonatal oxygen. Lungs of 78-week-old (A and B) or 8-week-old (C and D) mice exposed to room air or hyperoxia as neonates were perfused through the right ventricle with 1% low-melting agarose and 30% barium. The lungs were inflation fixed with formaldehyde and removed for three-dimensional analysis of the pulmonary vasculature using X-ray micro-computed tomography. For each lung, the number of vessels of a given size were determined and graphed. Images and graph are representative of findings seen with at least three mice per group (n = 3, aged animals; n = 4, room air, and n = 5, hyperoxia exposed at 8 weeks). Note that both the distribution and absolute number of microvessels are approximately similar in the 8-week-old mice (there was a trend toward more vessels in hyperoxia-exposed mice, not statistically different, see text). In the aged animals, there is a uniform reduction in vessels with a distinct loss in the 20 to 120 μm range, and this difference was statistically significant.
Although two-dimensional histology sections did not reveal an apparent vascular pathology,27 we had not previously explored vascular density with the micro-CT technique in mice 8 weeks after exposure to neonatal hyperoxia. We evaluated 8-week-old mice using the micro-CT technique to determine whether the vascular changes might be present at this early time point. Interestingly, using an identical methodology as in the aged mice, the young room air littermates had numerically fewer microvessels (Figure 5C, 2.7 × 107 ± 6.0 × 107, n = 4) than the aged adults although this difference was not statistically significant. As a group, the hyperoxia-exposed mice had numerically greater vessels than room air littermates 8 weeks after exposure (Figure 5D, 4.6 × 107 ± 2.4 × 107, n = 5; P = 0.14), and this vessel count was also numerically greater than the aged hyperoxia animals (not statistically different). Thus, the marked reduction in microvessels seen in aged mice after hyperoxia exposure was not present at 8 weeks.
This significant loss in microvessels in the aged mice, along with a reduction in type I epithelial cells, suggests that peripheral tissue hypoxia might be a cause of the mortality seen in these mice. However, peripheral oxygen saturations of 94-week-old mice exposed to room air (n = 3) or hyperoxia (n = 5) as neonates was 98% or higher, with the exception of one hyperoxia-treated mouse whose blood-oxygen saturation was 92%. That mouse died shortly after measurements were taken. Hematocrit levels in hyperoxia-exposed mice were modestly greater (47.2 ± 1.8 versus 44.7 ± 3.1%, P = 0.18), but that was not statistically significant. On the other hand, right ventricular weight, expressed as a percentage of full body weight, was significantly increased in hyperoxia-treated mice (Table 1). This was selective right ventricular hypertrophy because left ventricular plus septal weight when expressed as full body weight was not different. Thus, cardiac failure secondary to pulmonary hypertension may be a cause of the increased mortality seen in aged mice exposed to neonatal oxygen.
Table 1.
Body Weights and Heart Weights of Adult Mice Exposed to Room Air or Oxygen as Neonates
| Room air | Neonatal oxygen | |
|---|---|---|
| Full body weights, g | 31.2 ± 1.4 | 29.1 ± 0.7 |
| RV/FBW, ×10−5 | 75.1 ± 5.5 | 96.1 ± 4.2⁎ |
| LV+S/FBW, ×10−5 | 362.3 ± 13.6 | 384.8 ± 14.3 |
| RV/LV+S, % | 20.2 ± 1.5 | 26.2 ± 1.2⁎ |
Results are expressed as mean values ± SEM for nine mice exposed to room air and nine aged mice exposed to neonatal oxygen. All mice were 94 weeks of age except two room air–exposed mice that were 100 weeks of age.
FBW, full body weight; LV, left ventricle; RV, right ventricle; S, septum.
P < 0.02.
Neonatal Oxygen Suppresses Bone Morphogenetic Protein Signaling
Since VEGF protein and staining was not different between aged mice exposed to room air or hyperoxia as neonates, we considered alternative mechanisms that could cause the vascular defects observed in aged mice. We chose to investigate BMP and downstream phospho-Smad1/5/8 signaling in aged mice because mutations in the BMP receptor type II and activin receptor-like kinase 1 are associated with heritable pulmonary hypertension in humans.41,42 Intriguingly, phospho-Smad1/5/8, but not total Smad1/5/8, was significantly reduced in aged mice exposed to neonatal oxygen (Figure 6, A and B). To further investigate this loss in BMP signaling, expression of BMP type I and II receptors and Alk-1 were investigated by immunoblotting lung homogenates. Neonatal hyperoxia significantly reduced expression of BMPR1b and Alk-1, albeit there was more variability in the loss of BMPR1b than Alk-1 (Figure 6, A and B). Since antibodies against BMPR1a (Alk-3) or BMPR-II gave high backgrounds, semiquantitative RT-PCR was used to investigate mRNA expression. Not only did neonatal hyperoxia suppress mRNA levels of BMPR1b and Alk-1, it also suppressed expression of BMPR-II and BMPR1a (Figure 6C). Because pathological signs of PAH were not observed in young adult (8-week-old) mice,27,33 we also investigated whether BMP signaling was suppressed at this age. Consistent with the lack of overt vascular pathology and mortality at this time, phospho-Smad1/5/8 and expression of BMP receptors were similar between the two groups of mice (Figure 6, D–F). While these studies were under review, four additional mice exposed to hyperoxia reached 50 weeks of age. Interestingly, expression of BMPR1b, Alk-1, and phospho-Smad1/5/8 were suppressed in three of four oxygen-exposed mice, implying loss of BMP signaling occurs between 8 and 50 weeks of age (data not shown).
Figure 6.
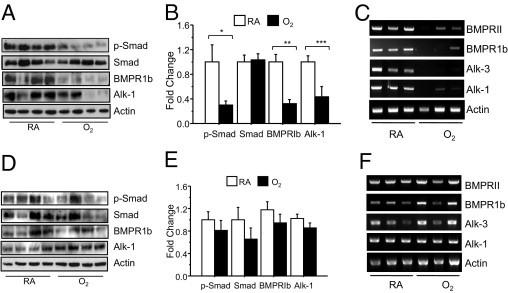
Neonatal oxygen suppresses BMP signaling in aged mice. Expression of phospho-Smad1/5/8, total Smad1/5/8, BMPR1b, Alk-1, Alk-3, and BMPRII were investigated in lungs of aged (A–C) and young adult (D–F) mice exposed to room air or 100% oxygen as neonates. Total lung homogenates from four mice per group were immunoblotted for phospho-Smad, total Smad, BMPR1b, and Alk-1 (A and D) and quantified (B and E) relative to expression of actin. Neonatal oxygen significantly reduced expression of phospho-Smad (*P < 0.05), BMPR1b (**P < 0.003), and Alk-1 (***P < 0.03) in aged, but not young mice. On the basis of an unpaired t-test, neonatal hyperoxia did not significantly affect expression of total Smad (P = 0.084) in aged mice. Semiquantitative RT-PCR of BMPR-II, Alk-6, Alk-3, and Alk-1 confirmed neonatal hyperoxia suppressed mRNA levels of these receptors in aged (C), but not young (F) mice. RA, room air.
Discussion
The current study provides evidence that brief neonatal hyperoxia profoundly shortens the life span of aging mice by promoting pulmonary microvascular rarefaction and associated right ventricular hypertrophy. The findings extend another study showing increased blood pressure, vascular dysfunction, and microvascular rarefaction in lungs and nephrons of 6-month-old rats exposed to 80% oxygen between postnatal days 3 and 10.43 Together, these two studies raise concern that the high blood pressure seen in very low birth weight individuals who reach late adolescence may persist and lead to premature mortality.44 If true, the therapeutic use of exogenous surfactant and milder ventilation practices, which have reduced the incidence of BPD and infant mortality, may have produced a new disease some people are aptly naming persistent pulmonary disease (PPD).
Despite emerging evidence that prematurity is a major risk factor for long-term deficits in lung function, there are few experimental models that can help us understand disease progression. By exposing newborn mice to hyperoxia at birth and recovering them in room air, we generated a mouse model of PPD that has many similarities with lung diseases reported in people born prematurely. Like children and adolescents born prematurely who suffer from abnormalities in airflow obstruction and air trapping,22–26 adult 8-week-old mice exposed to ≥60% oxygen between birth and postnatal day 4 have increased lung compliance attributed to alveolar simplification, increased elastin, and changes in the proportion of alveolar epithelial cells.27,32 Female C57Bl6/J females exposed to hyperoxia also show spontaneous airway reactivity in response to methacholine challenge, perhaps modeling the increased risk for asthma often seen in children born prematurely (unpublished observations). And like children born prematurely who are often rehospitalized when infected with respiratory syncytial virus,20,21 adult mice exposed to neonatal hyperoxia show increased susceptibility to influenza A virus infection.33 Since lung function in late adolescents born prematurely might be deteriorating faster than expected,24 we decided to investigate whether deficits in lung function and structure changed as oxygen-exposed mice aged. We found elevated lung compliance and reduced tissue elastance activity in aged mice, similar to the changes observed at 8 weeks following exposure to neonatal hyperoxia. Reduced airway resistance, levels of CCSP, and alveolar simplification seen at 8 weeks were not observed in aged mice, suggesting these deficits improve over time. On the other hand, levels of type II cell–specific proSP-C increased, and type I–specific T1α and vascular PECAM decreased, implying additional changes with aging are occurring in specific alveolar cell populations. Because alveolar compliance remained elevated despite reversal of alveolar size and further changes in alveolar cell populations, altered lung compliance cannot be explained solely by changes in alveolar structure. Increased elastin staining implies persistent changes in elastogenic molecules may be the cause of altered lung compliance. Intriguingly, increased proSP-C and reduced PECAM seen in aged lungs are also seen in the mouse model of BPD wherein newborn mice are exposed to neonatal hyperoxia for 10 to 14 days (unpublished observations) or in preterm baboons with BPD.9,45 Although this might suggest that aged animals with PPD are slowly developing BPD, the fact that VEGF is not reduced in the aged mice suggests the two diseases have different etiologies that still lead to pulmonary vascular disease and mortality.
Pulmonary hypertension in the neonate is attributed to impaired vascular development caused by loss of VEGF and nitric oxide signaling, with a simultaneous reduction in the number of circulating bone marrow–derived endothelial precursor cells.9–11,17 In contrast, the pathological features seen in aged mice exposed to neonatal oxygen appear to involve failure to maintain or properly densify existing microvasculature. As defined by expression of vascular-specific genes (PECAM, Tie1, Tie2, Flt1, Flt4, and thrombin receptor) and CT scans of the vasculature, vascular development in 8-week-old adult mice exposed to neonatal hyperoxia appeared to develop somewhat normally.27 Although not statistically significant, the microvascular density at 8 weeks in hyperoxia-exposed neonates was actually numerically higher than room air–exposed littermates; in contrast, room air animals had quadruple the number of microvessels at 78 weeks, whereas oxygen-exposed animals had a similar number of lung microvessels at 8 and 78 weeks. More detailed time course experiments are required to determine whether vessels developed and died or failed to fully develop in the hyperoxia-exposed animals. However, it seems reasonable to propose that an insufficient adult pulmonary vascular bed in the oxygen-exposed mice led to smooth muscle investment of the vasculature, right ventricular hypertrophy, and early mortality in aged mice.
We observed that levels of BMP receptors and phospho-Smad signaling were similar in 8-week-old mice but then declined by the time they were 67 weeks old. Decreased expression of PECAM, BMPR1b, and Alk-1 was also seen in four mice that were 50 weeks old (data not shown). This age-dependent loss of BMP signaling may cause or contribute to lung disease because loss of BMP signaling in precapillary arterioles and microvascular endothelial cells via mutations in BMPR-II is associated with heritable forms of pulmonary hypertension.42,46 Likewise, BMPR-II expression declines in hypoxia and monocrotaline models of pulmonary hypertension.47,48 Adenoviral-mediated gene delivery of BMPR-II protects against hypoxia-induced pulmonary hypertension.49 However, loss of BMPR-II by itself is not sufficient to cause pulmonary hypertension because there is only 20% disease penetrance despite autosomal dominant inheritance. A secondary environmental or genetic insult seems to be required for establishing progressive disease in mutation carriers,50 and similar observations have been made in BMPR2 heterozygous mice.51 We did not observe significant inflammation or other markers of oxidative stress in aged mice exposed to neonatal oxygen, nor did we see intimal hyperplasia or plexiform-like lesions. Hence, we speculate that the loss of BMP signaling in mice exposed to neonatal hyperoxia destabilized the pulmonary microvasculature as the mice aged. Further experiments will be required to understand the signaling that maintains the vasculature during aging before this hypothesis can be tested.
The appearance of distorted larger vessels we observed appear similar to hereditary hemorrhagic telangiectasia (HHT), an inherited disorder with systemic and pulmonary arteriovenous malformations. In HHT, individuals have loss of capillary networks and the formation of pulmonary arteriovenous malformations, a direct communication between arteries and veins. Pulmonary arteriovenous malformations can lead to shunting of deoxygenated blood from the right to the left heart. Since the capillary bed serves as a filter for blood clots, strokes and brain abscesses can also occur. Mutations in the TGF-β receptor Alk-1 and to a lesser extent endoglin are seen in patients with familial HHT.52 Although endothelial cell–specific ablation of Alk-1 in mice recapitulates HHT, loss of TGF-β receptor Alk-5 or the TGF-β type II receptor does not.53 This suggests that loss of Alk-1 contributes to HHT via a TGF-β–independent manner. But like people with mutations in BMPR-II, the incidence of HHT seen in heterozygous endoglin mice or in people with mutant Alk-1 or endoglin varies in age and degree. This implies that loss of Alk-1 signaling in aged mice exposed to neonatal hyperoxia contributes to, but is not sufficient to cause, HHT-like pathology. Based on the variable penetrance seen in affected humans and mice lacking BMPR-II, the vascular remodeling seen in aged mice is likely to involve a two-step process involving loss of BMP/TGF-β signaling and a second molecular hit that remains to be determined.
Exposure to oxygen during key developmental periods is clearly a major contributor to altered lung development. Hence, there is an urgent need to define targeted oxygen saturations that prevent hypoxemia in preterm infants without causing lung or peripheral tissue injury. The recent Surfactant, Positive Pressure, and Pulse Oximetry Randomized Trial (SUPPORT) compared the effects of early nasal continuous positive airway pressure (CPAP) versus intubation and surfactant treatment in infants born between 24 and 27 weeks.54,55 Infants were also randomized to test the effects of high (91% to 95%) and low (85% to 89%) oxygenation. Treatment with CPAP versus surfactant did not alter the incidence of BPD or mortality before discharge. However, the CPAP group required less intubation in the delivery room or postnatal steroids for treatment of BPD. The rate of retinopathy was significantly reduced in infants treated with lower oxygen tensions, but mortality was slightly increased. Although this study suggests starting CPAP at birth might be beneficial, it was unable to identify an optimum target for oxyhemoglobin saturations. On the other hand, quantifying the cumulative dose of oxygen as an area under the curve (oxygenAUC) for the first 3 days of treatment does predict the incidence of respiratory symptoms, use of medications, and need for health services at 1 year of corrected age.56 Our own studies in mice suggest that the response of the developing mouse lung to oxygen is not entirely dose-dependent because exposure to 60% or 80% oxygen caused mild, but similar, changes in alveolar simplification at 8 weeks of age, whereas exposure to 100% was more injurious.32 Whether lower levels of oxygen sufficient to alter epithelial development in young adult mice also affect vasculature in aged mice remains to be determined. The identification of BMP signaling as a cause or contributor for PPD in aged mice now provides an opportunity to test specific hypotheses relating to neonatal oxygen exposure, altered BMP signaling, and long-term pulmonary disease.
Interestingly, prolonged exposure of neonatal mice to 80% oxygen for 28 days inhibits BMP signaling,38 and conditional ablation of Alk-3, the BMP type I receptor in the respiratory epithelium, promotes BPD-like pathology in newborn mice.57 Taken together, these findings strongly suggest that aberrations in BMP/TGF-β–related signaling pathways cause or contribute to BPD in premature infants and PPD seen in survivors. A better understanding of how aberrations in BMP/TGF-β signaling contribute to lung disease might lead to novel treatment regimens. Until then, the data in the present study emphasize the need to continue surveillance of people with a history of neonatal exposure to hyperoxia, with early referral to appropriate specialists if signs of pulmonary hypertension develop.
Acknowledgments
We thank Sylvie Honnon for analysis of lung histopathology, Nate Miller and Debbie Haight for help running and analyzing small animal CT scans, and David Dean for access to the flexiVent machine.
Footnotes
Supported in part by March of Dimes Birth Defects Foundation grant #6-FY08-264, National Institutes of Health (NIH) grants HL-091968 (M.A.O.), American Heart Association National Scientist Development grant 073540N (R.J.W.), P50-AR054041 (H.A.A.), and the Grace Anne Dorney Fund (S.A.M.-M.). The animal inhalation facility was supported by NIH Center grant ES-01247.
None of the authors disclosed any relevant financial relationships.
References
- 1.Eber E., Zach M.S. Long term sequelae of bronchopulmonary dysplasia (chronic lung disease of infancy) Thorax. 2001;56:317–323. doi: 10.1136/thorax.56.4.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chess P.R., D'Angio C.T., Pryhuber G.S., Maniscalco W.M. Pathogenesis of bronchopulmonary dysplasia. Semin Perinatol. 2006;30:171–178. doi: 10.1053/j.semperi.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Askie L.M., Henderson-Smart D.J., Irwig L., Simpson J.M. Oxygen-saturation targets and outcomes in extremely preterm infants. N Engl J Med. 2003;349:959–967. doi: 10.1056/NEJMoa023080. [DOI] [PubMed] [Google Scholar]
- 4.Deulofeut R., Golde D., Augusto S. Treatment-by-gender effect when aiming to avoid hyperoxia in preterm infants in the NICU. Acta Paediatrica. 2007;96:990–994. doi: 10.1111/j.1651-2227.2007.00365.x. [DOI] [PubMed] [Google Scholar]
- 5.Madan A., Brozanski B.S., Cole C.H., Oden N.L., Cohen G., Phelps D.L. A pulmonary score for assessing the severity of neonatal chronic lung disease. Pediatrics. 2005;115:e450–e457. doi: 10.1542/peds.2004-1293. [DOI] [PubMed] [Google Scholar]
- 6.Bhatt A.J., Pryhuber G.S., Huyck H., Watkins R.H., Metlay L.A., Maniscalco W.M. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor: Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;164:1971–1980. doi: 10.1164/ajrccm.164.10.2101140. [DOI] [PubMed] [Google Scholar]
- 7.Warner B.B., Stuart L.A., Papes R.A., Wispe J.R. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol. 1998;275:L110–L117. doi: 10.1152/ajplung.1998.275.1.L110. [DOI] [PubMed] [Google Scholar]
- 8.Bonikos D.S., Bensch K.G., Ludwin S.K., Northway W.H., Jr Oxygen toxicity in the newborn: The effect of prolonged 100 per cent O2 exposure on the lungs of newborn mice. Lab Invest. 1975;32:619–635. [PubMed] [Google Scholar]
- 9.Maniscalco W.M., Watkins R.H., Pryhuber G.S., Bhatt A., Shea C., Huyck H. Angiogenic factors and alveolar vasculature: development and alterations by injury in very premature baboons. Am J Physiol Lung Cell Mol Physiol. 2002;282:L811–L823. doi: 10.1152/ajplung.00325.2001. [DOI] [PubMed] [Google Scholar]
- 10.Klekamp J.G., Jarzecka K., Perkett E.A. Exposure to hyperoxia decreases the expression of vascular endothelial growth factor and its receptors in adult rat lungs. Am J Pathol. 1999;154:823–831. doi: 10.1016/S0002-9440(10)65329-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maniscalco W.M., Watkins R.H., Roper J.M., Staversky R., O'Reilly M.A. Hyperoxic ventilated premature baboons have increased p53: Oxidant DNA damage and decreased VEGF expression. Pediatr Res. 2005;58:549–556. doi: 10.1203/01.pdr.0000176923.79584.f7. [DOI] [PubMed] [Google Scholar]
- 12.Le Cras T.D., Markham N.E., Tuder R.M., Voelkel N.F., Abman S.H. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol. 2002;283:L555–L562. doi: 10.1152/ajplung.00408.2001. [DOI] [PubMed] [Google Scholar]
- 13.McGrath-Morrow S.A., Cho C., Zhen L., Hicklin D.J., Tuder R.M. Vascular endothelial growth factor receptor 2 blockade disrupts postnatal lung development. Am J Respir Cell Mol Biol. 2005;32:420–427. doi: 10.1165/rcmb.2004-0287OC. [DOI] [PubMed] [Google Scholar]
- 14.Zhao L., Wang K., Ferrara N., Vu T.H. Vascular endothelial growth factor co-ordinates proper development of lung epithelium and vasculature. Mech Dev. 2005;122:877–886. doi: 10.1016/j.mod.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Kunig A.M., Balasubramaniam V., Markham N.E., Morgan D., Montgomery G., Grover T.R., Abman S.H. Recombinant human VEGF treatment enhances alveolarization after hyperoxic lung injury in neonatal rats. Am J Physiol Lung Cell Mol Physiol. 2005;289:L529–L535. doi: 10.1152/ajplung.00336.2004. [DOI] [PubMed] [Google Scholar]
- 16.Thebaud B., Ladha F., Michelakis E.D., Sawicka M., Thurston G., Eaton F., Hashimoto K., Harry G., Haromy A., Korbutt G., Archer S.L. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation. 2005;112:2477–2486. doi: 10.1161/CIRCULATIONAHA.105.541524. [DOI] [PubMed] [Google Scholar]
- 17.Balasubramaniam V., Mervis C.F., Maxey A.M., Markham N.E., Abman S.H. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1073–L1084. doi: 10.1152/ajplung.00347.2006. [DOI] [PubMed] [Google Scholar]
- 18.van Haaften T., Byrne R., Bonnet S., Rochefort G.Y., Akabutu J., Bouchentouf M., Rey-Parra G.J., Galipeau J., Haromy A., Eaton F., Chen M., Hashimoto K., Abley D., Korbutt G., Archer S.L., Thebaud B. Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am J Respir Crit Care Med. 2009;180:1131–1142. doi: 10.1164/rccm.200902-0179OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aslam M., Baveja R., Liang O.D., Fernandez-Gonzalez A., Lee C., Mitsialis S.A., Kourembanas S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med. 2009;180:1122–1130. doi: 10.1164/rccm.200902-0242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith V.C., Zupancic J.A., McCormick M.C., Croen L.A., Greene J., Escobar G.J., Richardson D.K. Rehospitalization in the first year of life among infants with bronchopulmonary dysplasia. J Pediatr. 2004;144:799–803. doi: 10.1016/j.jpeds.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 21.Weisman L.E. Populations at risk for developing respiratory syncytial virus and risk factors for respiratory syncytial virus severity: infants with predisposing conditions. Pediatr Infect Dis J. 2003;22:S33–S37. doi: 10.1097/01.inf.0000053883.08663.e5. discussion S37-S39. [DOI] [PubMed] [Google Scholar]
- 22.Bhandari A., Panitch H.B. Pulmonary outcomes in bronchopulmonary dysplasia. Semin Perinatol. 2006;30:219–226. doi: 10.1053/j.semperi.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Kennedy J.D., Edward L.J., Bates D.J., Martin A.J., Dip S.N., Haslam R.R., McPhee A.J., Staugas R.E., Baghurst P. Effects of birthweight and oxygen supplementation on lung function in late childhood in children of very low birth weight. Pediatr Pulmonol. 2000;30:32–40. doi: 10.1002/1099-0496(200007)30:1<32::aid-ppul6>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 24.Doyle L.W., Faber B., Callanan C., Freezer N., Ford G.W., Davis N.M. Bronchopulmonary dysplasia in very low birth weight subjects and lung function in late adolescence. Pediatrics. 2006;118:108–113. doi: 10.1542/peds.2005-2522. [DOI] [PubMed] [Google Scholar]
- 25.Doyle L.W. Respiratory function at age 8–9 years in extremely low birthweight/very preterm children born in Victoria in 1991–1992. Pediatr Pulmonol. 2006;41:570–576. doi: 10.1002/ppul.20412. [DOI] [PubMed] [Google Scholar]
- 26.Robin B., Kim Y.J., Huth J., Klocksieben J., Torres M., Tepper R.S., Castile R.G., Solway J., Hershenson M.B., Goldstein-Filbrun A. Pulmonary function in bronchopulmonary dysplasia. Pediatr Pulmonol. 2004;37:236–242. doi: 10.1002/ppul.10424. [DOI] [PubMed] [Google Scholar]
- 27.Yee M., Vitiello P.F., Roper J.M., Staversky R.J., Wright T.W., McGrath-Morrow S.A., Maniscalco W., Finkelstein J.N., O'Reilly M.A. Type II epithelial cells are a critical target for hyperoxia-mediated impairment of postnatal lung development. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1101–L1111. doi: 10.1152/ajplung.00126.2006. [DOI] [PubMed] [Google Scholar]
- 28.Schulman S.R., Canada A.T., Fryer A.D., Winsett D.W., Costa D.L. Airway hyperreactivity produced by short-term exposure to hyperoxia in neonatal guinea pigs. Am J Physiol. 1997;272:L1211–L1216. doi: 10.1152/ajplung.1997.272.6.L1211. [DOI] [PubMed] [Google Scholar]
- 29.McGrath-Morrow S.A., Cho C., Soutiere S., Mitzner W., Tuder R. The effect of neonatal hyperoxia on the lung of p21Waf1/Cip1/Sdi1-deficient mice. Am J Respir Cell Mol Biol. 2004;30:635–640. doi: 10.1165/rcmb.2003-0049OC. [DOI] [PubMed] [Google Scholar]
- 30.Denis D., Fayon M.J., Berger P., Molimard M., De Lara M.T., Roux E., Marthan R. Prolonged moderate hyperoxia induces hyperresponsiveness and airway inflammation in newborn rats. Pediatr Res. 2001;50:515–519. doi: 10.1203/00006450-200110000-00015. [DOI] [PubMed] [Google Scholar]
- 31.Dauger S., Ferkdadji L., Saumon G., Vardon G., Peuchmaur M., Gaultier C., Gallego J. Neonatal exposure to 65% oxygen durably impairs lung architecture and breathing pattern in adult mice. Chest. 2003;123:530–538. doi: 10.1378/chest.123.2.530. [DOI] [PubMed] [Google Scholar]
- 32.Yee M., Chess P.R., McGrath-Morrow S.A., Wang Z., Gelein R., Zhou R., Dean D.A., Notter R.H., O'Reilly M.A. Neonatal oxygen adversely affects lung function in adult mice without altering surfactant composition or activity. Am J Physiol Lung Cell Mol Physiol. 2009;297:L641–L649. doi: 10.1152/ajplung.00023.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Reilly M.A., Marr S.H., Yee M., Mcgrath-Morrow S.A., Lawrence B.P. Neonatal hyperoxia enhances the inflammatory response in adult mice infected with influenza A virus. Am J Respir Crit Care Med. 2008;177:1103–1110. doi: 10.1164/rccm.200712-1839OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mutlu G.M., Machado-Aranda D., Norton J.E., Bellmeyer A., Urich D., Zhou R., Dean D.A. Electroporation-mediated gene transfer of the Na+, K+ -ATPase rescues endotoxin-induced lung injury. Am J Respir Crit Care Med. 2007;176:582–590. doi: 10.1164/rccm.200608-1246OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pillow J.J., Korfhagen T.R., Ikegami M., Sly P.D. Overexpression of TGF-alpha increases lung tissue hysteresivity in transgenic mice. J Appl Physiol. 2001;91:2730–2734. doi: 10.1152/jappl.2001.91.6.2730. [DOI] [PubMed] [Google Scholar]
- 36.O'Reilly M.A., Staversky R.J., Stripp B.R., Finkelstein J.N. Exposure to hyperoxia induces p53 expression in mouse lung epithelium. Am J Respir Cell Mol Biol. 1998;18:43–50. doi: 10.1165/ajrcmb.18.1.2950m. [DOI] [PubMed] [Google Scholar]
- 37.Agrotis A., Samuel M., Prapas G., Bobik A. Vascular smooth muscle cells express multiple type I receptors for TGF-beta, activin, and bone morphogenetic proteins. Biochem Biophys Res Commun. 1996;219:613–618. doi: 10.1006/bbrc.1996.0282. [DOI] [PubMed] [Google Scholar]
- 38.Alejandre-Alcazar M.A., Kwapiszewska G., Reiss I., Amarie O.V., Marsh L.M., Sevilla-Perez J., Wygrecka M., Eul B., Kobrich S., Hesse M., Schermuly R.T., Seeger W., Eickelberg O., Morty R.E. Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L537–L549. doi: 10.1152/ajplung.00050.2006. [DOI] [PubMed] [Google Scholar]
- 39.Roper J.M., Staversky R.J., Finkelstein J.N., Keng P.C., O'Reilly M.A. Identification and isolation of mouse type II cells based upon intrinsic expression of enhanced green fluorescent protein. Am J Physiol Lung Cell Mol Physiol. 2003;285:L691–L700. doi: 10.1152/ajplung.00034.2003. [DOI] [PubMed] [Google Scholar]
- 40.Pang J., Hoefen R., Pryhuber G.S., Wang J., Yin G., White R.J., Xu X., O'Dell M.R., Mohan A., Michaloski H., Massett M.P., Yan C., Berk B.C. G-protein-coupled receptor kinase interacting protein-1 is required for pulmonary vascular development. Circulation. 2009;119:1524–1532. doi: 10.1161/CIRCULATIONAHA.108.823997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng Z., Haghighi F., Helleby L., Vanterpool K., Horn E.M., Barst R.J., Hodge S.E., Morse J.H., Knowles J.A. Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3-cM region on chromosome 2q33. Am J Respir Crit Care Med. 2000;161:1055–1059. doi: 10.1164/ajrccm.161.3.9906051. [DOI] [PubMed] [Google Scholar]
- 42.Lane K.B., Machado R.D., Pauciulo M.W., Thomson J.R., Phillips J.A., 3rd, Loyd J.E., Nichols W.C., Trembath R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium, Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 43.Yzydorczyk C., Comte B., Cambonie G., Lavoie J.C., Germain N., Ting Shun Y., Wolff J., Deschepper C., Touyz R.M., Lelievre-Pegorier M., Nuyt A.M. Neonatal oxygen exposure in rats leads to cardiovascular and renal alterations in adulthood. Hypertension. 2008;52:889–895. doi: 10.1161/HYPERTENSIONAHA.108.116251. [DOI] [PubMed] [Google Scholar]
- 44.Doyle L.W., Faber B., Callanan C., Morley R. Blood pressure in late adolescence and very low birth weight. Pediatrics. 2003;111:252–257. doi: 10.1542/peds.111.2.252. [DOI] [PubMed] [Google Scholar]
- 45.Maniscalco W.M., Watkins R.H., O'Reilly M.A., Shea C.P. Increased epithelial cell proliferation in very premature baboons with chronic lung disease. Am J Physiol Lung Cell Mol Physiol. 2002;283:L991–L1001. doi: 10.1152/ajplung.00050.2002. [DOI] [PubMed] [Google Scholar]
- 46.Machado R.D., Pauciulo M.W., Thomson J.R., Lane K.B., Morgan N.V., Wheeler L., Phillips J.A., 3rd, Newman J., Williams D., Galie N., Manes A., McNeil K., Yacoub M., Mikhail G., Rogers P., Corris P., Humbert M., Donnai D., Martensson G., Tranebjaerg L., Loyd J.E., Trembath R.C., Nichols W.C. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramos M.F., Lame M.W., Segall H.J., Wilson D.W. Smad signaling in the rat model of monocrotaline pulmonary hypertension. Toxicol Pathol. 2008;36:311–320. doi: 10.1177/0192623307311402. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi H., Goto N., Kojima Y., Tsuda Y., Morio Y., Muramatsu M., Fukuchi Y. Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;290:L450–L458. doi: 10.1152/ajplung.00206.2005. [DOI] [PubMed] [Google Scholar]
- 49.Reynolds A.M., Xia W., Holmes M.D., Hodge S.J., Danilov S., Curiel D.T., Morrell N.W., Reynolds P.N. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1182–L1192. doi: 10.1152/ajplung.00020.2006. [DOI] [PubMed] [Google Scholar]
- 50.Austin E.D., Loyd J.E. Genetics and mediators in pulmonary arterial hypertension. Clin Chest Med. 2007;28:43–57. doi: 10.1016/j.ccm.2006.11.007. vii-viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song Y., Jones J.E., Beppu H., Keaney J.F., Jr., Loscalzo J., Zhang Y.Y. Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation. 2005;112:553–562. doi: 10.1161/CIRCULATIONAHA.104.492488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harrison R.E., Flanagan J.A., Sankelo M., Abdalla S.A., Rowell J., Machado R.D., Elliott C.G., Robbins I.M., Olschewski H., McLaughlin V., Gruenig E., Kermeen F., Halme M., Raisanen-Sokolowski A., Laitinen T., Morrell N.W., Trembath R.C. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40:865–871. doi: 10.1136/jmg.40.12.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park S.O., Lee Y.J., Seki T., Hong K.H., Fliess N., Jiang Z., Park A., Wu X., Kaartinen V., Roman B.L., Oh S.P. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood. 2008;111:633–642. doi: 10.1182/blood-2007-08-107359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finer N.N., Carlo W.A., Walsh M.C., Rich W., Gantz M.G., Laptook A.R., Yoder B.A., Faix R.G., Das A., Poole W.K., Donovan E.F., Newman N.S., Ambalavanan N., Frantz I.D., 3rd, Buchter S., Sanchez P.J., Kennedy K.A., Laroia N., Poindexter B.B., Cotten C.M., Van Meurs K.P., Duara S., Narendran V., Sood B.G., O'Shea T.M., Bell E.F., Bhandari V., Watterberg K.L., Higgins R.D. Early CPAP versus surfactant in extremely preterm infants. N Engl J Med. 2010;362:1970–1979. doi: 10.1056/NEJMoa0911783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carlo W.A., Finer N.N., Walsh M.C., Rich W., Gantz M.G., Laptook A.R., Yoder B.A., Faix R.G., Das A., Poole W.K., Schibler K., Newman N.S., Ambalavanan N., Frantz I.D., 3rd, Piazza A.J., Sanchez P.J., Morris B.H., Laroia N., Phelps D.L., Poindexter B.B., Cotten C.M., Van Meurs K.P., Duara S., Narendran V., Sood B.G., O'Shea T.M., Bell E.F., Ehrenkranz R.A., Watterberg K.L., Higgins R.D. Target ranges of oxygen saturation in extremely preterm infants. N Engl J Med. 2010;362:1959–1969. doi: 10.1056/NEJMoa0911781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stevens T.P., Dylag A., Panthagani I., Pryhuber G., Halterman J. Effect of cumulative oxygen exposure on respiratory symptoms during infancy among VLBW infants without bronchopulmonary dysplasia. Pediatr Pulmonol. 2010;45:371–379. doi: 10.1002/ppul.21199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun J., Chen H., Chen C., Whitsett J.A., Mishina Y., Bringas P., Jr., Ma J.C., Warburton D., Shi W. Prenatal lung epithelial cell-specific abrogation of Alk3-bone morphogenetic protein signaling causes neonatal respiratory distress by disrupting distal airway formation. Am J Pathol. 2008;172:571–582. doi: 10.2353/ajpath.2008.070286. [DOI] [PMC free article] [PubMed] [Google Scholar]