

Abstract
Invasion of tumor cells into the local stroma is an important component in cancer progression. Here we report studies of the in vivo invasion of head and neck squamous cell carcinoma (HNSCC) cells in response to applied gradients of a growth factor [epidermal growth factor (EGF)] and a chemokine (CXCL12), using orthotopic floor-of-mouth models. Analysis of the invading cells indicated that >75% of them were tumor cells, about 15% macrophages, and <10% were unidentified. Surprisingly, although macrophages invaded together with tumor cells, macrophage contributions were not required for HNSCC invasion. CXCL12-induced in vivo invasion of HNSCC cells was also observed and found to occur via a unidirectional transactivation of epidermal growth factor receptor (EGFR) through CXCR4. Inhibition of tumor necrosis factor-α–converting enzyme using TNF-α protease inhibitor-2 selectively inhibited CXCL12-induced invasion but not EGF-induced invasion, consistent with CXCL12 activation of EGFR via release of EGFR ligands.
Head and neck squamous cell carcinoma (HNSCC) is one of the 10 most common types of cancer in the world, with over 500,000 new cases per year and 250,000 deaths worldwide as estimated by the World Health Organization. This includes 48,000 new cases and 11,260 deaths in 2009 from HNSCC in the United States.1 HNSCC originates from mucosal tissues of the upper aerodigestive tract and spans the oral cavity to the larynx. Despite some improvements in treatment methods, the 5-year survival rate remains just above 50%.1 Current treatments for HNSCC include single and multimodality therapies using surgical and nonsurgical approaches (chemotherapy and/or radiotherapy).2
A key constraint that limits the ability of surgical treatment to cure HNSCC is the location—adequate margins to guarantee removal of all tumor cells are difficult to achieve in many cases without severely compromising quality of life or survival. Thus, the degree to which tumor cells have locally spread from the primary tumor can impact the likelihood of recurrence. Indeed, morphological examination of HNSCC has revealed that the pattern of tumor invasion, presence of perineural invasion, and presence of inflammatory cells correlate with clinical outcome.3–6 Understanding the mechanisms underlying HNSCC invasion could provide an opportunity to reduce local invasion and improve patient outcome. The epidermal growth factor receptor (EGFR) is often overexpressed in HNSCC7,8 and correlated with poor prognosis.9 In addition to driving proliferation, the EGFR has the potential to drive invasion. EGFR ligands are chemoattractants, stimulating directly cell motility and HNSCC invasion in vitro.10–12 Studies of EGFR function in HNSCC in vivo have focused on tumor growth,13,14 and direct evaluation of EGFR-mediated invasion in vivo has been poorly explored.
In the primary tumor–host microenvironment, interactions between tumor cells and surrounding host stromal elements (including macrophages and fibroblasts) can also contribute to tumor cell invasion. Stromal cells are known to release chemotactic signals that drive invasion of tumor cells further into host stoma. For example, tumor-associated macrophages, fibroblasts, or platelets can produce EGFR ligands such as EGF,15–17 whereas tumor-associated fibroblasts can produce CXCL12.18 Macrophages express the CXCL12 receptor CXCR4, whereas tumor cells can express both EGFR and CXCR4. Macrophage infiltration into tumors as well as the tumor–host interface has been shown to correlate with poor prognosis of many malignancies,19–21 including HNSCC.6,22 In a study of 102 HNSCC patients, macrophage count at the primary tumor correlated positively with lymph node metastasis and stage, and was found to be an independent predictor of lymph node metastasis.22 We have previously demonstrated macrophage-dependent tumor invasion in breast cancer animal models15,23,24 based on an in vivo invasion assay. This assay collects invasive cells from primary xenograft and transgenic tumors in response to chemotactic cues.25 It was determined that macrophages aided breast cancer cell invasion into surrounding tissue by forming a paracrine communication loop between colony-stimulating factor 1 (CSF-1)-secreting cancer cells and EGF-secreting macrophages.15 Blockade of either EGF or CSF-1 signaling was able to inhibit this invasion. Invasion induced by other chemotactic stimuli such as CXCL12 and heregulin β1 (HRGβ1) also relied on this paracrine loop.23
Given the importance of local and regional invasion in HNSCC; the abundance of EGFR in HNSCC tumors; and the published evidence that macrophage infiltration correlates with poor prognosis in HNSCC, it is important to evaluate in vivo the contributions of macrophages to HNSCC invasion. In this paper, we directly evaluate the roles of EGFR and macrophages in HNSCC invasion in vivo using FaDu and UMSCC47 HNSCC cell lines in an orthotopic floor-of-mouth model.26 We characterize the in vivo invasion of HNSCC tumor cells using the chemoattractants EGF and CXCL12. Remarkably, macrophages are not required for HNSCC invasion. However, invasion in response to CXCL12 does depend on EGFR function, demonstrating the importance of the EGFR for HNSCC invasion in vivo.
Materials and Methods
Cell Lines and Animal Models
The cell lines used are FaDu (derived from a hypopharyngeal tumor, purchased from ATCC, Manassas, Virginia, HTB-43) and UMSCC47 (derived from an oral cavity tumor, kindly provided by Dr. Tom Carey, University of Michigan). They were cultured at 37°C and 5% CO2. FaDu cells were cultured in Eagle's minimum essential medium (Cellgro/Fisher Scientific, Pittsburgh, PA) with 1 mmol/L sodium pyruvate (Cellgro/Fisher) and 1× minimum essential medium nonessential amino acids (Sigma-Aldrich, St. Louis, MO). The UMSCC47 cell line was cultured in Dulbecco's minimum essential medium (Cellgro/Fisher) with 100 nmol/L nonessential amino acids and 2 mmol/L l-glutamine (Sigma-Aldrich). All culture media were supplemented with 10% fetal bovine serum [10082–147 (Gibco/Invitrogen, Carlsbad, CA) for UMSCC47 and SV300-1403 (Fisher) for FaDu cultures] and penicillin (100 U/mL)-streptomycin (0.1 mg/mL).
All procedures involving mice were conducted in accordance with the National Institutes of Health regulations concerning the use and care of experimental animals and approved by the Albert Einstein College of Medicine animal use committee. The injection protocol was adapted from Henson et al26 Cell lines were grown to 85% to 90% confluence. On the day of injection, cells were trypsinized (0.25% Trypsin; Gibco 25200), resuspended to a final concentration of 2 × 107 cells in 50% (v/v) medium and 50% (v/v) Matrigel (354234; BectonDickinson, San Jose, CA), and placed on ice. Six-to-eight-week-old athymic nude mice (NCI-Frederick, Frederick, MD) were then anesthetized using isoflurane.25,27 Mice were injected into the floor of the mouth with 50 μL of cell/Matrigel suspension, resulting in 1 × 106 cells per injection. Tumors were allowed to grow for 3 to 4 weeks before in vivo invasion assay and histopathology.
In Vivo Invasion Assay
The measurement of cell invasion into needles placed in the primary tumor of anesthetized mice was performed as described previously in detail.25 Invasive cells were collected into 33-gauge Hamilton needles (14-815-423; Fisher) filled with Matrigel (356234; Becton Dickinson) diluted 1:10 with L15–bovine serum antigen (BSA) ± chemoattractant [EGF (Life Technologies/Invitrogen) or CXCL12α (460-SD; R&DSystems, Minneapolis, MN)] for 4 hours. At the end of collection, the contents of the needles were extruded using approximately 30 μL of 0.5 μg/mL DAPI (in PBS) with a syringe onto a coverslip. To inhibit activity of the EGF receptor, Iressa (gefitinib; provided generously by AstraZeneca), a tyrosine kinase inhibitor specific for the EGF receptor, was used at 1 μmol/L. To block activation of CXCR4, AMD3100 (Sigma, A5602) was used at 500 nmol/L. Tumor necrosis factor-α–converting enzyme (TACE) inhibitor TNF-α protease inhibitor-2 (TAPI-2) (INH-3852-PI; Peptides International, Louisville, KY) was used at a final concentration of 0.5 μmol/L in the microneedles. To impair macrophages functionally in mice bearing tumors, PBS (control) and clodronate-containing liposomes were administered by tail vein into mice 48 and 24 hours before the in vivo invasion assay as described previously28 using clodronate at a concentration of 2.5 g/10 mL PBS. Clodronate (or Cl2MDP) was a gift of Roche Diagnostics GmbH (Penzberg, Germany). Phosphatidylcholine (Lipoid E PC) was obtained from LipoidGmbH (Ludwigshafen, Germany). Cholesterol was purchased from Sigma. Animals were injected with 100 μL of liposome solution per 10 g of weight.
Determination of Cell Types Collected in the in Vivo Invasion Assay
After a 4-hour collection, invasive cells were extruded from needles using 10% buffered formalin (SF100-20; Fisher Scientific) onto poly-l-lysine coated MatTek dishes and fixed for 1 hour at room temperature. To block nonspecific binding, the samples were incubated overnight at 4°C in Tris-buffered saline (TBS) with 1% FBS (TBS-FBS). The cells were permeabilized with 100 μL of TBS with 0.1% Triton ×100 for 10 minutes at room temperature, and washed three times with TBS-BSA, blocked overnight with TBS-FBS, and incubated with a primary antibody mixture of mouse anti–pan-cytokeratin antibody (SC15367; Santa Cruz Biotechnology, Santa Cruz, CA) for carcinoma cells and rat anti-F4/8029 for macrophages in TBS-BSA at a dilution of 1:50 and 1:25 respectively. After 1-hour incubation with the primary antibodies, cells were washed with TBS-BSA and incubated in a mixture of goat anti-mouse Cy3 and sheep anti-rat fluorescein isothiocyanate. Cells were rinsed and left in TBS-BSA with DAPI and counted using a fluorescence microscope.
Intravital Imaging of Texas Red Dextran Uptake by Macrophages in Primary Tumors and Spleens
Nude mice carrying 3- to 4-week-old tumors were injected via the tail vein with either control liposomes or clodronate-containing liposomes (100 μL of liposome solution per 10 g animal weight) 48 and 24 hours before the experiments. To visualize the effect of control and clodronate-containing liposomes on phagocytic activity of macrophages, 48 hours after liposome injection, animals were injected i.v. via the tail vein with 200 μL of 10 mg/mL Texas Red dextran (70 kDa, D1830; Molecular Probes/Invitrogen) in Dulbecco's phosphate buffered saline (14040; Gibco). Two hours after dextran injection, the animal was anesthetized, the skin over the tumor removed, and macrophage function was determined by the ability of the cells to take up dextran. Texas Red–labeled stromal cells were imaged using a Radiance 2000 MP multiphoton microscope (Bio-Rad, Hercules, CA) at an excitation wavelength of 870 nm using a 20 × 0.95 NA water objective. Multiple Z series were taken for tumor and spleen using 10-μm steps. Quantitation of images was done using ImageJ to obtain the pixel intensity of the dextran (red fluorescence) taken up by host cells. For image analysis, three fields were selected per spleen and per tumor, and at least three spleens and three tumors were imaged per clodronate liposome treatment or per control liposome treatment. Total pixel intensity was measured in the Texas Red dextran channel for each slice in a Z-stack (five slices 10-μm apart per Z-stack). Background intensity was determined by selecting an area without macrophages and measuring the average pixel intensity for that area. For each z-slice, the average Texas Red uptake was calculated by subtracting the average background pixel intensity from the average pixel intensity for each slice, and averaging the results for each Z-stack.
HNSCC in Vivo Model Histopathology
After needle cell collection and/or in vivo imaging, the submaxilla of the mouse containing the tumor xenograft and intact surrounding stromal tissues was carefully excised for histopathology. The slides were then stained using H&E or immunohistochemistry (IHC). For IHC, the sections were incubated for 60 minutes at room temperature with either rabbit anti–mouse-IBA1 (019–1974, 1:1000; Wako, Richmond, VA) or mouse anti–human-pan-cytokeratin (C2562, 1:1000; Sigma). This was followed by incubation with biotinylated secondary antibodies: goat anti-mouse (E0433; Dako, Carpinteria, CA) and goat anti-rabbit (E0432; Dako); both at a 1:500 dilution for 60 minutes at room temperature. The staining was visualized using a horseradishperoxidase–avidin-biotin complex reaction for 20 minutes (PK-6100;Vector,) followed by a diaminobenzidine reaction (SK-4100; VectorLaboratories, Burlingame, CA), and a hematoxylin counterstain. For negative control, primary antibody was omitted.
FACS Analysis
Cell were washed and detached with Accutase (AT-104, Innovative Cell Technologies, San Diego, CA) and resuspended to make a final concentration of 1 × 106 cells/mL. One hundred microliters of the cell suspension were then stained with 1 μL of primary antibody (500 μg/mL CXCR4 MAB172; R&D Systems) on ice for 60 minutes. The samples were then washed and stained with secondary antibody (anti–mouse-PE, 115-116-146; Jackson Immunoresearch, West Grove, PA) for 30 minutes. The samples were analyzed using a FACSCanto benchtop flow cytometer (Becton Dickinson).
Results
HNSCC in Vivo Mouse Model and Histopathology
Two HNSCC cell lines, FaDu and UMSCC47, were used for in vivo assays. Cells in 50% v/v Matrigel were injected into the floor of the mouth of nude mice to form an orthotopic tumor. H&E staining revealed invasion of the tumor cells into the host stroma (Figure 1, A and B). This was confirmed by IHC using a human-specific pan-cytokeratin antibody (Figure 1, C and D). Analysis using IHC to detect mouse-specific ionized calcium binding adaptor molecule-1 (IBA1) confirmed the presence of macrophages in our xenograft models. These macrophages were present in the tumors as well as in the surrounding stroma (Figure 1, E and F).
Figure 1.
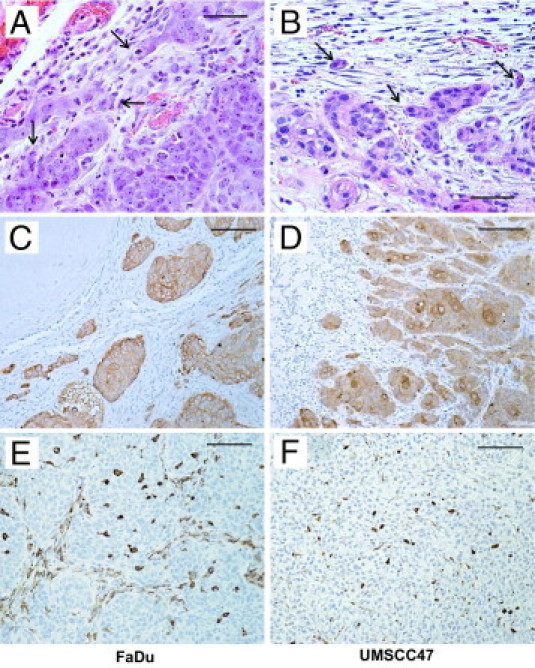
Patterns of invasion and tumoral macrophages of 3- to 4-week-old floor-of-mouth tumors. A and B: Host–tumor interface. Both FaDu and UMSCC47 grow as moderate- to well-differentiated large acini. They invade by budding out small groups of cells (arrows) from the acini. C and D: Invasive patterns. Tumor cells (brown) form acini that individually bud out into the surrounding host tissue. E and F: Macrophages (brown) within tumor acini are polygonal, whereas those within stroma between acini are elongated. A and B: H&E; scale bars, 50 μm; C and D, pancytokeratin immunoperoxidase, scale bars 100 μm; (E and F) IBA1 immunoperoxidase; scale bars = 100 μm.
EGF Stimulates in Vivo Invasion of Head and Neck Orthotopic Tumors
Three to 4 weeks postinjection, microneedles containing extracellular matrix components (10% Matrigel) with or without chemotactic signals were inserted into the primary tumors to collect invasive cells (Figure 2A). EGF induced in vivo invasion in FaDu tumors (Figure 2B) in a dose-dependent fashion, with invasion saturating at concentrations of EGF in the needle of 25 nmol/L or higher. We therefore selected 25 nmol/L EGF as the standard concentration to use. This concentration of EGF also induced UMSCC47 tumor cell invasion in vivo (Figure 2C).
Figure 2.
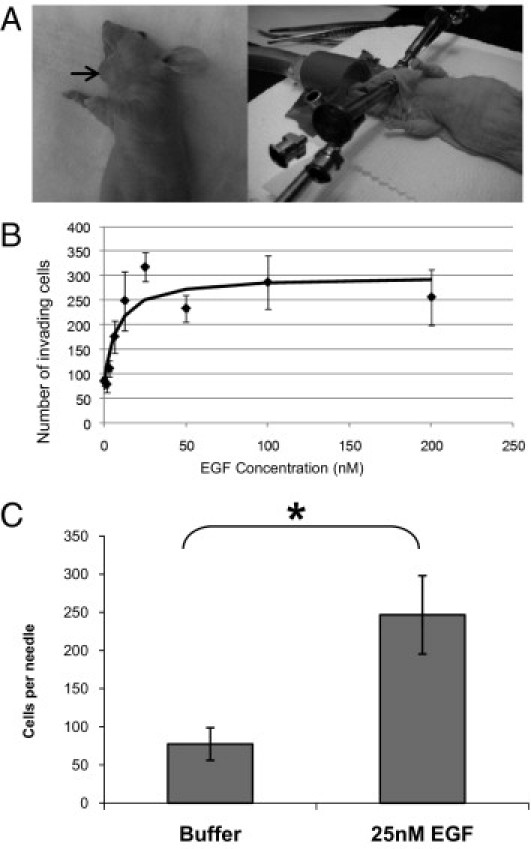
EGF stimulates in vivo invasion of head and neck cancer cells. A: The in vivo invasion assay is performed on floor-of-mouth tumors that are at least 0.5 cm in diameter (left). Typical assays involve two sets of three needles in specially designed holders25 inserted into either side of the tumor (right). B: Needles containing varying concentrations of EGF were inserted into FaDu floor-of-mouth tumors, and the number of cells invading the needles was determined. Data are means and standard errors of the mean of at least three measurements from at least two different tumors for all data points. The curve is a fit of the data to the equation I = I0 + I1*C/(C+C50), where C is the concentration of EGF in the needle, and I0 = 70, I1 = 230, and C50 = 7 nmol/L. C: Needles containing buffer alone or 25 nmol/L EGF were inserted into UMSCC47 orthotopic tumors, and the number of cells invading the needles was determined. Data are mean and SD of measurements from at least three different tumors. *P < 0.05
Previous studies with breast cancer models have shown that macrophages comigrate with cancer cells in response to EGF stimulation in this in vivo invasion assay.15,23,24 To assess macrophage comigration in HNSCC tumor models, the invasive cells collected from tumors were fixed and stained with a pan-cytokeratin antibody to detect tumor cells and an F4/80 antibody to detect macrophages (Figure 3A). The relatively weak F4/80 staining is consistent with our previous studies in which the permeabilization required for detection of cytokeratin may reduce the amount of F4/80 detected. Tumor cells represented approximately 76% of the invasive population and 16% of macrophages, with 8% of the cells with DAPI-labeled nuclei not being stained by either cytokeratin or F4/80 antibodies (Figure 3B). The cytokeratin-staining cells are unlikely to be normal epithelial cells since the needles collected the cells from the tumors, which formed at the site of injection in the floor of the mouth well below the normal oral mucosa (Figure 1). Thus, gradients of EGF in vivo can stimulate invasion of HNSCC tumor cells in concert with macrophages.
Figure 3.
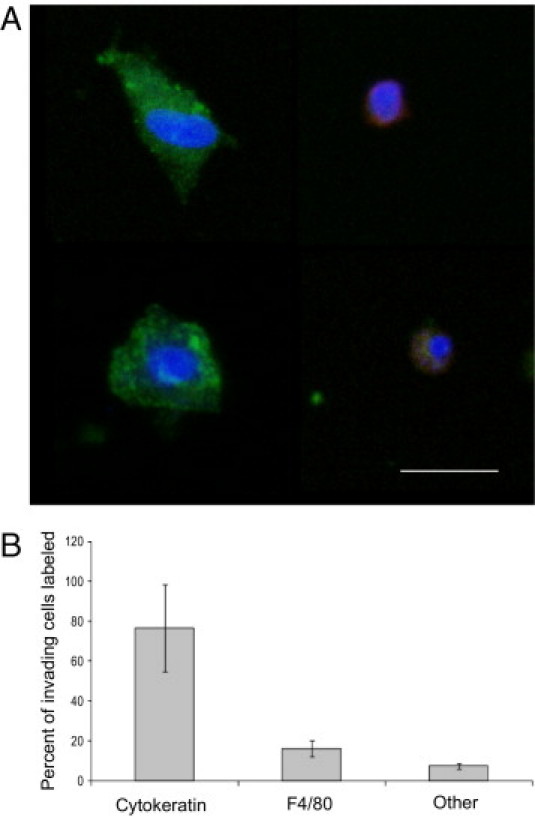
EGF stimulates co-invasion of head and neck cancer cells and host macrophages. A: Cell-typing imaging of UMSCC47 tumor cells migrating into needles containing 25 nmol/L EGF. A pan-cytokeratin antibody was used to stain tumor cells (green, left), whereas an F4/80 antibody was used to stain macrophages (red, right). Cell nuclei were stained with DAPI. Scale bar = 25 μm. B: Cell typing analysis by immunofluorescence of FADU tumor cells migrating into needles containing 25 nmol/L EGF. As in A, a pan-cytokeratin antibody was used to stain tumor cells, whereas an F4/80 antibody was used to stain macrophages. Cells that were positive for DAPI but did not stain with pan-cytokeratin or F4/80 antibodies were labeled as “other.” Similar results were seen for UMSCC47 tumors. Data are mean and SD of measurements from five tumors.
Macrophages Are Not Required for EGF-Induced in Vivo Invasion
Though it has been suggested that macrophages induce angiogenesis in head and neck tumors,30 the role of macrophages in HNSCC invasion is not well understood. To investigate the role of macrophages in head and neck tumor invasion, we examined the ability of the cells to invade when macrophages are functionally inactivated. We injected tumor-bearing mice i.v. with either clodronate-containing liposomes or PBS-containing liposomes (as control) 48 and 24 hours before measuring in vivo invasion. Clodronate has been found to suppress macrophage migration in vitro as well.31 As we had found previously,23 clodronate was effective in inhibiting macrophage function; uptake of Texas Red dextran was dramatically inhibited in both the primary tumor and the spleen (Figure 4, A–C). However, unlike the breast cancer models,23 functionally disabling macrophages by clodronate pretreatment did not affect in vivo invasion toward EGF in either FaDu- or UMSCC47-derived tumors (Figure 4D). This indicates that head and neck tumors do not depend on macrophages for EGF-stimulated invasion in vivo.
Figure 4.

Clodronate inhibition of macrophage function does not block EGF-induced in vivo invasion. A: Control liposomes (left columns) or clodronate-containing liposomes (right columns) were injected i.v. 48 hours and 24 hours before imaging into animals bearing FaDu (left panel) or UMSCC47 (right panel) floor-of-mouth tumors. Two hours before analysis, 70-kDa Texas Red dextran was injected i.v. The uptake of the dextran by phagocytic cells in the spleen (top rows) and tumor (bottom rows) was determined using multiphoton imaging. Representative images are shown from three experiments performed with FaDu and UMSCC47 tumors. Scale bar = 25 μm. B and C: Quantification of the amount of phagocytosed Texas Red dextran in multiple Z-stacks of tumors (B) and spleens (C) in animals treated with control liposomes (Liposomes) or clodronate liposomes (Clodronate). Data for FaDu (top, light gray) or UMSCC47 (bottom, dark gray) tumors are mean and SD of the mean of measurements from multiple Z-stacks from at least three different tumors and spleens for control liposome and clodronate liposome treated mice for each tumor model. *P < 0.05, **P < 0.005. D: Needles containing buffer alone or EGF were inserted into the tumors of animals that were treated with control liposomes (Liposomes) or clodronate liposomes (Clodronate) and the number of cells invading the needle determined. The invasion responses of FaDu (light gray) and UMSCC47 tumors (dark gray) were measured. Data are mean and SD of the mean of measurements from at least three different tumors for each condition and tumor model.
CXCL12-Induced in Vivo Invasion Is Dependent on EGFR Signaling
Given the importance of the chemokine CXCL12 and its receptor, CXCR4, in the progression of different malignancies,32 we evaluated the role of CXCL12 in HNSCC invasion in vivo. CXCR4 is expressed on FaDu and UMSCC47 (see Supplemental Figure S1 at http://ajp.amjpathol.org). CXCL12 induced in vivo invasion of both FaDu and UMSCC47 tumors, which were significantly reduced by Iressa, an EGFR inhibitor33 (Figure 5, A and B). We then asked whether invasion toward EGF was also dependent on CXCR4 signaling. Addition of AMD3100, a CXCR4 inhibitor,34 did not block the invasion of FaDu or UMSCC47 tumor cells toward EGF (Figure 5, C and D), although AMD3100 did block CXCL12-mediated invasion. These results show that although CXCL12-induced invasion depends on EGFR signaling, EGF-induced invasion does not depend on CXCR4. Thus, EGFR-mediated invasion of HNSCC can be activated either directly by EGF and/or by transactivation through CXCL12/CXCR4. The concentration and duration of Iressa and AMD3100 treatment did not affect cell viability (data not shown).
Figure 5.
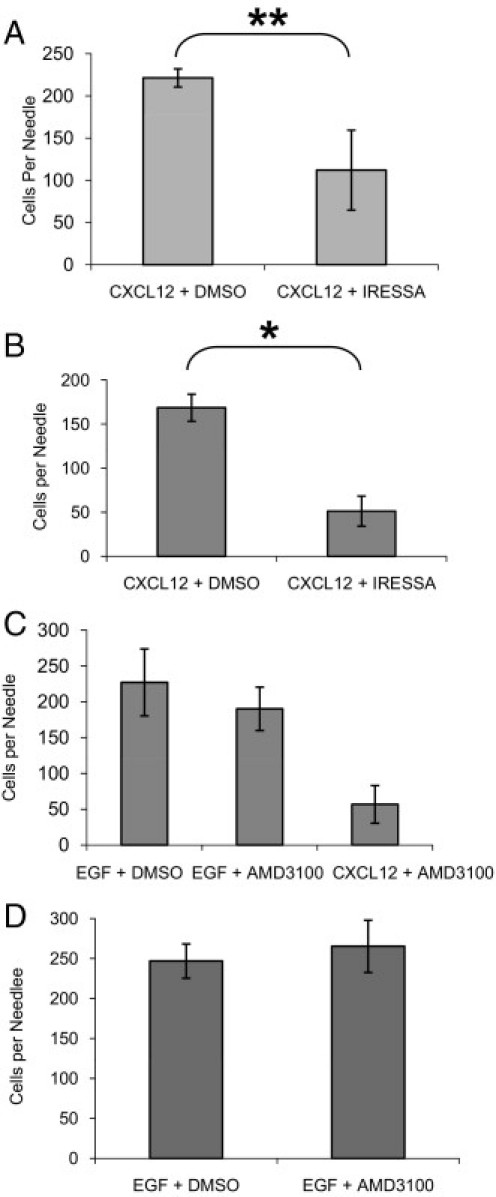
CXCL12-induced invasion is dependent on EGFR. A: Needles containing 15.6 nmol/L CXCL12 with DMSO or 5 μmol/L Iressa were inserted into FaDu floor-of-mouth tumors, and the number of cells invading the needle was determined. The invasion of FaDu tumor cells in response to CXCL12 was inhibited by Iressa 2 × 10−8. B: Needles containing 62.5 nmol/L CXCL12 with DMSO or 5 μmol/L Iressa were inserted into UMSCC47 floor-of-mouth tumors, and the number of cells invading the needle was determined. The invasion of UMSCC47 tumor cells induced by CXCL12 was inhibited by Iressa 2 × 10−5. C: Needles containing 25 nmol/L EGF with DMSO, 25 nmol/L EGF with 0.5 μmol/L AMD3100, or 15.6 nmol/L CXCL12 with 0.5 μmol/L AMD3100 were inserted into FaDu floor-of-mouth tumors, and the number of cells invading the needle was determined. FaDu tumor invasiveness to EGF is not significantly affected by AMD3100 (P < 0.1). D: Needles containing 25 nmol/L EGF with DMSO or 25 nmol/L EGF with 0.5 μmol/L AMD3100 were inserted into UMSCC47 floor-of-mouth tumors, and the number of cells invading the needle was determined. UMSCC47 tumor invasion in response to EGF was not significantly affected by AMD3100 (P < 0.35). Data are means and standard deviations of multiple measurements from at least three different tumors.
CXCL12-Induced in Vivo Invasion Is Dependent on ADAM17
Since CXCL12-induced invasion depends on EGFR signaling, we hypothesized that CXCR4 signaling could be activating the release of EGFR ligands that activate EGFR, thereby stimulating invasion. The metalloprotease ADAM17 (a disintegrin and metalloprotease domain 17), also known as TACE, has been known to release multiple ligands including transforming growth factor-α (TGF-α), amphiregulin (AREG), and heparin binding EGF (HB-EGF), and has been reported to play a role in the proliferation and invasion of multiple types of cancer.35 In HNSCC, it has been shown that G protein coupled receptors (GPCRs) are able to transactivate the EGFR by ADAM17-dependent release of EGFR ligands in vitro to enhance tumor growth and invasion.36–38 We found that TAPI-2, an inhibitor of ADAM17, significantly reduced the number of cells invading in response to CXCL12, whereas the number of cells invading in response to EGF was not affected (Figure 6, A and B). This result provides in vivo evidence that CXCL12 stimulates invasion via metalloprotease-mediated release of EGFR ligands, whereas EGF-induced invasion is not dependent on metalloprotease activation.
Figure 6.
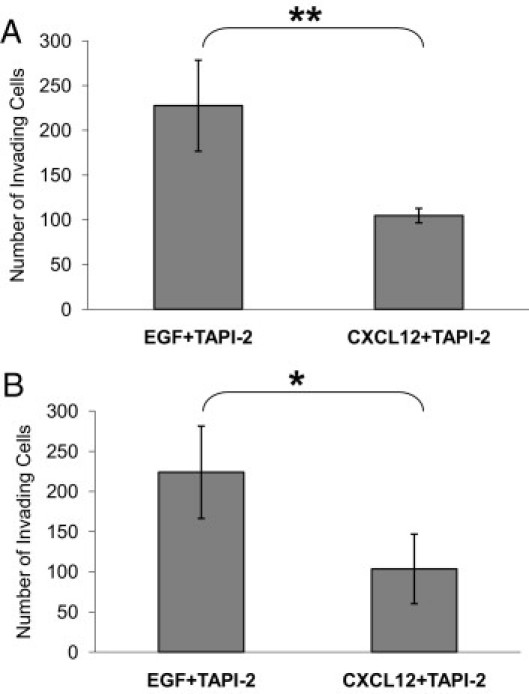
CXCL12-induced in vivo invasion is ADAM17 dependent. A: Needles containing 25 nmol/L EGF with 0.5 μmol/L TAPI-2 or 15.6 nmol/L CXCL12 with 0.5 μmol/L TAPI-2 were inserted into FaDu floor-of-mouth tumors, and the number of cells invading the needle was determined. TAPI-2 reduced invasion toward CXCL12 (**P ≤ 0.02), whereas EGF-stimulated invasion remained unaffected. B: Needles containing 25 nmol/L EGF with 0.5 μmol/L TAPI-2 or 62.5 nmol/L CXCL12 with 0.5 μmol/L TAPI-2 were inserted into UMSCC47 floor-of-mouth tumors, and the number of cells invading the needle was determined. TAPI-2 reduced UMSCC47 invasion toward CXCL12 (*P ≤ 0.0008), whereas EGF-stimulated invasion remained unaffected. Data are mean and standard deviations.
Discussion
Herein, we report the first in vivo analysis of invasion of HNSCC cells in response to applied gradients of a growth factor (EGF) and a chemokine (CXCL12). The studies used an in vivo invasion assay previously developed and established in orthotopic models of breast cancer.15,23,24 FaDu and UMSCC47 cells were injected into the floor of the mouth of nude mice as orthotopic models of HNSCC. We found that EGF stimulated in vivo invasion of both tumors. Surprisingly, macrophage function overall was not necessary for EGF-induced in vivo invasion, since inhibition of macrophage function using clodronate liposomes had no effect. CXCL12 was also found to stimulate in vivo invasion through EGFR function. Inhibition of the metalloprotease ADAM17 using TAPI-2 selectively inhibited CXCL12-induced invasion but not EGF-induced invasion, consistent with CXCL12 activation of EGFR via release of EGFR ligands.
EGFR is often overexpressed in HNSCC,7,8 and high levels of EGFR are present in the FaDu and UMSCC47 cell lines (data not shown). Our direct measurements of invasion in response to an EGF stimulus confirm that the EGFR is functional in vivo for mediating invasion in HNSCC. Maximal EGF-induced invasion occurred in response to 25 nmol/L EGF, similar to what we have previously reported for breast cancer.27 However, peak invasion occurred over a broad range of EGF concentrations (roughly 20 to 200 nmol/L in the needle), whereas in breast cancer, the response showed a decline at higher concentrations.27 This suggests differences in invasion characteristics in vivo between breast cancer and HNSCC.
We found additional differences between breast cancer and HNSCC in the roles played by macrophages during invasion in vivo. Stromal cells have been known to enhance the invasive potential of many types of tumors.20,39,40 In 2004, we described a synergistic interaction between macrophages and breast tumor cells during tumor cell invasion in vivo.24 This interaction relies on a paracrine communication loop between tumor cells and macrophages. In those studies, we found that breast tumor cells secrete CSF-1, which stimulates macrophages to secrete EGF, thereby enhancing invasion of breast tumor cells. Furthermore, blockade of either CSF-1 receptor or EGF receptor was able to prevent macrophage and tumor cell migration and invasion.15 Recently, we reported that other ligand/receptor systems such as CXCL12/CXCR4 were able to induce this EGF/CSF-1 paracrine loop–dependent invasion in vivo.23 Functionally disabling macrophages by injection of clodronate-containing liposomes did not affect EGF-dependent invasion in HNSCC. We conclude that unlike in breast cancer, macrophages are not directly involved in HNSCC invasion. This could explain why the invasion responses that we see with HNSCC cells appear to be roughly half as large as the invasion responses we find in breast cancer—the contribution of macrophages may increase the size of the response. Conversely, the optimal functioning of the paracrine loop involving macrophages may be more sensitive to specific stimulation parameters resulting in the more narrow optimal response range seen in breast cancer, unlike the broad range of concentrations we found in this study that stimulate in vivo invasion in HNSCC. Macrophages may be more important in other aspects of tumor progression in HNSCC, such as angiogenesis.30 However, it is important to note that this study was focused on determining the role of macrophages in HNSCC invasion, and we have not ruled out contributions by other stromal cell types to the in vivo invasion of HNSCC tumors.
We also evaluated the ability of CXCL12 to induce in vivo invasion of HNSCC cells since expression of the CXCL12 receptor, CXCR4, is correlated with poor prognosis, including increased lymph node metastasis.41–43 Our studies demonstrate that CXCL12 is an effective inducer of invasion in vivo in orthotopic HNSCC models. Furthermore, CXCL12-induced invasion was dependent on EGFR function, as shown using the EGFR-specific inhibitor Iressa. However, EGF-induced in vivo invasion was not dependent on CXCR4, since the CXCR4 inhibitor AMD3100 did not have a significant effect. CXCR7 is unlikely to be mediating the invasion response to CXCL12, since AMD3100 does not inhibit CXCR7 signaling as measured by β-arrestin recruitment.44 The mechanism by which CXCR4 transactivates EGFR for invasion in vivo is likely to be due to EGFR ligand release, since the ADAM17 inhibitor TAPI-2 blocked CXCL12-induced invasion. However, EGF-induced invasion was not inhibited by TAPI-2 at 0.5 μmol/L, indicating that EGF-induced invasion does not involve relay of EGFR ligand signaling. These results are consistent with previous in vitro analyses of GPCR-induced responses. In vitro studies of HNSCC cell lines have demonstrated that CXCL12 can stimulate proliferation and invasion.45 Further analysis of the mechanism of induction of proliferation indicates that CXCL12 induces ERK activation through transactivation of the EGFR and can occur through release of EGFR ligands.41,46,47 ADAM17/TACE has been associated with tumor progression in oral squamous cell carcinoma48 and other cancers,49,50 and can cleave multiple EGFR ligands, including TGF-α, HB-EGF, and AREG.51 It has been shown that GPCR activation by ligands such as LPA can lead to transactivation of EGFR in HNSCC cells37,52,53 and in normal prostate cells, CXCL12 induced EGFR transactivation as the result of shedding an EGFR ligand AREG in vitro.54 Our studies complement the in vitro data on ADAM17 by confirming an in vivo role for ADAM17 in CXCL12-induced invasion.
In conclusion, we have found that EGF and CXCL12 can induce invasion in vivo of HNSCC from an orthotopic site. These studies have used established tumor cell lines with a fully malignant phenotype. Thus we are evaluating the invasion events that occur after development of tumors to the invasive stage. Our results support a model of HNSCC invasion in which local sources of EGF or CXCL12 can enhance tumor cell invasion into the local stroma. For example, CXCL12 produced by tumor-associated fibroblasts18 outside of the tumor could result in gradients of CXCL12 directing tumor cell invasion out from the primary tumor in an EGFR-dependent fashion. There are multiple sources of EGF in the local microenvironment, such as macrophages,15 fibroblasts,17 or activated platelets,16,55 and we find that EGF gradients will also direct in vivo invasion. Thus, our results support treatment of HNSCC with EGFR inhibitors to inhibit local invasion independent of effects on tumor growth rate. Combination therapy using cytotoxic treatment together with EGFR inhibitors may be useful for reducing both tumor growth and tumor spread, and extending patient survival.
Acknowledgments
We thank Jeffrey Wyckoff for his help with the in vivo invasion assay and Pamela Boimel for her help with the tail-vain liposome injection. We thank Drs. E. Richard Stanley, Tom Carey, Orsi Giricz, Esther Peterson-Peguero, Paraic Kenny, Carl Manthey, and the Segall, Condeelis, and Cox labs for materials and suggestions.
Footnotes
Supported by grants from the NIH (CA77522 and CA1000324 to J.E.S.) and the Department of Pathology. J.E.S. is the Betty and Sheldon Feinberg Senior Faculty Scholar in Cancer Research. Iressa (gefitinib) was provided by AstraZeneca.
T.S. and A.A. contributed equally to this manuscript.
CME Disclosure: None of the authors disclosed any relevant financial relationships.
Supplemental material for this manuscript can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.02.030.
A guest editor acted as editor-in-chief for the manuscript. No person at Thomas Jefferson University or Albert Einstein College of Medicine was involved in the peer review process or final disposition of this article.
Contributor Information
Michael B. Prystowsky, Email: Michael.Prystowsky@einstein.yu.edu.
Jeffrey E. Segall, Email: jeffrey.segall@einstein.yu.edu.
Supplementary data
FaDu (left) and UMSCC47 (right) cells were stained with either secondary antibody alone (top) or with anti-CXCR4 primary antibody followed by secondary antibody (bottom). X axes show forward scattering, Y axes show amount of fluorescently labeled secondary antibody bound as the log of the fluorescence.
References
- 1.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Belbin T.J., Bergman A., Brandwein-Gensler M., Chen Q., Childs G., Garg M., Haigentz M., Hogue-Angeletti R., Moadel R., Negassa A., Owen R., Prystowsky M.B., Schiff B., Schlecht N.F., Shifteh K., Smith R.V., Zheng X. Head and neck cancer: reduce and integrate for optimal outcome. Cytogenet Genome Res. 2007;118:92–109. doi: 10.1159/000108290. [DOI] [PubMed] [Google Scholar]
- 3.Brandwein-Gensler M., Teixeira M.S., Lewis C.M., Lee B., Rolnitzky L., Hille J.J., Genden E., Urken M.L., Wang B.Y. Oral squamous cell carcinoma: histologic risk assessment, but not margin status, is strongly predictive of local disease-free and overall survival. Am J Surg Pathol. 2005;29:167–178. doi: 10.1097/01.pas.0000149687.90710.21. [DOI] [PubMed] [Google Scholar]
- 4.Bryne M., Koppang H.S., Lilleng R., Kjaerheim A. Malignancy grading of the deep invasive margins of oral squamous cell carcinomas has high prognostic value. J Pathol. 1992;166:375–381. doi: 10.1002/path.1711660409. [DOI] [PubMed] [Google Scholar]
- 5.Koide N., Nishio A., Sato T., Sugiyama A., Miyagawa S. Significance of macrophage chemoattractant protein-1 expression and macrophage infiltration in squamous cell carcinoma of the esophagus. Am J Gastroenterol. 2004;99:1667–1674. doi: 10.1111/j.1572-0241.2004.30733.x. [DOI] [PubMed] [Google Scholar]
- 6.Lu C.F., Huang C.S., Tjiu J.W., Chiang C.P. Infiltrating macrophage count: a significant predictor for the progression and prognosis of oral squamous cell carcinomas in Taiwan. Head Neck. 2010;32:18–25. doi: 10.1002/hed.21138. [DOI] [PubMed] [Google Scholar]
- 7.Bei R., Budillon A., Masuelli L., Cereda V., Vitolo D., Di Gennaro E., Ripavecchia V., Palumbo C., Ionna F., Losito S., Modesti A., Kraus M.H., Muraro R. Frequent overexpression of multiple ErbB receptors by head and neck squamous cell carcinoma contrasts with rare antibody immunity in patients. J Pathol. 2004;204:317–325. doi: 10.1002/path.1642. [DOI] [PubMed] [Google Scholar]
- 8.Grandis J.R., Tweardy D.J. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579–3584. [PubMed] [Google Scholar]
- 9.Lothaire P., de Azambuja E., Dequanter D., Lalami Y., Sotiriou C., Andry G., Castro G., Jr., Awada A. Molecular markers of head and neck squamous cell carcinoma: promising signs in need of prospective evaluation. Head Neck. 2006;28:256–269. doi: 10.1002/hed.20326. [DOI] [PubMed] [Google Scholar]
- 10.Egloff A.M., Rothstein M.E., Seethala R., Siegfried J.M., Grandis J.R., Stabile L.P. Cross-talk between estrogen receptor and epidermal growth factor receptor in head and neck squamous cell carcinoma. Clin Cancer Res. 2009;15:6529–6540. doi: 10.1158/1078-0432.CCR-09-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O-Charoenrat P., Rhys-Evans P., Eccles S. Expression and regulation of c-ERBB ligands in human head and neck squamous carcinoma cells. Int J Cancer. 2000;88:759–765. doi: 10.1002/1097-0215(20001201)88:5<759::aid-ijc12>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 12.Yeudall W.A., Miyazaki H., Ensley J.F., Cardinali M., Gutkind J.S., Patel V. Uncoupling of epidermal growth factor-dependent proliferation and invasion in a model of squamous carcinoma progression. Oral Oncol. 2005;41:698–708. doi: 10.1016/j.oraloncology.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Bruzzese F., Di Gennaro E., Avallone A., Pepe S., Arra C., Caraglia M., Tagliaferri P., Budillon A. Synergistic antitumor activity of epidermal growth factor receptor tyrosine kinase inhibitor gefitinib and IFN-alpha in head and neck cancer cells in vitro and in vivo. Clin Cancer Res. 2006;12:617–625. doi: 10.1158/1078-0432.CCR-05-1671. [DOI] [PubMed] [Google Scholar]
- 14.Johns T.G., Luwor R.B., Murone C., Walker F., Weinstock J., Vitali A.A., Perera R.M., Jungbluth A.A., Stockert E., Old L.J., Nice E.C., Burgess A.W., Scott A.M. Antitumor efficacy of cytotoxic drugs and the monoclonal antibody 806 is enhanced by the EGF receptor inhibitor AG1478. Proc Natl Acad Sci U S A. 2003;100:15871–15876. doi: 10.1073/pnas.2036503100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goswami S., Sahai E., Wyckoff J.B., Cammer M., Cox D., Pixley F.J., Stanley E.R., Segall J.E., Condeelis J.S. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005;65:5278–5283. doi: 10.1158/0008-5472.CAN-04-1853. [DOI] [PubMed] [Google Scholar]
- 16.Oka Y., Orth D.N. Human plasma epidermal growth factor/beta-urogastrone is associated with blood platelets. J Clin Invest. 1983;72:249–259. doi: 10.1172/JCI110964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurobe M., Furukawa S., Hayashi K. Synthesis and secretion of an epidermal growth factor (EGF) by human fibroblast cells in culture. Biochem Biophys Res Commun. 1985;131:1080–1085. doi: 10.1016/0006-291x(85)90201-3. [DOI] [PubMed] [Google Scholar]
- 18.Orimo A., Gupta P.B., Sgroi D.C., Arenzana-Seisdedos F., Delaunay T., Naeem R., Carey V.J., Richardson A.L., Weinberg R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 19.Coussens L.M., Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joyce J.A., Pollard J.W. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis C.E., Pollard J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 22.Marcus B., Arenberg D., Lee J., Kleer C., Chepeha D.B., Schmalbach C.E., Islam M., Paul S., Pan Q., Hanash S., Kuick R., Merajver S.D., Teknos T.N. Prognostic factors in oral cavity and oropharyngeal squamous cell carcinoma. Cancer. 2004;101:2779–2787. doi: 10.1002/cncr.20701. [DOI] [PubMed] [Google Scholar]
- 23.Hernandez L., Smirnova T., Kedrin D., Wyckoff J., Zhu L., Stanley E.R., Cox D., Muller W.J., Pollard J.W., Van Rooijen N., Segall J.E. The EGF/CSF-1 paracrine invasion loop can be triggered by heregulin beta1 and CXCL12. Cancer Res. 2009;69:3221–3227. doi: 10.1158/0008-5472.CAN-08-2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wyckoff J., Wang W., Lin E.Y., Wang Y., Pixley F., Stanley E.R., Graf T., Pollard J.W., Segall J., Condeelis J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 25.Hernandez L., Smirnova T., Wyckoff J., Condeelis J., Segall J.E. In vivo assay for tumor cell invasion. Methods Mol Biol. 2009;571:227–238. doi: 10.1007/978-1-60761-198-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henson B., Li F., Coatney D.D., Carey T.E., Mitra R.S., Kirkwood K.L., D'Silva N.J. An orthotopic floor-of-mouth model for locoregional growth and spread of human squamous cell carcinoma. J Oral Pathol Med. 2007;36:363–370. doi: 10.1111/j.1600-0714.2007.00549.x. [DOI] [PubMed] [Google Scholar]
- 27.Wyckoff J.B., Segall J.E., Condeelis J.S. The collection of the motile population of cells from a living tumor. Cancer Res. 2000;60:5401–5404. [PubMed] [Google Scholar]
- 28.van Rooijen N., van Kesteren-Hendrikx E. “In vivo” depletion of macrophages by liposome-mediated “suicide.”. Methods Enzymol. 2003;373:3–16. doi: 10.1016/s0076-6879(03)73001-8. [DOI] [PubMed] [Google Scholar]
- 29.Austyn J.M., Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11:805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 30.Liss C., Fekete M.J., Hasina R., Lam C.D., Lingen M.W. Paracrine angiogenic loop between head-and-neck squamous-cell carcinomas and macrophages. Int J Cancer. 2001;93:781–785. doi: 10.1002/ijc.1407. [DOI] [PubMed] [Google Scholar]
- 31.Stevenson P.H., Stevenson J.R. Cytotoxic and migration inhibitory effects of bisphosphonates on macrophages. Calcif Tissue Int. 1986;38:227–233. doi: 10.1007/BF02556715. [DOI] [PubMed] [Google Scholar]
- 32.Wang D.F., Lou N., Zeng C.G., Zhang X., Chen F.J. Expression of CXCL12/CXCR4 and its correlation to prognosis in esophageal squamous cell carcinoma. Chin J Cancer. 2009;28:154–158. [PubMed] [Google Scholar]
- 33.Cohen M.H., Williams G.A., Sridhara R., Chen G., McGuinn W.D., Jr., Morse D., Abraham S., Rahman A., Liang C., Lostritto R., Baird A., Pazdur R. United States Food and Drug Administration Drug Approval summary: gefitinib (ZD1839; Iressa) tablets. Clin Cancer Res. 2004;10:1212–1218. doi: 10.1158/1078-0432.ccr-03-0564. [DOI] [PubMed] [Google Scholar]
- 34.Hatse S., Princen K., Bridger G., De Clercq E., Schols D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 2002;527:255–262. doi: 10.1016/s0014-5793(02)03143-5. [DOI] [PubMed] [Google Scholar]
- 35.Bhola N.E., Grandis J.R. Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front Biosci. 2008;13:1857–1865. doi: 10.2741/2805. [DOI] [PubMed] [Google Scholar]
- 36.Kalyankrishna S., Grandis J.R. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24:2666–2672. doi: 10.1200/JCO.2005.04.8306. [DOI] [PubMed] [Google Scholar]
- 37.Morgan S., Grandis J.R. ErbB receptors in the biology and pathology of the aerodigestive tract. Exp Cell Res. 2009;315:572–582. doi: 10.1016/j.yexcr.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schafer B., Marg B., Gschwind A., Ullrich A. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J Biol Chem. 2004;279:47929–47938. doi: 10.1074/jbc.M400129200. [DOI] [PubMed] [Google Scholar]
- 39.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 40.Condeelis J., Pollard J.W. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 41.Lee J.I., Jin B.H., Kim M.A., Yoon H.J., Hong S.P., Hong S.D. Prognostic significance of CXCR-4 expression in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;107:678–684. doi: 10.1016/j.tripleo.2008.12.047. [DOI] [PubMed] [Google Scholar]
- 42.Oliveira-Neto H.H., Silva E.T., Leles C.R., Mendonca E.F., Alencar Rde C., Silva T.A., Batista A.C. Involvement of CXCL12 and CXCR4 in lymph node metastases and development of oral squamous cell carcinomas. Tumour Biol. 2008;29:262–271. doi: 10.1159/000152944. [DOI] [PubMed] [Google Scholar]
- 43.Sasaki K., Natsugoe S., Ishigami S., Matsumoto M., Okumura H., Setoyama T., Uchikado Y., Kita Y., Tamotsu K., Hanazono K., Owaki T., Aikou T. Expression of CXCL12 and its receptor CXCR4 in esophageal squamous cell carcinoma. Oncol Rep. 2009;21:65–71. [PubMed] [Google Scholar]
- 44.Zabel B.A., Wang Y., Lewen S., Berahovich R.D., Penfold M.E., Zhang P., Powers J., Summers B.C., Miao Z., Zhao B., Jalili A., Janowska-Wieczorek A., Jaen J.C., Schall T.J. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J Immunol. 2009;183:3204–3211. doi: 10.4049/jimmunol.0900269. [DOI] [PubMed] [Google Scholar]
- 45.Tan C.T., Chu C.Y., Lu Y.C., Chang C.C., Lin B.R., Wu H.H., Liu H.L., Cha S.T., Prakash E., Ko J.Y., Kuo M.L. CXCL12/CXCR4 promotes laryngeal and hypopharyngeal squamous cell carcinoma metastasis through MMP-13-dependent invasion via the ERK1/2/AP-1 pathway. Carcinogenesis. 2008;29:1519–1527. doi: 10.1093/carcin/bgn108. [DOI] [PubMed] [Google Scholar]
- 46.Porcile C., Bajetto A., Barbieri F., Barbero S., Bonavia R., Biglieri M., Pirani P., Florio T., Schettini G. Stromal cell-derived factor-1alpha (SDF-1alpha/CXCL12) stimulates ovarian cancer cell growth through the EGF receptor transactivation. Exp Cell Res. 2005;308:241–253. doi: 10.1016/j.yexcr.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Q., Bhola N.E., Lui V.W., Siwak D.R., Thomas S.M., Gubish C.T., Siegfried J.M., Mills G.B., Shin D., Grandis J.R. Antitumor mechanisms of combined gastrin-releasing peptide receptor and epidermal growth factor receptor targeting in head and neck cancer. Mol Cancer Ther. 2007;6:1414–1424. doi: 10.1158/1535-7163.MCT-06-0678. [DOI] [PubMed] [Google Scholar]
- 48.Takamune Y., Ikebe T., Nagano O., Nakayama H., Ota K., Obayashi T., Saya H., Shinohara M. ADAM-17 associated with CD44 cleavage and metastasis in oral squamous cell carcinoma. Virchows Arch. 2007;450:169–177. doi: 10.1007/s00428-006-0350-y. [DOI] [PubMed] [Google Scholar]
- 49.Borrell-Pages M., Rojo F., Albanell J., Baselga J., Arribas J. TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J. 2003;22:1114–1124. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kenny P.A. TACE: a new target in epidermal growth factor receptor dependent tumors. Differentiation. 2007;75:800–808. doi: 10.1111/j.1432-0436.2007.00198.x. [DOI] [PubMed] [Google Scholar]
- 51.Sahin U., Weskamp G., Kelly K., Zhou H.M., Higashiyama S., Peschon J., Hartmann D., Saftig P., Blobel C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gschwind A., Hart S., Fischer O.M., Ullrich A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 2003;22:2411–2421. doi: 10.1093/emboj/cdg231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas S.M., Bhola N.E., Zhang Q., Contrucci S.C., Wentzel A.L., Freilino M.L., Gooding W.E., Siegfried J.M., Chan D.C., Grandis J.R. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006;66:11831–11839. doi: 10.1158/0008-5472.CAN-06-2876. [DOI] [PubMed] [Google Scholar]
- 54.Kasina S., Scherle P.A., Hall C.L., Macoska J.A. ADAM-mediated amphiregulin shedding and EGFR transactivation. Cell Prolif. 2009;42:799–812. doi: 10.1111/j.1365-2184.2009.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ho-Tin-Noe B., Goerge T., Cifuni S.M., Duerschmied D., Wagner D.D. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008;68:6851–6858. doi: 10.1158/0008-5472.CAN-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FaDu (left) and UMSCC47 (right) cells were stained with either secondary antibody alone (top) or with anti-CXCR4 primary antibody followed by secondary antibody (bottom). X axes show forward scattering, Y axes show amount of fluorescently labeled secondary antibody bound as the log of the fluorescence.