

Abstract
Hyperplastic polyposis syndrome (HPS) is characterized by the presence of multiple colorectal serrated polyps and is associated with an increased colorectal cancer (CRC) risk. The mixture of distinct precursor lesion types and malignancies in HPS provides a unique model to study the canonical pathway and a proposed serrated CRC pathway in humans. To establish which CRC pathways play a role in HPS and to obtain new support for the serrated CRC pathway, we assessed the molecular characteristics of polyps (n = 84) and CRCs (n = 19) in 17 patients with HPS versus control groups of various sporadic polyps (n = 59) and sporadic microsatellite-stable CRCs (n = 16). In HPS and sporadic polyps, APC mutations were exclusively identified in adenomas, whereas BRAF mutations were confined to serrated polyps. Six of 19 HPS CRCs (32%) were identified in a serrated polyp. Mutation analysis performed in the CRC and the serrated component of these lesions showed identical BRAF mutations. One HPS CRC was located in an adenoma, both components harboring an identical APC mutation. Overall, 10 of 19 HPS CRCs (53%) carried a BRAF mutation versus none in control group CRCs (P = 0.001). Six BRAF-mutated HPS CRCs (60%) were microsatellite unstable owing to MLH1 methylation. These findings provide novel supporting evidence for the existence of a predominant serrated CRC pathway in HPS, generating microsatellite-stable and microsatellite-instable CRCs.
Colorectal cancer (CRC) ranks as the second most common cause of cancer-related death in the Western world.1 The classic model that describes CRC development is the adenoma-carcinoma sequence associated with activation of the WNT signaling pathway.2,3 This pathway is characterized by an initial bi-allelic inactivation of the adenomatous polyposis coli gene (APC) followed by mutations in key oncogenes and tumor suppressor genes, including KRAS, DCC, and TP53, resulting in adenoma initiation and progression to CRC. This multistep process of carcinogenesis has been elaborately studied, and much information has been derived from the familial adenomatous polyposis and MUTYH-associated polyposis syndromes.
In addition to the adenoma-carcinoma sequence, an alternative, microsatellite-instability (MSI) pathway exists that is characterized by deletion or inactivation of mismatch repair (MMR) genes. Loss of one of the MMR genes occurs in 10% to 15% of sporadic CRCs, whereas 1% to 2% of CRCs with MMR gene loss are due to a hereditary predisposition, ie, in patients with Lynch syndrome carrying a mono-allelic MMR gene defect in the germline.
Recently, a “serrated neoplasia pathway” has been proposed that involves the progression of serrated polyps, ie, hyperplastic polyps (HPs), sessile serrated adenomas (SSAs), and/or traditional serrated adenomas (TSAs), to CRC. The early genetic events of this route, as currently identified, are BRAF or KRAS mutations and an enhanced CPG island methylation status of multiple genes.4–11 There is evidence to suggest that a proportion of sporadic MSI CRCs originate from serrated polyps because these lesions commonly harbor hypermethylated MLH1 combined with BRAF mutations.12–16 In addition, clinico-histologic studies supporting a serrated CRC pathway include CRCs in close vicinity to large HPs,17,18 CRCs identified in mixed hyperplastic and adenomatous polyps,19 and increased incidence of serrated polyps in patients with sporadic microsatellite-unstable CRCs.4,10,20 Currently, however, proof of the existence of a serrated CRC pathway, demonstrated by the combined histologic findings of a serrated polyp directly adjacent to a CRC and concurrent molecular evidence of a sequential relationship, has not been delivered.
Hyperplastic polyposis syndrome (HPS) is a condition characterized by the presence of multiple colorectal serrated polyps. The genetic cause(s) of HPS is largely unknown. We recently demonstrated that HPS can occur in the context of MUTYH-associated polyposis,21 but MUTYH mutations seem to occur in only a small proportion of patients with HPS.22 HPS is associated with an increased CRC risk.5,7,19,22–27 Previously published case series report CRC at clinical presentation in up to 50% of patients with HPS and interval carcinomas, ie, carcinomas occurring after HPS diagnosis and during endoscopic surveillance, in up to 25% of patients.24,28 However, because HPS is a heterogeneous condition, comprising serrated polyps of different categories, (ie, HPs and SSAs but also coexistent conventional adenomas22,23,28–30) it is uncertain which polyps eventually lead to CRC in these patients and, thus, are clinically relevant. When the CRCs originate from the serrated polyps, HPS may prove to be a valuable model for studying the serrated CRC pathway.
In a cohort of 56 patients with HPS, we identified 17 patients with CRC. By combined histopathologic and molecular analyses of the polyps and CRCs in these patients, we obtained novel evidence of a serrated CRC pathway in HPS that predominates over the classic WNT pathway of carcinogenesis.
Materials and Methods
Patients
In a cohort of 56 patients with HPS undergoing endoscopic treatment and surveillance at the Academic Medical Center (Amsterdam, the Netherlands), 21 patients with HPS had CRC. Tissue samples (n = 19) from 17 of these patients were included in this study. HPS was defined as at least five histologically diagnosed HPs and/or SSAs proximal to the sigmoid colon, of which two were >10 mm in diameter, or >20 HPs and/or SSAs distributed throughout the colon.28 Because HPs and SSAs are common findings in HPS and have been shown to be difficult to differentiate microscopically, all serrated polyps were included in the criteria.31–34 Patients with a known germline APC mutation or a bi-allelic MUTYH mutation were excluded from the study. The study was conducted in accordance with the research code of the institutional medical ethical committee on human experimentation of the Academic Medical Center and in agreement with the Helsinki Declaration of 1975 as revised in 1983.
Specimens
All retrieved CRCs (n = 19) were formalin-fixed, paraffin-embedded. H&E-stained tissue sections were reevaluated by two pathologists (S.v.E. and C.J.M.v.N.). In addition, all CRCs were reevaluated for the presence of an adjacent serrated component and for features of a serrated adenocarcinoma as claimed by other researchers.10 A control group was selected consisting of sporadic microsatellite-stable (MSS) CRCs (n = 14) from patients without polyposis matched for age, sex, and CRC location. These CRCs were reevaluated as described previously herein.
In the case of a mutation identified in a CRC, ≥5 polyps in the closest proximity of the CRC were selected and reviewed by a single pathologist (C.J.M.v.N.) who was blinded to patient characteristics and original histologic diagnosis. Polyps were classified as HP, SSA, TSA, mixed polyp, or conventional adenoma based on the histologic features on H&E staining.35–37 Polyps with serrated morphologic features, ie, HPs, SSAs, TSAs, and mixed polyps, were collectively designated as “serrated polyps.” A polyp control group consisting of sporadic HPs (n = 24), SSAs (n = 18), and conventional adenomas (n = 17) was also selected from patients without polyposis. For this analysis, lesions from the cecum, ascending colon, transverse colon, and descending colon were regarded as proximal and those from the sigmoid colon and rectum were regarded as distal.
Somatic Mutation Analysis
Epithelial cells from polyps and CRCs were microdissected, and DNA was isolated as described previously.38,39 Using previously described primers and assays, DNA was analyzed for mutations in the APC–mutation cluster region (APC-MCR), KRAS (exon 2), BRAF (exon 15), and NRAS (exons 1 and 2).38,39 In the case of a CRC in a polyp, mutation analysis of TP53 (exons 4 to 10) was performed in an attempt to assess whether these two components were clonally related. In the case of an identified genetic mutation in a CRC, mutation analysis of surrounding polyps (≥5) was performed as described previously herein. Detected mutations were confirmed in a second independent experiment.
MSI Analysis
The microsatellite status of the CRCs of patients with HPS was determined using an international standard panel of five microsatellite markers (D17S250, D2S123, D5S346, BAT25, and BAT26) using standard techniques. A high degree of MSI (MSI-high) was defined as 2 (40%) unstable markers, MSI-low as one unstable marker, and MSS as no unstable markers.
IHC Analysis
Immunohistochemical (IHC) analysis was performed on CRCs and polyps of patients with HPS. Unstained 5-μm sections were cut from paraffin blocks, and the slides were deparaffinized. The primary monoclonal antibodies used were specific for MLH1 (1:50; BD Pharmingen, San Diego, CA); MSH2 (1:100; Oncogene Research Products, San Diego, CA); MSH6 (1:200; BD Transduction Laboratories, San Jose, CA); PMS2 (1:250; BD Transduction Laboratories); SMAD4 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA); CTNNB1 (1:10.000; BD Biosciences, San Diego, CA), and TP53 (1:2000; Neomarkers Inc., Fremont, CA). Slides were immersed in 0.3% hydrogen peroxide in methanol for 20 minutes. Subsequently, antigen retrieval was performed by 10 minutes of boiling in a solution of 10 mmol/L Tris and 1 mmol/L EDTA (pH 9) followed by incubation with the previously mentioned diluted primary antibodies during 1 hour at room temperature. Postantibody blocking (ImmunoLogic, Duiven, the Netherlands) in PBS was performed, followed by implementation of an antipolyvalent horseradish peroxidase detection system (ImmunoLogic) to visualize antibody binding sites with 3,3′-diaminobenzidine as a chromogen. Sections were counterstained with hematoxylin.
Immunoreactivity for CTNNB1 (β-catenin) was regarded as positive when strong nuclear staining was observed in >25% of the cells. Stains for TP53 were regarded to be indicative of TP53 dysfunction or deletion when >75% of the lesional nuclei were strongly positive or completely negative (absent staining). Stains for SMAD4, MLH1, MSH2, MSH6, and PMS2 were considered negative when there was complete absence of nuclear expression in all lesional cells. Negative staining in a part of a lesion or in a single crypt was registered separately.
Statistics
Statistical analyses were performed using a statistical software package (SPSS 12.0.2; SPSS Inc., Chicago, IL). Somatic mutations in CRCs and polyps of patients with HPS were compared with those of a control panel using a two-sided Fisher's exact test. A P value of <0.05 was considered statistically significant.
Results
Patients
The clinicopathologic features of patients with HPS are summarized in Table 1. The median age of this cohort of 17 patients with HPS at CRC diagnosis was 58 years (range, 41 to 75 years), with a male/female ratio of 8:9. In all the patients, germline APC and MUTYH mutation analyses were previously performed and were found to be negative. Surgical colonic resection was performed in 14 of these 17 patients (82%): 6 subtotal colectomies, 4 hemicolectomies (2 right sided), and 4 rectosigmoidal resections. Histologic evaluation of biopsy samples and surgical resection specimens from these patients revealed a median of 16 HPs, 7 SSAs, and 2 conventional adenomas per patient. All the patients fullfilled the criteria for HPS defined by the World Health Organization.30
Table 1.
Clinicopathologic Features of Patients with HPS and CRC
| Case no. | Age (years) | Sex | CRC location | CRC size (mm) (TNM) | Adjacent polyps |
|---|---|---|---|---|---|
| 1 | 59 | F | Cecum | 56 (T3N0M0) | None |
| 2 | 41 | F | Rectosigmoid | 25 (T2N0M0) | None |
| 3 | 75 | M | AC | 50 (T3N0M0) | None |
| 4 | 58 | M | Rectosigmoid | 4 (TisN0M0) | HP |
| 5 | 59 | M | AC | <20 (T1N0M0) | SSA |
| 6 | 68 | F | AC | 95 (T3N0M0) | None |
| 7 | 43 | F | DC | 40 (T4N1M0) | None |
| 8 | 49 | F | DC | Not stated (TisN0M0) | HP |
| 9 | 54 | M | DC | 50 (T3N0M0) | None |
| 9 | 54 | M | Rectosigmoid | 20 (T1N0M0) | None |
| 10 | 54 | F | AC | 16 (T1N0M0) | SSA |
| 11 | 56 | F | Rectosigmoid | 65 (T2N0M0) | Adenoma |
| 12 | 61 | F | Rectosigmoid | 20 (T1N0M0) | None |
| 13 | 52 | M | Rectosigmoid | 40 (T3N1M0) | None |
| 14 | 66 | M | AC | 8 (T1N0M0) | SSA |
| 15 | 63 | M | AC | 6 (T1N0M0) | SSA |
| 16 | 69 | M | TC | 120 (T3N1M1) | None |
| 17 | 68 | F | AC | 12 (TisNoM0) | None |
| 17 | 68 | F | Rectosigmoid | 29 (T2N0M0) | None |
F, female; M, male; AC, ascending colon; DC, descending colon; TC, transverse colon; HP, hyperplastic polyps; SSA, sessile serrated adenomas.
Mutation Analysis in HPS Polyps and Control Group Polyps
A total of 84 polyps, originating from 17 patients with HPS and CRC, were analyzed for pathogenic mutations in the APC-MCR, KRAS (codons 12 and 13), BRAF (codon 600), and NRAS (exons 1 and 2). The 84 HPS polyps consisted of 21 HPs, 38 SSAs, 3 TSAs, and 2 mixed polyps (64 serrated polyps) and 20 conventional adenomas. The control group of sporadic lesions (n = 59) consisted of 24 HPs and 18 SSAs (42 serrated polyps) and 17 conventional adenomas (Table 2).
Table 2.
Detected APC, KRAS, and BRAF Mutations in Serrated Polyps (HPs, SSAs, TSAs, and Mixed Polyps) and Adenomas (ADs) of Patients with HPS Compared with a Control Group
| Patients with HPS, No. (%) |
Control group, No. (%) |
||||
|---|---|---|---|---|---|
| Somatic mutation | Serrated polyps (n = 64) | AD (n = 20) | Serrated polyps (n = 42) | AD (n = 17) | P value |
| APC | 0 | 9 (45) | 0 | 7 (41) | NS |
| BRAF | 48 (75) | 0 | 20 (48) | 0 | 0.007⁎ |
| KRAS | 4 (6) | 0 | 7 (17) | 4 (24) | 0.029† |
NS, not significant.
Statistically significant P value for BRAF mutation frequency in serrated polyps of patients with HPS compared with the frequency in serrated polyps of the control group.
Statistically significant P value for KRAS mutation frequency in ADs of patients with HPS compared with the frequency in ADs of the control group.
Molecular analysis of the HPS polyps and control group polyps showed APC-MCR mutations exclusively in the conventional adenomas and BRAF mutations exclusively in the serrated polyps: BRAF mutations were detected in 48 of 64 HPS serrated polyps (75%), whereas 20 of 42 control group serrated polyps (48%) harbored a BRAF mutation (P = 0.007). Also, when evaluating patients with HPS individually, each patient harbored predominantly BRAF mutations in their serrated polyps. In four patients, besides BRAF mutations, a single KRAS mutation was identified in a distal serrated polyp. No significant difference in frequency of KRAS mutations in serrated polyps was seen between groups (6% versus 17%; P = 0.1), but in conventional adenomas of patients with HPS, no KRAS mutations were detected compared with 4 of 17 conventional adenomas (24%) in the control group (P = 0.029). In none of the polyps were KRAS and BRAF mutations found together.
When location in the colon was analyzed, no association was seen between the presence of a BRAF mutation and polyp location in the HPS and control groups (Table 3). BRAF mutations were observed in 36 of 46 proximal HPS serrated polyps (78%) and in 12 of 18 distal HPS serrated polyps (67%). In HPS, KRAS-mutated serrated polyps [4 of 18 (22%)] were exclusively detected in the distal colon (P = 0.005).
Table 3.
Serrated Polyp Location and BRAF and KRAS Mutations
|
BRAF mutation (%) |
KRAS mutation (%) |
|||||||
|---|---|---|---|---|---|---|---|---|
| HPS group |
Control group |
HPS group |
Control group |
|||||
| Polyp | Proximal | Distal | Proximal | Distal | Proximal | Distal | Proximal | Distal |
| HP | 8/13 (62) | 5/8 (63) | 1/2 (50) | 9/22 (41) | 0/13 | 3/8 (38) | 0/2 | 5/22 (23) |
| SSA | 25/30 (83) | 5/8 (63) | 10/18 (56) | 0/1 | 0/30 | 1/8 (13) | 2/17 (12) | 0/1 |
| TSA | 1/1 (100) | 2/2 (100) | 0/0 | 0/0 | 0/1 | 0/2 | 0/0 | 0/0 |
| MP | 2/2 (100) | 0/0 | 0/0 | 0/0 | 0/2 | 0/0 | 0/0 | 0/0 |
All values are expressed as number/total number (percentage).
HP, hyperplastic polyps; SSA, sessile serrated adenomas; TSA, traditional serrated adenomas; MP, mixed polyp.
IHC Analysis in Polyps
In none of the 84 HPS polyps, loss of expression of the MMR genes (MLH1, MSH2, MSH6, and PMS2) or SMAD4 was observed (data not shown). In addition, none of these polyps showed abnormal TP53 staining (either strong nuclear TP53 staining or complete TP53 loss; data not shown). In nine conventional HPS adenomas, we detected strong nuclear CTNNB1 staining (ie, in >25% of lesional cells; data not shown). In five of these adenomas, an APC-MCR mutation was identified. In four other APC-MCR–mutated adenomas, no abnormal nuclear CTNNB1 staining was detected. Nuclear CTNNB1 was not found in any of the serrated polyps of either group.
Histologic Characteristics of the CRCs
On histologic reevaluation of CRCs, 6 of 19 HPS CRCs (32%) were identified in or directly adjacent to a serrated polyp. In 1 of 19 cases (5%), the HPS CRC was identified in a conventional adenoma (Figure 1). One patient had two synchronous CRCs. Although six CRCs were identified in serrated polyps, no HPS CRCs displayed distinguishing morphologic features justifying the diagnosis of serrated adenocarcinoma.10
Figure 1.
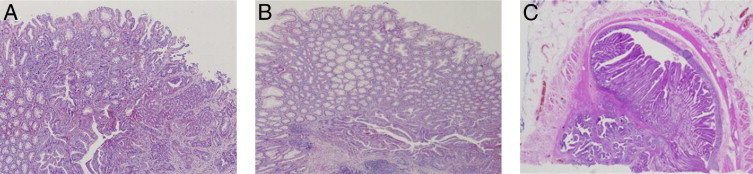
Three CRCs of three HPS patients; two CRCs located within two serrated polyps (A and B), and one CRC within a villous adenoma (C). Original magnification, ×50.
Mutation Analysis in HPS CRCs and Control Group CRCs
BRAF mutations were detected in 10 of 19 HPS CRCs (53%), whereas no BRAF mutations were detected in the CRCs of the control group (P = 0.001; Table 4 and Figure 2). All BRAF mutations involved the same thymine to adenine transversion at nucleotide 1796, resulting in a valine to glutamine substitution at codon 600 (V600E). We found no NRAS mutations in the CRCs of either group. Of the six CRCs identified in a serrated polyp, five (83%) carried the same BRAF mutation (GTG→GAG) in the polypous and tumor components (Table 5). However, considering that these are hotspot mutations, these findings do not prove a clonal relationship between these lesions. TP53 (exons 4 to 10) sequence analysis performed on these six CRCs, to further explore the clonal relationship between the carcinomas and precursor lesions, yielded no mutations.
Table 4.
Mutation Spectrum of CRCs in Patients with HPS Compared with Control Group CRCs
| Mutation | HPS carcinomas (n = 19) | Control group carcinomas (n = 14) | P value |
|---|---|---|---|
| APC | 2 (11) | 4 (29) | NS |
| KRAS | 1 (5) | 5 (36) | NS |
| BRAF | 10 (53) | 0 | 0.001⁎ |
| NRAS | 0 | 0 | NS |
| MSS | 12 (63) | 14 (100) | NA |
| MSI-low | 1 (5) | 0 | NA |
| MSI-high | 6 (32) | 0 | NA |
All values are expressed as number (percentage).
NS, not significant; NA, not applicable.
Statistically significant compared with the control group CRC.
Figure 2.
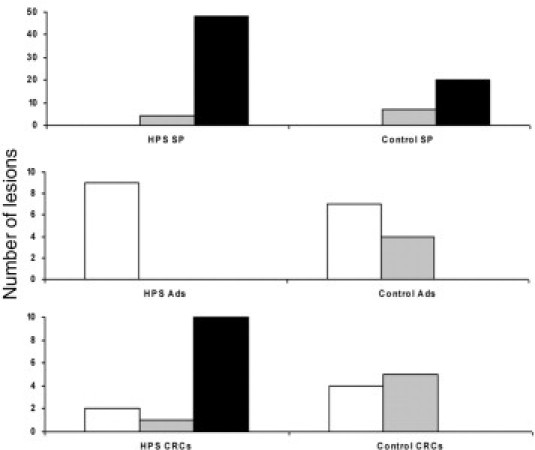
Mutation profiles of HPS polyps/CRCs compared with sporadic control group polyps/CRCs. White bars indicate APC; gray bars, KRAS; black bars, BRAF. SPs, serrated polyps; Ads, conventional adenomas.
Table 5.
Mutations in CRCs and Adjacent Polyps
| Carcinoma | Adjacent polyp | Mutation in CRC | Mutation in adjacent polyp |
|---|---|---|---|
| 4 | HP | None | None |
| 5 | SSA | BRAF (GTG→GAG) | BRAF (GTG→GAG) |
| 8 | HP | BRAF (GTG→GAG) | BRAF (GTG→GAG) |
| 10 | SSA | BRAF (GTG→GAG) | BRAF (GTG→GAG) |
| 11 | Adenoma | APC (insertion G 1354) | APC (insertion G 1354) |
| 15 | SSA | BRAF (GTG→GAG) | BRAF (GTG→GAG) |
| 16 | SSA | BRAF (GTG→GAG) | BRAF (GTG→GAG) |
APC-MCR mutations were identified in 2 of 19 HPS CRCs (11%) compared with in 4 of 14 CRCs in the control group (29%) (not significant), and 1 of 19 KRAS mutations (5%) were detected in HPS CRCs compared with 5 (36%) in the control group (not significant). In one HPS CRC (patient 6), a BRAF mutation and an APC-MCR mutation were detected. One of the HPS CRCs (patient 11), harboring an APC-MCR mutation (insertion 1364), was the single HPS CRC found in a villous adenoma (Figure 1C). Subsequent APC-MCR sequence analysis of the adenoma revealed the same mutation (insertion 1364), establishing a clonal relationship between these lesions. Accordingly, both the adenomatous and malignant components displayed evident nuclear CTNNB1 staining (Figure 3).
Figure 3.
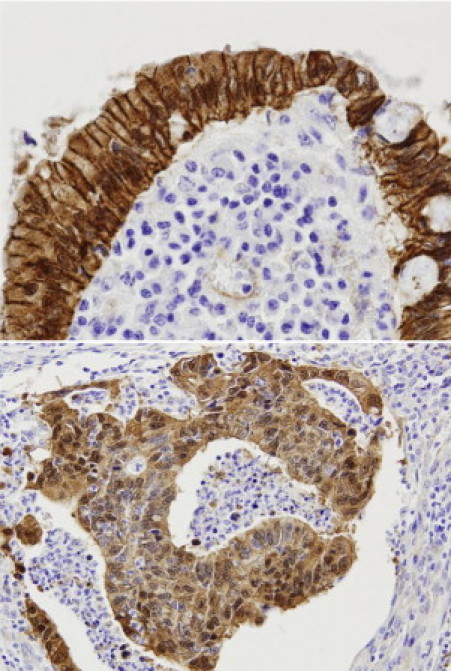
Nuclear CTNNB1 expression in the adenocarcinoma (top) and the adjacent adenoma (bottom) of patient 11, both harboring the same APC gene mutation. Original magnification, ×200.
When location in the colon was analyzed, all BRAF-mutated HPS CRCs were exclusively detected in the proximal colon (P = 0.007). No association was seen between location and KRAS mutations in the control group or APC mutations in both groups. The entire list of molecular analyses on a per-carcinoma basis can be found in Supplemental Table S1 (available at http://ajp.amjpathol.org).
Genetic Events Detected by IHC and Microsatellite Analyses in CRCs
In 6 of 19 HPS CRCs (32%), there was complete loss of expression of MLH1 and PMS2. All six of these HPS CRCs were MSI-high, were proximally located, and harbored a BRAF mutation. Two of the six MSI-high CRCs involved combined serrated polyp–CRC lesions. In these lesions, the serrated polyp components were MSS.
Strong nuclear CTNNB1 staining (>25%) was observed in 5 of 19 HPS CRCs (26%). In one of these CRCs, an APC-MCR mutation was identified, and none of these was BRAF mutated. In all five of these HPS CRCs, abnormal TP53 staining was observed (>75% nuclear staining or complete absence), indicative of loss of functional TP53. Three additional HPS CRCs displayed a perturbed TP53 status. In 2 of 19 CRCs (11%), loss of SMAD4, either focal or complete, was detected.
Discussion
In this first comprehensive cohort study of patients with HPS and CRCs and polyps, we demonstrated that the serrated polyps and conventional adenomas of patients with HPS not only morphologically resemble their respective sporadic counterparts but also have similar molecular profiles (Table 3 and Figure 2). We identified five combined serrated polyp–CRC lesions that showed identical BRAF mutations in both components, supporting the existence of a serrated CRC pathway. Overall, we demonstrated that MSS and MSI CRCs in HPS predominantly originate from the serrated polyps, thus confirming that patients with HPS provide a valuable model to analyze the molecular characteristics of the serrated CRC pathway.
In accord with previous studies,13,40,41 the HPS serrated polyps harbored significantly more BRAF mutations than did those of the control group (75% versus 48%; P = 0.007). The slight difference in the KRAS mutation frequencies between both groups (7% versus 17%) was not statistically significant. Thus, BRAF mutations correlate even stronger with serrated polyps in patients with HPS than in patients without HPS. This contrasts with serrated polyps in patients with MUTYH-associated polyposis and a germline MUTYH gene mutation, which contain significantly more KRAS mutations (70%) than BRAF mutations (4%).21 BRAF and KRAS have been identified as early or instigating events in the serrated pathway. It has to be determined, however, whether serrated lesions with these mutually exclusive mutations are biologically equivalent and have the same risk of developing into CRC.
The mutation profiles of the HPS CRCs were clearly more similar to those found in the serrated polyps than in the conventional adenomas (Table 3 and Figure 2). The profiles of the control group MSS CRCs, as expected, were comparable with the profiles of the conventional adenomas. As many as 10 of the included 19 HPS CRCs were BRAF mutated compared with none of the 14 control group CRCs (P = 0.001). All these BRAF-mutated CRCs were proximally located. Previous molecular studies in HPS CRCs encompassed small noncontrolled series of selected cases,23,42–44 and a comparison between the present results and these studies is not straightforward owing to variation in patient selection criteria and analysis of different or single genes. Overall, however, BRAF mutations were identified in 6 of 15 HPS-related CRC cases (40%) described in the literature until now. We identified no BRAF mutations in the control group of age- and sex-matched MSS CRCs from patients without polyposis. Concordantly, BRAF mutations have been reported in only 5% to 10% of sporadic MSS CRCs of patients without polyposis (n > 100).45–48 Considering that in the literature serrated polyps are suggested to be precursor lesions particularly of MSI CRCs, we chose MSS carcinomas in the control group to exclude MSI CRCs from patients potentially with unrecognized HPS. However, this strategy is arbitrary and eliminates sporadic MSI CRCs from patients without HPS. Hence, the statistically significant difference in BRAF mutation frequency between HPS CRCs and control group CRCs is higher compared with that in an unselected cohort of CRCs. We detected only one KRAS mutation in a distally located HPS CRC, which corroborates two previously published studies together analyzing 15 HPS CRCs.40,41 To our knowledge, only one other study has described the presence of a KRAS mutation in a single distally located HPS CRC, supporting the notion that KRAS plays a minor role in the carcinogenesis of HPS and is confined to the distal colon.23
In the present study, two patients had synchronous CRCs. Theoretically, a field effect may account for the development of synchronous polyps/CRCs.49 In patient 9, no mutations were identified in both CRCs. Patient 17 harbored one KRAS mutation and one BRAF mutation in each of the CRCs. Hence, although a field effect associated with a serrated CRC pathway may cause simultaneous CRCs with identical mutations, we did not demonstrate this in patient 9 owing to the lack of any mutations, and it seems excluded in patient 17. Of note, clonal markers are necessary to demonstrate a true field effect. Such clonal markers associated with the serrated pathway are currently not available.
The histologic characteristics of the HPS CRCs as a group, either BRAF mutated or not, were inconspicuous and not obviously different from those of control group CRCs. In particular, a serrated growth pattern, as has been reported,50,51 was not apparent in the present series of HPS CRCs. We found a remarkable number of HPS CRCs [6 of 19 (32%)] to be located in or directly adjacent to a serrated polyp (Figure 1). In five of six of these combined lesions, an identical hotspot mutation at codon 600 (GTG→GAG) of BRAF was detected in both components. These combined histologic and molecular data are strongly suggestive of a sequential relationship between serrated polyps and CRCs. A clonal relationship between the carcinomas and their assumed precursor lesions could not be further substantiated owing to a lack of appropriate clonal markers.
In the literature, it has been shown that 90% of sporadic MSI-high CRCs display loss of MLH1 expression as a result of methylation of this MMR gene. In these lesions, BRAF mutations, which are associated with serrated polyps, are also a common finding, suggesting that the serrated pathway can generate MSI-high CRCs.13,15 Six of 19 HPS CRCs were MSI-high owing to loss of MLH1 and PMS2, and all 6 carried BRAF mutations. Additional analysis in these CRCs also showed hypermethylation of MLH1 in all cases (data not shown). These findings suggest a causal relationship between this novel carcinogenic route and microsatellite-unstable CRCs. On the other hand, the present findings demonstrate that the serrated pathway, at least in HPS, also generates MSS CRCs.
Previous large cohort studies (>30 patients) have reported the presence of at least one conventional adenoma in 69% to 85% of patients with HPS and >5 conventional adenomas in 21% to 32% of patients.22,23,28–30 Concordantly, in the present study, APC-MCR mutations were detected in two HPS CRCs (13%). The identification of an HPS CRC (patient 11) located in a conventional adenoma of the same clonal origin, reflected by the identical APC-MCR mutation in both components, is significant because this proves that the classic adenoma-carcinoma pathway52 is also operational in patients with HPS.
The apparent dominance of the serrated over the nonserrated CRC pathway in HPS may be due to a greater intrinsic risk of tumor progression of the serrated polyps or may be simply a reflection of the numerical prevalence of serrated polyps over adenomas. To our knowledge, the only study that has addressed this issue in two HPS CRC cases reported no APC mutations.23 In that study, however, patients with >10 conventional adenomas were excluded. In the present study, all cases satisfied the World Health Organization criteria for HPS in which the presence or number of conventional adenomas is not an issue. In patient 11 with HPS and the classic CRC, a relative abundance of conventional adenomas was observed (12 adenomas versus 17 serrated polyps), suggesting a stochastic rather than an intrinsically biased process of carcinogenesis. In the other APC-MCR–mutated HPS CRC (patient 6), a BRAF mutation was also identified. Assuming that this CRC is of monoclonal origin, it may be the outcome of an elusive “fusion” pathway that combines mechanisms associated with both conventional adenomas and serrated polyps, as proposed previously.36
We observed four HPS CRCs with nuclear CTNNB1 staining associated with the WNT pathway. Although in these CRCs no APC-MCR mutations or CTNNB1 exon 4 to 10 hotspot mutations were identified, alternative mechanisms may be operational to activate the WNT pathway, eg, methylation of the APC promotor regions and MSI-related frameshift mutations in WNT pathway regulators, such as AXIN2.53 In these patients with HPS, a median of only one adenoma (range, zero to five) was identified, suggesting that the WNT pathway may, indeed, be involved at some stage of carcinogenesis via the serrated pathway. Considering that no BRAF mutations or directly adjacent serrated polyps were found, it seems unlikely that these CRCs are the outcome of a proposed fusion pathway, but formally this cannot be excluded.
Based on these observations, we conclude that distinct APC- and non-APC–mediated CRC pathways are functional in HPS. A serrated pathway, skewed toward initial BRAF mutations and proximal localization, however, seems to predominate, most likely owing to the numerical prevalence of serrated polyps in these patients. From this finding it is inferred that all polyp types in HPS should be considered clinically relevant and should be removed.
Footnotes
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.02.023.
Supplementary data
References
- 1.Jemal A., Thun M.J., Ries L.A., Howe H.L., Weir H.K., Center M.M., Ward E., Wu X.C., Eheman C., Anderson R., Ajani U.A., Kohler B., Edwards B.K. Annual report to the nation on the status of cancer, 1975–2005, featuring trends in lung cancer, tobacco use, and tobacco control. J Natl Cancer Inst. 2008;100:1672–1694. doi: 10.1093/jnci/djn389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fearon E.R., Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B., Fearon E.R., Hamilton S.R., Kern S.E., Preisinger A.C., Leppert M., Nakamura Y., White R., Smits A.M., Bos J.L. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 4.Hawkins N.J., Ward R.L. Sporadic colorectal cancers with microsatellite instability and their possible origin in hyperplastic polyps and serrated adenomas. J Natl Cancer Inst. 2001;93:1307–1313. doi: 10.1093/jnci/93.17.1307. [DOI] [PubMed] [Google Scholar]
- 5.Iino H., Jass J.R., Simms L.A., Young J., Leggett B., Ajioka Y., Watanabe H. DNA microsatellite instability in hyperplastic polyps, serrated adenomas, and mixed polyps: a mild mutator pathway for colorectal cancer? J Clin Pathol. 1999;52:5–9. doi: 10.1136/jcp.52.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jass J.R., Biden K.G., Cummings M.C., Simms L.A., Walsh M., Schoch E., Meltzer S.J., Wright C., Searle J., Young J., Leggett B.A. Characterisation of a subtype of colorectal cancer combining features of the suppressor and mild mutator pathways. J Clin Pathol. 1999;52:455–460. doi: 10.1136/jcp.52.6.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jass J.R., Iino H., Ruszkiewicz A., Painter D., Solomon M.J., Koorey D.J., Cohn D., Furlong K.L., Walsh M.D., Palazzo J., Edmonston T.B., Fishel R., Young J., Leggett B.A. Neoplastic progression occurs through mutator pathways in hyperplastic polyposis of the colorectum. Gut. 2000;47:43–49. doi: 10.1136/gut.47.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jass J.R., Young J., Leggett B.A. Hyperplastic polyps and DNA microsatellite unstable cancers of the colorectum. Histopathology. 2000;37:295–301. doi: 10.1046/j.1365-2559.2000.01028.x. [DOI] [PubMed] [Google Scholar]
- 9.Jass J.R. Serrated route to colorectal cancer: back street or super highway? J Pathol. 2001;193:283–285. doi: 10.1002/1096-9896(200103)193:3<283::AID-PATH799>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 10.Makinen M.J., George S.M., Jernvall P., Makela J., Vihko P., Karttunen T.J. Colorectal carcinoma associated with serrated adenoma: prevalence, histological features, and prognosis. J Pathol. 2001;193:286–294. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH800>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Yao T., Nishiyama K., Oya M., Kouzuki T., Kajiwara M., Tsuneyoshi M. Multiple “serrated adenocarcinomas” of the colon with a cell lineage common to metaplastic polyp and serrated adenoma: case report of a new subtype of colonic adenocarcinoma with gastric differentiation. J Pathol. 2000;190:444–449. doi: 10.1002/(SICI)1096-9896(200003)190:4<444::AID-PATH520>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 12.Jass J.R. Serrated adenoma of the colorectum and the DNA-methylator phenotype. Nat Clin Pract Oncol. 2005;2:398–405. doi: 10.1038/ncponc0248. [DOI] [PubMed] [Google Scholar]
- 13.Kambara T., Simms L.A., Whitehall V.L., Spring K.J., Wynter C.V., Walsh M.D., Barker M.A., Arnold S., McGivern A., Matsubara N., Tanaka N., Higuchi T., Young J., Jass J.R., Leggett B.A. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut. 2004;53:1137–1144. doi: 10.1136/gut.2003.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGivern A., Wynter C.V., Whitehall V.L., Kambara T., Spring K.J., Walsh M.D., Barker M.A., Arnold S., Simms L.A., Leggett B.A., Young J., Jass J.R. Promoter hypermethylation frequency and BRAF mutations distinguish hereditary non-polyposis colon cancer from sporadic MSI-H colon cancer. Fam Cancer. 2004;3:101–107. doi: 10.1023/B:FAME.0000039861.30651.c8. [DOI] [PubMed] [Google Scholar]
- 15.Rajagopalan H., Bardelli A., Lengauer C., Kinzler K.W., Vogelstein B., Velculescu V.E. Tumorigenesis: rAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 16.Young J., Simms L.A., Biden K.G., Wynter C., Whitehall V., Karamatic R., George J., Goldblatt J., Walpole I., Robin S.A., Borten M.M., Stitz R., Searle J., McKeone D., Fraser L., Purdie D.R., Podger K., Price R., Buttenshaw R., Walsh M.D., Barker M., Leggett B.A., Jass J.R. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: parallel pathways of tumorigenesis. Am J Pathol. 2001;159:2107–2116. doi: 10.1016/S0002-9440(10)63062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Azimuddin K., Stasik J.J., Khubchandani I.T., Rosen L., Riether R.D., Scarlatto M. Hyperplastic polyps: “more than meets the eye”? report of sixteen cases. Dis Colon Rectum. 2000;43:1309–1313. doi: 10.1007/BF02237443. [DOI] [PubMed] [Google Scholar]
- 18.Warner A.S., Glick M.E., Fogt F. Multiple large hyperplastic polyps of the colon coincident with adenocarcinoma. Am J Gastroenterol. 1994;89:123–125. [PubMed] [Google Scholar]
- 19.Urbanski S.J., Kossakowska A.E., Marcon N., Bruce W.R. Mixed hyperplastic adenomatous polyps: an underdiagnosed entity: report of a case of adenocarcinoma arising within a mixed hyperplastic adenomatous polyp. Am J Surg Pathol. 1984;8:551–556. [PubMed] [Google Scholar]
- 20.Goldstein N.S., Bhanot P., Odish E., Hunter S. Hyperplastic-like colon polyps that preceded microsatellite-unstable adenocarcinomas. Am J Clin Pathol. 2003;119:778–796. doi: 10.1309/DRFQ-0WFU-F1G1-3CTK. [DOI] [PubMed] [Google Scholar]
- 21.Boparai K.S., Dekker E., van E.S., Polak M.M., Bartelsman J.F., Mathus-Vliegen E.M., Keller J.J., van Noesel C.J. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology. 2008;135:2014–2018. doi: 10.1053/j.gastro.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Chow E., Lipton L., Lynch E., D'Souza R., Aragona C., Hodgkin L., Brown G., Winship I., Barker M., Buchanan D., Cowie S., Nasioulas S., du S.D., Young J., Leggett B., Jass J., Macrae F. Hyperplastic polyposis syndrome: phenotypic presentations and the role of MBD4 and MYH. Gastroenterology. 2006;131:30–39. doi: 10.1053/j.gastro.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 23.Carvajal-Carmona L., Howarth K., Lockett M., Polanco-Echeverry G., Volikos E., Gorman M., Barclay E., Martin L., Jones A., Saunders B., Guenther T., Donaldson A., Paterson J., Frayling I., Novelli M., Phillips R., Thomas H., Silver A., Atkin W., Tomlinson I. Molecular classification and genetic pathways in hyperplastic polyposis syndrome. J Pathol. 2007;212:378–385. doi: 10.1002/path.2187. [DOI] [PubMed] [Google Scholar]
- 24.Hyman N.H., Anderson P., Blasyk H. Hyperplastic polyposis and the risk of colorectal cancer. Dis Colon Rectum. 2004;47:2101–2104. doi: 10.1007/s10350-004-0709-6. [DOI] [PubMed] [Google Scholar]
- 25.Rashid A., Houlihan P.S., Booker S., Petersen G.M., Giardiello F.M., Hamilton S.R. Phenotypic and molecular characteristics of hyperplastic polyposis. Gastroenterology. 2000;119:323–332. doi: 10.1053/gast.2000.9361. [DOI] [PubMed] [Google Scholar]
- 26.Rubio C.A., Stemme S., Jaramillo E., Lindblom A. Hyperplastic polyposis coli syndrome and colorectal carcinoma. Endoscopy. 2006;38:266–270. doi: 10.1055/s-2006-925026. [DOI] [PubMed] [Google Scholar]
- 27.Oono Y., Fu K., Nakamura H., Iriguchi Y., Yamamura A., Tomino Y., Oda J., Mizutani M., Takayanagi S., Kishi D., Shinohara T., Yamada K., Matumoto J., Imamura K. Progression of a sessile serrated adenoma to an early invasive cancer within 8 months. Dig Dis Sci. 2009;54:906–909. doi: 10.1007/s10620-008-0407-7. [DOI] [PubMed] [Google Scholar]
- 28.Boparai K.S., Mathus-Vliegen E.M., Koornstra J.J., Nagengast F.M., van L.M., van Noesel C.J., Houben M., Cats A., van Hest L.P., Fockens P., Dekker E. Increased colorectal cancer risk during follow-up in patients with hyperplastic polyposis syndrome: a multicentre cohort study. Gut. 2010;59:1094–1100. doi: 10.1136/gut.2009.185884. [DOI] [PubMed] [Google Scholar]
- 29.Buchanan D.D., Sweet K., Drini M., Jenkins M.A., Win A.K., Gattas M., Walsh M.D., Clendenning M., McKeone D., Walters R., Roberts A., Young A., Hampel H., Hopper J.L., Goldblatt J., George J., Suthers G.K., Phillips K., Young G.P., Chow E., Parry S., Woodall S., Tucker K., Muir A., Field M., Greening S., Gallinger S., Green J., Woods M.O., Spaetgens R., de la Chapelle A., Macrae F., Walker N.I., Jass J.R., Young J.P. Phenotypic diversity in patients with multiple serrated polyps: a genetics clinic study. Int J Colorectal Dis. 2010;25:703–712. doi: 10.1007/s00384-010-0907-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burt R.W., Jass J. Hyperplastic polyposis. In: Hamilton S.R., Aaltonen L.A., editors. World Health Organisation Classification of Tumours: Pathology and Genetics. Springer-Verlag; Berlin: 2000. pp. 135–136. [Google Scholar]
- 31.Khalid O., Radaideh S., Cummings O.W., O'Brien M.J., Goldblum J.R., Rex D.K. Reinterpretation of histology of proximal colon polyps called hyperplastic in 2001. World J Gastroenterol. 2009;15:3767–3770. doi: 10.3748/wjg.15.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandmeier D., Seelentag W., Bouzourene H. Serrated polyps of the colorectum: is sessile serrated adenoma distinguishable from hyperplastic polyp in a daily practice? Virchows Arch. 2007;450:613–618. doi: 10.1007/s00428-007-0413-8. [DOI] [PubMed] [Google Scholar]
- 33.Sandmeier D., Benhattar J., Martin P., Bouzourene H. Serrated polyps of the large intestine: a molecular study comparing sessile serrated adenomas and hyperplastic polyps. Histopathology. 2009;55:206–213. doi: 10.1111/j.1365-2559.2009.03356.x. [DOI] [PubMed] [Google Scholar]
- 34.Wong N.A., Hunt L.P., Novelli M.R., Shepherd N.A., Warren B.F. Observer agreement in the diagnosis of serrated polyps of the large bowel. Histopathology. 2009;55:63–66. doi: 10.1111/j.1365-2559.2009.03329.x. [DOI] [PubMed] [Google Scholar]
- 35.Hamilton S.R., Vogelstein B., Kudo S. IARC Press; Lyon, France: 2000. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of the Digestive System; pp. 104–119. [Google Scholar]
- 36.Jass J.R., Baker K., Zlobec I., Higuchi T., Barker M., Buchanan D., Young J. Advanced colorectal polyps with the molecular and morphological features of serrated polyps and adenomas: concept of a “fusion” pathway to colorectal cancer. Histopathology. 2006;49:121–131. doi: 10.1111/j.1365-2559.2006.02466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snover D.C., Jass J.R., Fenoglio-Preiser C., Batts K.P. Serrated polyps of the large intestine: a morphologic and molecular review of an evolving concept. Am J Clin Pathol. 2005;124:380–391. doi: 10.1309/V2EP-TPLJ-RB3F-GHJL. [DOI] [PubMed] [Google Scholar]
- 38.de Leng W.W., Keller J.J., Luiten S., Musler A.R., Jansen M., Baas A.F., de Rooij F.W., Gille J.J., Menko F.H., Offerhaus G.J., Weterman M.A. STRAD in Peutz-Jeghers syndrome and sporadic cancers. J Clin Pathol. 2005;58:1091–1095. doi: 10.1136/jcp.2005.026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Leng W.W., Westerman A.M., Weterman M.A., de Rooij F.W., Dekken H.H., De Goeij A.F., Gruber S.B., Wilson J.H., Offerhaus G.J., Giardiello F.M., Keller J.J. Cyclooxygenase 2 expression and molecular alterations in Peutz-Jeghers hamartomas and carcinomas. Clin Cancer Res. 2003;9:3065–3072. [PubMed] [Google Scholar]
- 40.Beach R., Chan A.O., Wu T.T., White J.A., Morris J.S., Lunagomez S., Broaddus R.R., Issa J.P., Hamilton S.R., Rashid A. BRAF mutations in aberrant crypt foci and hyperplastic polyposis. Am J Pathol. 2005;166:1069–1075. doi: 10.1016/S0002-9440(10)62327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan A.O., Issa J.P., Morris J.S., Hamilton S.R., Rashid A. Concordant CpG island methylation in hyperplastic polyposis. Am J Pathol. 2002;160:529–536. doi: 10.1016/S0002-9440(10)64872-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beach R., Chan A.O., Wu T.T., White J.A., Morris J.S., Lunagomez S., Broaddus R.R., Issa J.P., Hamilton S.R., Rashid A. BRAF mutations in aberrant crypt foci and hyperplastic polyposis. Am J Pathol. 2005;166:1069–1075. doi: 10.1016/S0002-9440(10)62327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan T.L., Zhao W., Leung S.Y., Yuen S.T. BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res. 2003;63:4878–4881. [PubMed] [Google Scholar]
- 44.Minoo P., Baker K., Goswami R., Chong G., Foulkes W.D., Ruszkiewicz A.R., Barker M., Buchanan D., Young J., Jass J.R. Extensive DNA methylation in normal colorectal mucosa in hyperplastic polyposis. Gut. 2006;55:1467–1474. doi: 10.1136/gut.2005.082859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samowitz W.S., Sweeney C., Herrick J., Albertsen H., Levin T.R., Murtaugh M.A., Wolff R.K., Slattery M.L. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65:6063–6069. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 46.Barault L., Charon-Barra C., Jooste V., de la Vega M.F., Martin L., Roignot P., Rat P., Bouvier A.M., Laurent-Puig P., Faivre J., Chapusot C., Piard F. Hypermethylator phenotype in sporadic colon cancer: study on a population-based series of 582 cases. Cancer Res. 2008;68:8541–8546. doi: 10.1158/0008-5472.CAN-08-1171. [DOI] [PubMed] [Google Scholar]
- 47.Ogino S., Nosho K., Kirkner G.J., Kawasaki T., Meyerhardt J.A., Loda M., Giovannucci E.L., Fuchs C.S. CpG island methylator phenotype, microsatellite instability: BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58:90–96. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Vogel S., Weijenberg M.P., Herman J.G., Wouters K.A., de Goeij A.F., van den Brandt P.A., de Bruine A.P., van Engeland M. MGMT and MLH1 promoter methylation versus APC, KRAS and BRAF gene mutations in colorectal cancer: indications for distinct pathways and sequence of events. Ann Oncol. 2009;20:1216–1222. doi: 10.1093/annonc/mdn782. [DOI] [PubMed] [Google Scholar]
- 49.Nosho K., Kure S., Irahara N., Shima K., Baba Y., Spiegelman D., Meyerhardt J.A., Giovannucci E.L., Fuchs C.S., Ogino S. A prospective cohort study shows unique epigenetic, genetic, and prognostic features of synchronous colorectal cancers. Gastroenterology. 2009;137:1609–1620. doi: 10.1053/j.gastro.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Makinen J.M., Makinen M.J., Karttunen T.J. Serrated adenocarcinoma of the rectum associated with perianal Paget's disease: a case report. Histopathology. 2002;41:177–179. doi: 10.1046/j.1365-2559.2002.01424_7.x. [DOI] [PubMed] [Google Scholar]
- 51.Makinen M.J. Colorectal serrated adenocarcinoma. Histopathology. 2007;50:131–150. doi: 10.1111/j.1365-2559.2006.02548.x. [DOI] [PubMed] [Google Scholar]
- 52.Vogelstein B., Fearon E.R., Hamilton S.R., Kern S.E., Preisinger A.C., Leppert M., Nakamura Y., White R., Smits A.M., Bos J.L. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 53.Thorstensen L., Lind G.E., Lovig T., Diep C.B., Meling G.I., Rognum T.O., Lothe R.A. Genetic and epigenetic changes of components affecting the WNT pathway in colorectal carcinomas stratified by microsatellite instability. Neoplasia. 2005;7:99–108. doi: 10.1593/neo.04448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.