

Abstract
In 2000, approximately 10 million women were receiving hormone replacement therapy (HRT) for alleviation of menopausal symptoms. A number of prior animal studies suggested that HRT may be neuroprotective and cardioprotective. Then, in 2003, reports from the Women's Health Initiative (WHI) indicated that long-term estrogen/progestin supplementation led to increased incidence of stroke. A second branch of the WHI in women with prior hysterectomy found an even stronger correlation between estrogen supplementation alone and stroke incidence. Follow-up analyses of the data, as well as data from other smaller clinical trials, have also demonstrated increased stroke severity in women receiving HRT or estrogen alone. This review examines the studies indicating that estrogen is neuroprotectant in animal models and explores potential reasons why this may not be true in postmenopausal women. Specifically, age-related differences in estrogen receptors and estrogenic actions in the brain are discussed, with the conclusion that animal models of disease must closely mimic human disease to produce clinically relevant results.
Age is the single greatest risk factor for stroke; yet, most stroke research utilizes young animals. Failure to account for age-related changes in the brain may, in part, explain the failure of successful neuroprotectants in animal studies to translate into clinically effective therapies in humans. Estrogen has been repeatedly demonstrated to confer neuroprotection from neural injury in young animals. Clinical trials, however, show increased incidence and severity of stroke in women taking hormone replacement therapy (HRT). Although an effective therapy for menopausal symptoms, current FDA guidelines for the use of HRT have been altered to limit patient exposure and thereby reduce cardiovascular risk. This review discusses age-related differences in estrogenic activity in the brain which may account for discrepancies between preclinical and clinical data.
Age-Related Differences in Brain Injury
Stroke is the leading cause of long-term disability and the third leading cause of death in the United States. Traumatic brain injury (TBI) is the leading cause of disability for children and young adults and the primary cause of death for individuals between 18 and 45 years of age. These forms of neural injury have similar cellular and subcellular events that occur after the primary insult, but the typical age of individuals affected by these injuries is very different. Most TBI patients are younger than 35 years; however, a second peak of incidence exists for those older than 65 years, in whom falls account for most TBIs. According to the American Heart Association, approximately 72% of people who have a stroke are older than 65 years, almost 25% of the population older than 75 years will have a stroke, and for people older than 55 years, the incidence of stroke more than doubles for each successive decade. Because of the age distribution for the two main types of neurologic insult, young animal models have a role in understanding the mechanisms involved in the injured brain, particularly in the traumatically injured brain. However, this review argues that young animal models do not accurately represent the response to injury unique to the aged brain; thus, aged animals must have a place in the study of neural injury, particularly stroke.
Both basic science1 and clinical studies2 indicate that older age decreases the ability of the brain to recover from neural injury. Neural injury causes an initial region of cell death followed by a secondary phase of injury characterized by release of intracellular glutamate and calcium, an inflammatory response, activation of apoptotic pathways, formation of reactive oxygen species (ROS), and blood brain barrier (BBB) disruption. Animal studies of stroke have shown unique, age-related differences in both phases of ischemic injury.3,4 Specifically, aged rats have larger infarct volume1 and less functional recovery5 than young adult rats after transient focal cerebral ischemia. Morphologic changes in the brain of aged rats after neural injury include massive infiltration of polymorphonuclear neutrophils, petechiae, and necrosis formation.6 These markers of inflammation are reduced in the postischemic brain of young adult rats.7 Clinically, age-related differences in functional recovery from neural injury are well established. Results from a recent clinical study assessing more than 400 TBI patients (ages ranging from 16 to 85 years and divided into three groups: 16 to 26, 27 to 39, and 40 to 85 years old) at 1 and 4 years after injury showed similar improvements initially between age groups but vast differences at the 5-year assessment.2 These findings, along with a number of other investigations, cite age as a predictive factor in functional outcome from TBI.8 Older age also leads to greater disability after cerebral ischemia, including decreased ability to perform activities of daily living, higher percentage of use of assistive devices for ambulation, and increased use of institutionalized care facilities.9
Several hypotheses exist to explain the mechanisms at work in the aging brain: increased inflammation, decreased reserve, and accumulation of ROS. The inflammation hypothesis of aging suggests that increased inflammation during the aging process results from dysregulation of the immune system and a progressive inability to handle pathologic stimuli.10 Aging in the brain is characterized by cortical atrophy and an increased number of activated astrocytes and microglia.11 Another prominent hypothesis is that aged brains have an accumulation of ROS along with reductions in the enzymes to control them. Specific genes (ie, age-1, daf-2, methuselah, and shc) have been identified that demonstrate a definitive connection between accumulation of ROS and longevity.12 Although aging is a complex process influenced by many factors, the aged brain is clearly unique from its younger counterpart in both health and disease.
Age-Related Differences in Estrogen Activity in the Brain
The secondary phase of neural injury activates numerous inflammatory, proapoptotic and antiapoptotic, and angiogenic factors. These processes respond to estrogen differentially, depending on age. There is substantial evidence that estrogen is an effective neuroprotectant. Studies performed by a variety of investigators clearly show that estrogen reduces infarct size,13 neuronal cell death,14 and mortality15 after neural injury. One common variable in almost all studies showing neuroprotection by estrogen is the use of young animals. The few studies that have examined age-related differences in estrogenic effects have used models of injury that do not mimic stroke or methods of estrogen supplementation that do not mimic its clinical use. One study that investigated age-related differences in estrogenic actions used a 10-minute period of global ischemia—an injury that mimics cardiac arrest rather than stroke.16 A study concluding that estrogen supplementation attenuates stroke injury in aged female rats used only 1 week of estrogen exposure before middle cerebral artery occlusion by intraluminal filament.17 Clinical trials, however, have shown a clear relationship between increased stroke incidence and severity with estrogen supplementation in postmenopausal women.18,19
Clinical Trials Show Estrogen Increases Stroke Risk and Stroke Severity
The Women's Health Initiative (WHI) randomized controlled trials investigated the benefits and risks of 0.625 mg/d of conjugated equine estrogen (CEE) plus 2.5 mg/d of medroxyprogesterone acetate (MPA) in 16,608 women. The trials were terminated before the planned end date because of elevated breast cancer risk along with other risks defined in the global index as time to incident coronary heart disease, stroke, pulmonary embolism, hip fracture, or death from other causes.18 The average follow-up of women in the CEE/MPA trial was 5.6 years. In that time, the CEE/MPA group experienced 151 strokes (1.8%), whereas the placebo-treated group had 107 strokes (1.3%). The hazard ratio (HR) for all stroke subtypes was 1.31 [95% confidence interval (CI), 1.02 to 1.68]. For ischemic stroke, however, the HR was 1.44 (95% CI, 1.09 to 1.90), indicating an approximate 31% increase in stroke risk with CEE/MPA compared with placebo. The elevated risk of stroke with CEE/MPA could not be attributed to any other risk factor and did not appear until after the first year of treatment.
A second branch of the WHI simultaneously studied the effects of CEE alone in 10,739 women with prior hysterectomy.19 Researchers also found an increased risk of ischemic stroke with CEE, warranting the early termination of those trials as well. When the CEE trial was stopped, the participants had an average of 6.8 years of follow-up. The HR for stroke in those taking CEE versus placebo was 1.39 (95% CI, 1.10 to 1.77), with an absolute excess risk of 12 additional strokes per 10,000 person-years. Although this elevation of stroke risk may be associated with an observed increase in systolic blood pressure, there are likely more global effects of CEE requiring further investigation.
Smaller-scaled clinical trials have shown increased stroke severity with estrogen treatment in women with known vascular disease. The Women's Estrogen for Stroke Trial was a randomized, double-blind, placebo-controlled trial of 664 postmenopausal women that demonstrated increased risk of fatal stroke and worse neurologic outcome from stroke in women taking estrogen (17β-estradiol, 1 mg/d) after a first cerebral ischemic event.20 The Heart and Estrogen/progestin Replacement Study (HERS) investigated the use of CEE/MPA (0.625 mg/d of CEE and 2.5 mg/d of MPA) in 2763 women with coronary heart disease and found that treatment was not significantly associated with risk of stroke (HR, 1.23; 95% CI, 0.89 to 1.70), but it also did not reduce the incidence of coronary or cerebrovascular events as expected.21 Interestingly, HERS22 and the unblinded follow-up study (HERSII) found a significant increase in the risk of thromboembolic events (deep vein thrombosis and pulmonary embolism) in women taking HRT compared with those taking placebo (HR, 2.08; 95% CI, 1.28 to 3.40),23 indicating a possible role of estrogen in thrombus creation. Two separate meta-analyses combining data from each of these three clinical trials and many other smaller trials have shown a clearly increased risk of stroke incidence and severity with HRT.24,25
Timing of Initiation Hypothesis
Researchers have responded to these reports by arguing that variations in the timing of initiation of estrogen treatment is the probable reason for differences between animal studies and the clinical trial data. They contend that a period of hypoestrogenicity after menopause leads to irreversible vascular changes that are not mimicked in ovariectomized animals immediately supplemented with estrogen. Participants in the WHI trials were between 50 and 79 years old, with an average age of 63 years (approximately 12 years after the average age of menopause). However, subsequent analyses of WHI data of women with prior CEE and/or CEE/MPA use that began within 5 years of menopause have not supported the timing of initiation hypothesis.26 However, basic science research from proponents of the timing of initiation hypothesis has supported the idea that a period of hypoestrogenicity negates the neuroprotection afforded by constant estrogen exposure. In particular, one study examining the timing of initiation hypothesis showed a significant difference between infarct volumes of mice supplemented with estrogen immediately after ovariectomy and a placebo-treated control group. A second comparison was made between mice undergoing a 10-week period of no estrogen exposure compared with a second control group, showing no significant differences in infarct volumes.27 The key comparison, however, was not presented in that study, namely, differences in infarct volumes between mice immediately supplemented with estrogen and those undergoing a period of hypoestrogenicity. By comparing each treatment group to a control (placebo-treated) group rather than to each other, no conclusions regarding the timing of initiation hypothesis can be made. Furthermore, these studies, among others, using young animals cannot be used to predict the role estrogen plays in the aged brain.
Age, Not Timing, Determines Response to Estrogen
Age is the likely variable accounting for the contrast between animal studies and clinical trial findings. Most basic science studies in neural injury, including studies on the effects of estrogen, use young animals. This research is based on the assumption that aging is a linear process, whereas studies on aging do not support that claim.28 Therefore, assessment of the response to neural injury in young or middle-aged rats cannot be used to extrapolate data to predict a postinjury response in an aged animal or, more importantly, in an aged patient. Together, these factors indicate that more studies on estrogenic effects on stroke, using clinically relevant animal models, are warranted. Evidence presented in this review suggests that estrogenic down-regulation of the postischemic inflammatory response may contribute to worsened stroke injury by attenuating beneficial downstream antiapoptotic and angiogenic pathway activation.
Estrogen Receptors within the Central Nervous System
Estrogen mediates many of its actions in the periphery via two forms of the estrogen receptor (ER), ERα and ERβ, encoded on chromosomes 6 and 14, respectively. There are at least three known ERα isoforms and five known ERβ isoforms, with varying degrees of transactivation function.29 Some ER agonists bind preferentially to one form of the ER. Estrone binds with greater affinity to ERα, whereas estriol and raloxifene preferentially bind ERβ. 17β-Estradiol binds equally well to both forms of the ER; thus, it is used as the most common form of estrogen supplementation in research. On activation, these receptors act as ligand-dependent transcription factors by either binding directly to estrogen response elements on target DNA sequences or interacting with nuclear proteins to alter gene expression. Estrogen-induced genomic actions are well understood and occur in hours or days.30
Neuroprotection by estrogen in young animals is thought to be partially mediated through ERα-dependent activity. ERα and ERβ are constitutively expressed in cortical31 and hippocampal32 neurons in addition to cells in many other brain regions; yet, only ERα is up-regulated in astrocytes and microglia after injury.33 Estrogen-induced neuronal sparing from ischemia is abolished in ERα knockout mice compared with exacerbated infarcts in ERβ knockout mice.34 However, studies using both the ERα and ERβ knockout mice are likely confounded by the fact that ERβ knockout mice have been shown to be in a state of systemic hypoxia.35 ERβ is highly expressed in type I and type II pneumocytes, and the deletion of this gene leads to fewer alveoli36 and reduced lung volume.37 In treadmill tests, ERβ knockout mice are highly susceptible to ischemic injury compared with wild-type littermates,35 thus making this transgenic model less than ideal for the study of cerebral ischemia.
The G-protein coupled ER 30 has also been found and cited as the possible mediator of rapidly occurring, nongenomic estrogenic action.38 Specifically, estrogen has been shown to directly stimulate calcium mobilization, activate cAMP-mediated signaling, and activate extracellular signal–regulated kinase 1/2 (ERK1/2) activity in multiple cell types.39 However, debate still exists regarding the role of the G-protein coupled ER 30. One study found that the receptor was not activated by radioactive estradiol and that estradiol exposure did not lead to elevations in intracellular cAMP or calcium levels.40 Another hypothesis is that rapidly occurring, nongenomic actions of estrogen are mediated through membrane-bound ER. Some reports have shown evidence of an association between a membrane-bound ER and insulin-like growth factor 1 (IGF-1) receptors. Colocalization studies indicate that IGF-1 is present in neurons expressing ERα and in neurons expressing ERβ in the hypothalamus, hippocampus, and cerebral cortex.41 Although these studies provide important information on ER-independent activity, to date a complete understanding of the mechanisms of estrogenic effects on the brain are still unclear and warrant further investigation.
ERs Differ in the Aged Brain
Evidence of a differential effect of estrogen with age has been demonstrated in the brain and other tissues. Animal models show decreased nuclear ER concentrations in selected brain regions of middle-aged rats compared with young adult counterparts.42 Further investigation has shown differential response to estrogen supplementation with age in the hypothalamus and pituitary, although it is unknown whether the same age-related changes occur in other brain regions. Specifically, ovariectomized, young animals supplemented with estrogen showed increased ER expression, whereas middle-aged and aged animals had little or no increase in ER in cytosolic or nuclear fractions of hypothalamus and pituitary, despite similar increments in plasma estrogen levels in all age groups.43
More recently, postmortem analysis of specific human brain regions has demonstrated age-related changes in levels of a certain ERα splice variant known as mamillary body exon 1.44 Discovered in 2005, this receptor protein variant lacks exon 1, giving it an expected fourfold decrease in transactivation function, which would alter its response to estrogen.29 Similarly, aging has been associated with reduced expression and activity of ERα-transactivation domain interacting protein, metastasis-associated protein 1, an important regulator of histone deacetylation and transcriptional control.45 These findings suggest that the aged brain may respond differently to circulating estrogen (Table 1). Although these studies do not provide a complete understanding of age-related differences in estrogenic activity in the brain, emerging evidence supports the idea that estrogen has differential effects, depending on age both in health and after neurological insult.
Table 1.
Reported Age-Related Differences in Estrogenic Actions and Receptors in the Brain
| Model | Treatment | Injury | Age-related difference | Reference |
|---|---|---|---|---|
| Postmortem human | Increased MB1 splice variant of ERα in hypothalamus of aged women compared with younger women | 44 | ||
| Aged mouse | Decreased expression of MTA1 and decreased interaction with ERα transactivation domain compared with young adult female mice | 45 | ||
| Aged female mouse | 60-day time release pellet of 1.7 mg of 17β-estradiol | Decreased astrocytes and microglial cells in dentate gyrus and CA1 compared with young adult female mice | 46 | |
| Aged female rat | 6 μg/kg of 17β-estradiol (i.v.) | Decreased ER levels in nucleus in multiple brain regions compared with young adult female rats | 42 | |
| Aged female mouse | 0.2 μg of 17β-estradiol (s.c.) | Decreased ER and decreased binding kinetics of ER in hypothalamus/pituitary compared with young adult female mice | 43 | |
| Aged female rat | 60-day time release pellet of 1.0 mg of 17β-estradiol | Forebrain excitotoxic injury | Increased IL-1β expression and decreased nerve growth factor compared with placebo-treated, aged female rats | 47 |
| Aged female gerbil | Single, high-dose 4-mg/kg pretreatment | Global ischemia | Improved performance on memory tasks, fewer TUNEL and caspase 3 positive cells in CA1 and CA2 compared with placebo-treated, aged female gerbils | 16 |
| Aged female rat | 3-week time release pellet of 25 μg of 17β-estradiol for 7 days | Filament occlusion of MCA | Reduced cortical and striatal infarct volume and no changes in cerebral blood flow compared with placebo-treated, aged female rats | 17 |
CA, cornu ammonis; MB1, mamillary body exon 1; MCA, middle cerebral artery; MTA1, metastasis associated protein 1; TUNEL, terminal deoxynucleotidyl transferase mediated dUTP nick end labeling.
Estrogen Produced Endogenously by Aromatase
Local production of estrogen by aromatase may play an important role in recovery from neural injury, particularly in postmenopausal females and males. Aromatase is a cytochrome P450 enzyme that converts testosterone to estradiol and androstenedione to estrone and provides the only source of estrogen in men. Aromatase is bound to the endoplasmic reticulum and under physiologic conditions is expressed at low levels in the brain after the perinatal period.48 Studies have shown the presence of aromatase in both neurons49 and, after insult, in glia.50 Testosterone, administered before or immediately after penetrating brain lesion, has been shown to decrease expression of vimentin immunoreactive astrocytes in male orchidectomized rats51; however, this effect is likely due to a local conversion of testosterone to estrogen by aromatase. Both in vitro52 and in vivo53 studies with male aromatase knockout mice and cell cultures from these transgenic animals show greater cell death from experimental neural injury compared with wild-type counterparts. These reports suggest a significant role for the endogenously produced estrogen that arises in response to neural injury. Levels and activity of neural aromatase have not been reported in aged animals.
Estrogen Reduces Inflammation
Neuropoietic Cytokines and the JAK/STAT Pathway
Inflammation contributes to neural damage in ischemic injury models. Injury leads to increased NF-κB activation, which causes induction of many mediators of inflammation. TBI causes elevation of IL-6, IL-1β, and tumor necrosis factor-α (TNF-α). IL-1 is also a key component of the inflammatory response to neural injury and has been shown to cause neuronal cell death.54 IL-1 receptor-1 null mice subjected to neural injury demonstrate decreased edema, leukocyte infiltration, and overall cell death, as well as decreased astrocytic reactivity,55 indicating that IL-1 receptor activation may be detrimental to recovery from neural injury.
Yet, not all inflammatory factors have adverse effects. In some types of neural injury, such as cerebral ischemia, the inflammatory response contributes to the induction of genes that aid in microvessel regrowth and the salvage of cells through activation of antiapoptotic pathways.56 Specifically, IL-6 and other neuropoietic cytokines act on type 1 cytokine receptors, composed of a ligand-binding domain, CD126, and a signal-transducing domain, glycoprotein 130 (gp130). Intracellular regions of the transmembrane gp130 molecule are coupled to the Janus kinase (JAK)/signaling transducer and STAT pathway, most notably STAT3.57 STAT3 is activated by phosphorylation on a tyrosine residue on its C terminus. On phosphorylation, STAT3 forms a heterodimer or homodimer and translocates to the nucleus, where it activates transcription of target genes, including proteins that are involved in cell survival and proliferation, such as bcl-2, bcl-xL, mcl-1, Fas, survivin, cyclin D1, cyclin E1, and p21.58 In addition, other transcription factors, including c-myc, c-jun, and c-fos, are STAT3 targets.59 Vascular endothelial growth factor and brain-derived neurotrophic factor have also been shown to be targets of STAT3 and contribute to angiogenesis.60
The IL-6 signaling pathways, specifically JAK2/STAT3, play a major role in modulating the complex relationship between aging and disease. Research demonstrates that not all actions mediated by IL-6 are beneficial. Elevated central nervous system levels of IL-6 lead to increased astrogliosis and BBB permeability, and IL-6 activation of the JAK2/STAT3 pathway contributes to neuronal loss after transient focal cerebral ischemia.61 Unpublished data from our laboratory indicate that dysfunction of the JAK2/STAT3 pathway plays a significant role in the increased injury and decreased functional recovery observed in studies of ischemic injury in aged animals. Specifically, during the acute phase after injury, JAK2/STAT3 signaling contributes to a robust proinflammatory response that leads to an early, more severe BBB disruption. As the injury response continues, JAK2/STAT3 signaling is attenuated by desensitization or increased negative regulation by the suppressor of cytokine signaling 3 (SOCS3), resulting in decreased angiogenesis and increased apoptosis. Data from clinical studies confirm that an elevated plasma IL-6 level is predictive of greater stroke severity and worsened functional outcome in older adults,62 indicating a connection among the inflammatory response, stroke severity, and aging. However, studies on aging show that STAT3 expression is decreased in the rodent brain with increasing age. The counterpart of STAT3, known as STAT1, is associated with detrimental effects after neural injury and remains unchanged with age.63 Studies have shown that estrogen increases activation of STAT3 in young adult rats after ischemic injury,56 but it is unknown whether this occurs in aged rats after neural insult. Other studies indicate that estrogen also increases inhibitors of the JAK2/STAT3 pathway.64 Figure 1 indicates known effects of estrogen on gp130 signaling through JAK2/STAT3 pathway. It is unknown what the ultimate results of estrogenic actions are on STAT3 activation in aged animals after neural injury.
Figure 1.
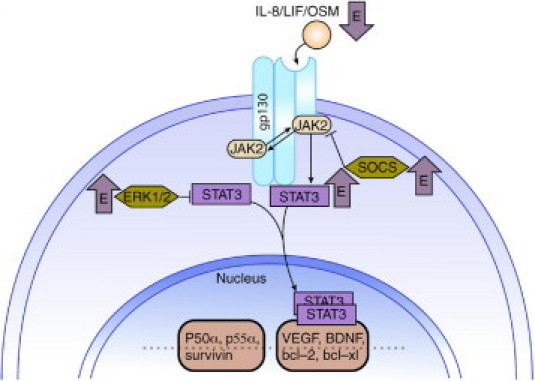
Estrogenic effects on gp130 signaling. gp130 signaling plays an important role in the injured brain. Although quiescent in the healthy brain, on injury, STAT3 activation leads to transcription of genes that promote cell survival. STAT3 gene deletion studies show enhanced neuronal apoptosis and increased infarct size after middle cerebral artery occlusion. Estrogen has been shown to increase levels of activated STAT354; however, estrogen also increases inhibitors of the pathway, ERK1/2 and SOCS3.62 BDNF, brain-derived neurotrophic factor; VEFG, vascular endothelial growth factor.
Other neuropoietic cytokines also use gp130 and activate STAT3. Leukemia inhibitory factor (LIF) and oncostatin M (OsM) are two neuropoietic cytokines of particular interest in the injured brain. LIF has a myriad of actions throughout the body, including enhancing migration of inflammatory cells to damaged neuronal tissue.65 During mammary gland involution, LIF induces expression of the two phosphatidylinositol-3-kinase (PI3K) regulatory subunits p50 and p55 via STAT3, resulting in diminished levels of Akt activity and eventually leading to apoptosis.66 OsM reduces excitotoxic damage, similar to that occurring in the peri-infarct region after ischemia/reperfusion injury,67 and activates STAT3 to induce apoptosis and cell cycle arrest in certain tumor cells. Unpublished data from our laboratory suggest that aged rats have a more robust postischemic increase in LIF, OsM, and SOCS3 compared with young adult rats, indicating a potential role of these neuropoietic cytokines and SOCS3-mediated inhibition of STAT3 in the worsened recovery from experimental stroke observed in aged animals.
Estrogen Attenuates Inflammation in Vivo and in Vitro
Estrogen plays a significant role in regulation of inflammatory pathways.68 In young animals treated with estrogen before middle cerebral artery occlusion, there are reductions in IL-6 and monocyte chemoattractant protein-1.27 Further evidence of estrogen-mediated attenuation of neuroinflammation indicates specific estrogenic effects on microglia after exposure to lipopolysaccharide (LPS) in female ovariectomized rats.69 It is unknown whether such a reduction in microglia reactivity occurs in aged animals after neural injury. The anti-inflammatory action of estrogen is also supported in cell culture models of neural injury. Primary rat microglia cell lines exposed to LPS show reduced microglial superoxide release and phagocytic activity when pretreated with estrogen. These effects are abolished by both ER inhibitor and mitogen-activated protein kinase (MAPK) inhibitor.70 However, no effect is seen in NF-κB levels, suggesting that estrogen-induced anti-inflammatory actions in microglia are likely dependent on ER-mediated activation of MAPK. MAPK promotes cell survival through cAMP response element binding protein transcription pathways.
Microglial cell cultures treated with estrogen also have reduced basal levels of TNF-α and reduced TNF-α after exposure to LPS.71 However, in animal models using N-methyl-d-aspartic acid–induced cytotoxic injury, the effects on IL-1β expression are dependent on age and hormonal status.47 Therefore, the ability of estrogen to attenuate inflammation may only occur in young animals or may occur to a lesser extent or by different mechanisms in aged animals. In aged brains, the inflammatory response may serve a vital purpose that becomes weakened by anti-inflammatory activity of estrogen. After ischemic injury, gp130-mediated up-regulation of angiogenic and antiapoptotic factors may serve to protect the peri-infarct region from cell death and aid in regrowth of microvessels to the damaged area. If these processes are inhibited by estrogen in aged animals, perhaps due to differential expression of ERα splice variants and their responsiveness to circulating estrogen, then exacerbated injury may result.
Estrogen Attenuates Reactive Gliosis
Estrogen may also exert effects on astrocytes and their response to neural injury. Reactive astrocytes undergo gliosis after neural injury, which involves a continuum of specific molecular and morphologic changes. In patient studies of ischemic stroke, there is severe reactive astrogliosis accompanied by proliferation of astrocytes, forming a glial scar at the infarct border. Although some evidence suggests that glial scar formation serves to limit and contain postischemic neuronal death, gliosis has also been shown to inhibit axonal regeneration and remyelination, thereby hindering recovery from injury.72 Likewise, astrocytes release a host of proinflammatory cytokines that may contribute to neuronal damage. Astrocytic response to neural injury may be directly affected by estrogen because studies have shown that astrocytes express both nuclear ER and membrane-associated ER.73 Several in vivo models of neural injury, including penetrating brain lesion, excitotoxic-induced neurodegeneration, and cholinergic basal forebrain lesion, demonstrate estrogen-mediated reduction in glial fibrillary acidic protein and vimentin expression, indicating estrogenic attenuation of postinjury activation of astrocytes.33,74 This activity appears to be preserved with age because long-term estrogen treatment also attenuates age-related astrocyte activation in hippocampus of 20-month-old mice.46
Estrogen Affects the BBB
The BBB is a functional element of the neurovascular unit, an extensive network composed of the endothelium, glia, pericytes, neurons, and extracellular matrix. The BBB partitions the systemic circulation from brain parenchyma and serves to establish, maintain, and regulate discrete microenvironments within the brain for optimal neuronal function.75 The neurovascular unit undergoes a number of morphologic and functional changes during the aging process that make the BBB more vulnerable to insult.76
After injury, there are two principle mechanisms by which brain edema develops: cytotoxic and vasogenic. Cytotoxic edema results from a loss of ionic homeostasis and can occur as astrocytes increase glutamate uptake in an attempt to limit excitotoxicity, which requires glucose use and leads to intracellular water and sodium accumulation. Neurons can undergo cytotoxic swelling when ionic exchangers dysfunction or as part of the metabolic consequences of excitotoxicity. Vasogenic edema occurs when there is a breakdown in the tight junctions between the endothelial cells of the BBB or up-regulation of water transport channels in perivascular cells, allowing fluid to enter the brain parenchyma. Although relatively little is known about the specific mechanisms by which estrogen attenuates postinjury edema, evidence suggests it inhibits BBB breakdown after neural insult.77
Estrogen Inhibits Postinjury Edema via Multiple Mechanisms
Some evidence suggests that estrogen-mediated attenuation of BBB breakdown may be related to inhibition of matrix metalloproteinases (MMPs), the enzymes responsible for extracellular matrix remodeling.78 Although the effects of estrogen on the family of MMPs have long been studied in regard to reproductive physiology and diseases of the endometrium, recent research has focused on the effects of estrogen on centrally located MMPs, particularly in relation to BBB disruption. This research has led to potential mechanisms by which estrogen helps maintain functional integrity of the BBB after insult. In particular, estrogen has been shown to significantly decrease Evans blue extravasation into the brain,78 a common assay for assessment of BBB disruption. Evans blue complexes to albumin, making it a 68-kDa marker, the passage of which through the BBB indicates marked disruption. More subtle changes are better assayed with dextrans (10 to 60 kDa), inulin (5 kDa), or sodium fluorescein (376 Da), but the effects of estrogen on BBB permeability to those markers have not been measured to date. Estrogen also decreases local MMP2 and MMP9 activity after ischemia/reperfusion injury.78 By inhibiting the enzymes responsible for injury-induced BBB breakdown, estrogen indirectly preserves BBB integrity. Several reports have also indicated that estrogen can modulate injury-induced ROS generation,79,80 which may contribute to the maintenance of BBB functional integrity after insult. Because ROS contribute to BBB breakdown through lipid membrane peroxidation, attenuation of ROS production should protect BBB integrity. F(2)-isoprostanes result from free radical oxidation of arachidonic acid and are lower in cerebrospinal fluid from female patients with severe TBI than in male counterparts81; thus, antioxidant activity may account for estrogen-mediated protection of the BBB. Estrogen decreases ROS-related cell death through a nongenomic mechanism of action in organotypic hippocampal slice cultures.79 Alternatively, this BBB-sparing effect may be attributable to antioxidant capacity of progesterone, which decreases F(2)-isoprostanes in vitro.82
In addition to the mechanisms by which estrogen inhibits BBB disruption, the hormone is also involved in formation and clearance of the subsequent edema through its regulation of the major water transport channel in the brain, aquaporin 4. In LPS-induced BBB disruption models, estrogen attenuates the induction of aquaporin 4 in perivascular glial cell processes, thereby decreasing the associated edema.83 Together, these studies demonstrate that estrogen has potential use in the short-term treatment of neural injury. Some studies on estrogenic neuroprotection in young animal models have used short-term treatment after neural insult and found promising results.84,85 In terms of treatment for stroke, however, aged animal models must also have a place in validating the results.
ERs in Cerebral Vasculature
After TBI, signs of postinjury neuronal hypoxia, including hypoperfusion and jugular venous desaturation, have been observed. Estrogen may have a role in reducing injury-induced hypoxia through actions on cerebral vasculature because the vasorelaxing property of estrogen has been demonstrated. Middle cerebral arteries isolated from female ovariectomized rats supplemented with estrogen show greater increase in diameter in response to elevated transmural pressure compared with untreated and male rats; yet, differences in vascular responsiveness between groups are abolished by removal of the vascular endothelium or by inhibition of both nitric oxide synthase and cyclooxygenase.86 These studies and others suggest that activation of ERα leads to cyclooxygenase-1 (COX-1) induction of prostacyclin86 and enhanced nitric oxide synthesis in cerebral vasculature87 (Figure 2). Cerebral blood vessels isolated from estrogen-treated rats have increased COX-1, prostacyclin synthase, and prostacyclin expression.88 Prostacyclin is a prostanoid product of COX-1 that acts on both platelets and endothelial cells and activates the protein kinase A pathway, resulting in smooth muscle relaxation and subsequent vasodilation. Estrogen-treated animals also show elevated levels of endothelial-derived nitric oxide synthase (eNOS) mRNA and protein, suggesting a role for eNOS-dependent vasodilation.89 This mechanism of estrogen-mediated vasodilation is further supported by studies using mouse cerebral arteries that demonstrate a reversal of estrogen-mediated changes in vascular tone when eNOS is inhibited86 (Figure 2). Similar results are found in cultured cerebral vessels, where eNOS levels are significantly depressed by ER antagonist treatment.90 Modulation in local eNOS levels may be the predominant vascular effect of estrogen. Evidence from eNOS knockout mice show elevated COX-1 and prostacyclin compared with control, estrogen-treated animals, although the vascular responses in both groups are similar.91 However, in studies using young adult female rats, ERα immunoreactivity is not present in only vascular smooth muscle cells; rather, ERα immunoreactivity is also found in the nucleus, in the mitochondria, and on the membrane of endothelial cells.92 Any potential differences in the effects of estrogen on cerebral blood vessels in the aged brain have not been elucidated but may be an interesting area for future investigation because of the vasogenic actions of estrogen and the known age-related changes in cerebral vasculature.
Figure 2.
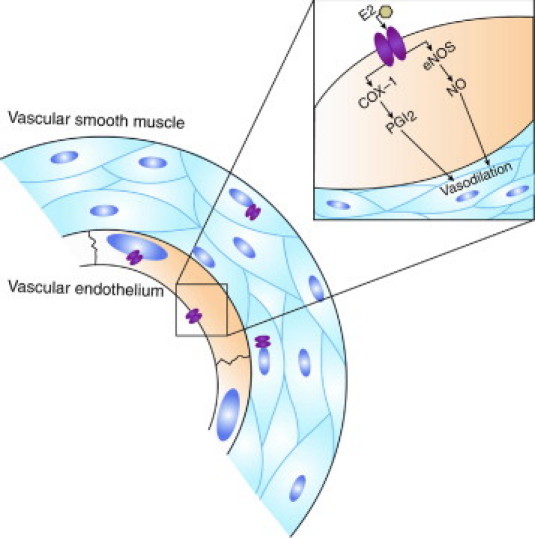
Estrogenic effects on cerebral vasculature. Although ERα immunoreactivity has been found in the nucleus, in the mitochondria, and on the membrane of endothelial cells from cerebral blood vessels, the role of these receptors in estrogen-mediated vasodilation is unclear. Evidence suggests that estrogenic alteration in myogenic tone occurs via activation of eNOS and COX-1, which result in vasodilation. E2, 17β-estradiol; NO, nitric oxide; PGI2, prostaglandin I2.
Effects of Estrogen on Excitotoxicity
Most data regarding the effects of estrogen on excitotoxicity rely on cell culture models. Evidence from these models suggests that estrogen affects the extent of excitotoxic cell death by modulating glutamate-induced cellular influx of calcium.93,94 Research demonstrates that estrogen acts directly on N-methyl-d-aspartic acid receptors to enhance excitatory amino acid activity,95 which would enhance glutamate toxicity; however, in cultured hippocampal neurons, estrogen can decrease excitoxicity.96 Other evidence indicates that estrogen down-regulates ionotropic glutamate receptor subunits in neurons, which would attenuate the effects of excess extracellular glutamate like that occurring after neural injury.94 Estrogen also acts to increase glutamate uptake by astrocytes, thus limiting excitotoxic insult to neurons,97 although this effect was observed in cultured human astrocytes of Alzheimer's disease patients, so it is unknown whether the same response to estrogen would occur in acute neural injury.
Estrogen Modulates Apoptotic Signaling Pathways
After direct, excitotoxic, oxidative, or hypoxic injury, cells in the brain parenchyma may undergo apoptosis or necrosis. Research has shown that estrogen inhibits apoptotic cell death after neural injury in young animal models. Specifically, estrogen activates MAPK by triggering calcium influx through L-type calcium channels98 and increases levels of activated bcl-2 present in the nucleus of neurons in the peri-ischemic area of young adult rats subjected to focal cerebral ischemia.56 Further evidence of neuronal sparing by estrogen comes from immortalized hippocampal cell lines, which show rapid estrogen-induced cAMP response element binding protein phosphorylation that is blocked by both ER inhibitors and MAPK inhibitors.99
Estrogen also modulates levels of IGF-1,98 which is a potent activator of the Akt signaling pathway. Akt inhibits apoptosis by inactivating proapoptotic proteins, such as bcl-xl/bcl-2–associated death promoter, and activating NF-κB for transcription of survival-promoting genes. Studies demonstrate estrogenic activation of Akt and the family of MAPK known as ERK1/2 independent of IGF-1 signaling,31 although estrogen-mediated increases in IGF-1 may bolster this response. Furthermore, estrogen activates the PI3K signaling cascade through an ER-mediated binding with PI3K regulator, p85.31 The interaction between ER and p85 leads to activation of both Akt and ERK1/2 in cortical neurons in a time-dependent manner.31 Overall, estrogenic effects on apoptotic pathways are complex, a complete understanding of which has yet to be elucidated.
Conclusion
The idea of estrogen as a neuroprotectant first emerged with evidence that premenopausal females experience less damage and greater functional and cognitive recovery from neurologic insult than males, with this disparity disappearing around the age of menopause. In hypoxia/ischemia models of neural injury, many studies have shown estrogen to be neuroprotective,15,17,80,100 with an equal number of mechanisms postulated to explain these results. Estrogen attenuates BBB disruption from neurologic insult, reduces edema, lowers levels of inflammatory mediators,68 activates antiapoptotic pathways,56 and has antioxidant capabilities79 in young animals and cell culture models. Some of these actions are receptor mediated, whereas others have nongenomic mechanisms.
However, many questions regarding the mechanisms by which estrogen acts remain unanswered. Specifically, why is estrogen neuroprotective in young animals but not in postmenopausal women? An estimated 10 million women were receiving HRT in 2000 for alleviation of menopausal symptoms. Then, after the release of reports from the WHI and subsequent analyses of that data showing that estrogen increases the incidence and severity of stroke,18,19,24,25 widespread use of estrogen therapy is no longer recommended. By more fully understanding the mechanisms of estrogenic action in the aged, injured brain, parameters for the safe and effective use of female gonadal hormones may be possible. However, most basic science research in neural injury is conducted on healthy, young animals, despite the fact that there are age-related differences in estrogenic activity in the brain (Table 1). Animal studies show age-dependent alterations in ER levels after estrogen treatment,43,45 whereas studies from human cadavers demonstrate age-related changes in the specific splice variant of the ER,44 indicating that aged individuals may have greater levels of a less active form of ER in specific brain regions. This disregard of age-dependent differences in the actions of female gonadal hormones has led to unnecessary patient morbidity and mortality19; thus, research models of disease that account for age-related differences must have a place in future investigations of estrogenic activity.
Footnotes
Supported by National Institutes of Health, National Institutes of Neurological Disorders and Stroke grant RO1 061954 (J.D.H). Activase was provided by Genentech (San Francisco, California).
References
- 1.Popa-Wagner A., Badan I., Walker L., Groppa S., Patrana N., Kessler C. Accelerated infarct development, cytogenesis and apoptosis following transient cerebral ischemia in aged rats. Acta Neuropathol. 2007;113:277–293. doi: 10.1007/s00401-006-0164-7. [DOI] [PubMed] [Google Scholar]
- 2.Marquez de la Plata C.D., Hart T., Hammond F.M., Frol A.B., Hudak A., Harper C.R., O'Neil-Pirozzi T.M., Whyte J., Carlile M., Diaz-Arrastia R. Impact of age on long-term recovery from traumatic brain injury. Arch Phys Med Rehabil. 2008;89:896–903. doi: 10.1016/j.apmr.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baltan S., Besancon E.F., Mbow B., Ye Z., Hamner M.A., Ransom B.R. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J Neurosci. 2008;28:1479–1489. doi: 10.1523/JNEUROSCI.5137-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badan I., Platt D., Kessler C., Popa-Wagner A. Temporal dynamics of degenerative and regenerative events associated with cerebral ischemia in aged rats. Gerontology. 2003;49:356–365. doi: 10.1159/000073763. [DOI] [PubMed] [Google Scholar]
- 5.Badan I., Buchhold B., Hamm A., Gratz M., Walker L.C., Platt D., Kessler C., Popa-Wagner A. Accelerated glial reactivity to stroke in aged rats correlates with reduced functional recovery. J Cereb Blood Flow Metab. 2003;23:845–854. doi: 10.1097/01.WCB.0000071883.63724.A7. [DOI] [PubMed] [Google Scholar]
- 6.Rosen C.L., Dinapoli V.A., Nagamine T., Crocco T. Influence of age on stroke outcome following transient focal ischemia. J Neurosurg. 2005;103:687–694. doi: 10.3171/jns.2005.103.4.0687. [DOI] [PubMed] [Google Scholar]
- 7.Dinapoli V.A., Rosen C.L., Nagamine T., Crocco T. Selective MCA occlusion: a precise embolic stroke model. J Neurosci Methods. 2006;154:233–238. doi: 10.1016/j.jneumeth.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 8.Flanagan S.R., Hibbard M.R., Gordon W.A. The impact of age on traumatic brain injury. Phys Med Rehabil Clin N Am. 2005;16:163–177. doi: 10.1016/j.pmr.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 9.Kelly-Hayes M., Beiser A., Kase C.S., Scaramucci A., D'Agostino R.B., Wolf P.A. The influence of gender and age on disability following ischemic stroke: the Framingham study. J Stroke Cerebrovasc Dis. 2003;12:119–126. doi: 10.1016/S1052-3057(03)00042-9. [DOI] [PubMed] [Google Scholar]
- 10.Chung H.Y., Kim H.J., Kim J.W., Yu B.P. The inflammation hypothesis of aging: molecular modulation by calorie restriction. Ann N Y Acad Sci. 2001;928:327–335. [PubMed] [Google Scholar]
- 11.Rosenberg P.B. Clinical aspects of inflammation in Alzheimer's disease. Int Rev Psychiatry. 2005;17:503–514. doi: 10.1080/02646830500382037. [DOI] [PubMed] [Google Scholar]
- 12.Finkel T., Holbrook N.J. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 13.Dubal D.B., Kashon M.L., Pettigrew L.C., Ren J.M., Finklestein S.P., Rau S.W., Wise P.M. Estradiol protects against ischemic injury. J Cereb Blood Flow Metab. 1998;18:1253–1258. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- 14.Rau S.W., Dubal D.B., Bottner M., Gerhold L.M., Wise P.M. Estradiol attenuates programmed cell death after stroke-like injury. J Neurosci. 2003;23:11420–11426. doi: 10.1523/JNEUROSCI.23-36-11420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simpkins J.W., Rajakumar G., Zhang Y.Q., Simpkins C.E., Greenwald D., Yu C.J., Bodor N., Day A.L. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;87:724–730. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- 16.Wappler EA, Felszeghy K, Szilagyi G, Gal A, Skopal J, Mehra RD, Nyakas C, Nagy Z: Neuroprotective effects of estrogen treatment on ischemia-induced behavioural deficits in ovariectomized gerbils at different ages. Behav Brain Res 209:42–48 [DOI] [PubMed]
- 17.Alkayed N.J., Murphy S.J., Traystman R.J., Hurn P.D., Miller V.M. Neuroprotective effects of female gonadal steroids in reproductively senescent female rats. Stroke. 2000;31:161–168. doi: 10.1161/01.str.31.1.161. [DOI] [PubMed] [Google Scholar]
- 18.Wassertheil-Smoller S., Hendrix S.L., Limacher M., Heiss G., Kooperberg C., Baird A., Kotchen T., Curb J.D., Black H., Rossouw J.E., Aragaki A., Safford M., Stein E., Laowattana S., Mysiw W.J. Effect of estrogen plus progestin on stroke in postmenopausal women: the Women's Health Initiative: a randomized trial. JAMA. 2003;289:2673–2684. doi: 10.1001/jama.289.20.2673. [DOI] [PubMed] [Google Scholar]
- 19.Anderson G.L., Limacher M., Assaf A.R., Bassford T., Beresford S.A., Black H., Bonds D., Brunner R., Brzyski R., Caan B., Chlebowski R., Curb D., Gass M., Hays J., Heiss G., Hendrix S., Howard B.V., Hsia J., Hubbell A., Jackson R., Johnson K.C., Judd H., Kotchen J.M., Kuller L., LaCroix A.Z., Lane D., Langer R.D., Lasser N., Lewis C.E., Manson J., Margolis K., Ockene J., O'Sullivan M.J., Phillips L., Prentice R.L., Ritenbaugh C., Robbins J., Rossouw J.E., Sarto G., Stefanick M.L., Van Horn L., Wactawski-Wende J., Wallace R., Wassertheil-Smoller S. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 20.Viscoli C.M., Brass L.M., Kernan W.N., Sarrel P.M., Suissa S., Horwitz R.I. A clinical trial of estrogen-replacement therapy after ischemic stroke. N Engl J Med. 2001;345:1243–1249. doi: 10.1056/NEJMoa010534. [DOI] [PubMed] [Google Scholar]
- 21.Hulley S., Grady D., Bush T., Furberg C., Herrington D., Riggs B., Vittinghoff E., Heart and Estrogen/progestin Replacement Study (HERS) Research Group Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. JAMA. 1998;280:605–613. doi: 10.1001/jama.280.7.605. [DOI] [PubMed] [Google Scholar]
- 22.Grady D., Wenger N.K., Herrington D., Khan S., Furberg C., Hunninghake D., Vittinghoff E., Hulley S. Postmenopausal hormone therapy increases risk for venous thromboembolic disease: The Heart and Estrogen/progestin Replacement Study. Ann Intern Med. 2000;132:689–696. doi: 10.7326/0003-4819-132-9-200005020-00002. [DOI] [PubMed] [Google Scholar]
- 23.Hulley S., Furberg C., Barrett-Connor E., Cauley J., Grady D., Haskell W., Knopp R., Lowery M., Satterfield S., Schrott H., Vittinghoff E., Hunninghake D. Noncardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II) JAMA. 2002;288:58–66. doi: 10.1001/jama.288.1.58. [DOI] [PubMed] [Google Scholar]
- 24.Sare G.M., Gray L.J., Bath P.M. Association between hormone replacement therapy and subsequent arterial and venous vascular events: a meta-analysis. Eur Heart J. 2008;29:2031–2041. doi: 10.1093/eurheartj/ehn299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bath P.M., Gray L.J. Association between hormone replacement therapy and subsequent stroke: a meta-analysis. BMJ. 2005;330:342. doi: 10.1136/bmj.38331.655347.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prentice R.L., Manson J.E., Langer R.D., Anderson G.L., Pettinger M., Jackson R.D., Johnson K.C., Kuller L.H., Lane D.S., Wactawski-Wende J., Brzyski R., Allison M., Ockene J., Sarto G., Rossouw J.E. Benefits and risks of postmenopausal hormone therapy when it is initiated soon after menopause. Am J Epidemiol. 2009;170:12–23. doi: 10.1093/aje/kwp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki S., Brown C.M., Dela Cruz C.D., Yang E., Bridwell D.A., Wise P.M. Timing of estrogen therapy after ovariectomy dictates the efficacy of its neuroprotective and antiinflammatory actions. Proc Natl Acad Sci U S A. 2007;104:6013–6018. doi: 10.1073/pnas.0610394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bishop NA, Lu T, Yankner BA: Neural mechanisms of ageing and cognitive decline. Nature 464:529–535 [DOI] [PMC free article] [PubMed]
- 29.Ishunina T.A., Swaab D.F., Fischer D.F. Estrogen receptor-alpha splice variants in the medial mamillary nucleus of Alzheimer's disease patients: identification of a novel MB1 isoform. J Clin Endocrinol Metab. 2005;90:3757–3765. doi: 10.1210/jc.2004-1858. [DOI] [PubMed] [Google Scholar]
- 30.McDonnell D.P., Norris J.D. Connections and regulation of the human estrogen receptor. Science. 2002;296:1642–1644. doi: 10.1126/science.1071884. [DOI] [PubMed] [Google Scholar]
- 31.Mannella P., Brinton R.D. Estrogen receptor protein interaction with phosphatidylinositol 3-kinase leads to activation of phosphorylated Akt and extracellular signal-regulated kinase 1/2 in the same population of cortical neurons: a unified mechanism of estrogen action. J Neurosci. 2006;26:9439–9447. doi: 10.1523/JNEUROSCI.1443-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehra R.D., Sharma K., Nyakas C., Vij U. Estrogen receptor alpha and beta immunoreactive neurons in normal adult and aged female rat hippocampus: a qualitative and quantitative study. Brain Res. 2005;1056:22–35. doi: 10.1016/j.brainres.2005.06.073. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Ovejero D., Veiga S., Garcia-Segura L.M., Doncarlos L.L. Glial expression of estrogen and androgen receptors after rat brain injury. J Comp Neurol. 2002;450:256–271. doi: 10.1002/cne.10325. [DOI] [PubMed] [Google Scholar]
- 34.Dubal D.B., Zhu H., Yu J., Rau S.W., Shughrue P.J., Merchenthaler I., Kindy M.S., Wise P.M. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morani A., Barros R.P., Imamov O., Hultenby K., Arner A., Warner M., Gustafsson J.A. Lung dysfunction causes systemic hypoxia in estrogen receptor beta knockout (ERbeta−/−) mice. Proc Natl Acad Sci U S A. 2006;103:7165–7169. doi: 10.1073/pnas.0602194103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patrone C., Cassel T.N., Pettersson K., Piao Y.S., Cheng G., Ciana P., Maggi A., Warner M., Gustafsson J.A., Nord M. Regulation of postnatal lung development and homeostasis by estrogen receptor beta. Mol Cell Biol. 2003;23:8542–8552. doi: 10.1128/MCB.23.23.8542-8552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Massaro D., Massaro G.D. Estrogen receptor regulation of pulmonary alveolar dimensions: alveolar sexual dimorphism in mice. Am J Physiol Lung Cell Mol Physiol. 2006;290:L866–L870. doi: 10.1152/ajplung.00396.2005. [DOI] [PubMed] [Google Scholar]
- 38.Revankar C.M., Cimino D.F., Sklar L.A., Arterburn J.B., Prossnitz E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 39.Prossnitz E.R., Oprea T.I., Sklar L.A., Arterburn J.B. The ins and outs of GPR30: a transmembrane estrogen receptor. J Steroid Biochem Mol Biol. 2008;109:350–353. doi: 10.1016/j.jsbmb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otto C., Rohde-Schulz B., Schwarz G., Fuchs I., Klewer M., Brittain D., Langer G., Bader B., Prelle K., Nubbemeyer R., Fritzemeier K.H. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology. 2008;149:4846–4856. doi: 10.1210/en.2008-0269. [DOI] [PubMed] [Google Scholar]
- 41.Cardona-Gomez G.P., DonCarlos L., Garcia-Segura L.M. Insulin-like growth factor I receptors and estrogen receptors colocalize in female rat brain. Neuroscience. 2000;99:751–760. doi: 10.1016/s0306-4522(00)00228-1. [DOI] [PubMed] [Google Scholar]
- 42.Wise P.M., McEwen B.S., Parsons B., Rainbow T.C. Age-related changes in cytoplasmic estradiol receptor concentrations in microdissected brain nuclei: correlations with changes in steroid-induced sexual behavior. Brain Res. 1984;321:119–126. doi: 10.1016/0006-8993(84)90687-5. [DOI] [PubMed] [Google Scholar]
- 43.Belisle S., Bellabarba D., Lehoux J.G. Age-dependent, ovary-independent decrease in the nuclear binding kinetics of estrogen receptors in the brain of the C57BL/6J mouse. Am J Obstet Gynecol. 1985;153:394–401. doi: 10.1016/0002-9378(85)90077-8. [DOI] [PubMed] [Google Scholar]
- 44.Ishunina T.A., Swaab D.F. Age-dependent ERalpha MB1 splice variant expression in discrete areas of the human brain. Neurobiol Aging. 2008;29:1177–1189. doi: 10.1016/j.neurobiolaging.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 45.Thakur M.K., Ghosh S. Interaction of estrogen receptor alpha transactivation domain with MTA1 decreases in old mouse brain. J Mol Neurosci. 2009;37:269–273. doi: 10.1007/s12031-008-9131-1. [DOI] [PubMed] [Google Scholar]
- 46.Lei D.L., Long J.M., Hengemihle J., O’Neill J., Manaye K.F., Ingram D.K., Mouton P.R. Effects of estrogen and raloxifene on neuroglia number and morphology in the hippocampus of aged female mice. Neuroscience. 2003;121:659–666. doi: 10.1016/s0306-4522(03)00245-8. [DOI] [PubMed] [Google Scholar]
- 47.Nordell V.L., Scarborough M.M., Buchanan A.K., Sohrabji F. Differential effects of estrogen in the injured forebrain of young adult and reproductive senescent animals. Neurobiol Aging. 2003;24:733–743. doi: 10.1016/s0197-4580(02)00193-8. [DOI] [PubMed] [Google Scholar]
- 48.Tsuruo Y., Ishimura K., Fujita H., Osawa Y. Immunocytochemical localization of aromatase-containing neurons in the rat brain during pre- and postnatal development. Cell Tissue Res. 1994;278:29–39. doi: 10.1007/BF00305775. [DOI] [PubMed] [Google Scholar]
- 49.Celotti F., Melcangi R.C., Martini L. The 5 alpha-reductase in the brain: molecular aspects and relation to brain function. Front Neuroendocrinol. 1992;13:163–215. [PubMed] [Google Scholar]
- 50.Saldanha C.J., Rohmann K.N., Coomaralingam L., Wynne R.D. Estrogen provision by reactive glia decreases apoptosis in the zebra finch (Taeniopygia guttata) J Neurobiol. 2005;64:192–201. doi: 10.1002/neu.20147. [DOI] [PubMed] [Google Scholar]
- 51.Barreto G., Veiga S., Azcoitia I., Garcia-Segura L.M., Garcia-Ovejero D. Testosterone decreases reactive astroglia and reactive microglia after brain injury in male rats: role of its metabolites, oestradiol and dihydrotestosterone. Eur J Neurosci. 2007;25:3039–3046. doi: 10.1111/j.1460-9568.2007.05563.x. [DOI] [PubMed] [Google Scholar]
- 52.Liu M., Oyarzabal E.A., Yang R., Murphy S.J., Hurn P.D. A novel method for assessing sex-specific and genotype-specific response to injury in astrocyte culture. J Neurosci Methods. 2008;171:214–217. doi: 10.1016/j.jneumeth.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCullough L.D., Blizzard K., Simpson E.R., Oz O.K., Hurn P.D. Aromatase cytochrome P450 and extragonadal estrogen play a role in ischemic neuroprotection. J Neurosci. 2003;23:8701–8705. doi: 10.1523/JNEUROSCI.23-25-08701.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Basu A., Krady J.K., Levison S.W. Interleukin-1: a master regulator of neuroinflammation. J Neurosci Res. 2004;78:151–156. doi: 10.1002/jnr.20266. [DOI] [PubMed] [Google Scholar]
- 55.Lin H.W., Basu A., Druckman C., Cicchese M., Krady J.K., Levison S.W. Astrogliosis is delayed in type 1 interleukin-1 receptor-null mice following a penetrating brain injury. J Neuroinflammation. 2006;3:15. doi: 10.1186/1742-2094-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dziennis S., Jia T., Ronnekleiv O.K., Hurn P.D., Alkayed N.J. Role of signal transducer and activator of transcription-3 in estradiol-mediated neuroprotection. J Neurosci. 2007;27:7268–7274. doi: 10.1523/JNEUROSCI.1558-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirano T., Ishihara K., Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–2556. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- 58.Hodge D.R., Hurt E.M., Farrar W.L. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2512. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 59.Yang E., Lerner L., Besser D., Darnell J.E., Jr. Independent and cooperative activation of chromosomal c-fos promoter by STAT3. J Biol Chem. 2003;278:15794–15799. doi: 10.1074/jbc.M213073200. [DOI] [PubMed] [Google Scholar]
- 60.Xu Q., Briggs J., Park S., Niu G., Kortylewski M., Zhang S., Gritsko T., Turkson J., Kay H., Semenza G.L., Cheng J.Q., Jove R., Yu H. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene. 2005;24:5552–5560. doi: 10.1038/sj.onc.1208719. [DOI] [PubMed] [Google Scholar]
- 61.Satriotomo I., Bowen K.K., Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J Neurochem. 2006;98:1353–1368. doi: 10.1111/j.1471-4159.2006.04051.x. [DOI] [PubMed] [Google Scholar]
- 62.Nakase T., Yamazaki T., Ogura N., Suzuki A., Nagata K. The impact of inflammation on the pathogenesis and prognosis of ischemic stroke. J Neurol Sci. 2008;271:104–109. doi: 10.1016/j.jns.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 63.De-Fraja C., Conti L., Govoni S., Battaini F., Cattaneo E. STAT signalling in the mature and aging brain. Int J Dev Neurosci. 2000;18:439–446. doi: 10.1016/s0736-5748(00)00007-1. [DOI] [PubMed] [Google Scholar]
- 64.Wong J.K., Le H.H., Zsarnovszky A., Belcher S.M. Estrogens and ICI182,780 (Faslodex) modulate mitosis and cell death in immature cerebellar neurons via rapid activation of p44/p42 mitogen-activated protein kinase. J Neurosci. 2003;23:4984–4995. doi: 10.1523/JNEUROSCI.23-12-04984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sugiura S., Lahav R., Han J., Kou S.Y., Banner L.R., de Pablo F., Patterson P.H. Leukaemia inhibitory factor is required for normal inflammatory responses to injury in the peripheral and central nervous systems in vivo and is chemotactic for macrophages in vitro. Eur J Neurosci. 2000;12:457–466. doi: 10.1046/j.1460-9568.2000.00922.x. [DOI] [PubMed] [Google Scholar]
- 66.Abell K., Bilancio A., Clarkson R.W., Tiffen P.G., Altaparmakov A.I., Burdon T.G., Asano T., Vanhaesebroeck B., Watson C.J. Stat3-induced apoptosis requires a molecular switch in PI(3)K subunit composition. Nat Cell Biol. 2005;7:392–398. doi: 10.1038/ncb1242. [DOI] [PubMed] [Google Scholar]
- 67.Weiss T.W., Samson A.L., Niego B., Daniel P.B., Medcalf R.L. Oncostatin M is a neuroprotective cytokine that inhibits excitotoxic injury in vitro and in vivo. FASEB J. 2006;20:2369–2371. doi: 10.1096/fj.06-5850fje. [DOI] [PubMed] [Google Scholar]
- 68.Maggi A., Ciana P., Belcredito S., Vegeto E. Estrogens in the nervous system: mechanisms and nonreproductive functions. Annu Rev Physiol. 2004;66:291–313. doi: 10.1146/annurev.physiol.66.032802.154945. [DOI] [PubMed] [Google Scholar]
- 69.Vegeto E., Belcredito S., Ghisletti S., Meda C., Etteri S., Maggi A. The endogenous estrogen status regulates microglia reactivity in animal models of neuroinflammation. Endocrinology. 2006;147:2263–2272. doi: 10.1210/en.2005-1330. [DOI] [PubMed] [Google Scholar]
- 70.Bruce-Keller A.J., Keeling J.L., Keller J.N., Huang F.F., Camondola S., Mattson M.P. Antiinflammatory effects of estrogen on microglial activation. Endocrinology. 2000;141:3646–3656. doi: 10.1210/endo.141.10.7693. [DOI] [PubMed] [Google Scholar]
- 71.Dimayuga F.O., Reed J.L., Carnero G.A., Wang C., Dimayuga E.R., Dimayuga V.M., Perger A., Wilson M.E., Keller J.N., Bruce-Keller A.J. Estrogen and brain inflammation: effects on microglial expression of MHC, costimulatory molecules and cytokines. J Neuroimmunol. 2005;161:123–136. doi: 10.1016/j.jneuroim.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 72.Rosen C.L., Bunge R.P., Ard M.D., Wood P.M. Type 1 astrocytes inhibit myelination by adult rat oligodendrocytes in vitro. J Neurosci. 1989;9:3371–3379. doi: 10.1523/JNEUROSCI.09-10-03371.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuo J., Hariri O.R., Bondar G., Ogi J., Micevych P. Membrane estrogen receptor-alpha interacts with metabotropic glutamate receptor type 1a to mobilize intracellular calcium in hypothalamic astrocytes. Endocrinology. 2009;150:1369–1376. doi: 10.1210/en.2008-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Djebaili M., Guo Q., Pettus E.H., Hoffman S.W., Stein D.G. The neurosteroids progesterone and allopregnanolone reduce cell death, gliosis, and functional deficits after traumatic brain injury in rats. J Neurotrauma. 2005;22:106–118. doi: 10.1089/neu.2005.22.106. [DOI] [PubMed] [Google Scholar]
- 75.Huber J.D., Witt K.A., Hom S., Egleton R.D., Mark K.S., Davis T.P. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2001;280:H1241–H1248. doi: 10.1152/ajpheart.2001.280.3.H1241. [DOI] [PubMed] [Google Scholar]
- 76.DiNapoli V.A., Huber J.D., Houser K., Li X., Rosen C.L. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. 2008;29:753–764. doi: 10.1016/j.neurobiolaging.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cipolla M.J., Godfrey J.A., Wiegman M.J. The effect of ovariectomy and estrogen on penetrating brain arterioles and blood-brain barrier permeability. Microcirculation. 2009;16:685–693. doi: 10.3109/10739680903164131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu R., Wen Y., Perez E., Wang X., Day A.L., Simpkins J.W., Yang S.H. 17beta-Estradiol attenuates blood-brain barrier disruption induced by cerebral ischemia-reperfusion injury in female rats. Brain Res. 2005;1060:55–61. doi: 10.1016/j.brainres.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 79.Behl C., Skutella T., Lezoualc'h F., Post A., Widmann M., Newton C.J., Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- 80.Culmsee C., Vedder H., Ravati A., Junker V., Otto D., Ahlemeyer B., Krieg J.C., Krieglstein J. Neuroprotection by estrogens in a mouse model of focal cerebral ischemia and in cultured neurons: evidence for a receptor-independent antioxidative mechanism. J Cereb Blood Flow Metab. 1999;19:1263–1269. doi: 10.1097/00004647-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 81.Bayir H., Marion D.W., Puccio A.M., Wisniewski S.R., Janesko K.L., Clark R.S., Kochanek P.M. Marked gender effect on lipid peroxidation after severe traumatic brain injury in adult patients. J Neurotrauma. 2004;21:1–8. doi: 10.1089/089771504772695896. [DOI] [PubMed] [Google Scholar]
- 82.Roof R.L., Hoffman S.W., Stein D.G. Progesterone protects against lipid peroxidation following traumatic brain injury in rats. Mol Chem Neuropathol. 1997;31:1–11. doi: 10.1007/BF02815156. [DOI] [PubMed] [Google Scholar]
- 83.Tomas-Camardiel M., Venero J.L., Herrera A.J., De Pablos R.M., Pintor-Toro J.A., Machado A., Cano J. Blood-brain barrier disruption highly induces aquaporin-4 mRNA and protein in perivascular and parenchymal astrocytes: protective effect by estradiol treatment in ovariectomized animals. J Neurosci Res. 2005;80:235–246. doi: 10.1002/jnr.20443. [DOI] [PubMed] [Google Scholar]
- 84.Liu R, Liu Q, He S, Simpkins JW, Yang SH: Combination therapy of 17beta-estradiol and recombinant tissue plasminogen activator for experimental ischemic stroke. J Pharmacol Exp Ther 332:1006–1012 [DOI] [PMC free article] [PubMed]
- 85.Lebesgue D., Chevaleyre V., Zukin R.S., Etgen A.M. Estradiol rescues neurons from global ischemia-induced cell death: multiple cellular pathways of neuroprotection. Steroids. 2009;74:555–561. doi: 10.1016/j.steroids.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Geary G.G., Krause D.N., Duckles S.P. Estrogen reduces mouse cerebral artery tone through endothelial NOS- and cyclooxygenase-dependent mechanisms. Am J Physiol Heart Circ Physiol. 2000;279:H511–H519. doi: 10.1152/ajpheart.2000.279.2.H511. [DOI] [PubMed] [Google Scholar]
- 87.Stirone C., Boroujerdi A., Duckles S.P., Krause D.N. Estrogen receptor activation of phosphoinositide-3 kinase, akt, and nitric oxide signaling in cerebral blood vessels: rapid and long-term effects. Mol Pharmacol. 2005;67:105–113. doi: 10.1124/mol.104.004465. [DOI] [PubMed] [Google Scholar]
- 88.Ospina J.A., Krause D.N., Duckles S.P. 17beta-estradiol increases rat cerebrovascular prostacyclin synthesis by elevating cyclooxygenase-1 and prostacyclin synthase. Stroke. 2002;33:600–605. doi: 10.1161/hs0202.102732. [DOI] [PubMed] [Google Scholar]
- 89.Stirone C., Chu Y., Sunday L., Duckles S.P., Krause D.N. 17 Beta-estradiol increases endothelial nitric oxide synthase mRNA copy number in cerebral blood vessels: quantification by real-time polymerase chain reaction. Eur J Pharmacol. 2003;478:35–38. doi: 10.1016/j.ejphar.2003.08.037. [DOI] [PubMed] [Google Scholar]
- 90.McNeill A.M., Zhang C., Stanczyk F.Z., Duckles S.P., Krause D.N. Estrogen increases endothelial nitric oxide synthase via estrogen receptors in rat cerebral blood vessels: effect preserved after concurrent treatment with medroxyprogesterone acetate or progesterone. Stroke. 2002;33:1685–1691. doi: 10.1161/01.str.0000016325.54374.93. [DOI] [PubMed] [Google Scholar]
- 91.Li X., Geary G.G., Gonzales R.J., Krause D.N., Duckles S.P. Effect of estrogen on cerebrovascular prostaglandins is amplified in mice with dysfunctional NOS. Am J Physiol Heart Circ Physiol. 2004;287:H588–H594. doi: 10.1152/ajpheart.01176.2003. [DOI] [PubMed] [Google Scholar]
- 92.Stirone C., Duckles S.P., Krause D.N., Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005;68:959–965. doi: 10.1124/mol.105.014662. [DOI] [PubMed] [Google Scholar]
- 93.Ciriza I., Azcoitia I., Garcia-Segura L.M. Reduced progesterone metabolites protect rat hippocampal neurones from kainic acid excitotoxicity in vivo. J Neuroendocrinol. 2004;16:58–63. doi: 10.1111/j.1365-2826.2004.01121.x. [DOI] [PubMed] [Google Scholar]
- 94.Numakawa Y., Matsumoto T., Yokomaku D., Taguchi T., Niki E., Hatanaka H., Kunugi H., Numakawa T. 17beta-estradiol protects cortical neurons against oxidative stress-induced cell death through reduction in the activity of mitogen-activated protein kinase and in the accumulation of intracellular calcium. Endocrinology. 2007;148:627–637. doi: 10.1210/en.2006-1210. [DOI] [PubMed] [Google Scholar]
- 95.Smith S.S. Estrogen administration increases neuronal responses to excitatory amino acids as a long-term effect. Brain Res. 1989;503:354–357. doi: 10.1016/0006-8993(89)91691-0. [DOI] [PubMed] [Google Scholar]
- 96.Goodman Y., Bruce A.J., Cheng B., Mattson M.P. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 97.Liang Z., Valla J., Sefidvash-Hockley S., Rogers J., Li R. Effects of estrogen treatment on glutamate uptake in cultured human astrocytes derived from cortex of Alzheimer's disease patients. J Neurochem. 2002;80:807–814. doi: 10.1046/j.0022-3042.2002.00779.x. [DOI] [PubMed] [Google Scholar]
- 98.Cheng C.M., Cohen M., Wang J., Bondy C.A. Estrogen augments glucose transporter and IGF1 expression in primate cerebral cortex. FASEB J. 2001;15:907–915. doi: 10.1096/fj.00-0398com. [DOI] [PubMed] [Google Scholar]
- 99.Wade C.B., Dorsa D.M. Estrogen activation of cyclic adenosine 5′-monophosphate response element-mediated transcription requires the extracellularly regulated kinase/mitogen-activated protein kinase pathway. Endocrinology. 2003;144:832–838. doi: 10.1210/en.2002-220899. [DOI] [PubMed] [Google Scholar]
- 100.Toung T.J., Traystman R.J., Hurn P.D. Estrogen-mediated neuroprotection after experimental stroke in male rats. Stroke. 1998;29:1666–1670. doi: 10.1161/01.str.29.8.1666. [DOI] [PubMed] [Google Scholar]