

Abstract
A defining characteristic of most human cancers is heterogeneity, resulting from the somatic acquisition of a complex array of genetic and genomic alterations. Dissecting this heterogeneity is critical to developing an understanding of the underlying mechanisms of disease and to paving the way toward personalized treatments of the disease. We used gene expression data sets from the analysis of primary and metastatic melanomas to develop a molecular description of the heterogeneity that characterizes this disease. Unsupervised hierarchical clustering, gene set enrichment analyses, and pathway activity analyses were used to describe the genetic heterogeneity of melanomas. Patterns of gene expression that revealed two distinct classes of primary melanoma, two distinct classes of in-transit melanoma, and at least three subgroups of metastatic melanoma were identified. Expression signatures developed to predict the status of oncogenic signaling pathways were used to explore the biological basis underlying these differential patterns of expression. This analysis of activities revealed unique pathways that distinguished the primary and metastatic subgroups of melanoma. Distinct patterns of gene expression across primary, in-transit, and metastatic melanomas underline the genetic heterogeneity of this disease. This heterogeneity can be described in terms of deregulation of signaling pathways, thus increasing the knowledge of the biological features underlying individual melanomas and potentially directing therapeutic opportunities to individual patients with melanoma.
A dominant characteristic of virtually all cancers is heterogeneity. For instance, breast cancer is a collection of diseases, each with distinct underlying molecular mechanisms and clinical characteristics.1–3 The importance of dissecting the heterogeneity is illustrated with the example of trastuzumab (Herceptin), an important drug for the treatment of breast cancer, but only in the few patients who are Her2 positive.4 This challenge is further compounded by the evident complexity of most cancers, involving multiple mutations and alterations that generate the cancer phenotype and, thus, requiring therapeutic strategies that can match the complexity with equally complex combination regimens.5–7 Clearly, it is critical to develop methods to stratify cancers into homogeneous subgroups, representing common mechanisms of disease, to then allow development of combination therapeutics that target these mechanisms.
Melanoma is no exception to this paradigm, with previous work highlighting substantial heterogeneity in the disease. Multiple studies have documented chromosomal copy number alterations, loss of heterozygosity, mutations in oncogenes, and differences in gene expression patterns in melanomas. A total of 14 regions of copy number gains and 13 regions of copy number losses are significantly present in a large collection of cultured melanoma cells and in primary melanomas.8 From a hierarchical clustering analysis of the cultured melanomas, six main groups and two major subgroups reflective of copy number alterations and mutational status of particular oncogenes can be identified. Significant differences in DNA copy numbers and mutational status of particular oncogenes have also been documented in melanomas exposed to different degrees of UV light.9–11 These distinctions are further amplified in analyses of differences in gene expression patterns in melanomas. Differential gene expression patterns and the distinct biological processes associated with such patterns have been documented in normal skin, common nevi, dysplastic nevi, radial and vertical growth phase melanomas, metastatic melanomas, and thin versus intermediate and thick tumors.12–16 In addition, a subtype of melanomas exhibiting differential regulation of genes involved in the ability of melanomas to form primitive tubular networks in vitro, a characteristic of highly aggressive metastatic melanomas, has been identified.17
Previous work18,19 has focused on the development of genomic signatures that can measure various aspects of cancer biology, including the deregulation of various oncogenic pathways. More important, we showed that these genomic signatures accurately predict responses to various targeted therapeutics in vitro, in vivo, and in human studies, a result seen in other independent studies.20 The opportunity to use these genomic signatures to dissect the complexity of melanoma provides a more in-depth understanding of the disease subtypes compared with an analysis of global gene expression data obtained from each patient. In short, we propose that gene expression signatures predicting activation of cell signaling pathways known to be critical for defining the oncogenic state can serve to dissect the heterogeneity of melanoma and to provide opportunities to develop novel therapeutic strategies. Given the 5-year survival rates of patients with later-stage melanomas and the lack of a complete understanding of the molecular mechanisms involved in melanoma progression, genomic profiling of melanoma samples has the potential to improve both the effectiveness of therapeutics and our understanding of the biological features of melanoma.21 Specifically, genomic profiling has the potential to guide physicians to the therapeutic agents that would be most effective in the treatment of particular melanomas. In addition, genomic profiling has the potential to assimilate the biological characteristics of melanoma with its clinicopathological characteristics. Therefore, we characterized melanoma gene expression data from eight independent studies publicly available through Gene Expression Omnibus (GEO). We characterized gene expression patterns, biological processes, and the activity of oncogenic signaling pathways that are associated with specific melanomas.
Materials and Methods
Human Melanoma Samples
Melanoma gene expression data from eight independent studies publicly available through GEO were characterized. A total of 303 samples comprising benign nevi, primary melanoma, and metastatic melanoma were available from six independent data sets (GSE3189, GSE4570, GSE4587, GSE4845, GSE7553, and GSE8401), and a total of 76 samples comprising in-transit melanoma were available from two independent data sets (GSE10282 and GSE19293). The samples comprising the GSE3189 data set had a melanocyte content of >50%, with no mixed histological characteristics, and the samples comprising the GSE7553 data set contained >95% melanoma cells. The percentage of tumor in the samples comprising the other GEO data sets is not known. All gene expression data used in the study were arrayed on the Affymetrix HG-U133 platforms (Santa Clara, CA), and probes were filtered to include only those probes present on the HG-U133A array (Affymetrix, Santa Clara, CA).
Computer Hardware
Analyses were performed using a Dell desktop computer with a 2.8 GHz Intel Pentium D CPU processor, 2 Gb RAM, and running Microsoft Windows XP Professional 2002.
Preprocessing of Microarray Data of Human Melanoma Samples
MAS5.0 normalized data for each sample were downloaded from GEO from previously published studies (GSE3189, GSE4570, GSE4587, GSE4845, GSE7553, GSE8401, GSE10282, and GSE19293). Only benign nevi and primary, in-transit, and metastatic melanoma samples were used in the analyses. The MAS5.0 data were filtered, resulting in data sets composed of only probes present on Affymetrix Human Genome U133A 2.0 arrays. The MAS5.0 normalized filtered data were then log2 transformed in MATLAB R2008a with the Bioinformatics and Statistical toolboxes installed using the following code:
To normalize these samples, bayesian factor regression modeling (BFRM) was used to normalize the data against 69 Affymetrix probes for human maintenance genes using 15 principal components.22,23 BFRM was performed as previously described to generate a normalized log2 MAS5.0 version of the data set.19 More details of this software are available at http://www.stat.duke.edu/research/software/west/bfrm/index.html.
Preprocessing of Microarray Data of Pathway Signatures
The methods used to generate the oncogene and tumor suppressor pathway signatures have been described in great detail,19 and the training data are publicly available at http://data.genome.duke.edu/breast_subgroups. The Binreg ver2 program and tutorial are available at http://www.duke.edu/∼dinbarry/BINREG. By using Affymetrix Expression Console version 1.0, .CEL files were MAS5.0 normalized. The MAS5.0-normalized data were then log2 transformed in MATLABR2008a with the Bioinformatics and Statistical toolboxes installed using the following code:
All microarray statistical analyses were performed using R/Bioconductor software.
Pathway Analysis
The training data and conditions used to generate patterns of pathway activity in the human melanomas were performed as previously described.18,19 All training data comprised only probes present on Affymetrix Human Genome U133A 2.0 arrays and were MAS5.0 log transformed.
The signatures were applied to the human melanoma samples using the parameters in Table 1.
Table 1.
Parameters Used in the Application of Oncogenic Pathway Signatures to the Human Melanoma Samples
| Signature | AKT | βCAT | EGFR | ER | E2F1 | E2F3 | HER2 | MYC | p53 | p63 | PI3K | PR | RAS | SRC | STAT3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of genes | 250 | 85 | 500 | 125 | 150 | 350 | 250 | 500 | 250 | 75 | 250 | 250 | 350 | 85 | 125 |
| No. of metagenes | 3 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 3 | 3 | 2 | 3 | 2 | 3 | 3 |
| Burn-in | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 |
| Iterations | 5000 | 5000 | 5000 | 5000 | 5000 | 5000 | 5000 | 5000 | 5000 | 5000 | 5000 | 5000 | 5000 | 5000 | 5000 |
| Skips | 1 | 1 | 1 | 1 | 3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| CI | 95% | 95% | 95% | 95% | 95% | 95% | 95% | 95% | 95% | 95% | 95% | 95% | 95% | 95% | 95% |
| RMA or MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 | MAS5.0 |
| Quantile normalized | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Shift scale | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Binreg Ver | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
CAT, catenin; EGFR, epidermal growth factor receptor; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; PI3K, phosphatidylinositol 3-kinase; PR, progesterone receptor.
Unsupervised Hierarchical Clustering
Unsupervised hierarchical clustering was performed using Cluster 3.0. The MAS5.0 log-transformed BFRM-normalized data were imported into Cluster 3.0. Data were filtered using the SD (Gene Vector) property, resulting in a data set containing 1000 genes. The filtered data were then mean centered for genes and arrays. Hierarchical clustering of the adjusted data, genes, and arrays was performed using the correlation (uncentered) similarity metric and average linkage clustering. Data matrices and array trees were viewed using Java TreeView 1.0.12.
Gene Set Enrichment Analysis
The GenePattern gene set enrichment analysis (GSEA) module was used to run analyses on a server at the Broad Institute.24,25 MAS5.0 log-transformed BFRM-normalized gene expression data for the appropriate subgroup of human melanoma samples were uploaded. The c2.all.v2.5.symbols.gmt [Curated] gene set database and the HG_U133A.chip chip platform file from the GSEA website were used. A total of 1000 permutations were performed. All other default parameters were used. Gene sets significantly enriched at a nominal P value <1% were noted and used for subsequent GATHER analyses.
GATHER Analysis
Genes composing gene sets significantly enriched at a nominal P value <1% were identified using the c2.all.v2.5.symbols.gmt [Curated] gene set file from the GSEA website. Identified genes were annotated for gene ontology codes using GATHER.26 For analyses of in-transit melanoma, the most positively expressed probe identifiers that define the in-transit melanoma subgroups in the unsupervised hierarchical clustering analysis were annotated for gene ontology codes using GATHER.26
BRAF and NRAS Mutation Status
In-transit melanoma tumor samples were homogenized using a miniature bead beater (Biospec Products, Bartlesville, OK) and lysing matrix A (MP Biomedicals, Solon, OH), total RNA isolated (RNeasy; Qiagen, Valencia, CA), and cDNA synthesized (first-strand cDNA synthesis; Roche, Indianapolis, IN). PCR amplification of BRAF and NRAS mutation sites (exons 15 and 3, respectively; primer sequences are given later) was performed on a Stratagene Robocycler 96 using HotStart TaqDNA polymerase (Qiagen) in a 50-μL reaction volume (reaction settings are given later). Purified PCR products (Qiaquick PCR purification kit; Qiagen) were sequenced by the Duke University DNA Analysis Facility using the Applied Biosystems Dye Terminator Cycle Sequencing system with AmpliTaq DNA Polymerase and ABI 377 PRISM DNA sequencing instruments and analysis software (Applied Biosystems, Carlsbad, CA). PCR amplification of BRAF was performed using the following primer sequences (208 bp product spanning nt 118 to 325 of exon 15): 5′-TCATAATGCTTGCTCTGATAGGA-3′ (forward) and 5′-AGTGGAAAAATAGCCTCAA-3′ (reverse). The following reaction settings were used: 1 cycle at 95°C for 15 min; 2 cycles at 94°C for 45 s, 70°C for 1.5 min, and 72°C for 1.5 min; 2 cycles at 94°C for 45 s, 68°C for 1.5 min, and 72°C for 1.5 min; 2 cycles at 94°C for 45 s, 66°C for 1.5 min, and 72°C for 1.5 min; 2 cycles at 94°C for 45 s, 64°C for 1.5 min, and 72°C for 1.5 min; 25 cycles at 94°C for 45 s, 62°C for 1.5 min, and 72°C for 1.5 min; and 1 cycle at 72°C for 10 min. PCR amplification of NRAS was performed using the following primer sequences (150 bp product spanning nt 170 to 320 of exon 3): 5′-CACACCCCCAGGATTCTTAC-3′ (forward) and 5′-TGGCAAATACACAGAGGAAGC-3′ (reverse). The following reaction settings were used: 1 cycle at 95°C for 15 min; 38 cycles at 94°C for 45 s, 57°C for 1.5 min, and 72°C for 1.5 min; and 1 cycle at 72°C for 10 min.
Results
Current classification systems for melanoma are morphology based, with the main prognostic factors being Breslow tumor thickness, tumor ulceration, and metastatic involvement of the regional lymph nodes.27 Previous work28 has identified various cell signaling networks in melanoma tumorigenesis, and several genetic alterations have been identified in melanoma. Nevertheless, the extent to which these studies fully detail the complexity of the disease process has been limited. In contrast, other work18,19 has clearly shown the power of gene expression profiling to both dissect the complexity and reveal functional information regarding the consequences of the various genetic alterations. In addition, these studies have shown the potential to link these expression profiles with therapeutic opportunities. As such, we have sought to make use of available melanoma expression data to provide a characterization of the disease.
Identification of Distinct Subgroups of Primary and Metastatic Melanomas
Given previous work in the study of other cancers that suggest substantial heterogeneity of disease, an ability to examine many samples is critical to develop an understanding of the full range of the heterogeneity. Thus, we initially characterized gene expression data from benign nevi and primary and metastatic melanomas from six independent studies publicly available through GEO. A total of 303 samples were available from six independent data sets (Table 2).
Table 2.
Data Sets Used for Expression Data Analysis
| GEO data set | Benign nevi samples | Primary melanoma samples | Metastatic melanoma samples |
|---|---|---|---|
| GSE3189 | 18 | 45 | 0 |
| GSE4570 | 0 | 0 | 6 |
| GSE4587 | 2 | 4 | 4 |
| GSE4845 | 0 | 4 | 83 |
| GSE7553 | 0 | 14 | 40 |
| GSE8401 | 0 | 31 | 52 |
Data are given as the number of each sample in each data set. Six independent data sets (publicly available through GEO) were used; melanoma gene expression data were obtained from these data sets for the current study.
A major challenge in making use of these combined data is the inherent laboratory-specific batch effects that confound DNA microarray data, a characteristic clearly evident in the principal component analysis of the overall gene expression characteristics of these data sets (see Supplemental Figure S1A at http://ajp.amjpathol.org). Clearly, there are substantial batch differences in these data sets that would confound any attempt to identify meaningful biological variation. To address this challenge and normalize the data from the independent studies, we log transformed the MAS5.0 data and used BFRM to normalize the data against 69 Affymetrix probes for human maintenance genes using 15 principal components.22,23 A principal component analysis after the adjustment shows that the variability present between the initial data sets has been eliminated (see Supplemental Figure S1B at http://ajp.amjpathol.org).
As a starting point in the analysis of patterns of gene expression that characterize melanoma, we performed an unsupervised hierarchical clustering analysis of the gene expression data encompassing the 303 samples. As shown in Figure 1, a complex series of patterns of gene expression was evident and coincided with the three distinct forms of disease state in this collection of samples: benign nevi, primary melanoma, and metastatic melanoma.
Figure 1.
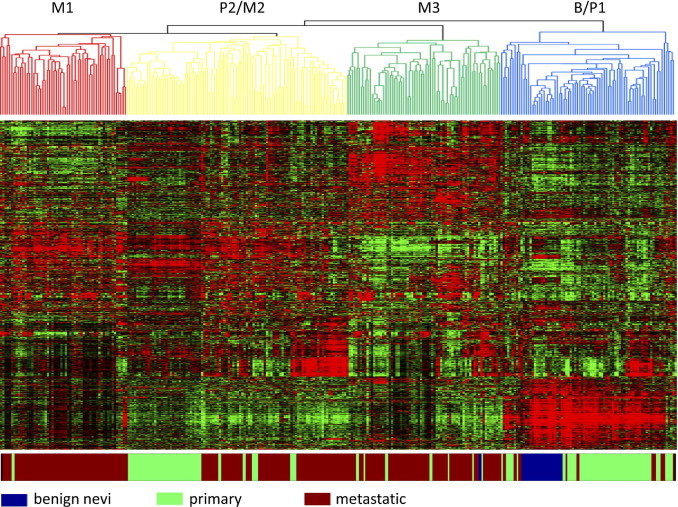
Unsupervised hierarchical clustering analysis of melanoma expression data. MAS5.0 gene expression data were log transformed and normalized, using BFRM. Expression data were filtered in Cluster (see Materials and Methods) to 1000 genes that are represented in the data matrix. Within the data matrix, columns represent samples; rows, genes; red, higher expression values; and green, lower expression values. The color bar below the data matrix defines the sample in each column. Subgroups of samples exhibiting similar gene expression patterns, as defined by the nodes of the array tree, are color coded within the array tree.
Based on patterns of gene expression, the primary melanoma samples could be subdivided into two distinct groups (P1 and P2 in Figure 1). The metastatic samples were distinct and clustered into three distinct groups based on patterns of gene expression (M1, M2, and M3). The benign samples clustered together with the P1 subgroup of primary tumor samples. The other primary tumor subgroup (P2) was distinct and more similar to the M2 metastatic cluster (P2/M2).
Taken together, this analysis emphasizes the heterogeneity of melanoma, both primary and metastatic, and suggests that one class of the primary tumors has characteristics in common with metastatic disease, suggesting the possibility that these tumors might represent a more advanced stage of the primary disease.
Biological Distinctions in Primary and Metastatic Melanoma Subgroups
To gain insight into the biological processes represented in the gene expression patterns associated with the primary and metastatic melanoma subsets, we used two approaches. First, we used GSEA to identify sets of genes showing a statistically significant difference between subsets.24,25 To facilitate the interpretation of the biological processes associated with the identified sets of genes, we used an annotation tool called GATHER.26 An analysis of the genes in the P2 subset of primary melanomas compared with the P1 subset reveals enrichment for cell proliferation ontology terms (see Supplemental Table S1, A and B, at http://ajp.amjpathol.org), perhaps consistent with the observation that these tumors may represent a more advanced or aggressive form with characteristics similar to metastatic disease.
As a second approach to characterize the distinctions in the melanoma subgroups, we used expression signatures developed to measure the activity of various signaling pathways. These signatures were previously used to measure the activity of the signaling pathways in various tumor samples.18,19 At the same time, these pathway signatures have predicted sensitivity to various cancer drugs that target components of the relevant pathway and, thus, provide the further benefit of potentially identifying therapeutic options for subgroups of patients. We predicted the activity of these pathways in the collection of melanoma samples, leading to the generation of probability measures that has previously reflected the state of pathway activity, as measured by various biochemical assays. These probability scores are then used in a manner similar to gene expression values to provide a picture of patterns of pathway activity in the samples (Figure 2).
Figure 2.
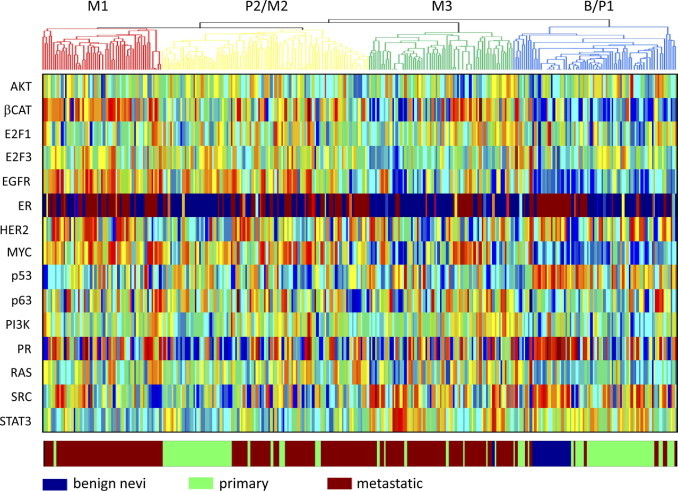
The status of oncogenic signaling pathway activities analyzed in the benign nevi, primary melanoma, and metastatic melanoma samples. Within the heat map, columns represent samples; and rows, the oncogenic signaling pathway analyzed. Pathway activities on a low to high continuum are represented by a blue to red continuum, respectively. The color bar below the heatmap defines the sample in each column. The samples are ordered according to the defined subgroups, as shown in Figure 1 (the array tree above the heat map defines the subgroups of melanoma samples exhibiting similar gene expression patterns). CAT indicates catenin; EGFR, epidermal growth factor receptor; ER, estrogen receptor; HER, 2 human epidermal growth factor receptor 2; PI3k, phosphatidylinositol 3-kinase; PR, progesterone receptor.
It is evident from this analysis that the various subgroups of primary and metastatic melanoma exhibit distinct patterns of pathway activity. In particular, the analysis of the distinctions in the P1 and P2 subgroups of primary melanoma reveals an increase in MYC pathway activity in the P2 samples versus the P1 samples (Figure 3A). In light of the enrichment for cell proliferation gene sets in the P2 subgroup, as previously described, this result suggests that a primary distinction in these two forms of primary tumor is proliferative capacity, with the subgroup exhibiting an increase in this phenotype also being similar to one of the metastatic subgroups.29,30 The P1 subgroup exhibits an increase in p53 pathway activity and a decreased proliferative phenotype; other work31 has shown that wild-type levels of p53 have been associated with melanomas, exhibiting improved prognoses.
Figure 3.
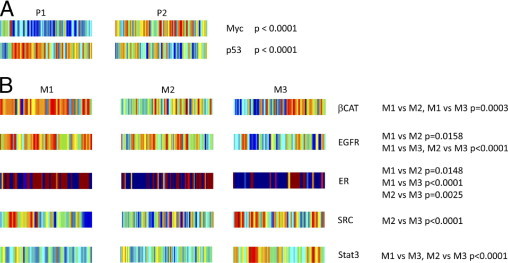
Statistically significant differences in the distribution of activities of particular oncogenic signaling pathways identified between subgroups of melanoma samples. A: Oncogenic signaling pathways, as indicated, determined to exhibit statistically significantly distinct levels of activity between the two primary melanoma subgroups (P1 and P2). B: Oncogenic signaling pathways, as indicated, determined to exhibit statistically significantly distinct levels of activity among the three metastatic melanoma subgroups (M1, M2, and M3). Relevant sections of the heat map from Figure 2 are reproduced. The differences in the distribution of activities of particular oncogenic signaling pathways within samples composing one subgroup of melanomas compared with another subgroup of melanomas, as indicated, were quantified with a Mann-Whitney U-test. CAT indicates catenin; EGFR, epidermal growth factor receptor; ER, estrogen receptor; STAT3, signal transducer and activator of transcription.
Distinct distributions of the activity of certain oncogenic signaling pathways can also be seen among the three subgroups of metastatic melanoma (Figure 3B). The M1 subset exhibits higher activity of the β-catenin pathway. Consistent with these results, nuclear accumulation of β-catenin has been shown in melanoma.32 Two other pathways exhibiting subset-specific increases in activity are the epidermal growth factor receptor and estrogen receptor pathways, with the M1 and M2 subsets exhibiting higher activity of these two pathways. Consistent with these results, epidermal growth factor receptor expression has been shown in metastatic melanoma.33 Interestingly, one study34 has shown clinical activity of tamoxifen in metastatic melanoma, with dacarbazine plus tamoxifen treatment resulting in improved response rates and survival compared with dacarbazine treatment alone among women. The M1 subset also exhibits elevated levels of the SRC pathway. Consistent with these results, c-Src expression has been shown in malignant melanoma.35 The M3 subset exhibits elevated levels of the SRC and STAT3 pathways. Interestingly, dasatinib has inhibited migration and invasiveness of melanoma cells in vitro and reduced metastases of melanoma in vivo.36,37 STAT3 has also played a role in the metastatic potential of melanoma, with the presence of constitutively activated STAT3 being able to change poorly metastatic melanoma cells into highly metastatic tumor cells.38 Thus, perhaps the M3 subset of metastatic melanomas, which exhibit elevated levels of activity of SRC and STAT3, is a more highly aggressive subset. There are three distinct forms of metastatic melanomas revealed by this analysis.
Classification and Biological Features of In-Transit Melanoma Metastases
Approximately 20% of patients experiencing a recurrence of melanoma will present with in-transit metastases, multiple lesions in the epidermal, dermal, or subcutaneous layers of tissue of an extremity between the primary melanoma site and the regional lymph nodes. Many of these in-transit melanoma metastases will ultimately metastasize to distant sites. Although in-transit melanoma is referred to as a type of metastatic melanoma, the relationship between the gene expression patterns of in-transit melanoma and other melanoma states is not known.
By using the framework of gene expression patterns established from the unsupervised hierarchical clustering analysis of benign nevi, primary melanoma, and metastatic melanoma, we investigated the similarities and differences in gene expression patterns of in-transit melanoma samples in relation to the other tumors. To eliminate any inherent laboratory-specific batch effects between the data sets composed of benign nevi, primary melanoma, and metastatic melanoma and those composed of in-transit melanoma metastases, we log transformed the MAS5.0 data and used BFRM to normalize the data against 69 Affymetrix probes for human maintenance genes using 15 principal components22,23 (see Supplemental Figure S2 at http://ajp.amjpathol.org). Unsupervised hierarchical clustering of the gene expression data from the in-transit melanoma metastases revealed that in-transit melanoma metastases from a given patient clustered together and exhibited similar patterns of gene expression (data not shown).39 As shown in Figure 4, the in-transit melanoma metastases could be subdivided into two distinct groups based on patterns of gene expression (IT1 and IT2 in Figure 4). One of the subgroups of in-transit melanomas (IT1) was similar to one of the classes of primary tumor and the benign nevi samples (B/P1 in Figure 1), whereas the other subgroup (IT2) was distinct and more similar to one of the metastatic clusters (M3 in Figure 1).
Figure 4.
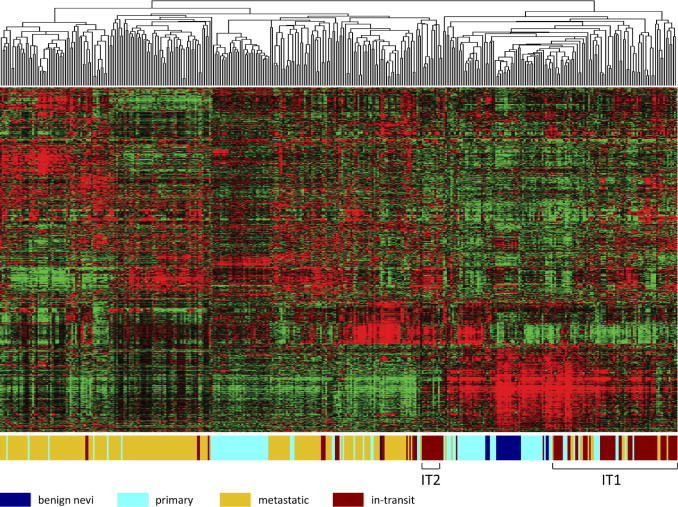
Unsupervised hierarchical clustering analysis of in-transit melanoma expression data. MAS5.0 gene expression data were log transformed and normalized, using BFRM. Expression data were filtered in Cluster (see Materials and Methods) to 1000 genes that are represented in the data matrix. Within the data matrix, columns represent samples; rows, genes; red, higher expression values; and green, lower expression values. The color bar below the data matrix defines the sample in each column.
To gain insight into the biological processes represented in the gene expression patterns associated with the in-transit melanoma subgroups, we again took two approaches. First, an analysis of the gene expression pattern characteristic of the IT1 subset of in-transit melanoma reveals an enrichment for ontology terms associated with development, whereas positively expressed genes of the IT2 subset of in-transit melanoma reveal an enrichment for immune response ontology terms, further emphasizing the distinct biological characteristics underlying the two subsets of in-transit melanoma (see Supplemental Table S2, A and B, at http://ajp.amjpathol.org). Second, we used the gene expression signatures developed to measure the activity of various oncogenic signaling pathways to further characterize the in-transit melanoma subgroups. Consistent with a high degree of genetic heterogeneity and complexity within in-transit melanoma, no single oncogenic signaling pathway is deregulated in all in-transit melanoma metastases; rather, several oncogenic signaling pathways are deregulated in individual in-transit melanoma metastases (see Supplemental Figure S3 at http://ajp.amjpathol.org). Although many oncogenic signaling pathways exhibit deregulation in individual in-transit melanoma metastases, deregulation of particular oncogenic signaling pathways is associated with in-transit melanoma metastases of given BRAF or NRAS mutation status (see Supplemental Figure S4 at http://ajp.amjpathol.org). In particular, deregulation of the E2F3 and p53 pathways is more commonly associated with in-transit melanoma of mutant BRAF status; deregulation of the Her2, p53, and progesterone receptor pathways is more commonly associated with in-transit melanoma of mutant NRAS status; and deregulation of the E2F1 and ER pathways is more commonly associated with in-transit melanoma of wild-type BRAF and NRAS status.
Discussion
The advances in genomic technologies have led to the recognition that human cancers, including melanoma, exhibit enormous complexity of somatic gene alterations that produce a substantial heterogeneity of disease. This realization has profound implications for the treatment of patients with cancer because it is clear that distinct mechanisms and, thus, disease biological features are associated with this complexity and heterogeneity. As such, an ability to dissect and understand the distinct forms of various cancers is a key in the development of strategies for personalized cancer treatment.
Dissecting and understanding the distinct forms of various cancers from data generated from biochemical assays or genomewide expression analyses are challenging. Biochemical assays for the mutation status of individual genes give limited insight into the underlying biological features of tumors. Although genomewide expression analyses yield gene expression data for thousands of genes, interpreting the underlying biological characteristics of tumors from such information is difficult. From gene expression profiling, it is difficult to interpret the impact that functions associated with certain genes relevant to a profile have within the context of the whole genome. In addition, it is difficult to assess the biological activity of relevant functions across individual tumors composing a data set.
In light of these challenges, there are several advantages to classifying the distinct forms of various cancers based on alterations in signaling pathways that have contributed to oncogenesis. This basis for classification can do the following: i) separate tumors into more homogeneous groups with common underlying biological features, ii) identify coderegulation of multiple oncogenic pathways that have combined to contribute to transformation, and iii) provide a potential strategy to couple therapeutic options with distinct subgroups of disease. Previous work18,19 has demonstrated that, in many instances, the prediction of pathway activation using an expression signature can, at the same time, predict sensitivity to therapeutic agents that target a component of that pathway.
The work we describe herein identifies at least two distinct forms of primary melanoma, two distinct forms of in-transit melanoma, and three distinct forms of metastatic melanoma on the basis of patterns of gene expression and pathway activity, providing a starting point toward dissecting the heterogeneity of the disease. Moreover, we suggest that the characterization of distinct forms of melanoma on the basis of patterns of pathway activity provides a potential approach to further understand the biological features underlying individual melanomas and to develop new therapeutic options for subgroups of patients. The approach of matching targeted therapies in rational ways with characteristics of patient melanomas has shown promise because most patients with metastatic melanoma with a BRAF V600E mutation exhibited a complete or partial response to treatment with PLX4032, an inhibitor of mutant BRAF kinase.40,41 In addition to pathway activity, several immune response genes have been identified as being differentially expressed in tumor samples.42,43 This observation has led our group to initiate a study to immunologically characterize patient in-transit tumors and lymph nodes using polychromatic flow cytometry to evaluate immune response, microarray-based analysis of gene expression, and histological measurements to determine the role of the immune system in defining a tumor's biological features and predicting response to therapy. Furthermore, a therapeutic phase 1 regional isolated limb infusion trial using melphalan as the chemotherapeutic agent, followed by systemic therapy with ipilimumab (an anti–cytotoxic T-lymphocyte antigen 4 antibody), has been initiated at Memorial Sloan-Kettering Cancer Center to examine how manipulation of the immune system can affect regional tumor response and augment the potential systemic immune response generated by a regional cytotoxic chemotherapeutic agent. Although our results cannot identify which subgroups of melanomas would be most likely to be effectively treated by ipilimumab, an analysis of the gene expression pattern characteristic of the IT2 subset of in-transit melanoma reveals an enrichment for ontology terms associated with immune response. In future work, it will be informative to stratify melanomas based on patterns of oncogenic signaling pathway deregulation using prospectively collected samples in a clinical trial to ensure that results are obtained from samples containing equivalent tumor content. In addition, samples from a clinical trial would allow examination of clinical outcome data, including patient survival, recurrence, and response to therapy, within the context of the subgroups of melanoma, thus enabling better classification. This approach to classifying melanoma has the potential to yield a new diagnostic tool that could be used to optimize treatment selections for subgroups of patients with melanoma.
Although surgery is usually effective in treating earlier-stage melanomas, patients with later-stage melanomas have poor prognoses because the current therapeutic options exhibit limited efficacy.44 Single-agent chemotherapies, most commonly dacarbazine, have clinical activity in melanoma, but response rates for patients treated with such agents are low (ie, <25%). Response rates for patients treated with dacarbazine along with another class of chemotherapy agent are higher (ie, 14% to 37%); however, in randomized clinical trials, such combination chemotherapy regimens have not demonstrated superior results when compared with single-agent chemotherapy regimens. Three-drug regimens (ie, cisplatin, vinblastine, and dacarbazine) and four-drug regimens (ie, cisplatin, dacarbazine, carmustine, and tamoxifen) exhibit 30% to 50% response rates. However, it is unclear whether combination chemotherapy regimens are superior treatment regimens compared with single-agent chemotherapy treatment regimens because these combination chemotherapy regimens are associated with increased toxicity in patients.
Clearly, new approaches are needed to improve response rates and overall survival rates for patients with later-stage melanomas. A focus on targeted therapies that carry reduced toxicity is appealing but only if they can be matched in a rational way with the characteristics of the patient's tumor. The approach we describe herein, using biological characterizations that distinguish different forms of melanoma, is one method toward that goal.
Acknowledgments
We thank members of the Nevins laboratory for valuable input throughout this study and for comments on the manuscript and Kaye Culler for assistance with preparation of the manuscript.
Footnotes
Supported by awards from the National Cancer Institute (RO1CA104663, RO1CA106520, and U54CA112952 to J.R.N.).
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Disclosures: D.S.T. has received commercial research grants of greater than $10,000 from Scherring-Plough and Adherex and honorarium from the speaker's bureau at Novartis and is a member of the Scientific Advisory Board at Genetech; J.R.N. has ownership interest of greater than $10,000 in Expression Analysis and is a member of the Scientific Advisory Board at Millenium Pharmaceuticals and Qiagen.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.02.037.
Supplementary data
Principal component (Prin.Comp.) analysis of benign nevi, primary melanoma, and metastatic melanoma samples. A: MAS5.0 gene expression data from the six GEO datasets. B: MAS5.0 gene expression data from the six GEO datasets, log transformed and normalized, using BFRM. Black filled squares represent GSE3189 samples; red filled circles, GSE4570 samples; green filled triangles, GSE4587 samples; blue filled diamonds, GSE4845 samples processed on Affymetrix Human Genome U133A 2.0 arrays; light blue filled circles, GSE4845 samples processed on Affymetrix Human Genome U133 Plus 2.0 arrays; pink filled small circles, GSE7553 samples; and yellow open circles, GSE8401 samples.
Principal component (Prin.Comp.) analysis of in-transit melanoma samples. A: MAS5.0 gene expression data from the eight GEO datasets. B: MAS5.0 gene expression data from the eight GEO datasets, log transformed and normalized, using BFRM. Black filled squares represent GSE3189 samples; red filled circles, GSE4570 samples; green filled triangles, GSE4587 samples; blue filled diamonds, GSE4845 samples processed on Affymetrix Human Genome U133A 2.0 arrays; light blue filled circles, GSE4845 samples processed on Affymetrix Human Genome U133 Plus 2.0 arrays; pink filled small circles, GSE7553 samples; yellow open circles, GSE8401 samples; and gray open squares, GSE10282 and GSE19293 samples.
The status of oncogenic signaling pathway activities analyzed in the in-transit melanoma samples. The percentage of in-transit melanoma samples exhibiting a predicted probability >0.5 of deregulated signaling of the oncogenic signaling pathway, as indicated.
The status of oncogenic signaling pathway activities analyzed in BRAF-mutant, NRAS-mutant, and BRAF/NRAS wild-type in-transit melanoma samples. The percentage of BRAF-mutant (A), NRAS-mutant (B), and BRAF/NRAS wild-type in-transit (C) melanoma samples exhibiting a predicted probability >0.5 of deregulated signaling of the oncogenic signaling pathway, as indicated.
References
- 1.Sjoblom T., Jones S., Wood L.D., Parsons D.W., Lin J., Barber T.D., Mandelker D., Leary R.J., Ptak J., Silliman N., Szabo S., Buckhaults P., Farrell C., Meeh P., Markowitz S.D., Willis J., Dawson D., Willson J.K., Gazdar A.F., Hartigan J., Wu L., Liu C.C., Parmigiani G., Park B.H., Bachman K.E., Papadopoulos N., Vogelstein B., Kinzler K.W., Velculescu V.E. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 2.Wood L.D., Parsons D.W., Jones S., Lin J., Sjoblom T., Leary R.J., Shen D., Boca S.M., Barber T., Ptak J., Silliman N., Szabo S., Dezso Z., Ustyanksky V., Nikolskaya T., Nikolsky Y., Karchin R., Wilson P.A., Kaminker J.S., Zhang Z., Croshaw R., Willis J., Dawson D., Shipitsin M., Willson J.K., Sukumar S., Polyak K., Park B.H., Pethiyagoda C.L., Pant P.V., Ballinger D.G., Sparks A.B., Hartigan J., Smith D.R., Suh E., Papadopoulos N., Buckhaults P., Markowitz S.D., Parmigiani G., Kinzler K.W., Velculescu V.E., Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 3.Andrechek E.R., Cardiff R.D., Chang J.T., Gatza M.L., Acharya C.R., Potti A., Nevins J.R. Genetic heterogeneity of Myc-induced mammary tumors reflecting diverse phenotypes including metastatic potential. Proc Natl Acad Sci U S A. 2009;106:16387–16392. doi: 10.1073/pnas.0901250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hudis C.A. Trastuzumab: mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- 5.Mullighan C.G., Goorha S., Radtke I., Miller C.B., Coustan-Smith E., Dalton J.D., Girtman K., Mathew S., Ma J., Pounds S.B., Su X., Pui C.H., Relling M.V., Evans W.E., Shurtleff S.A., Downing J.R. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 6.Weir B.A., Woo M.S., Getz G., Perner S., Ding L., Beroukhim R., Lin W.M., Province M.A., Kraja A., Johnson L.A., Shah K., Sato M., Thomas R.K., Barletta J.A., Borecki I.B., Broderick S., Chang A.C., Chiang D.Y., Chirieac L.R., Cho J., Fujii Y., Gazdar A.F., Giordano T., Greulich H., Hanna M., Johnson B.E., Kris M.G., Lash A., Lin L., Lindeman N., Mardis E.R., McPherson J.D., Minna J.D., Morgan M.B., Nadel M., Orringer M.B., Osborne J.R., Ozenberger B., Ramos A.H., Robinson J., Roth J.A., Rusch V., Sasaki H., Shepherd F., Sougnez C., Spitz M.R., Tsao M.S., Twomey D., Verhaak R.G., Weinstock G.M., Wheeler D.A., Winckler W., Yoshizawa A., Yu S., Zakowski M.F., Zhang Q., Beer D.G., Wistuba I.I., Watson M.A., Garraway L.A., Ladanyi M., Travis W.D., Pao W., Rubin M.A., Gabriel S.B., Gibbs R.A., Varmus H.E., Wilson R.K., Lander E.S., Meyerson M. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding L., Getz G., Wheeler D.A., Mardis E.R., McLellan M., Cibulskis K., Sougnez C., Wilson R.K. Somatic mutations effect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin W.M., Baker A.C., Beroukhim R., Winckler W., Feng W., Marmion J.M., Laine E., Greulich H., Tseng H., Gates C., Hodi F.S., Dranoff G., Sellers W.R., Thomas R.K., Meyerson M., Golub T.R., Dummer R., Herlyn M., Getz G., Garraway L.A. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res. 2008;68:664–673. doi: 10.1158/0008-5472.CAN-07-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curtin J.A., Fridlyand J., Kageshita T., Patel H.N., Busam K.J., Kutzner H., Cho K.H., Aiba S., Brocker E.B., LeBoit P.E., Pinkel D., Bastian B.C. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 10.Bastian B.C., Olshen A.B., LeBoit P.E., Pinkel D. Classifying melanocytic tumors based on DNA copy number changes. Am J Pathol. 2003;163:1765–1770. doi: 10.1016/S0002-9440(10)63536-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maldonado J.L., Fridlyand J., Patel H.N., Jain A.N., Busam K.J., Kageshita T., Ono T., Albertson D.G., Pinkel D., Bastian B.C. Determinants of BRAF mutations in primary melanomas. J Natl Canc Inst. 2003;95:1878–1890. doi: 10.1093/jnci/djg123. [DOI] [PubMed] [Google Scholar]
- 12.Scatolini M., Grand M.M., Grosso E., Venesio T., Pisacane A., Balsamo A., Sirovich R., Risio M., Chiorino G. Altered molecular pathways in melanocytic lesions. Int J Cancer. 2010;126:1869–1881. doi: 10.1002/ijc.24899. [DOI] [PubMed] [Google Scholar]
- 13.Haqq C., Nosrati M., Sudilovsky D., Crothers J., Khodabakhsh D., Pulliam B.L., Federman S., Miller J.R., 3rd, Allen R.E., Singer M.I., Leong S.P., Ljung B.M., Sagebiel R.W., Kashani-Sabet M. The gene expression signatures of melanoma progression. Proc Natl Acad Sci U S A. 2005;102:6092–6097. doi: 10.1073/pnas.0501564102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith A.P., Hoek K., Becker D. Whole-genome expression profiling of the melanoma progression pathway reveals marked molecular differences between nevi/melanoma in situ and advanced-stage melanomas. Cancer Biol Ther. 2005;4:1018–1029. doi: 10.4161/cbt.4.9.2165. [DOI] [PubMed] [Google Scholar]
- 15.Jaeger J., Koczan D., Thiesen H.J., Ibrahim S.M., Gross G., Spang R., Kunz M. Gene expression signatures for tumor progression, tumor subtype, and tumor thickness in laser-microdissected melanoma tissues. Clin Cancer Res. 2007;13:806–815. doi: 10.1158/1078-0432.CCR-06-1820. [DOI] [PubMed] [Google Scholar]
- 16.Lewis T.B., Robison J.E., Bastien R., Milash B., Coucher K., Samlowski W.E., Leachman S.A., Dirk Noyes R., Wittwer C.T., Perreard L., Bernard P.S. Molecular classification of melanoma using real-time quantitative reverse transcriptase-polymerase chain reaction. Cancer. 2005;104:1678–1686. doi: 10.1002/cncr.21372. [DOI] [PubMed] [Google Scholar]
- 17.Bittner M., Meltzer P., Chen Y., Jiang Y., Seftor E., Hendrix M., Radmacher M., Simon R., Yakhini Z., Ben-Dor A., Sampas N., Dougherty E., Wang E., Marincola F., Gooden C., Lueders J., Glatfelter A., Pollock P., Carpten J., GiDietrich K., Beaudry C., Berens M., Alberts D., Sondak V., Hayward N., Trent J. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- 18.Bild A., Yao G., Chang J.T., Wang Q., Potti A., Chasse D., Joshi M.-B., Harpole D., Lancaster J.M., Berchuck A., Olson J.A., Marks J.R., Dressman H.K., West M., Nevins J.R. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 19.Gatza M.L., Lucas J.E., Barry W.T., Kim J.W., Wang Q., Crawford M.D., Datto M.B., Kelley M., Mathey-Prevot B., Potti A., Nevins J.R. A pathway-based classification of human breast cancer. Proc Natl Acad Sci U S A. 2010;107:6994–6999. doi: 10.1073/pnas.0912708107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loboda A., Nebozhyn M., Klinghoffer R., Frazier J., Chastain M., Arthur W., Roberts B., Zhang T., Chenard M., Haines B., Andersen J., Nagashima K., Paweletz C., Lynch B., Feldman I., Dai H., Huang P., Watters J. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genomics. 2010;3:26. doi: 10.1186/1755-8794-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greene F.L., Page D.L., Fleming I.D., Fritz A.G., Balch C.M., Haller D.G., Morrow M. Springer; New York: 2002. Melanoma of the Skin; pp. 209–217. [Google Scholar]
- 22.Carvalho C., Chang J., Lucas J., Nevins J.R., Wang Q., West M. High-dimensional sparse factor modelling: applications in gene expression genomics. J Am Stat Assoc. 2008;103:1438–1456. doi: 10.1198/016214508000000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucas J., Carvalho C., West M. A bayesian analysis strategy for cross-study translation of gene expression biomarkers. Stat Appl Genet Mol Biol. 2009;8:8. doi: 10.2202/1544-6115.1436. Article 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Houstis N., Daly M.J., Patterson N., Mesirov J.P., Golub T.R., Tamyo P., Spiegelman B.M., Lander E.S., Hirschhorn J.N., Altshuler D., Groop L.C. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 26.Chang J.T., Nevins J.R. GATHER: a systems approach to interpreting genomic signatures. Bioinformatics. 2006;22:2926–2933. doi: 10.1093/bioinformatics/btl483. [DOI] [PubMed] [Google Scholar]
- 27.Palmieri G., Casula M., Sini M.C., Ascierto P.A., Cossu A. Issues affecting molecular staging in the management of patients with melanoma. J Cell Mol Med. 2007;11:1052–1068. doi: 10.1111/j.1582-4934.2007.00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hocker T.L., Singh M.K., Tsao H. Melanoma genetics and therapeutic approaches in the 21st century: moving from the benchside to the bedside. J Invest Dermatol. 2008;128:2575–2595. doi: 10.1038/jid.2008.226. [DOI] [PubMed] [Google Scholar]
- 29.Adhikary S., Eilers M. Transcription regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 30.Dang C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Box N.F., Terzian T. The role of p53 in pigmentation, tanning and melanoma. Pigment Cell Melanoma Res. 2008;21:525–533. doi: 10.1111/j.1755-148X.2008.00495.x. [DOI] [PubMed] [Google Scholar]
- 32.Rimm D.L., Caca K., Hu G., Harrison F.B., Fearon E.R. Frequent nuclear/cytoplasmic localization of beta-catenin without exon 3 mutations in malignant melanoma. Am J Pathol. 1999;154:325–329. doi: 10.1016/s0002-9440(10)65278-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kraehn G.M., Schartl M., Peter R.U. Human malignant melanoma: a genetic disease. Cancer. 1995;75:1228–1237. doi: 10.1002/1097-0142(19950315)75:6<1228::aid-cncr2820750604>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 34.Cocconi G., Bella M., Calabresi F., Tonato M., Canaletti R., Boni C., Buzzi F., Ceci G., Corgna E., Costa P., Lottici R., Papadia F., Sofra M.C., Bacchi M. Treatment of metastatic malignant melanoma with dacarbazine plus tamoxifen. N Engl J Med. 1992;327:516–523. doi: 10.1056/NEJM199208203270803. [DOI] [PubMed] [Google Scholar]
- 35.Lee J.K., Coutant C., Kim Y.-C., Qi Y., Theodorescu D., Symmans W.F., Baggerly K.A., Rouzier R., Pusztai L. Prospective comparison of clinical and genomic multivariate predictors of response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res. 2010;16:711–718. doi: 10.1158/1078-0432.CCR-09-2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buettner R., Mesa T., Vultur A., Lee F., Jove R. Inhibition of Src family kinases with dasatinib blocks migration and invasion of human melanoma cells. Mol Cancer Res. 2008;6:1766–1774. doi: 10.1158/1541-7786.MCR-08-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fraser C.K., Lousberg E.L., Guerin L.R., Hunges T.P., Brown M.P., Diener K.R., Hayball J.D. Dasatinib alters the metastatic phenotype of B16-OVA melanoma in vivo. Cancer Biol Ther. 2010;10:715–727. doi: 10.4161/cbt.10.7.12926. [DOI] [PubMed] [Google Scholar]
- 38.Xie T.X., Wei D., Liu M., Gao A.C., Ali-Osman F., Sawaya R., Huang S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23:3550–3560. doi: 10.1038/sj.onc.1207383. [DOI] [PubMed] [Google Scholar]
- 39.Augustine C.K., Jung S.H., Sohn I., Yoo J.S., Yoshimoto Y., Olson J.A., Jr, Friedman H.S., Ali-Osman F., Tyler D.S. Gene expression signatures as a guide to treatment strategies for in-transit metastatic melanoma. Mol Cancer Ther. 2010;9:779–790. doi: 10.1158/1535-7163.MCT-09-0764. [DOI] [PubMed] [Google Scholar]
- 40.Bollag G., Hirth P., Tsai J., Zhang J., Ibrahim P.N., Cho H., Spevak W., Zhang C., Zhang Y., Habets G., Burton E.A., Wong B., Tsang G., West B.L., Powell B., Shellooe R., Marimuthu A., Nguyen H., Zhang K.Y., Artis D.R., Schlessinger J., Su F., Higgins B., Iyer R., D'Andrea K., Koehler A., Stumm M., Lin P.S., Lee R.J., Grippo J., Puzanov I., Kim K.B., Ribas A., McArthur G.A., Sosman J.A., Chapman P.B., Flaherty K.T., Xu X., Nathanson K.L., Nolop K. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flaherty K.T., Puzanov I., Kim K.B., Ribas A., McArthur G.A., Sosman J.A., O'Dwyer P.J., Lee R.J., Grippo J.F., Nolop K., Chapman P.B. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarhini A.A., Kirkwood J.M. Clinical and immunologic basis of interferon therapy in melanoma. Ann N Y Acad Sci. 2009;1182:47–57. doi: 10.1111/j.1749-6632.2009.05073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonsson G., Busch C.M.C., Knappskog S., Geisler J., Miletic H., Ringner M., Lillehaug J.R., Borg A., Lonning P.E. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin Cancer Res. 2010;16:3356–3367. doi: 10.1158/1078-0432.CCR-09-2509. [DOI] [PubMed] [Google Scholar]
- 44.Jilaveanu L.B., Aziz S.A., Kluger H.M. Chemotherapy and biologic therapies for melanoma: do they work. Clin Dermatol. 2009;27:614–625. doi: 10.1016/j.clindermatol.2008.09.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Principal component (Prin.Comp.) analysis of benign nevi, primary melanoma, and metastatic melanoma samples. A: MAS5.0 gene expression data from the six GEO datasets. B: MAS5.0 gene expression data from the six GEO datasets, log transformed and normalized, using BFRM. Black filled squares represent GSE3189 samples; red filled circles, GSE4570 samples; green filled triangles, GSE4587 samples; blue filled diamonds, GSE4845 samples processed on Affymetrix Human Genome U133A 2.0 arrays; light blue filled circles, GSE4845 samples processed on Affymetrix Human Genome U133 Plus 2.0 arrays; pink filled small circles, GSE7553 samples; and yellow open circles, GSE8401 samples.
Principal component (Prin.Comp.) analysis of in-transit melanoma samples. A: MAS5.0 gene expression data from the eight GEO datasets. B: MAS5.0 gene expression data from the eight GEO datasets, log transformed and normalized, using BFRM. Black filled squares represent GSE3189 samples; red filled circles, GSE4570 samples; green filled triangles, GSE4587 samples; blue filled diamonds, GSE4845 samples processed on Affymetrix Human Genome U133A 2.0 arrays; light blue filled circles, GSE4845 samples processed on Affymetrix Human Genome U133 Plus 2.0 arrays; pink filled small circles, GSE7553 samples; yellow open circles, GSE8401 samples; and gray open squares, GSE10282 and GSE19293 samples.
The status of oncogenic signaling pathway activities analyzed in the in-transit melanoma samples. The percentage of in-transit melanoma samples exhibiting a predicted probability >0.5 of deregulated signaling of the oncogenic signaling pathway, as indicated.
The status of oncogenic signaling pathway activities analyzed in BRAF-mutant, NRAS-mutant, and BRAF/NRAS wild-type in-transit melanoma samples. The percentage of BRAF-mutant (A), NRAS-mutant (B), and BRAF/NRAS wild-type in-transit (C) melanoma samples exhibiting a predicted probability >0.5 of deregulated signaling of the oncogenic signaling pathway, as indicated.