

Abstract
Among the exotoxins produced by Staphylococcus aureus and Streptococcus pyogenes, the superantigens (SAgs) are the most potent T-cell activators known to date. SAgs are implicated in several serious diseases including toxic shock syndrome (TSS), Kawasaki disease, and sepsis. However, the immunopathogenesis of TSS and other diseases involving SAgs are still not completely understood. The commonly used conventional laboratory mouse strains do not respond robustly to SAgs in vivo. Therefore, they must be artificially rendered susceptible to TSS by using sensitizing agents such as d-galactosamine (d-galN), which skews the disease exclusively to the liver and, hence, is not representative of the disease in humans. SAg-induced TSS was characterized using transgenic mice expressing HLA class II molecules that are extremely susceptible to TSS without d-galN. HLA-DR3 transgenic mice recapitulated TSS in humans with extensive multiple-organ inflammation affecting the lung, liver, kidneys, heart, and small intestines. Heavy infiltration with T lymphocytes (both CD4+ and CD8+), neutrophils, and macrophages was noted. In particular, the pathologic changes in the small intestines were extensive and accompanied by significantly altered absorptive functions of the enterocytes. In contrast to massive liver failure alone in the d-galN sensitization model of TSS, findings of the present study suggest that gut dysfunction might be a key pathogenic event that leads to high morbidity and mortality in humans with TSS.
The gram-positive cocci Staphylococcus aureus and Streptococcus pyogenes cause a spectrum of diseases ranging from localized minor skin infections to life-threatening systemic illnesses.1 The virulence of these organisms and the nature and outcome of diseases caused by these bacteria are determined by their many exotoxins.2 Important among them are the superantigen exotoxins.3,4 Superantigens (SAgs) are the most potent naturally occurring biological activators of T lymphocytes. Unlike conventional antigens, unprocessed SAgs bind directly to cell surface major histocompatibility complex class II (MHC-II) molecules outside of the peptide-binding groove. Subsequently, they activate both CD4+ and CD8+ T cells by binding to certain variable-region β-chain families of the T-cell receptor (TCR Vβ) and cross-linking the TCR. The broad specificity of SAgs only to the TCR Vβ families rather than to the paratope of TCR results in activation of a large pool of T cells (30% to 70% of total T cells). Therefore, unlike conventional antigens, SAgs cause MHC-II–dependent, MHC-unrestricted, CD4 and CD8 co-receptor–independent, TCR Vβ–specific but antigen-nonspecific T-cell activation.3,5 Activated T cells produce a variety of cytokines and chemokines, which both directly and indirectly amplify this inflammatory cascade and trigger production of additional cytokines and chemokines from a variety of other sources. This process results in a massive cytokine/chemokine storm called systemic inflammatory response syndrome (SIRS), which ultimately leads to multiple-organ dysfunction syndrome (MODS) and culminates in high mortality if intervention is not prompt.6,7
S. aureus and S. pyogenes produce nearly 30 different SAgs.3,4 While the mechanisms that regulate production of these exotoxins in vivo are not fully understood, several SAgs are produced in vivo after infection, and SAgs contribute to the pathogenesis of the associated disease.8-10 The severity of disease is probably determined by the quantitative and qualitative characteristics of the SAg. Toxic shock syndrome (TSS) is one of the most serious clinical syndromes caused by S. aureus and S. pyogenes and their SAgs. TSS can be either menstrual or nonmenstrual.11 The incidence of menstrual TSS is declining worldwide as a result of discontinuation of the tampons associated with it and increased awareness among tampon users. However, menstrual TSS continues to be a significant health concern.12,13 In addition, nonmenstrual TSS, sepsis, and other serious infectious diseases caused by these bacteria continue to pose substantial problems.7,14-16 Exposure to SAgs produced ex vivo by organisms growing in surgical tampons also can cause TSS.17 Additional systemic diseases believed to be caused by SAgs and that share immunopathogenesis similar to TSS include Kawasaki disease18 and neonatal TSS-like exanthematous disease,19 among others. In addition, SAgs are highly expressed by the methicillin-resistant S. aureus strains associated with community and hospital settings.9,20-22
In addition to their clinical significance, SAgs are also important from a biodefense initiative. One of the SAg exotoxins released by S. aureus, staphylococcal enterotoxin B (SEB), is a designated biological weapon. Weaponized SEB is intended for aerosol use or for contaminating food and drinking water.23,24 Many countries including the United States have developed bioweapons containing SEB. Although development of bioweapons containing SEB has been disbanded in the United States, the possibility that SEB might be used as one of several bioweapons remains. Realizing the potential for malicious use of SEB in biological warfare or bioterrorism, the NIH has included SEB in category B of priority pathogens in its biodefense initiative. Malicious use of weapon-grade SEB can cause significant morbidity and mortality in defense personnel, emergency health care workers, and the general public. Exposure to weaponized SEB causes symptoms similar to TSS because weaponized SEB also causes potent immune activation.
Despite the clinical significance of staphylococcal SAgs, no specific therapies are available to treat staphylococcal TSS. However, intravenously administered immunoglobulins are effective in treating streptococcal TSS,25 and recently, antibiotics such as linezolid have been effective in treatment of staphylococcal TSS in some patients.26 Nevertheless, the lack of effective therapies could be attributed to incomplete understanding of the immunopathogenesis of TSS. This could be due in part to lack of good animal models. Rabbits have been used in staphylococcal toxin research including staphylococcal enterotoxins even before their superantigenicity was established.27 However, even the rabbit model of TSS seems to require sensitization with lipopolysaccharide to induce death,28,29 or require continuous infusion of SAg for 7 days.30 Moreover, even in the rabbit model of TSS, extensive investigation of the temporal changes in systemic cytokine and chemokine concentrations, an in-depth immunopathologic characterization of multiple-organ disease, and ultimately the correlation between systemic inflammatory response, multiple-organ disease, and the outcome of TSS have not been performed. Additional inherent drawbacks associated with rabbit or other animal models in biomedical research such as husbandry difficulties, availability of reagents and antibodies, and ease of gene manipulation led to development of mouse models of TSS.
Existing knowledge of the pathogenesis of TSS caused by SAgs is based on studies using conventional mice such as BALB/c or C57BL/6. However, poor binding of SAgs to murine MHC-II molecules results in weak activation of T cells in mice.31 As a result, SIRS induced by SAgs is substantially subdued in conventional mice, rendering them resistant to TSS.11,31 Therefore, conventional mice are artificially made susceptible to TSS by using sensitizing agents such as d-galactosamine (d-galN), actinomycin, or lipopolysaccharide.32 d-galN, the commonly used sensitizing agent for inducing TSS, is extremely toxic even when administered alone,33 and induces liver injury mimicking viral hepatitis.34,35 d-galN and actinomycin profoundly sensitize hepatocytes to tumor necrosis factor-α (TNF-α)–mediated injury.36 Therefore, murine TSS is characterized by massive liver failure alone, with dramatically elevated serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) concentrations, whereas TSS in humans is characterized by MODS. Therefore, TSS in conventional mouse models is not truly reflective of the disease in humans and cannot be extrapolated to humans. For example, in studies using d-galN–sensitized mouse models, TNF-α blockade completely prevented SAg-induced TSS and death in conventional mice.37 However, TNF-α blockade is ineffective or even counterproductive in S. aureus–induced sepsis in humans.38 These discrepancies warrant an animal model that better recapitulates the disease in humans.
SAgs bind to human MHC [called human leukocyte antigen (HLA)]–II molecules with much higher affinity than to murine MHC-II molecules.39 Therefore, substitution of endogenous mouse MHC-II molecules, which are poor binders of SAgs, with HLA class II (HLA-II) molecules, which are more efficient binders of SAgs, converts the otherwise poor responders to robust responders to SAgs. Challenging HLA-II transgenic mice with SAgs thorough several different routes elicits robust SIRS and renders them highly susceptible to TSS.40-49 One of our HLA-II transgenic lines that expresses HLA-DR3 is extremely sensitive to SEB. HLA-DR3 transgenic mice readily die of SEB-induced TSS without d-galN sensitization. This provides a unique opportunity to dissect the immunopathogenesis of SAg-induced TSS without d-galN sensitization, recapitulating the situation in humans. Further, HLA-DR3 transgenic mice are susceptible also to d-galN–sensitized TSS. Therefore, HLA-DR3 transgenic mice provide a unique tool for comparison of the immunopathogenesis of TSS with and without d-galN sensitization. The present study demonstrates marked differences between the immunopathogenesis of TSS induced with and without d-galN sensitization and how d-galN–sensitized models have likely confounded understanding of the pathogenesis of TSS.
Materials and Methods
Mice
AE.HLA-DR3 transgenic mice expressing functional HLA-DRA1*0101 and HLA-DRB1*0301 transgenes on the complete mouse MHC-II–deficient background were used in the present study.42,50,51 HLA-DR3 transgenic mice lacking CD4+ T cells (HLA-DR3.CD4−/−) and CD8+ T cells (HLA-DR3.CD8−/−) were also used. AE.HLA-DR3 mice transgenically expressing the luciferase (Luc) gene under the nuclear factor κB (NF-κB) promoter were generated by mating AE.HLA-DR3 mice and NF-κB-Luc mice [B10.Cg-H2k Tg (NF-κB/Fos-Luc); 26Rinc/J; stock No. 006100 (Jackson Laboratory, Bar Harbor, ME)].52 Endogenous MHC-II–null (AE), HLA-DR3+, and NF-κB–Luc+ offspring were selected and backcrossed to AE.HLA-DR3 transgenic mice to establish the AE.HLA-DR3.NF-κB–Luc line (hereafter referred to as DR3.Luc mice). Mice were bred within the barrier facility of the Mayo Clinic Immunogenetics Mouse Colony (Rochester, MN) and were moved to a conventional facility after weaning. All of the experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee. The Association for Assessment and Accreditation of Laboratory Animal Care accreditation number is 000717, and the Office of Laboratory Animal Welfare assurance number is A3291-01.
Reagents
Endotoxin-reduced highly purified SEB (Toxin Technology Inc., Sarasota, FL) was dissolved in 1 mg/mL PBS and stored frozen at −80°C in aliquots. The purity of SEB was verified using SDS-PAGE and a staphylococcal enterotoxin identification visual immunoassay (SET VIA; TECRA International Pty Ltd., New South Wales, Australia). d-galN and fluorescein isothiocyanate (FITC)–conjugated DEAE-dextran (4 kDa) were obtained from Sigma-Aldrich Corp. (St. Louis, MO). d-Luciferin was obtained from Gold Biotechnology, Inc. (St. Louis, MO).
Induction of TSS
Mice were assigned to one of three treatment groups: 10 μg SEB alone (low dose), 10 μg SEB immediately followed by 30 mg dgalN (SEB+d-galN), or 50 μg SEB alone (high dose) or 30 mg d-galN alone. All agents were dissolved in 200 μL PBS and administered via intraperitoneal injection. Mice were monitored frequently for symptoms of TSS as recommended by the Institutional Animal Care and Use Committee. As recommended, moribund animals were removed from the study. At the end of the experiment, survival curves were plotted.
Serum Biochemical Studies, Cytokine Quantification, and Histopathologic Analysis
Groups of HLA-DR3 transgenic mice were challenged with SEB as described and were sacrificed at 3, 6, 9, 12, 24, 48, or 72 hours. At the time of sacrifice, blood from experimental mice was collected in serum separation tubes (BD Biosciences, Franklin Lakes, NJ), and serum samples were separated and stored frozen at −80°C in aliquots. Serum ALT, AST, and creatinine concentrations were determined by the Mayo Clinic clinical chemistry laboratory. Serum samples from three or four mice from each group were pooled for biochemical studies. The cytokine concentrations in individual serum samples were determined using a multiplex bead assay (Bio-Plex; Bio-Rad Laboratories, Inc., Hercules, CA) per the manufacturer's protocol and using their software and hardware. Tissues collected in buffered formalin were paraffin-embedded, cut, and stained with H&E per standard procedure for histopathologic analysis.
Immunofluorescence
Immediately after mice were sacrificed, tissues were also collected in optimal cutting compound (OCT) (Tissue-Tek; Sakura Finetek USA, Inc., Torrance, CA) and stored frozen at −80°C. Five-μm sections were cut using a cryostat, fixed in cold acetone, and stained with fluorochrome-conjugated antibodies using standard techniques. Sections were mounted using the SlowFade Gold Antifade Reagent with DAPI (Invitrogen Corp., Carlsbad, CA) and were analyzed the next day.
Detection of Apoptosis using TUNEL Assay
Apoptosis in tissue preparations was ascertained in thin sections of paraffin-embedded blocks on microscopic glass slides using the In Situ Cell Death Detection kit following the manufacturer's directions (Roche Diagnostics Corp., Indianapolis, IN). As a positive control for the TUNEL reaction, sections were incubated with DNase I to introduce DNA strand breaks before adding the reaction mix. Negative control sections were treated the same as samples except that terminal deoxyribonucleotidyl transferase was omitted from the reaction mix. After staining, sections were evaluated using an Olympus AX70 research microscope (Olympus America Inc., Center Valley, PA). Images were acquired using an Olympus DP70 camera.
Gut Permeability
Changes in intestinal permeability were determined using 4-KDa FITC-labeled dextran.53 Two hours after injection with low-dose SEB, d-galN, or low-dose SEB+d-galN, mice were deprived of food for 3 hours. In the high-dose SEB group, 24, 48, and 72 hours after SEB challenge, mice were deprived of food for 3 hours. Naïve mice were also deprived of food for 3 hours. All mice then underwent gavage with FITC–labeled dextran (0.6 mg/g body weight). Subsequently, all mice were sacrificed 3 hours after gavage, and blood was collected via cardiac puncture. (Note: Because mice treated with low-dose SEB+d-galN rapidly die of TSS, gut permeability status was determined at 8 hours after SEB injection in this group). Serum samples were separated using centrifugation and stored frozen at −80°C until analysis. Serum FITC-dextran content was determined indirectly by measuring FITC fluorescence using a microplate reader (excitation, 490 nm; emission, 525 nm).
In Vivo Imaging of Luciferase Activity
Naive and SEB-challenged (50 μg per mouse) DR3.Luc transgenic mice were injected intraperitoneally with 200 μL d-luciferin (15 mg/mL stock) at indicated times. After 12 to 15 minutes, mice were placed in the anesthesia chamber delivering isoflurane. While under anesthesia, mice were transferred to the imaging chamber and imaged using the Xenogen IVIS Imaging System 200 Series with the XGI-8 Gas Anesthesia System using Living Image software, version 2.6 (Xenogen Corp., Alameda, CA). Fur was also removed before imaging. In some experiments, mice were sacrificed 10 minutes after d-luciferin injection. Individual organs were harvested and placed in small Petri dishes for imaging.
Statistical Analysis
Survival curves and statistical significance of other results were determined using PRISM version 3.0a software (GraphPad Software Inc., San Diego, CA).
Results
Temporal Differences between Occurrence of TSS with and without d-galN Sensitization
Age-matched HLA-DR3 transgenic mice were challenged with 10 μg SEB alone (low dose), 30 mg D-galN alone, 10 μg SEB+D-galN, or 50 μg SEB alone (high dose). Mice were observed daily for a week. Mice challenged with low-dose SEB alone seldom died of TSS (Figure 1). Similarly, d-galN alone was rarely lethal. However, a combination of low-dose SEB+d-galN was uniformly lethal. Although the animals that received low-dose SEB+d-galN remained healthy during the first 4 to 5 hours, thereafter they rapidly became hypothermic and lethargic, and invariably died of TSS by 9 hours. In the d-galN sensitization model, even 5 and 2 μg SEB were uniformly lethal, with similar time kinetics (data not shown). In contrast, mice that received 50 μg SEB alone (high dose) gradually exhibited hypothermia, diarrhea, and body weight loss.54 In the high-dose SEB model, mice generally began dying after 48 hours, and most of the animals died by 72 hours. The remaining few animals continued to lose weight, and seldom survived beyond 6 days. Death almost always occurred during the overnight hours. No significant difference was observed in patterns of death between male and female mice. There was a significant difference (P < 0.0001) in the survival pattern induced by high-dose SEB without d-galN sensitization and low-dose SEB with d-galN sensitization.
Figure 1.
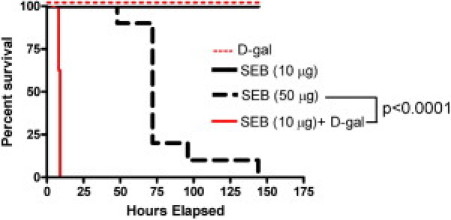
Incidence of SEB-induced mortality in HLA-DR3 transgenic mice with and without d-galN sensitization. Twelve- to 14-week-old HLA-DR3 transgenic mice (6 to 11 mice per group) were treated as indicated. Mice were monitored once every 3 hours through 9 hours, and daily thereafter. Survival curves and statistical significance were generated using PRISM software (version 3.0a; GraphPad Software Inc., San Diego, CA).
Distinct Serum Biochemical Profiles between TSS with and without d-galN Sensitization
Serum samples for biochemical studies were collected from treated mice. Because mice treated with low-dose SEB+d-galN were dead by 9 hours, serum samples were collected every 3 hours through 9 hours, using different groups of mice for every 3-hour interval. Because mice treated with high-dose SEB without d-galN survived for up to 72 hours, serum samples were collected every 3 hours through 12 hours and then every 24 hours through 72 hours. ALT and AST concentrations in mice that received d-gal alone or low-dose SEB alone were not significantly different from those in naïve mice (Figure 2). In contrast, mice that received low-dose SEB with d-galN sensitization demonstrated the highest elevation in serum ALT and AST concentrations. There was a sudden spike in ALT and AST concentrations at 9 hours, at which time most of the animals had died of TSS. Serum ALT and AST concentrations in mice that received high-dose SEB were not elevated and were comparable to those in mice challenged with low-dose SEB alone or d-galN alone. This suggested that only the combination of SEB and D-galN is highly hepatotoxic. Serum creatinine concentrations were comparable in all SEB-treated groups irrespective of d-galN sensitization status (Figure 2). Even the nonlethal low-dose SEB alone and the lethal high-dose SEB groups demonstrated similar serum creatinine concentrations at all time points (additional data not shown). This excludes the possibility of kidney dysfunction as a likely cause of death in the high-dose SEB group. Conversely, inasmuch as only SEB+d-galN–sensitized mice had highly elevated ALT and AST concentrations and rapidly died of SEB-induced TSS, liver dysfunction could be a possible cause of death in this group of mice.
Figure 2.
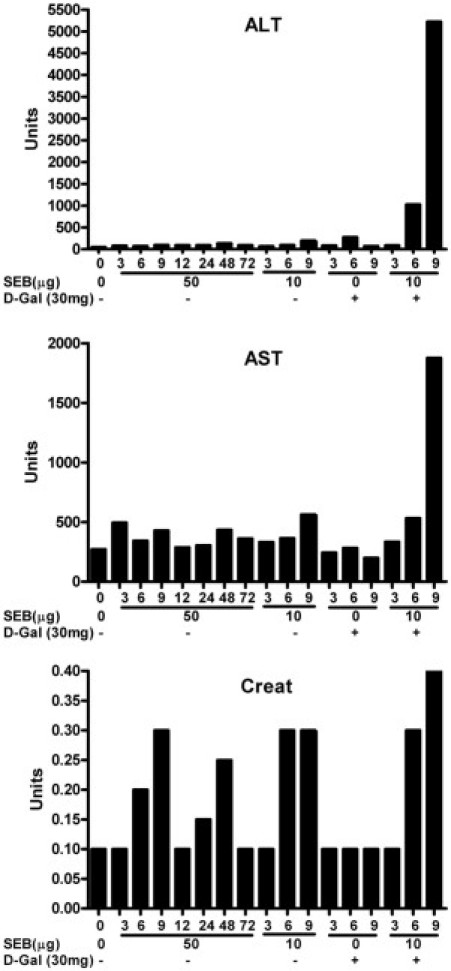
Serum biochemical changes in HLA-DR3 transgenic mice undergoing SEB-induced TSS with and without d-galN sensitization.Twelve- to 14-week-old HLA-DR3 transgenic mice were challenged as indicated. Groups of mice (three or four mice per group) were sacrificed at indicated times. Serum ALT, AST, and creatinine concentrations were determined in serum samples pooled from each group. The entire experiment was repeated at least three times, with similar results.
Distinct Cytokine Profiles between TSS with and without d-galN Sensitization
Serum concentrations of a panel of cytokines and chemokines were measured. Serum cytokine and chemokine concentrations in mice challenged with d-galN alone at 3 hours were not very different from those of naïve mice, which suggests that d-galN by itself does not possess immune activating potential (Figures 3 and 4). Compared with naïve mice, mice challenged with a nonlethal low dose of SEB alone demonstrated significantly elevated concentrations of almost all cytokines and chemokines by 3 hours, as has been demonstrated previously.50,55 The serum concentrations of various cytokines and chemokines changed over time (Figures 3 and 4). In mice that received the lethal combination of D-galN and low-dose SEB, the serum concentrations of various cytokines and chemokines were not significantly different from those in mice that received low-dose SEB alone (except IL-4, which was reduced in the combination group compared with the low-dose SEB alone group) at 3 hours (unpaired Student's t-test). However, some cytokines were significantly changed at 6 hours (unpaired Student's t-test). For example, inclusion of d-galN significantly increased the serum concentrations of IL-2, IL-17, and interferon-γ (IFN-γ). At 9 hours, there were no significant differences in the concentrations of these and other cytokines and chemokines between the low-dose SEB+d-galN and the low-dose SEB alone groups except for IL-5, which was reduced in the combination group. Thus, inclusion of d-galN reduced the serum concentrations of Th2-type cytokines such as IL-4 and IL-5, and at the same time increased serum concentrations of proinflammatory-type cytokines such as IL-2, IL-17, and IFN-γ. There were no significant differences in serum concentrations of TNF-α between the low-dose SEB+d-galN and the low-dose SEB alone groups (unpaired Student's t-test). Serum concentrations of cytokines and chemokines in HLA-DR3 transgenic mice challenged with the lethal high-dose SEB were not significantly different from those in mice challenged with the nonlethal low-dose SEB except for IFN-γ at 9 hours. However, when compared with mice challenged with low-dose SEB+d-galN, serum concentrations of IL-2, IL-6, IL-17, and IFN-γ were significantly lower in mice challenged with high-dose SEB, and serum concentrations of IL-4 and IL-5 were higher. These observations indicated that in the high-dose SEB model, even though death generally occurred after 48 hours, serum cytokine and chemokine concentrations were not persistently elevated and that inclusion of d-galN reduced the serum concentrations of Th2-type cytokines such as IL-4 and IL-5 and at the same time increased the serum concentrations of the proinflammatory-type cytokines such as IL-2, IL-17, and IFN-γ. Overall, there were some differences in serum cytokine and chemokine profiles between TSS caused by SEB with and without d-galN sensitization, which implies possible differences in immunopathogenesis.
Figure 3.
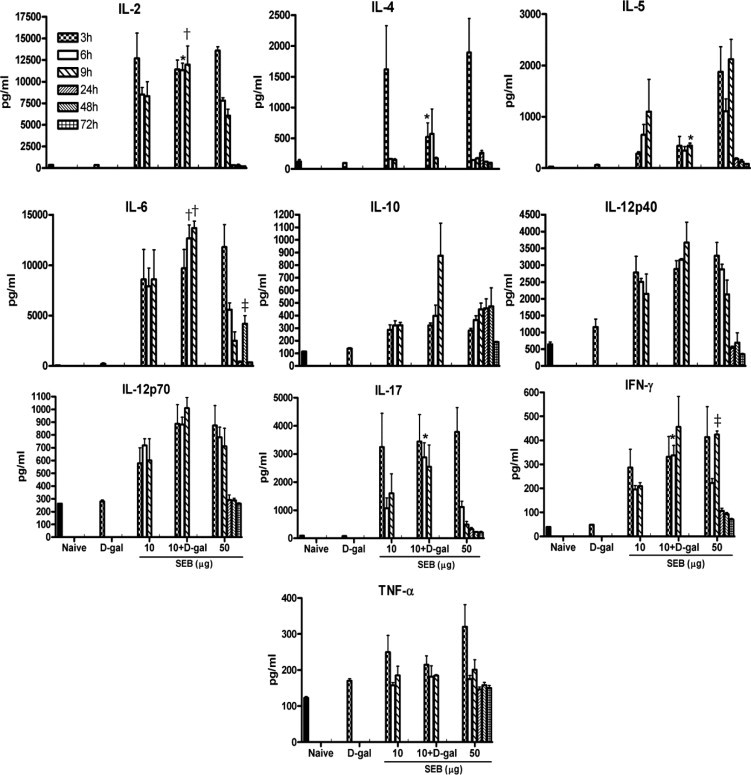
Temporal changes in systemic cytokine levels during SEB-induced TSS. Twelve- to 14-week-old HLA-DR3 transgenic mice were challenged as indicated. Groups of mice (four to six mice per group) were sacrificed at indicated times, and serum cytokine levels were determined using a multiplex suspension array system (Bio-Plex; Bio-Rad Laboratories, Inc., Hercules, CA). Bars represent mean ± SE values for four to six mice. Unpaired Student's t-test was used to determine statistical significance using PRISM software (version 3.0a; GraphPad Software Inc., San Diego, CA). *P < 0.05 with 10 and 50 µg SEB; †P < 0.05 with 50 µg SEB; ‡P < 0.05 with 10 µg SEB.
Figure 4.
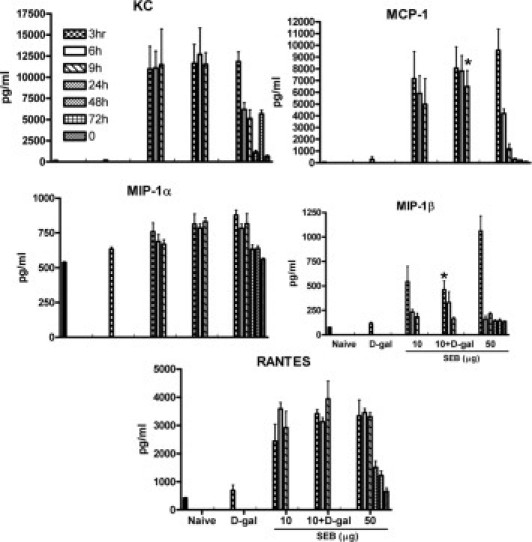
Temporal changes in systemic chemokine levels during SEB-induced TSS. Twelve- to 14-week-old HLA-DR3 transgenic mice were challenged as indicated. Groups of mice (four to six mice per group) were sacrificed at indicated times, and serum cytokine levels were determined using a multiplex suspension array system (Bio-Plex; Bio-Rad Laboratories, Inc., Hercules, CA). Bars represent mean ± SE values for four to six mice. Unpaired Student's t-test was used to determine statistical significance using PRISM software (version 3.0a; GraphPad Software Inc., San Diego, CA). *P < 0.05 with SEC 50 µg.
CD4+ and CD8+ T-Cell Subsets in Pathogenesis of SEB-Induced TSS without d-galN Sensitization
Whether both CD4+ and CD8+ T-cell subsets are required for precipitating TSS with and without d-galN sensitization was investigated. In the presence of d-galN sensitization, both HLA-DR3.CD4−/− and HLA-DR3.CD8−/− mice were equally susceptible (mortality, 6 of 6 in each group) to TSS induced by low-dose SEB. The disease course was identical to that observed in HLA-DR3 transgenic mice with both CD4+ and CD8+ T-cell subsets present (data not shown). However, in the absence of d-galN sensitization in the mice with high-dose SEB-induced TSS, none of six HLA-DR3.CD4−/− mice and only 1 of 6 HLA-DR3.CD8−/− mice were susceptible. This further confirmed that the pathogenesis of TSS with and without d-galN sensitization differs.
Distinct Histopathologic Profiles between TSS with and without d-galN Sensitization
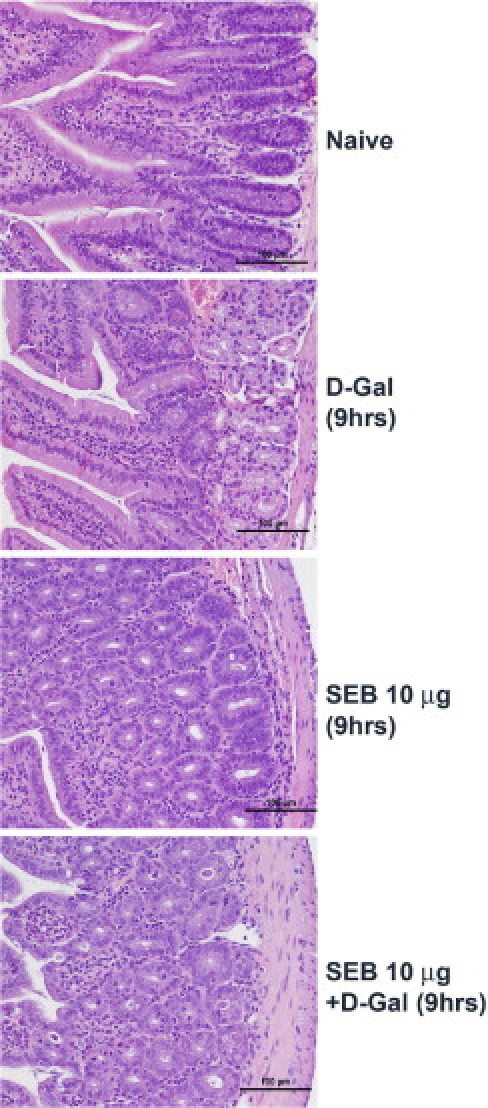
Histopathologic changes associated with TSS caused by SEB with and without d-galN sensitization were studied. Inasmuch as mice treated with SEB+d-galN rarely survived longer than 10 hours, histopathologic analyses were performed at 9 hours in the d-galN–sensitized mice. Compared with the normal liver sections from naïve mice, liver sections from mice that received d-galN alone exhibited mild hepatitis with focal areas of infiltration with mononuclear cells, as had been demonstrated in previous studies34,35 (Figure 5). Mice that received SEB alone also demonstrated perivascular infiltration with mononuclear cells. However, mice that received both SEB and d-galN demonstrated extensive hemorrhagic hepatitis with large areas of mononuclear cell infiltration (Figure 5). This correlated with markedly elevated serum ALT and AST concentrations. Whereas lung sections from naïve mice were normal, lungs from mice treated with d-GalN alone, SEB alone, and SEB+d-galN demonstrated comparable minor inflammatory changes with perivascular cuffing (Figure 5). Kidney sections from mice from all groups were relatively normal (Figure 5). These histopathologic findings along with the serum biochemical analyses reiterate that massive liver failure is probably the major cause of death in mice treated with low-dose SEB+d-galN. Because death in the SEB alone (high-dose) group always occurred after 72 hours, histopathologic analyses were performed at 24, 48, and 72 hours in this cohort.
Figure 5.
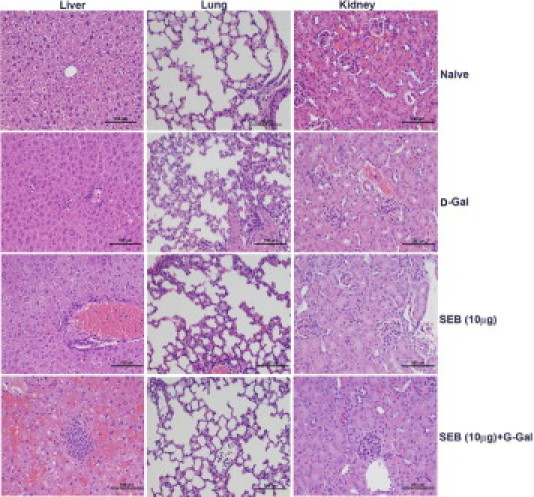
Immunopathology in d-galN–sensitized model of TSS with low-dose SEB. Twelve- to 14-week-old HLA-DR3 transgenic mice were challenged as indicated, with SEB alone, d-galN alone, or SEB+d-galN. Mice were sacrificed at 9 hours, and organs were collected in buffered formalin and processed for H&E staining. Scale bars = 100 μm.
Compared with normal lung sections from naïve mice, lung sections from mice that received 50 μg SEB began to exhibit inflammatory changes at 24 hours and thereafter (Figure 6). There was visible perivascular cuffing and mononuclear cell infiltration in the alveolar spaces at 24 hours. Immunofluorescence staining revealed infiltration of both CD4+ and CD8+ T lymphocytes. The degree of inflammatory infiltrates increased over time, and by 72 hours, the lungs exhibited extensive inflammatory changes. The alveoli were collapsed, with widespread mononuclear cell infiltration largely composed of CD4+ and CD8+ T cells (Figure 6). The kidneys also demonstrated similar temporal changes in inflammation and mononuclear cell infiltration (Figure 7). Perivascular and periglomerular infiltration with CD4+ and CD8+ T cells were present (Figure 7). In the liver, infiltration with mononuclear cells was minimal at 24 hours (Figure 8). At 48 and 72 hours, there was extensive infiltration with CD4+ and CD8+ T cells, largely around the central vein (Figure 8). However, parenchymal infiltration with T cells was also observed (Figure 8). Most important, unlike the liver sections from mice treated with low-dose SEB+d-galN, liver sections from mice receiving 50 μg SEB alone did not demonstrate massive hemorrhagic hepatitis. The presence of macrophages (CD11b+) and neutrophils (7/4+) was readily detected in the inflammatory infiltrates in the lung and liver but not in the kidneys (see Supplemental Figure S1 at http://ajp.amjpathol.org). Large-scale apoptosis was not detected in liver, lungs, or kidneys of mice receiving high-dose SEB (see Supplemental Figure S2 at http://ajp.amjpathol.org). In general, tissues collected from the high-dose SEB group at 9 hours demonstrated minimal inflammatory changes (data not shown). Inflammatory changes were also demonstrated at 72 hours in heart tissue in mice treated with high-dose SEB but not in other groups (Figure 9) (additional data not shown). Infiltration of the myocardium with CD4+ T lymphocytes was detected (Figure 9). Overall, SEB-induced TSS without d-galN sensitization in HLA-II transgenic mice resulted in multiple-organ inflammation similar to that described in TSS in humans. Whereas in the SEB+d-galN murine model of SEB-induced TSS, the disease was restricted largely to the liver.
Figure 6.
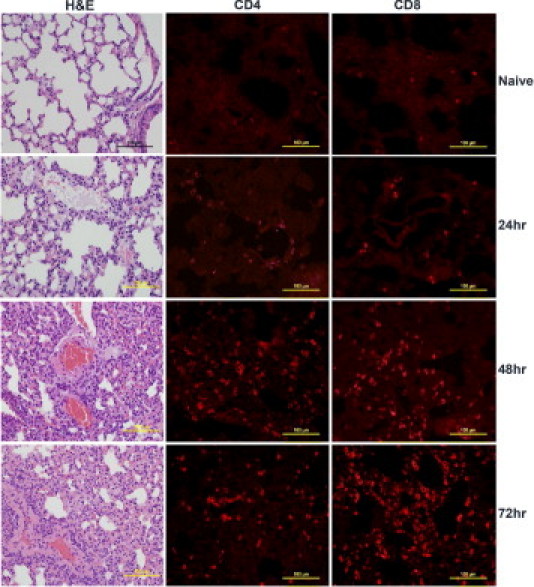
Pulmonary immunopathology in high-dose SEB-induced TSS without d-galN sensitization. Twelve- to 14-week-old HLA-DR3 transgenic mice were either untreated or challenged with 50 μg SEB. Mice were sacrificed at indicated times, and lungs were collected in either buffered formalin or OCT compound and processed for H&E or immunostaining, respectively. Scale bars = 100 μm.
Figure 7.
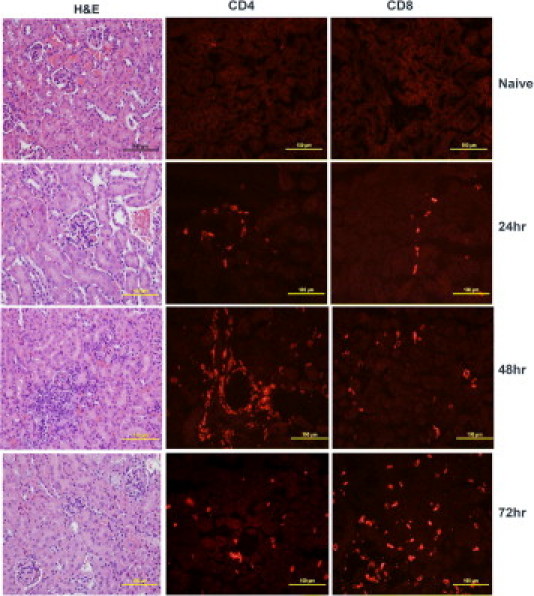
Renal immunopathology in high-dose SEB-induced TSS without d-galN sensitization. Twelve- to 14-week-old HLA-DR3 transgenic mice were either untreated or challenged with 50 μg SEB. Mice were sacrificed at indicated times, and kidneys were collected in either buffered formalin or OCT compound and processed for H&E or immunostaining, respectively. Scale bars = 100 μm.
Figure 8.
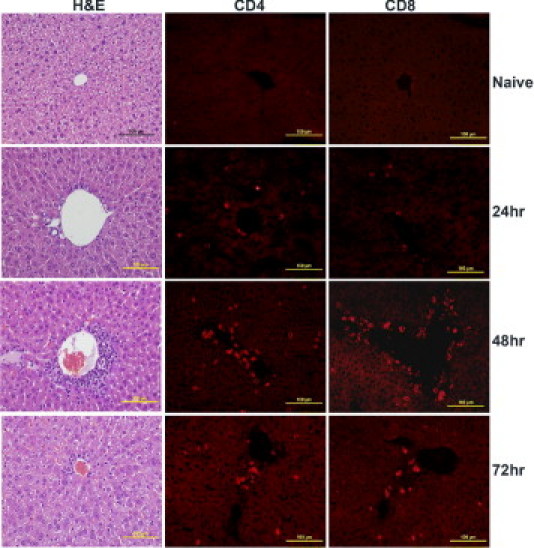
Hepatic immunopathology in high-dose SEB-induced TSS without d-galN sensitization. Twelve- to 14-week-old HLA-DR3 transgenic mice were either untreated or challenged with 50 μg SEB. Mice were sacrificed at indicated times, and livers were collected in either buffered formalin or OCT compound and processed for H&E or immunostaining, respectively. Scale bars = 100 μm.
Figure 9.
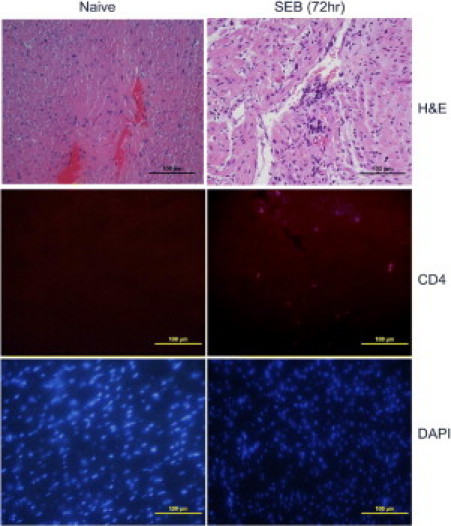
Cardiac immunopathology in high-dose SEB-induced TSS without d-galN sensitization. Twelve- to 14-week-old HLA-DR3 transgenic mice were either untreated or challenged with 50 μg SEB. Mice were sacrificed at various time, and hearts were collected in either buffered formalin or OCT compound and processed for H&E or immunostaining, respectively. Figure shows heart collected at 72 hours. Scale bars = 100 μm.
Gut Disease in TSS with and without d-galN Sensitization
The gut is a major secondary lymphoid organ rich in T lymphocytes and antigen-presenting cells. As a result, systemic exposure to SAgs would directly stimulate T cells in the gut. Thus, the gut might also experience pathologic changes. However, to our knowledge, gut disease has not been reported in d-galN–sensitized models of TSS. Therefore, the extent of gut disease was studied in the high- and low-dose models of SEB-induced TSS. Small and large intestines from naïve mice and mice treated with D-galN alone, low-dose SEB alone, and SEB+d-galN were relatively normal, with occasional apoptotic cells in mice treated with SEB alone and SEB+d-galN (Figure 10). In contrast, small intestines from mice receiving high-dose SEB alone exhibited extensive pathologic changes. The villi demonstrated extensive degenerative changes. At 24 hours, degenerative changes were observed in the epithelial layer. Apoptotic cells were clearly visible (inset, Figure 11). At 48 hours, extensive cellular debris was observed in the intestinal lumen, and the epithelial integrity was lost. At 72 hours, the height of the villi was dramatically reduced, and blunting of the villi was readily noticeable (Figure 11). Infiltration with CD4+ and CD8+ T cells was evident (Figure 11). Macrophage and neutrophil infiltration was also detected (see Supplemental Figure S3A at http://ajp.amjpathol.org). TUNEL staining confirmed the presence of apoptosis at 24 hours but not at 48 and 72 hours (Figure S3B), which suggests early apoptosis followed by necrosis. This suggests that the small intestines are particularly affected during TSS.
Figure 10.
Intestinal disease in d-galN–sensitized model of TSS with low-dose SEB. Twelve- to 14-week-old HLA-DR3 transgenic mice were challenged as indicated with SEB alone, d-galN alone, or SEB+d-galN. Mice were sacrificed at 9 hours, and small intestines were collected in buffered formalin and processed for H&E staining. Scale bars = 100 μm.
Figure 11.
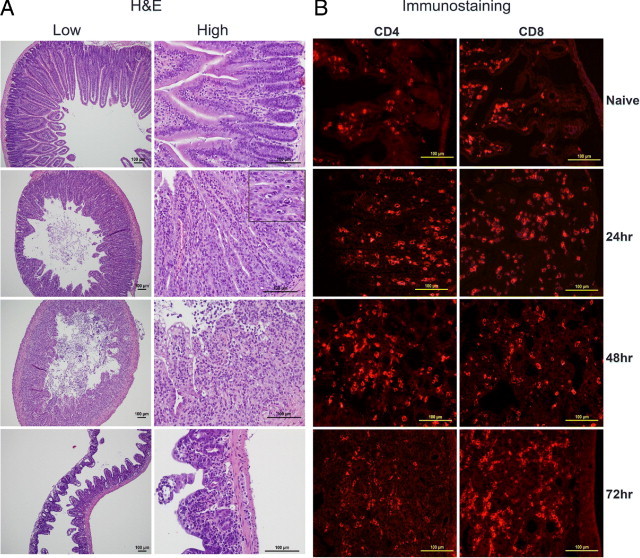
Intestinal immunopathology in high-dose SEB-induced TSS without d-galN sensitization. Twelve- to 14-week-old HLA-DR3 transgenic mice were either untreated or challenged with 50 μg SEB. Mice were sacrificed at indicated times, and small intestines were collected in either buffered formalin or OCT compound and processed for H&E (A) or immunostaining (B). Scale bars = 100 μm.
Altered Gut Permeability in High-Dose SEB Model
Given the extensive pathologic changes in the small intestine, the alteration in gut permeability was studied using FITC-conjugated dextran molecules (see Materials and Methods). There were no significant differences in the fluorescence activity in serum samples between naïve mice and those treated with d-galN alone, low-dose SEB alone, or SEB+d-galN (Figure 12). However, serum samples from mice treated with high-dose SEB exhibited high levels of fluorescent activity, demonstrating increased permeability in these mice. Permeability reverted to baseline levels by 72 hours.
Figure 12.
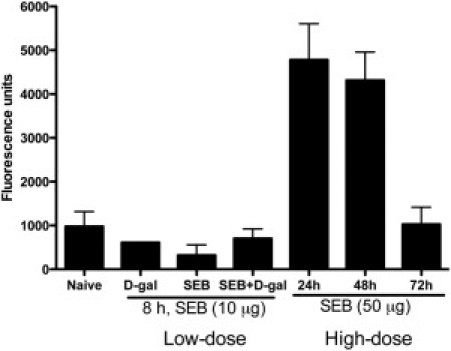
Altered gut permeability in HLA-DR3 transgenic mice during high-dose SEB-induced TSS without d-galN sensitization. Twelve- to 14-week-old HLA-DR3 transgenic mice were either untreated or treated as indicated. Mice underwent gavage using FITC-dextran (see Materials and Methods). Three hours after gavage, mice were sacrificed, serum samples were collected, and the fluorescence in the serum was determined at 490 nm excitation and 525 nm emission in duplicate. Representative results from one of three similar experiments are shown. Scale bars represent mean ± SE values for two mice in each group.
Real-Time in Vivo Imaging of TSS using DR3.Luc Transgenic Mice Predicts Gut-Directed Disease
Next documented was multiple-organ disease in real-time as it unfolds in living animals. Because NF-κB has an important role in several inflammatory processes, transgenic mice expressing the Luc gene under the NF-κB promoter facilitates in vivo imaging of inflammatory processes in living mice in real time.56 DR3.Luc transgenic mice were challenged with high-dose SEB and subjected to in vivo imaging (see Materials and Methods). Naïve DR3.Luc mice exhibit little basal luciferase activity (Figure 13, A and B). In SEB-challenged mice, luciferase activity was largely confined to the abdominal region, with little or no activity in the thoracic region. The abdominal activity rapidly increased, peaked at 6 hours, and later returned to baseline levels. This correlated well with serum cytokine and chemokine concentrations. The diffuse luciferase activity in the abdominal region precluded localizing the activity to any particular organ such as the spleen, liver, kidneys, or intestines. Therefore, the organ largely responsible for this activity was localized. To achieve this, 3 hours after SEB challenge, DR3.Luc transgenic mice were injected with luciferin. After 10 minutes, mice were sacrificed, and their vital organs were removed, placed in Petri dishes, and imaged. Little or no luciferase activity was detected in lungs, liver, kidney, heart, spleen, or pancreas (Figure 13C). However, intense activity was observed in the intestine and in the uterus in female mice.
Figure 13.
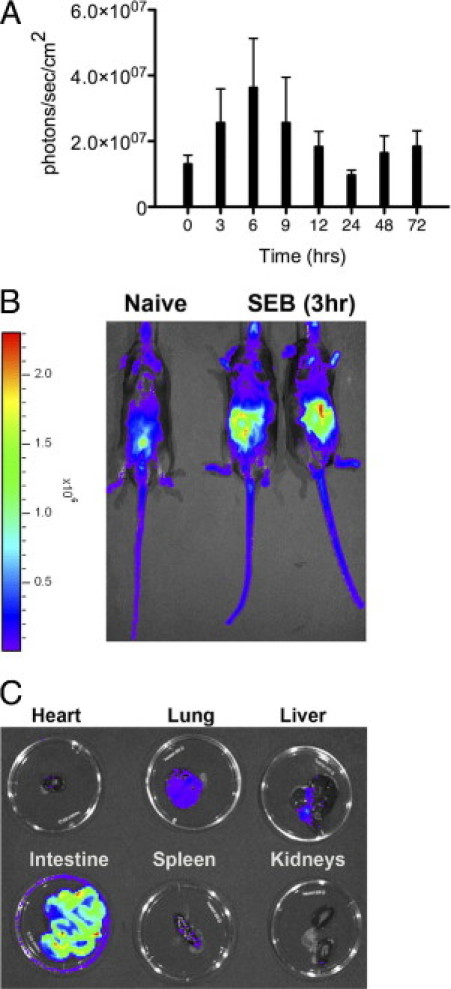
In vivo imaging of inflammatory changes in real-time during TSS using Luc transgenic mice. Twelve- to 14-week-old DR3.Luc transgenic mice were either untreated or challenged with 50 μg SEB. At indicated times, mice were injected intraperitoneally with d-luciferin. After 12 to 15 minutes, anesthetized mice were transferred to the imaging chamber and imaged. A: Mean ± SE values for four mice. Results are from one of three similar experiments. B: Whole mouse. C: Individual organs were harvested at 3 hours and placed in small Petri dishes for imaging.
Discussion
SAg exotoxins produced by S. aureus and S. pyogenes bind directly to MHC-II molecules without undergoing intracellular antigen processing. MHC-II–bound SAgs can vigorously activate large numbers of CD4+ and CD8+ T cells expressing certain TCR Vβ families irrespective of their antigen specificities. Exposure to SAgs that are either preformed (as in intentional exposure following attacks with biological weapons containing SAgs) or produced in vivo after bacterial infection can cause potent immune activation. In certain conditions, this immune activation can be robust, leading to a severe systemic cytokine storm (eg, TSS, Kawasaki disease, and neonatal TSS-like exanthematous disease4,10,11). While SIRS leads to MODS and death, in the absence of prompt intervention,3 the immunopathogenesis of this process is poorly understood because of lack of a robust animal model. The lack of a reliable animal model has also led to incomplete cellular and molecular characterization of TSS. Numerous technical and ethical concerns have precluded extensive use of primates and rabbits in SAg studies. Inadequate activation of the murine immune system with SAgs has led to use of artificial compounds to render them susceptible to TSS, although these strategies have little or no clinical relevance. The use of artificial agents to potentiate SAg-induced TSS has led to misunderstanding of the pathogenesis of TSS.
One of the most widely utilized mouse models of TSS involves the use of d-galN.57 d-galN is converted to uridine diphosphate–galN in vivo, in particular in hepatocytes. At high concentrations of d-galN, such as 15 to 30 mg used for sensitization of mice to TSS, uridine becomes sequestered as uridine diphosphate–galN. This results in profound reduction in the ability of cells to produce uridine triphosphate, thereby affecting RNA and protein synthesis.33,34,36 Although d-galN by itself can cause hepatocyte apoptosis and necrosis,33,34 hepatocytes are also rendered extremely susceptible to the cytotoxic actions of cytokines such as TNF-α by d-galN. SAgs induce TNF-α production in vivo. Normally, binding of TNF-α to its receptor on hepatocytes induces survival signals.58,59 However, in the presence of d-galN, TNF-α fails to elicit the NF-κB–dependent survival signals.60 Conversely, cytotoxic signals are transduced into the hepatocytes, and massive hepatocyte death ensues.57,61 Therefore, liver dysfunction characterized by significantly elevated serum ALT and AST concentrations and rapid death within 9 hours are the typical findings in d-galN sensitization models of shock caused by SAgs.
Similar observations were made in SEB+d-galN–induced TSS in HLA-DR3 transgenic mice. Death in the d-galN sensitization model occurred rapidly. Because HLA-DR3 transgenic mice are extremely responsive to SEB, even 2 μg SEB was associated with death with d-galN sensitization. However, in the high-dose SEB model of TSS, even though heavy infiltration of CD4+ and CD8+ T cells was observed in the liver, no evidence of significant hepatocyte damage was noted at histologic analysis. As corroborating evidence, even the ALT and AST concentrations were not significantly elevated in mice treated with high-dose SEB. This suggests that the immunopathogenesis of TSS caused by SEB alone is likely different from that caused by SEB with d-galN sensitization. Using a similar approach, Mignon et al62 demonstrated differences between lipopolysaccharide-induced shock in the presence or absence of d-galN sensitization. In the high-dose lipopolysaccharide model of shock without d-galN sensitization, those authors failed to observe any major histopathologic changes in the liver, whereas they observed multiple-organ inflammation. However, in the low-dose lipopolysaccharide model of shock with d-galN sensitization, they observed massive hepatocyte apoptosis and necrosis, with highly elevated serum ALT and AST concentrations and 100% mortality. Thus, d-galN sensitizes hepatocytes to death, and, therefore, liver failure is the primary cause of death in d-galN sensitization models of TSS and shock. More important, because death in the d-galN sensitization models ensues rapidly, there were no opportunities to investigate the immunopathogenesis of MODS, which is a characteristic feature of TSS. In the present study, the HLA-DR3 transgenic mouse model facilitated such a study.
The serum cytokine profiles reflected the ability of SAgs to activate T cells. In general, cytokines characteristic of activation of the innate immune system such as IL-6 and IL-12 and chemokines such as KC, MCP-1, macrophage inflammatory protein (MIP)-1β, and RANTES are elevated in models of sepsis. However, in addition to these cytokines and chemokines, profound elevation was observed in classic T-cell–derived cytokines such as IL-2, IL-4, IL-5, IL-17, and IFN-γ in SAg-induced TSS in HLA-DR3 transgenic mice. This underscores the characteristic feature of SAgs, ie, their ability to activate T cells. TNF-α has long been considered the crucial cytokine involved in the immunopathogenesis of TSS in the d-galN sensitization model for reasons discussed in detail earlier under the Discussion section. As a result, blocking TNF-α is protective in mouse models of TSS with d-galN sensitization. Administration of anti-TNF-α has not been protective in bacterial shock including that caused by S. aureus in humans38 and in mouse models of S. aureus sepsis,63 possibly because other cytokines also have an important role. For example, it was observed that the systemic levels of the relatively new cytokine IL-17 was profoundly elevated in HLA-DR3 transgenic mice with SAg-induced TSS. Moreover, the ratio between IL-17 and IFN-γ was high (10 at 3 hours). These observations coupled with the low serum TNF-α concentrations in HLA-DR3 transgenic mice suggest that IL-17 could have a far more important role in the immunopathogenesis of TSS than does TNF-α. Our mouse model will be uniquely suited to unravel the roles of selected cytokines, chemokines, or molecules in the immunopathogenesis of TSS. The other important observation is that while the serum cytokine concentration generally peaked by 6 hours, it was rarely elevated at 48 or 72 hours, by which time death generally ensued. This indicates that SIRS is not immediately followed by MODS and death. There was an appreciable interval during which organ failure occurred, similar to that in SIRS or MODS in humans. It remains to be tested whether the animals could be rescued from death during this period. This warrants a separate detailed study to understand the correlation between systemic cytokine and chemokine levels and death, and the need to explore a wider array of cytokines and chemokines.
To our knowledge, no other study has closely studied the immunopathogenesis of TSS without d-galN sensitization to the extent described herein. As a result, the existence of any temporal correlation between multiple-organ disease and death is unknown. The striking observation was that multiple-organ inflammation characteristic of TSS in humans could be recapitulated in the HLA-DR3 transgenic mouse model. Consistent with the ability of SAgs to activate both CD4+ and CD8+ T cells, temporal changes were observed in infiltration of multiple organs with T lymphocytes. Infiltration of organs with both of the T-cell subsets suggested that both CD4+ and CD8+ T cells are required for TSS. This explains why deficiency of either of these T-cell subsets protected against TSS. However, relatively normal serum ALT, AST, and creatinine concentrations and the absence of large numbers of apoptotic cells in the lungs, liver, or kidneys indicated the absence of extensive cell death in these organs. Nonetheless, extensive infiltration of these organs including heart and lung suggest that their functions might be compromised, leading to MODS.
The most dramatic pathologic changes were observed in the small intestines. While the staphylococcal SAgs, excluding TSST-1, are known enterotoxins and cause gastrointestinal tract toxicity when ingested, the gastrointestinal effects of systemically delivered SAgs has not been investigated in depth. Diarrhea and vomiting are common symptoms associated with many clinical conditions including TSS. The present study provides some insight into this phenomenon. McKay et al,64 using BALB/c mice, demonstrated that systemically administered SAgs (SEB) can cause pathologic changes in the gut. They demonstrated some changes in villous architecture, with alteration in the villous/crypt ratio and altered ion transportation in jejunum in mice challenged with SEB.65 Similarly, a study by Stone and Schlievert66 in rabbits suggested that altered gut permeability could have an important role in the pathogenesis of TSS. Nonetheless, such extensive necrotic and apoptotic changes in the small intestine, with significant infiltration with CD4+ and CD8+ T cells accompanied by profound alteration in gut permeability, as observed in HLA-DR3 transgenic mice, have not been previously reported in other models of TSS. Coinciding with the pathologic changes in the epithelium of the small bowel, gut permeability was increased at 24 and 48 hours. However, at 72 hours, permeability was completely shut down. The reason for these changes in permeability over time is not known. However, considering the extensive pathologic findings at 72 hours, it is believed that the absorptive functions of the intestines are severely compromised, as evidenced by weight loss and diarrhea in these mice.
In vivo imaging studies using luciferase transgenic mice also support the significance of gut disease. Several inflammatory signaling pathways use NF-κB.67 Therefore, transgenic mice expressing the Luc gene under the control of NF-κB have been widely used to study the kinetics of inflammation in real time.56 Consistent with the rapid elevation in inflammatory cytokines and chemokines at 3 to 4 hours in the present model, luciferase activity also increased over time. However, the intestines seemed to be a major contributor to luciferase activity in vivo. This observation correlated with the extensive gut disease observed in mice in the present study. Significant luciferase activity was not observed in other vital organs, whereas in female mice, the uterus exhibited enhanced luciferase activity ex vivo. The significance of this observation is unclear and warrants detailed investigation. We are currently exploring the kinetics of up-regulation of genes regulated by NF-κB and other transcription factors by gene expression profiling using microarrays.
Overall, inasmuch as the serum ALT, AST, and creatinine concentrations were not significantly elevated in HLA-DR3 transgenic mice undergoing SAg-induced TSS, it is unlikely that liver or kidney dysfunction was the primary cause of death. However, given that the heart, lungs, and small intestines exhibited signs of extensive inflammation, failure of these organs could have precipitated death. Increased gut permeability is well documented in humans during endotoxemia and other systemic inflammatory conditions such as pancreatitis.68,69 Similarly, altered gut permeability could have an important role in the pathogenesis of TSS in humans. Further studies are needed to address these issues, and the described HLA-II transgenic mouse model will be a valuable tool for dissection of the immunopathogenesis of TSS.
Acknowledgments
We thank Julie Hanson and Michele Smart for mouse husbandry and mouse genotyping.
Footnotes
Supported by NIH grant 1R01AI068741.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.02.033.
Supplementary data
Macrophage and neutrophil infiltration in lung, liver, and kidneys at 48 hours in high-dose SEB-induced TSS.
Apoptosis in high-dose SEB-induced TSS without d-galN sensitization. Heart, liver, and kidneys from HLA-DR3 transgenic mice challenged with 50 μg SEB (high dose) were collected at 48 hours and stained using the TUNEL technique. Positive and negative controls for TUNEL were set per the manufacturer's protocol.
Immunopathology and apoptosis in high-dose SEB-induced TSS without d-galN sensitization. Pieces of small intestine from HLA-DR3 transgenic mice challenged with 50 μg SEB were collected at 48 hours and stained for CD11b and 7/4 by immunostaining (A) using the TUNEL technique. B: TUNEL staining in small intestine. Positive and negative controls for TUNEL were set per the manufacturer's protocol.
References
- 1.Brooks G.F., Carroll K.C., Butel J.S., Morse S.A., editors. The staphylococci. Jawetz, Melnick, & Adelberg’s Medical Microbiology. ed 25. The McGraw-Hill Companies, Inc; New York: 2010. ch 13. [Google Scholar]
- 2.Manders S.M. Toxin-mediated streptococcal and staphylococcal disease. J Am Acad Dermatol. 1998;39:383–398. doi: 10.1016/s0190-9622(98)70314-7. [DOI] [PubMed] [Google Scholar]
- 3.Fraser D.J., Proft T. The bacterial superantigen and superantigen-like proteins. Immunol Rev. 2008;225:226–243. doi: 10.1111/j.1600-065X.2008.00681.x. [DOI] [PubMed] [Google Scholar]
- 4.Dinges M.M., Orwin P.M., Schlievert P.M. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13:16–34. doi: 10.1128/cmr.13.1.16-34.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H., Llera A., Malchiodi E.L., Mariuzza R.A. The structural basis of T cell activation by superantigens. Annu Rev Immunol. 1999;17:435–466. doi: 10.1146/annurev.immunol.17.1.435. [DOI] [PubMed] [Google Scholar]
- 6.Murray R.J. Recognition and management of Staphylococcus aureus toxin-mediated disease. Int Med J. 2005;35:S106–S119. doi: 10.1111/j.1444-0903.2005.00984.x. [DOI] [PubMed] [Google Scholar]
- 7.Lappin E., Ferguson A.J. Gram-positive toxic shock syndromes. Lancet Infect Dis. 2009;9:281–290. doi: 10.1016/S1473-3099(09)70066-0. [DOI] [PubMed] [Google Scholar]
- 8.Schlievert P.M. Staphylococcal toxic shock syndrome: still a problem. Med J Aust. 2005;182:651–652. doi: 10.5694/j.1326-5377.2005.tb06859.x. [DOI] [PubMed] [Google Scholar]
- 9.Schlievert P.M., Strandberg K.L., Lin Y.-C., Peterson M.L., Leung D.Y.M. Secreted virulence factor comparison between methicillin-resistant and methicillin-sensitive Staphylococcus aureus, and its relevance to atopic dermatitis. J Allergy Clin Immunol. 2010;125:39–49. doi: 10.1016/j.jaci.2009.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.John C.C., Niermann M., Sharon B., Peterson M.L., Kranz D.M., Schlievert P.M. Staphylococcal toxic shock syndrome erythroderma is associated with superantigenicity and hypersensitivity. Clin Infect Dis. 2009;49:1893–1896. doi: 10.1086/648441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCormick J.K., Yarwood J.M., Schlievert P.M. Toxic shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol. 2001;55:77–104. doi: 10.1146/annurev.micro.55.1.77. [DOI] [PubMed] [Google Scholar]
- 12.Tang Y.-W., Himmelfarb E., Wills M., Stratton C.W. Characterization of three Staphylococcus aureus isolates from a 17-year-old female who died of tampon-related toxic shock syndrome. J Clin Microbiol. 2010;48:1974–1977. doi: 10.1128/JCM.00224-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klug C.D., Keay C.R., Ginde A.A. Fatal toxic shock syndrome from an intrauterine device. Ann Emerg Med. 2009;54:701–703. doi: 10.1016/j.annemergmed.2009.05.030. [DOI] [PubMed] [Google Scholar]
- 14.Wasserzug O., Valinsky L., Klement E., Bar-Zeev Y., Davidovitch N., Orr N., Korenman Z., Kayouf R., Sela T., Ambar R., Derazne E., Dagan R., Zarka S. A cluster of ecthyma outbreaks caused by a single clone of invasive and highly infective Streptococcus pyogenes. Clin Infect Dis. 2009;48:1213–1219. doi: 10.1086/597770. [DOI] [PubMed] [Google Scholar]
- 15.Honda H., Krauss M.J., Jones J.C., Olsen M.A., Warren D.K. The value of infectious diseases consultation in Staphylococcus aureus bacteremia. Am J Med. 2010;123:631–637. doi: 10.1016/j.amjmed.2010.01.015. [Epub ahead of press May 20, 2010] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang Y.-W., Stratton C.W. Staphylococcus aureus: an old pathogen with new weapons. Clin Lab Med. 2010;30:179–208. doi: 10.1016/j.cll.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Abram A.C., Bellian K.T., Giles W.J., Gross C.W. Toxic shock syndrome after functional endonasal sinus surgery: an all or none phenomenon? Laryngoscope. 1994;104:927–931. doi: 10.1288/00005537-199408000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Pinna G.S., Kafetzis D.A., Tselkas O.I., Skevaki C.L. Kawasaki disease: an overview. Curr Opin Infect Dis. 2008;21:263–270. doi: 10.1097/QCO.0b013e3282fbf9cd. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi N., Nishida H., Kato H., Imanishi Ki, Sakata Y., Uchiyama T. Exanthematous disease induced by toxic shock syndrome toxin 1 in the early neonatal period. Lancet. 1998;351:1614–1619. doi: 10.1016/S0140-6736(97)11125-4. [DOI] [PubMed] [Google Scholar]
- 20.Baba T., Takeuchi F., Kuroda M., Yuzawa H., Aoki K.-I., Oguchi A., Nagai Y., Iwama N., Asano K., Naimi T., Kuroda H., Cui L., Yamamoto K., Hiramatsu K. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359:1819–1827. doi: 10.1016/s0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 21.Gbaguidi-Haore H., Thouverez M., Couetdic G., Cholley P., Talon D., Bertrand X. Usefulness of antimicrobial resistance pattern for detecting PVL- or TSST-1–producing methicillin-resistant Staphylococcus aureus in a French university hospital. J Med Microbiol. 2009;58:1337–1340. doi: 10.1099/jmm.0.010116-0. [DOI] [PubMed] [Google Scholar]
- 22.Kuroda M., Ohta T., Uchiyama I., Baba T., Yuzawa H., Kobayashi I., Cui L., Oguchi A., Aoki K.-I., Nagai Y., Lian J., Ito T., Kanamori M., Matsumaru H., Maruyama A., Murakami H., Hosoyama A., Mizutani U.Y., Takahashi N.K., Sawano T., Inoue R- I., Kaito C., Sekimizu K., Hirakawa H., Kuhara S., Goto S., Yabuzaki J., Kanehisa M., Yamashita A., Oshima K., Furuya K., Yoshino C., Shiba T., Hattori M., Ogasawara N., Hayashi H., Hiramatsu K. Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet. 2001;357:1225–1240. doi: 10.1016/s0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- 23.Madsen J.M. Toxins as weapons of mass destruction: a comparison and contrast with biological-warfare and chemical-warfare agents. Clin Lab Med. 2001;21:593–605. [PubMed] [Google Scholar]
- 24.Ulrich R., Sidell S., Taylor T.J., Wilhelmsen C.L., Franz D.R. Staphylococcal Enterotoxin B and Related Pyrogenic Toxins. In: Zajtchuk R., editor. Office of the Surgeon General, Department of the Army, United States of America; Bethesda, MD: 1997. pp. 621–630. [Google Scholar]
- 25.Kaul R., McGeer A., Norrby-Teglund A., Malak K., Schwartz B., O'Rourke K., Talbot J., Low D.E., Canadian Streptococcal Study Group Intravenous immunoglobulin therapy for streptococcal toxic shock syndrome: a comparative observational study. Clin Infect Dis. 1999;28:800–807. doi: 10.1086/515199. [DOI] [PubMed] [Google Scholar]
- 26.Stevens D.L., Wallace R.J., Hamilton S.M., Bryant A.E. Successful treatment of staphylococcal toxic shock syndrome with linezolid: a case report and in vitro evaluation of the production of toxic shock syndrome toxin type 1 in the presence of antibiotics. Clin Infect Dis. 2006;42:729–730. doi: 10.1086/500265. [DOI] [PubMed] [Google Scholar]
- 27.Sugiyama H., McKissic E.M., Jr, Bergdoll M.S., Heller B. Enhancement of Bacterial endotoxin lethality by staphylococcal enterotoxin. J Infect Dis. 1964;114:111–118. doi: 10.1093/infdis/114.2.111. [DOI] [PubMed] [Google Scholar]
- 28.Schlievert P.M., Schoettle D.J., Watson D.W. Purification and physicochemical and biological characterization of a staphylococcal pyrogenic exotoxin. Infect Immun. 1979;23:609–617. doi: 10.1128/iai.23.3.609-617.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buonpane R.A., Churchill H.R.O., Moza B., Sundberg E.J., Peterson M.L., Schlievert P.M., Kranz D.M. Neutralization of staphylococcal enterotoxin B by soluble, high-affinity receptor antagonists. Nature Med. 2007;13:725–729. doi: 10.1038/nm1584. [DOI] [PubMed] [Google Scholar]
- 30.Dinges M.M., Gregerson D.S., Tripp T.J., McCormick J.K., Schlievert P.M. Effects of total body irradiation and cyclosporin A on the lethality of toxic shock syndrome toxin-1 in a rabbit model of toxic shock syndrome. J Infect Dis. 2003;188:1142–1145. doi: 10.1086/378514. [DOI] [PubMed] [Google Scholar]
- 31.Lavoie P.M., Thibodeau J., Erard F., Sekaly R.P. Understanding the mechanism of action of bacterial superantigens from a decade of research. Immunol Rev. 1999;168:257–269. doi: 10.1111/j.1600-065x.1999.tb01297.x. [DOI] [PubMed] [Google Scholar]
- 32.Chen J.Y., Qiao Y., Komisar J.L., Baze W.B., Hsu I.C., Tseng J. Increased susceptibility to staphylococcal enterotoxin B intoxication in mice primed with actinomycin D. Infect Immun. 1994;62:4626–4631. doi: 10.1128/iai.62.10.4626-4631.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keppler D., Decker K. Studies on the mechanism of galactosamine-1-phosphate and its inhibition of UDP–glucose pyrophosphorylase. Eur J Biochem. 1969;10:219–225. doi: 10.1111/j.1432-1033.1969.tb00677.x. [DOI] [PubMed] [Google Scholar]
- 34.Keppler D., Lesch R., Reutter W., Decker K. Experimental hepatitis induced by d-galactosamine. Exp Mol Pathol. 1968;9:279–290. doi: 10.1016/0014-4800(68)90042-7. [DOI] [PubMed] [Google Scholar]
- 35.Record C.O., Alberti K.G., Williamson D.H. Metabolic studies in experimental liver disease resulting from D(+)-galactosamine administration. Biochem J. 1972;130:37–44. doi: 10.1042/bj1300037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silverstein R. D-galactosamine lethality model: scope and limitations. J Endotoxin Res. 2004;10:147–162. doi: 10.1179/096805104225004879. [DOI] [PubMed] [Google Scholar]
- 37.Miethke T., Wahl C., Heeg K., Echtenacher B., Krammer P.H., Wagner H. T cell–mediated lethal shock triggered in mice by the superantigen staphylococcal enterotoxin B: critical role of tumor necrosis factor. J Exp Med. 1992;175:91–98. doi: 10.1084/jem.175.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher C.J., Jr, Agosti J.M., Opal S.M., Lowry S.F., Balk R.A., Sadoff J.C., Abraham E., Schein R.M., Benjamin E., Soluble TNF Receptor Sepsis Study Group Treatment of septic shock with the tumor necrosis factor receptor: Fc fusion protein. N Engl J Med. 1996;334:1697–1702. doi: 10.1056/NEJM199606273342603. [DOI] [PubMed] [Google Scholar]
- 39.Seth A., Stern L.J., Ottenhoff T.H., Engel I., Owen M.J., Lamb J.R., Klausner R.D., Wiley D.C. Binary and ternary complexes between T-cell receptor, class II MHC and superantigen in vitro. Nature. 1994;369:324–327. doi: 10.1038/369324a0. [DOI] [PubMed] [Google Scholar]
- 40.Rajagopalan G., Sen M., David C.S. In vitro and in vivo evaluation of staphylococcal superantigen peptide antagonists. Infect Immun. 2004;72:6733–6737. doi: 10.1128/IAI.72.11.6733-6737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rajagopalan G., Smart M.K., Marietta E.V., David C.S. Staphylococcal enterotoxin B–induced activation and concomitant resistance to cell death in CD28-deficient HLA-DQ8 transgenic mice. Int Immunol. 2002;14:801–812. doi: 10.1093/intimm/dxf047. [DOI] [PubMed] [Google Scholar]
- 42.Rajagopalan G., Smart M.K., Cheng S., Krco C.J., Johnson K.L., David C.S. Expression and function of HLA-DR3 and DQ8 in transgenic mice lacking functional H2-M. Tissue Antigens. 2003;62:149–161. doi: 10.1034/j.1399-0039.2003.00088.x. [DOI] [PubMed] [Google Scholar]
- 43.Rajagopalan G., Smart M.K., Krco C.J., David C.S. Expression and function of transgenic HLA-DQ molecules and lymphocyte development in mice lacking invariant chain. J Immunol. 2002;169:1774–1783. doi: 10.4049/jimmunol.169.4.1774. [DOI] [PubMed] [Google Scholar]
- 44.Sriskandan S., Unnikrishnan M., Krausz T., Dewchand H., Van Noorden S., Cohen J., Altmann D.M. Enhanced susceptibility to superantigen-associated streptococcal sepsis in human leukocyte antigen-DQ transgenic mice. J Infect Dis. 2001;184:166–173. doi: 10.1086/322018. [DOI] [PubMed] [Google Scholar]
- 45.DaSilva L., Welcher B.C., Ulrich R.G., Aman M.J., David C.S., Bavari S. Humanlike immune response of human leukocyte antigen-DR3 transgenic mice to staphylococcal enterotoxins: a novel model for superantigen vaccines. J Infect Dis. 2002;185:1754–1760. doi: 10.1086/340828. [DOI] [PubMed] [Google Scholar]
- 46.Roy C.J., Warfield K.L., Welcher B.C., Gonzales R.F., Larsen T., Hanson J., David C.S., Krakauer T., Bavari S. Human leukocyte antigen-DQ8 transgenic mice: a model to examine the toxicity of aerosolized staphylococcal enterotoxin B. Infect Immun. 2005;73:2452–2460. doi: 10.1128/IAI.73.4.2452-2460.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Welcher B.C., Carra J.H., DaSilva L., Hanson J., David C.S., Aman M.J., Bavari S. Lethal shock induced by streptococcal pyrogenic exotoxin A in mice transgenic for human leukocyte antigen-DQ8 and human CD4 receptors: implications for development of vaccines and therapeutics. J Infect Dis. 2002;186:501–510. doi: 10.1086/341828. [DOI] [PubMed] [Google Scholar]
- 48.Rajagopalan G., Iijima K., Singh M., Kita H., Patel R., David C.S. Intranasal exposure to bacterial superantigens induces airway inflammation in HLA class II transgenic mice. Infect Immun. 2006;74:1284–1296. doi: 10.1128/IAI.74.2.1284-1296.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajagopalan G., Sen M., Singh M., Murali N., Nath K.A., Iijima K., Kita H., Leontovich A.A., Unnikrishnan G., Patel R., David C.S. Intranasal exposure to staphylococcal enterotoxin b elicits an acute systemic inflammatory response. Shock. 2006;25:647–656. doi: 10.1097/01.shk.0000209565.92445.7d. [DOI] [PubMed] [Google Scholar]
- 50.Rajagopalan G., Tilahun A.Y., Asmann Y.W., David C.S. Early gene expression changes induced by the bacterial superantigen, staphylococcal enterotoxin B and its modulation by a proteasome inhibitor. Physiol Genomics. 2009;37:279–293. doi: 10.1152/physiolgenomics.90385.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; [Epub ahead of print March 2009]
- 51.Cheng S., Smart M., Hanson J., David C.S. Characterization of HLA DR2 and DQ8 transgenic mouse with a new engineered mouse class II deletion, which lacks all endogenous class II genes. J Autoimmun. 2003;21:195–199. doi: 10.1016/s0896-8411(03)00120-3. [DOI] [PubMed] [Google Scholar]
- 52.Voll R.E., Jimi E., Phillips R.J., Barber D.F., Rincon M., Hayday A.C., Flavell R.A., Ghosh S. NF-kappa B activation by the pre–T cell receptor serves as a selective survival signal in T lymphocyte development. Immunity. 2000;13:677–689. doi: 10.1016/s1074-7613(00)00067-4. [DOI] [PubMed] [Google Scholar]
- 53.Wang Q., Fang C.H., Hasselgren P.-O. Intestinal permeability is reduced and IL-10 levels are increased in septic IL-6 knockout mice. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1013–R1023. doi: 10.1152/ajpregu.2001.281.3.R1013. [DOI] [PubMed] [Google Scholar]
- 54.Nemzek J.A., Xiao H.Y., Minard A.E., Bolgos G.L., Remick D.G. Humane endpoints in shock research. Shock. 2004;21:17–25. doi: 10.1097/01.shk.0000101667.49265.fd. [DOI] [PubMed] [Google Scholar]
- 55.Rajagopalan G., Asmann Y.W., Lytle A.K., Tilahun A.Y., Theuer J.E., Smart M.K., Patel R., David C.S. Cyclooxygenase 2 pathway and its therapeutic inhibition in superantigen-induced toxic shock. Shock. 2008;30:721–728. doi: 10.1097/SHK.0b013e31817048f7. [DOI] [PubMed] [Google Scholar]
- 56.Carlsen H., Moskaug J.O., Fromm S.H., Blomhoff R. In vivo imaging of NF-κB activity. J Immunol. 2002;168:1441–1446. doi: 10.4049/jimmunol.168.3.1441. [DOI] [PubMed] [Google Scholar]
- 57.Galanos C., Freudenberg M.A., Reutter W. Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc Natl Acad Sci USA. 1979;76:5939–5943. doi: 10.1073/pnas.76.11.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harmeet M., Gregory J.G., John J.L. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43:S31–S44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 59.Wullaert A., Heyninck K., Beyaert R. Mechanisms of crosstalk between TNF-induced NF-kappa B and JNK activation in hepatocytes. Biochem Pharmacol. 2006;72:1090–1101. doi: 10.1016/j.bcp.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 60.Andy W., Ben W., Sofie Van H., Veerle B., Bart De G., Peggy P., Peter S., El Karim B., Karen H., Claude L., Rudi B. Adenoviral gene transfer of ABIN-1 protects mice from TNF/galactosamine-induced acute liver failure and lethality. Hepatology. 2005;42:381–389. doi: 10.1002/hep.20785. [DOI] [PubMed] [Google Scholar]
- 61.Bahjat F.R., Dharnidharka V.R., Fukuzuka K., Morel L., Crawford J.M., Clare-Salzler M.J., Moldawer L.L. Reduced susceptibility of nonobese diabetic mice to TNF-alpha and d-galactosamine–mediated hepatocellular apoptosis and lethality. J Immunol. 2000;165:6559–6567. doi: 10.4049/jimmunol.165.11.6559. [DOI] [PubMed] [Google Scholar]
- 62.Mignon A., Rouquet N., Fabre M., Martin S., Pagès J.C., Dhainaut Jean F., Kahn A., Briand P., Joulin V. LPS challenge in D-galactosamine–sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am J Respir Crit Care Med. 1999;159:1308–1315. doi: 10.1164/ajrccm.159.4.9712012. [DOI] [PubMed] [Google Scholar]
- 63.Silverstein R., Norimatsu M., Morrison D. Fundamental differences during gram-positive versus gram-negative sepsis become apparent during bacterial challenge of D-galactosamine–treated mice. J Endotoxin Res. 1997;4:173–181. [Google Scholar]
- 64.Benjamin M.A., Lu J., Donnelly G., Dureja P., McKay D.M. Changes in murine jejunal morphology evoked by the bacterial superantigen Staphylococcus aureus enterotoxin B are mediated by CD4+ T cells. Infect Immun. 1998;66:2193–2199. doi: 10.1128/iai.66.5.2193-2199.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McKay D.M., Benjamin M.A., Lu J. CD4+ T cells mediate superantigen-induced abnormalities in murine jejunal ion transport. Am J Physiol. 1998;275:G29–G38. doi: 10.1152/ajpgi.1998.275.1.G29. [DOI] [PubMed] [Google Scholar]
- 66.Stone R.L., Schlievert P.M. Evidence for the involvement of endotoxin in toxic shock syndrome. J Infect Dis. 1987;155:682–689. doi: 10.1093/infdis/155.4.682. [DOI] [PubMed] [Google Scholar]
- 67.Liu S.F., Malik A.B. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L622–L645. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- 68.Hietbrink F., Besselink M.G., Renooij W., de Smet M.B., Draisma A., van der Hoeven H., Pickkers P. Systemic inflammation increases intestinal permeability during experimental human endotoxemia. Shock. 2009;32:374–378. doi: 10.1097/SHK.0b013e3181a2bcd6. [DOI] [PubMed] [Google Scholar]
- 69.Fink M.P., Delude R.L. Epithelial barrier dysfunction: a unifying theme to explain the pathogenesis of multiple organ dysfunction at the cellular level. Crit Care Clin. 2005;21:177–196. doi: 10.1016/j.ccc.2005.01.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Macrophage and neutrophil infiltration in lung, liver, and kidneys at 48 hours in high-dose SEB-induced TSS.
Apoptosis in high-dose SEB-induced TSS without d-galN sensitization. Heart, liver, and kidneys from HLA-DR3 transgenic mice challenged with 50 μg SEB (high dose) were collected at 48 hours and stained using the TUNEL technique. Positive and negative controls for TUNEL were set per the manufacturer's protocol.
Immunopathology and apoptosis in high-dose SEB-induced TSS without d-galN sensitization. Pieces of small intestine from HLA-DR3 transgenic mice challenged with 50 μg SEB were collected at 48 hours and stained for CD11b and 7/4 by immunostaining (A) using the TUNEL technique. B: TUNEL staining in small intestine. Positive and negative controls for TUNEL were set per the manufacturer's protocol.