

Abstract
Connexin 37 (Cx37) profoundly suppresses the proliferation of rat insulinoma (Rin) cells by unknown mechanisms. To determine whether a functional pore domain is necessary for Cx37-mediated growth suppression, we introduced a mutation that converted threonine 154 into alanine (T154A). Like other connexins mutated at the homologous site, Cx37-T154A localized to appositional membrane but failed to form functional channels and exerted a dominant-negative effect on coexpressed wild-type Cx37 or Cx43. Unlike the wild-type protein, Cx37-T154A did not suppress the proliferation of Rin cells and did not, with serum deprivation, result in cell cycle arrest. Furthermore, progression through the cell cycle was unaffected by expression of Cx37-T154A. These results indicate that a pore-forming domain that is able to form functional channels is essential for the anti-proliferative, cell-cycle arrest and serum-sensitivity effects of Cx37, and furthermore that the normally localized C-terminal domain is not sufficient for these effects of Cx37.
Key words: Gap junction, Connexin 37, Cell cycle
Introduction
Gap junction channels are composed of one or more of the 21 members of the connexin (Cx) gene family (Sohl and Willecke, 2004). These channels and proteins are thought to support coordinated tissue function in three ways. First, by forming gap junction channels, connexins support intercellular diffusion of the electrical and chemical signals (Harris, 2001), which are the foundation of syncitial tissue behavior. Second, by forming functional hemichannels (connexons) that mediate transmembrane diffusion of small signaling molecules, connexins support paracrine and autocrine responses, especially during cellular responses to injury and disease (Goodenough and Paul, 2003). Finally, through interactions with other intracellular proteins, connexins regulate and are regulated by intracellular signaling cascades (Kardami et al., 2007). Nearly all cells express multiple connexin isoforms (Sohl and Willecke, 2004). Not surprisingly, all isoforms share some functional parameters, for example, they all form electrically conductive channels, but other functions are often isoform specific. As such, each connexin isoform is thought to contribute to cell and tissue function in a connexin-specific manner (Kardami et al., 2007), a view supported by the unique phenotypes characteristic of animals with connexin-isoform-specific deletions or substitutions (Kruger et al., 2000; Scherer et al., 1998; Simon et al., 1997). Indeed, such studies emphasize the unique contributions of connexins to embryonic and post-natal development, cell and tissue homeostasis and response to injury and disease (Alcolea et al., 2004; Plum et al., 2000; Reaume et al., 1995; White, 2002).
Multiple members of the connexin gene family have been demonstrated to exert growth-regulatory effects (Kardami et al., 2007). The mechanisms underlying connexin-mediated growth regulation are both specific to the connexin isoform, reflecting the unique permselective properties of the channels formed by each connexin or the specific protein partners with which each connexin interacts, and specific to the cell type, reflecting the unique metabolic and signaling milieu of different cell types. Of the 21 members of the connexin gene family, growth regulation mediated by Cx43 has received the most attention because this protein is expressed by a diverse array of cell types. In some of these cell types, growth suppression by Cx43 appeared to require formation of functional gap junction channels, whereas in (most) other cell types such channel function appeared unnecessary. In the latter cell types, the C-terminal (CT) domain of Cx43 was both necessary and sufficient for growth suppression (Johnstone et al., 2010; Zhang et al., 2003a; Zhang et al., 2003b). Whether channel function is necessary or not, it appears that specific serine and tyrosine residues in the CT domain must be available for phosphorylation for Cx43 to exert its growth regulatory effects (Dang et al., 2006; Doble et al., 2004; Solan and Lampe, 2009). This latter observation highlights the significance of interactions between connexins and growth-factor-activated kinases. Importantly, in cell types where Cx43 had no obvious growth suppressive effect (Burt et al., 2008; Kardami et al., 2007), other connexins were effective at suppressing cell growth, thus highlighting the connexin- and cell-specificity of growth suppression, as well as raising the interesting possibility that one beneficial consequence of coexpressing multiple connexins might be to increase the strategies available to cells to regulate growth.
Recently, we reported that Cx37, unlike either Cx40 or Cx43, suppressed the proliferation of rat insulinoma (Rin) cells (Burt et al., 2008). Although the mechanisms underlying this effect of Cx37 are currently unknown, our previous studies showed that Cx37 significantly prolonged all phases of the cell cycle, increasing cell cycle time by approximately fourfold (Burt et al., 2008). Serum deprivation of Cx37-expressing Rin cells resulted in an accumulation of cells at the G1–S check point, an effect not seen in serum-deprived Cx-deficient Rin cells or Cx43-expressing Rin cells. Although much of the expressed Cx37 was cytosolic, some localized to appositional membrane to form functional gap junction channels, raising the question of whether channel function was incidental to or necessary for the growth suppressive effects of Cx37.
Here, we aimed to determine whether a functional channel-forming pore domain was necessary for the anti-proliferative effects of Cx37 in Rin cells. Beahm and colleagues demonstrated that Cx26, Cx43 and Cx50 when mutated at a highly conserved threonine (T135 in Cx26, T154 in Cx43, T157 in Cx50) in the third transmembrane domain formed non-functional gap junctions that were indistinguishable from those formed by the wild-type proteins (Beahm et al., 2006). They demonstrated further that the mutated protein exerted a dominant-negative effect on channel function when coexpressed with wild-type protein. The corresponding threonine residue in Cx37 is T154, which in the current study we have mutated to an alanine residue to determine whether a functional pore is necessary for the growth suppressive effects of Cx37 in Rin cells. We demonstrate that Cx37-T154A, like similarly mutated Cx43, Cx26 and Cx50, localized at appositional membrane to form non-functional gap junctions and exerted a dominant-negative effect on Cx43-mediated dye coupling and Cx37-mediated electrical coupling. However, Cx37-T154A did not suppress the proliferation of Rin cells nor did it support accumulation of cells in G1 upon serum deprivation. These results suggest that a functional pore domain is necessary and that the CT domain is insufficient for growth suppression by Cx37 in Rin cells.
Results
To determine whether channel function is necessary for the growth suppressive function of Cx37, we needed a mutation that would render the channel non-functional but preserve other properties of the Cx37 protein, including localization to appositional membrane so it can interact with the same array of proteins as the wild-type Cx37 and exert dominant-negative properties. Previously published work (Beahm et al., 2006) has suggested that T154 (supplementary material Fig. S1), which is conserved across the alpha and beta connexins, might satisfy these criteria. Therefore, we mutated Cx37-T154 to alanine and stably transfected iRin cells with the mutated sequence to establish cell lines comparable to the iRin37 cell lines already in hand. Six clonal cell lines were evaluated for doxycycline-induced expression of Cx37-T154A (supplementary material Fig. S2A), and two of these cell lines, with relatively high (iRin37-T154A-3C3) or low (iRin37-T154A-3E4) levels of Cx37-T154A expression, were selected for further study. Each clone expressed Cx37-T154A upon induction with doxycycline in a dose-dependent manner, with substantial expression observed at 24 hours with 2 and 4 μg/ml doxycycline and maximal expression at all doxycycline doses at 48 hours (supplementary material Fig. S2B, data from the 3C3 clone is shown). Cx37-T154A expression in the 3C3 clone (after 24 hours in 2 μg/ml doxycycline) was comparable (1.3×10−3 pmole Cx37-T154A per μg total cell protein) to that observed in the iRin37F clone that was characterized previously (supplementary material Fig. S2C,D).
Three approaches were used to determine whether Cx37-T154A localized to appositional membrane to form gap junction plaques. First, we determined whether Cx37-T154A isolated into the Triton-X-100-insoluble fraction of Rin cell protein (Larson et al., 2000; Musil and Goodenough, 1991). Fig. 1A shows that Cx37-T154A, like Cx37, was readily detected in the Triton-X-100-insoluble fraction isolated from both the 3C3 and 3E4 (high and low expressing) iRin37-T154A clones, consistent with the mutant protein participating in plaque formation. Second, we used immunocytochemistry and confocal microscopy to determine the localization of Cx37 in Rin cells stably expressing Cx37 wild-type or the T154A mutant, and in connexin-deficient MDCK cells transiently transfected to express either Cx37 wild-type or T154A. Both the wild-type and mutant proteins localized to appositional membranes in both cell types (Fig. 1B–E), consistent with comparable gap junction plaque formation by both proteins. Finally, because Cx37 was previously demonstrated to form functional heteromeric channels with Cx43 (Brink et al., 1997), we determined, using reciprocal co-immunoprecipitation techniques, whether Cx37-T154A would co-immunoprecipitate with Cx43 when coexpressed with this protein in MDCK43 cells. To verify that MDCK43 cells did not express Cx37, total protein was isolated from these cells, immunoprecipitated with either anti-Cx37 or anti-Cx43 antibody, and the immunoprecipitates blotted for these proteins. Cx37 was not detected in the MDCK43 cells (supplementary material Fig. S3). MDCK43 cells were then transiently transfected with Cx37 or Cx37-T154A, and total protein was isolated under non-denaturing conditions 24 hours later. As shown in Fig. 2A,B, Cx37 (wild-type and T154A) and Cx43 were both detected in samples derived from coexpressing cells, irrespective of the immunoprecipitating antibody. Total protein isolated from cells expressing only Cx43 or Cx37 and mixed after isolation showed no evidence of co-immunoprecipitation (Fig. 2C). Taken together, these results indicate that Cx37-T154A, like Cx37, oligomerizes and traffics to the plasma membrane where it forms gap junctions in regions of cell–cell contact.
Fig. 1.

Cx37-T154A, like Cx37, appears in the Triton-X-100-insoluble fraction of cellular protein and forms plaques at points of cell contact. (A) Western blot of whole-cell (WC) and Triton-insoluble (TX) fractions of cellular homogenates of iRin37 (non-induced: 37wt −; induced: 37wt +) or induced iRin37-T154A-3C3 or -3E4 cells (37-T154A-3E4; 37-T154A-3C3). Both proteins appear in the Triton-insoluble fraction, as well as the whole-cell homogenate. Each lane is loaded with 30 μg of protein, with the exception of 37wt+ WC and TX lanes, which contain 18.69 and 24.77 μg. (B,C) Confocal images of induced iRin37 (B) and iRin37-T154A-3C3 (C) cells immunostained for Cx37 (green) and surface proteins (red) reveal significant Cx37 expression and formation of plaques at cell–cell appositions (arrowheads). (D,E) Confocal images of MDCK cells transfected with Cx37 (D) or Cx37-T154A (E) revealed intracellular and appositional Cx37. The localization of wild-type and mutant proteins is comparable in both cell types. Scale bars: 10 μm. The optical section thickness was 0.56 μm.
Fig. 2.

Cx37 and Cx43 co-immunoprecipitate only when coexpressed. Total protein isolated under non-denaturing conditions from MDCK43 cells coexpressing Cx43 and either Cx37 wild-type (37-wt) or Cx37-T154A (37-T154A) was immunoprecipitated with an antibody against Cx37 (A) or an antibody against Cx43 (B), and the immunoprecipitate (IP) was probed with antibody for either Cx43 (upper blots, IB43) or Cx37 (lower blots, IB37). Irrespective of the immunoprecipitating antibody, immunoprecipitates contained both connexins, indicating interaction of these connexins when coexpressed. Each pair of lanes in A and B derive from the same blot, with molecular-mass-markers shown on the right-hand side of each blot and the positions of the bands corresponding to IgG (IGG, dash), Cx43 (solid arrowhead) and Cx37 (open arrowhead) indicated. In contrast to these coexpression results, Cx37 and Cx43 did not co-immunoprecipitate when total protein from MDCK43 cells was mixed after isolation with total protein isolated from iRin37 cells (C). These samples were from the same blots shown in B, thus the same molecular-mass-marker lanes apply to both B and C. Note in these mixed samples that Cx43 is not detected when the mixed sample was immunoprecipitated with Cx37 antibody (C: upper left) and Cx37 is not detected when the mixed sample was immunoprecipitated with Cx43 antibody (C: lower right).
We next determined whether Cx37-T154A-expressing cells were electrically coupled. Using dual whole-cell voltage clamp techniques, both the incidence (number of pairs exhibiting coupling) and extent of electrical coupling (mean junctional conductance for all pairs) were measured in iRin37 and iRin37-T154A-3C3 cells. As shown in Fig. 3A, the mean junctional conductance in iRin37 cell pairs was 1.6±0.56 nS (n=19), whereas no coupling could be detected in iRin37-T154A cell pairs (n=18). This result suggests that Cx37-T154A, like the homologous mutant forms of Cx43, Cx26 and Cx50, does not form functional channels despite formation of apparently normal plaques. As a further test of the non-functionality of Cx37-T154A, we transfected the iRin37 and iRin37-T154A-3C3 cells with Cx43 and used dual whole-cell voltage clamp to determine the incidence and extent of electrical coupling. As shown in Fig. 3A, introduction of Cx43 to iRin37 cells increased the incidence of electrical coupling from 53 to 80% (4 of 5 pairs) and the extent of coupling from 1.6±0.56 nS to 8.9±3.8 nS (the variability in coupling levels presumably reflects variable levels of Cx43 expression in the transiently transfected cells). By contrast, introduction of Cx43 into iRin37-T154A-3C3 cells increased the incidence of coupling from 0 to only 22% (2 of 9 pairs) and the extent of coupling from 0 to 1.2±0.9 nS (that any coupling is detected likely reflects the formation of some homomeric–homotypic Cx43 channels in the transiently transfected cells). To determine the effects of wild-type and mutant Cx37 in a setting where Cx43 expression levels were more consistent from cell pair to cell pair, we examined dye coupling in MDCK43 cells and MDCK43 cells transiently transfected with either Cx37 or Cx37-T154A. Dye coupling was determined using the small negatively charged dye Alexa Fluor 350, as this dye permeates Cx43 junctions quite readily but Cx37 junctions less well (Weber et al., 2004). All MDCK43 cell pairs (n=6) were dye-coupled (Fig. 3B). By contrast, dye coupling was observed in only 50% of MDCK43 cells coexpressing Cx37 (n=6) and was not observed in MDCK43 cells coexpressing Cx37-T154A (n=6), even allowing 20 minutes for detectable coupling to occur (Fig. 3B). Together, these electrical and dye coupling results suggest that Cx37-T154A forms heteromeric channels with Cx43 and exerts a dominant-negative effect on Cx43 channel function. These data strongly suggest that Cx37-T154A, like connexins 26, 43 and 50 mutated at the homologous site, does not form a functional channel and exerts a dominant-negative effect on channel function when coexpressed with the wild-type connexins with which it oligomerizes.
Fig. 3.
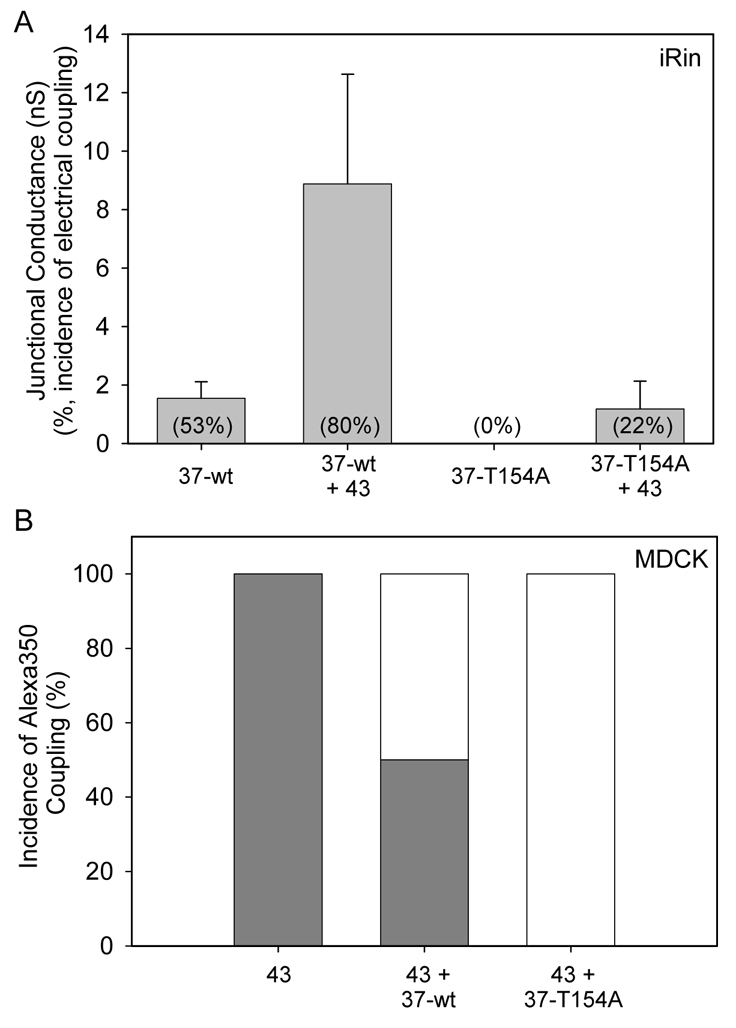
Cx37-T154A forms a non-functional channel and exerts dominant-negative activity when coexpressed with Cx43. (A) iRin cells expressing Cx37 (37-wt) were electrically coupled in 53% of cases (n=19), whereas iRin cells expressing Cx37-T154A (37-T154A) were never detectably coupled, not even with a single channel (n=18). Introduction of Cx43 into the iRin37 cells by transient transfection (37-wt + 43) increased the extent of electrical coupling by ~sixfold and the incidence of electrical coupling by 27%. Introduction of Cx43 into the iRin37-T154A cells (3C3 clone) also increased the incidence of electrical coupling (by 22%) and the extent of electrical coupling. (B) The incidence of Alexa Fluor 350 dye coupling in MDCK43 cells (43) is reduced by transient introduction of Cx37 (43 + 37-wt) and eliminated by transient introduction of Cx37-T154A (43 + 37-T154A), consistent with formation of heteromeric channels and a dominant-negative effect of the Cx37-T154A (n=6 for each group; gray indicates the percentage of cell pairs that were dye coupled).
We next determined whether expression of Cx37-T154A slowed the proliferation of Rin cells to the same extent as Cx37. As shown in Fig. 4, expression of Cx37-T154A in the two different clones did not delay or slow cell proliferation in the same manner as wild-type Cx37. Moreover, the induced iRin37-T154A cells of both clones (3C3, n=4; 3E4, n=3) proliferated comparably to the connexin-deficient parental iRin cell line (n=11) (each experiment was performed in triplicate). Periods of exponential growth (linear regions on a semi-logarithmic plot) were determined for each of these data sets (Fig. 4, insets). Cell cycle time was calculated for periods of exponential growth as (t2–t1)*(log(2)/log(q2/q1)) (where t is time and q is number of cells) using a 3- or 6-day window of analysis (combined mean reported) as previously described (Burt et al., 2008). For Cx37-expressing cells, cell cycle time was between 8 and 9 days before the transition point at day 12 and ~2.5 days after this time point (Fig. 5). For Cx-deficient and Cx37-T154A-expressing cells, cell cycle time was ~1.5 days before the transition at day 12. These data demonstrate that, unlike the effects of Cx37, Cx37-T154A expression did not lead to an increase in cell cycle time, which strongly suggests that a pore domain capable of functioning is crucial to growth suppression by Cx37 and that the CT-domain is not sufficient for growth suppression.
Fig. 4.

Proliferation of iRin cells is suppressed by expression of Cx37 but not Cx37-T154A. (A,B) Like the iRin parental cells (○), the iRin37-T154A-3C3 and 3E4 cells grow comparably in the presence and absence of the inducer doxycycline [compare ▵ (3C3) and ▿ (3E4) curves in B with those in A]. By contrast, induction of Cx37 expression in the iRin37 cells delayed proliferation considerably (compare the □ curves in B with those in A). Insets in both panels show the logarithm of cell number plotted versus time for the same data; note exponential growth (linear on logarithmic scale) between day 3 and day 9 for all induced and non-induced cell lines.
Fig. 5.
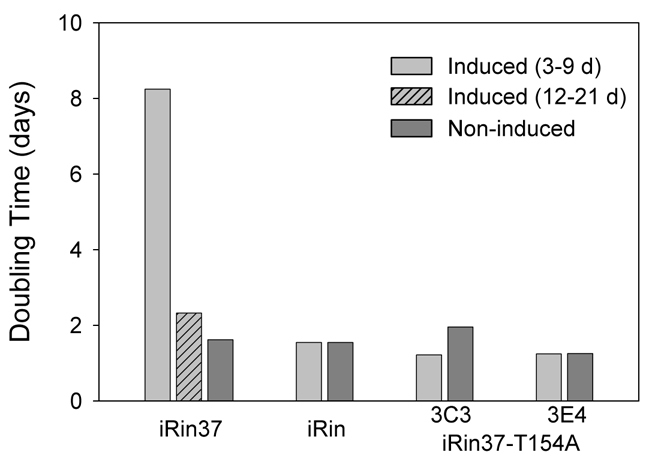
Doubling time is not increased by Cx37-T154A expression. The doubling time for induced and non-induced cells was calculated for all periods of exponential growth (see Fig. 4, insets), day 3 to day 9 for all cell lines, as well as from day 12 to day 21 for induced iRin37 cells (hatched bars). Cx37-T154A expression did not result in increased doubling time at low cell densities.
In addition to increasing doubling time, we showed previously that Cx37 expression also conferred onto Rin cells sensitivity to serum deprivation (Burt et al., 2008). Consequently, we next determined whether Cx37-T154A expression also conferred serum sensitivity, which would indicate that the CT-domain might be sufficient and the functional pore domain unnecessary for this effect of Cx37. Rin 1046, iRin, and iRin37-T154A cells were deprived (or not) of serum for 72 hours, with doxycycline added during the last 24 hours (to induce Cx37-T154A expression). Cells were then harvested, labeled with propidium iodide and subjected to FACS analysis. As expected, serum deprivation had a minimal effect on the cell cycle distribution of Cx-deficient Rin cells, demonstrating their relative insensitivity to serum (Table 1). Cx37 expression resulted in a significant increase in the percentage of cells in G1 and decrease in the percentage in S phase. Cx37-T154A expression resulted in a small decrease in the percentage of cells in G1 and increase in the percentage in S phase. When the data from the two clones expressing Cx37-T154A are combined, the percentage of cells in G1 is significantly decreased and the percentage in S phase significantly increased. Thus, it appears that Cx37-T154A confers serum sensitivity on the iRin cells, but the effect is opposite to that of the wild-type protein wherein both pore and CT domains are functional.
Table 1.
Serum deprivation affects cell-cycle distribution oppositely in Cx37-T154A-compared with Cx37-expressing Rin cells.

To determine whether progression through the cell cycle after release from serum deprivation might differ between Cx37- and Cx37-T154A-expressing Rin cells, cell cycle distribution as a function of time after release was examined for Rin1046, iRin37 and the iRin-37-T154A cell lines. The differential effect of serum deprivation on the cell cycle distribution of Cx37-compared with Cx37-T154A-expressing cells is evident at the zero hour time-point (Fig. 6) and throughout the first 24 hours. iRin37 cells proceeded from G1 to S phase by 48 hours after serum restoration; by contrast, cell cycle progression following release from serum deprivation in the Rin1046 and iRin37-T154A cell lines required only 24 hours (Fig. 6), suggesting Cx37-T154A had little effect on cell cycle progression compared with the effect of Cx37, which delayed arrival at S phase by an additional 24 hours.
Fig. 6.

Cell cycle position of Cx37-T154A-expressing iRin cells was unaffected by serum deprivation, and cell cycle progression was unaffected with serum restoration. Cx37-T154A-expressing cells did not accumulate in G1 as a consequence of serum deprivation (time 0 on plot). The timecourse of cell cycle progression following release from serum deprivation did not differ between Cx37-T154A-expressing iRin and Cx-deficient iRin cells, with G1 and S phase normalizing at 24 hours (n=5 for each of iRin, 3C3 and 3E4 cell lines). By contrast, Cx37-expressing cells (n=10) accumulated in G1 during the serum deprivation period and required 48 hours to emerge from G1. Asterisks denote time points where Cx37-expressing cells differed significantly (P<0.05) from one or both of the Cx37-T154A-expressing cell lines. Where error bars are not obvious, they are less than the size of the data point.
Discussion
Gap junctions and their comprising proteins, the connexins, have long been recognized to act as tumor suppressors, although their mechanism(s) of action in this regard remain blurred (Kardami et al., 2007; King and Bertram, 2005). Initially, their shared ability to form intercellular channels was thought to be central to their growth-suppressive properties. However, it is now clear that gap-junction-mediated growth suppression can occur by channel-dependent or -independent mechanisms, depending on the cell type and the expressed connexin. Whether channel dependent or independent, the connexin-specificity of growth suppression indicates that these proteins differentially influence the complex array of proteins required for cell cycle progression and growth. We had demonstrated previously that Cx37, but not Cx43 or Cx40, exerted growth suppressive effects when expressed in Rin cells (Burt et al., 2008). Cx37-mediated growth suppression involved prolongation of all phases of the cell cycle with a significant accumulation of cells in the G1 phase, particularly when the cells were also deprived of serum. We hypothesized that Cx37 channel function would not be required for growth suppression by Cx37 for two reasons: first, because neither Cx43 nor Cx40 exerted a growth suppressive effect in Rin cells, despite forming functional channels with similar (Cx40) or less selective (Cx43) properties, and second, because Cx43 can suppress growth in a channel-independent and CT domain-dependent manner in a diverse array of cell types (Fu et al., 2004; Gellhaus et al., 2004; Johnstone et al., 2010; Kardami et al., 2007; Zhang et al., 2003a; Zhang et al., 2003b). We show here that, contrary to this expectation, Cx37-mediated growth suppression relies on the channel being able to adopt an open channel configuration and, further, that the presence of an otherwise normal Cx37 protein is not sufficient to mediate growth suppression.
Cx43, Cx26 and Cx50 mutated at the site homologous to T154 in Cx37 form gap junction plaques that appear normal, but the channels are non-functional and the protein exerts a dominant-negative effect on coexpressed wild-type protein (Beahm et al., 2006). Using multiple approaches, we have demonstrated that Cx37-T154A forms a similarly non-functional gap junction channel and plaque. Both wild-type Cx37 and Cx37-T154A were found in the Triton-insoluble fraction of Rin cells, consistent with plaque formation. Immunocytochemistry and confocal microscopy revealed punctate labeling at points of cell–cell apposition for both the wild-type and mutant protein in both Rin and MDCK cells. In cells coexpressing Cx43 and either Cx37 or Cx37-T154A, reciprocal co-immunoprecipitation experiments revealed heteromerization of the coexpressed proteins. Finally, we could find no evidence that Cx37-T154A formed even one functional gap junction channel; and Cx37-T154A exerted a dominant-negative effect on coexpressed Cx43, as demonstrated using dye and electrical coupling techniques. Thus, as expected, the Cx37-T154A mutant protein appears to properly assemble connexons that traffic to the membrane and ultimately form gap junction channels and plaques, which appear normal, at points of cell–cell contact, but the channels fail to adopt an open configuration.
Despite the similarity in assembly and localization of wild-type Cx37 and Cx37-T154A, the mutant protein fails to exert a growth suppressive effect on Rin cell proliferation. Whereas doubling time in iRin37 cells increased from 2 to 9 days upon induction of Cx37 expression, doubling time in iRin37-T154A cells was unaffected by induction of connexin expression. Furthermore, unlike the effects of Cx37, no phase of the cell cycle was prolonged and no accumulation of cells in G1 occurred upon induction of Cx37-T154A expression, whether or not the cells were also deprived of serum. Cell cycle progression following release from serum deprivation was also unaffected by Cx37-T154A expression. These data strongly suggest that a functional channel, or at least the ability to adopt an open configuration, is required for Cx37-mediated growth suppression. Furthermore, the data indicate that the normal localization of a wild-type CT domain is not sufficient to mediate growth suppression.
That a functional pore domain is required for Cx37-mediated growth suppression raises the question of how a functional channel can modify the behavior of the machinery responsible for cell cycle progression. Clearly, one possible explanation is that Cx37 gap junction channels mediate intercellular exchange of some substance that inhibits cell cycle progression in coupled cells. Because neither Cx43 nor Cx40 similarly inhibited cell cycle progression in Rin cells (Burt et al., 2008), this same substance cannot be exchanged by these channel types. Although the permselective properties of Cx37 channels differ from both Cx43 and Cx40 channels (Ek-Vitorin and Burt, 2005; Weber et al., 2004), no permeants for which there might be a transjunctional diffusional gradient have been identified that would be expected to slow all phases of the cell cycle and promote G1 arrest. A second possibility is that Cx37 hemichannels mediate transmembrane exchange of such a substance. Three observations support this second possibility. First, gradients for numerous signaling molecules (e.g. calcium, ATP and chalones) exist across the plasma membrane. Thus, if the hemichannel opens, both influx and efflux of such molecules could occur and in both cases activate signaling cascades (through surface receptors and intracellular mediators) that could modulate simultaneously multiple elements of the cell cycle machinery. Second, the Cx37 hemichannel is active in at least some cell types under physiological conditions (Wong et al., 2006). And third, Cx37 exerts its growth suppressive effect at low cell densities where cell–cell contact, and consequently gap junction channel formation, is infrequent. A third possibility for how a functional Cx37 channel might influence the cell cycle machinery is that the closed configuration of Cx37, or at least the configuration of Cx37-T154A, precludes or favors interaction of Cx37 with a crucial cell cycle regulatory element. This latter possibility is particularly interesting to consider in the context of the proteins comprising the gap junction proteome (Laird, 2010) and whether these interacting proteins rely on specific configurations of the channel protein.
Connexins participate in multiple types of protein–protein interactions, many of which are already known to influence channel function. Most of the data on how such interactions influence gap junction channel gating derive from studies of Cx43, Cx32 or Cx26, but given similarities in connexin, connexon and gap junction structure it is generally assumed that other members of the connexin family will follow a common set of rules. It appears that both the N-terminal (NT) and CT domains of connexins are able to participate in channel gating (Kyle et al., 2008; Liu et al., 1993; Maeda et al., 2009; Paulauskas et al., 2009; Sorgen et al., 2004). Gating is proposed to involve interactions with self [NT–NT (Maeda et al., 2009); CT–CT (Sorgen et al., 2004)] or with the cytoplasmic loop (CL) domain [e.g. CT–CL (Liu et al., 1993)]. The CT also interacts with a variety of other proteins [e.g. kinases, phosphatases, scaffolding and cytoskeletal proteins and transcription factors (for a review, see Laird, 2010)], but it is not clear whether these interactions influence gating or whether they rely on a specific conformation of the protein (e.g. open or closed channels). Although connexins mutated at the T154 site form gap junction plaques that are normal in appearance, it is not clear whether the interactions with other cytosolic proteins are preserved or whether the tertiary structure and, consequently, plaque stability is comparable to that of the wild type (Ambrosi et al., 2010). This issue is further complicated by the absence of a clear understanding of gating stoichiometry – for example, can a gap junction channel (or hemichannel) be closed by a single CT interacting with the CL, or is interaction of all six CTs required (Bao et al., 2007)?
In the case of Cx37, comparatively little is known about its binding partners and their possible effects on channel and growth suppressive function. The CT confers some pH-dependent gating sensitivity (Stergiopoulos et al., 1999), consistent with CT–CL interaction. Cx37 also interacts with Cx43 to form functional heteromeric channels whose modified gating properties (Brink et al., 1997) are also consistent with possible CT–CL interactions. As described for other connexins, the structure of the NT of Cx37 (and probable NT–NT interaction) is also crucial for channel function (Kyle et al., 2008). Cx37 is a phosphoprotein (Larson et al., 2000; Traub et al., 1998); consensus sequence analysis of the CT predicts this protein has a high probability of being phosphorylated by growth related kinases, a prediction confirmed by in vitro phosphorylation experiments and kinase-blocking studies (J.M.B., unpublished data). Differential phosphorylation of these sites might explain the variety of open state conductances displayed by Cx37 in Rin cells (Burt et al., 2008; Kumari et al., 2001; Larson et al., 2000; Reed et al., 1993), and represent sites for control of growth related functions (Morel et al., 2010). Cx37 might also interact with elements of signaling cascades activated by cytokines and growth factors, such as endothelial nitric oxide synthase (eNOS) (Pfenniger et al., 2010). None of the currently identified members of the Cx37 interactome provide an obvious explanation for how expression of functional Cx37 can prolong the durations of all phases of the cell cycle and confer, onto an otherwise insensitive cell type, sensitivity to serum deprivation and G1–S growth arrest. Thus, despite the sufficiency of the CT domains of other connexins in conferring their growth suppressive effects, our data show that the CT of Cx37 is not sufficient for growth suppression in Rin cells. Moreover, despite the potential for its interaction with other entities involved in growth regulation, it remains unclear as to whether the CT domain is even necessary for Cx37-mediated growth suppression and, if so, whether its phosphorylation might modulate its effects on cell cycle progression and growth.
In summary, our data indicate that growth suppression of Rin cells by Cx37 requires that Cx37 form channels that are able to adopt an open configuration; the closed channel, even though it localizes comparably to the wild-type channel and has a normal CT domain, does not suppress proliferation of Rin cells. Future studies will need to address whether the CT domain is necessary for growth suppression, which seems likely, but our data clearly indicate that the CT domain is not sufficient to mediate growth suppression. It will also be interesting to determine whether the hemichannel is growth suppressive and, if so, whether permeants leaving or entering through the channel are crucial to Cx37-mediated growth suppression.
Materials and Methods
Antibodies and reagents
All general chemicals, unless otherwise noted, were purchased from Sigma–Aldrich. Anti-Cx37 antibody [αCx37-18264 (Simon et al., 2006)] was used for both immunoblots and immunocytochemistry. Anti-Cx43 antibody was from Sigma (C6219); Cy3-conjugated anti-rabbit-IgG (Jackson ImmunoResearch) was used as the secondary antibody in immunocytochemistry; horseradish peroxidase (HRP)-conjugated secondary antibodies (Amersham) and enhanced chemiluminescence strategies were used in immunoblotting (SuperSignal West Dura and Femto Systems mixed at 2:1 ratio; Pierce). Cy5-conjugated streptavidin was from Jackson ImmunoResearch.
Cell culture and expression vectors
Communication-deficient Rin cells [Rin1046-38 (Clark et al., 1990; Gazdar et al., 1980)] obtained from R. Lynch (University of Arizona) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) or 10% FetalPlex (FP) (both from Gemini Bioproducts, Sacramento, CA) and antibiotics (300 μg/ml penicillin and 500 μg/ml streptomycin) at 37°C in a humidified, 5% CO2 incubator. This RPMI medium was further supplemented with 300 μg/ml of G418 (GIBCO-Invitrogen) and 100 μg/ml hygromycin, as appropriate, for culturing iRin cells, which constitutively express rTetR (Tet-On; Clontech, Mountain View, CA), iRin37 cells, which inducibly express Cx37, and iRin37-T154A cells, which inducibly express Cx37-T154A. Connexin-deficient MDCK cells and MDCK43 cells [MDCK and MDCK43 (A2 clone) were obtained from Paul Lampe (Solan and Lampe, 2007)] were cultured in DMEM supplemented with 10% FBS and 200 μg/ml hygromycin, as appropriate, and maintained at 37°C in a humidified, 5% CO2 incubator.
Generation of mutant connexins
The T154A mutation was introduced into pTRE2h-mCx37 (Burt et al., 2008) using the QuikChange Site-Directed Mutagenesis kit (Stratagene, San Diego, CA) and oligonucleotide primers: 5′-GCGCTCATGGGTGCCTATGTGGTCAG-3′ and 5′-CTGACCACATAGGCACCCATGAGCGC-3′ (Operon Biotechnologies, Huntsville, AL) following the manufacturer's instructions and the correct sequence confirmed (Genomic Analysis and Technology Core at University of Arizona). iRin cells were stably transfected with the resulting pTRE2h-mCx37-T154A plasmid using Lipofectamine (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Stably transfected cells were selected in 100 μg/ml hygromycin and dilution subcloned. Cx37-T154A was also cloned into pcDNA3.1 hygromycin vector (pcDNA3.1h-mCx37-T154A) using the QuikChange Site-Directed Mutagenesis kit with the above oligonucleotide plasmids. pcDNA3.1h-mCx37-T154A was used for transient transfection of MDCK and MDCK43 cells.
Immunoblotting
Total cell protein and Triton X-100-insoluble protein were isolated as previously described (Burt et al., 2008) and their protein content determined using the BCA assay (Pierce Chemical). Samples (15–30 μg total protein, 25–30 μg triton insoluble protein) were electrophoresed on 12% SDS-PAGE gels (Bio-Rad) and transferred onto nitrocellulose. Blots were blocked with 5% NFDM and expressed connexins detected using Kodak Imagestation 2000.
Dual whole-cell voltage clamping
iRin37 and iRin37-T154A cells were plated at low density on coverslips and in some cases transfected with plasmids encoding GFP and mCx43. After 24 hours, cells were treated for 24 hours with doxycycline (2 μg/ml). Coverslips were mounted in a custom-made chamber and the cells bathed in external solution containing (in mM): 142.5 NaCl, 4 KCl, 1 MgCl2, 5 glucose, 2 sodium pyruvate, 10 HEPES, 15 CsCl, 10 TEA-Cl, 1 BaCl2, and 1 CaCl2, pH 7.2 and osmolarity of 330 mosM (external solution). Junctional conductance was determined as previously described (Burt et al., 2008; Kurjiaka et al., 1998) using dual whole-cell voltage-clamp techniques and patch pipets containing (in mM) 124 KCl, 14 CsCl, 9 HEPES, 9 EGTA, 0.5 CaCl2, 5 glucose, 9 TEA-Cl, 3 MgCl2, and 5 disodium ATP, pH 7.2 and osmolarity of 326 mosM (internal solution). Transjunctional voltages as large as 25 mV, sufficient to detect Cx37-comprised channels without inducing their closure, were applied to reveal the activity of single channels when present.
Immunocytochemistry
Cx37 was stained and localized relative to the plasma membrane in Rin cells after labeling proteins in the membrane with a lysine-biotinylation reagent. iRin37 and iRin37-T154A cells were plated at low density onto glass coverslips and 24 hours later treated for 24 hours with doxycycline. MDCK parental cells were plated at low density onto glass coverslips and 24 hours later transfected with either pcDNA3.1h-mCx37 or pcDNA3.1h-mCx37-T154A. Twenty-four hours after induction (iRin) or transfection (MDCK), coverslips with Rin cells were washed with Dulbecco's phosphate buffered saline containing divalent cations (D-PBS) and then treated for 1 hour at 4°C with 1 mM NHS-SS-Biotin in D-PBS (Thermo Fisher Scientific, Waltham MA) to label surface proteins. Excess biotin was quenched by washing with 100 mM glycine in D-PBS. Cells (Rin and MDCK) were then fixed in 100% methanol (−20°C, 5 minutes), exposed to 0.2% Triton X-100 in divalent cation free PBS (DCF-PBS) for 30 minutes, rinsed with DCF-PBS, washed with 0.5 M NH4Cl for 15 minutes, rinsed with DCF-PBS and finally exposed to blocking reagent (4% fish skin gelatin, 1% normal goat serum, and 0.1% Triton X-100 in PBS) for 1 hour at room temperature. For immunofluorescent detection of cell membrane and connexin expression, cells were incubated with primary antibody (1–2 hours, room temperature) followed by secondary antibody (1 hour, room temperature) with intervening washes with 0.1% Triton X-100 in DCF-PBS. Cy3-conjugated secondary antibody (diluted 1:200 in blocking reagent) was used to detect Cx37 and Cy5-conjugated Streptavidin (diluted 1:250 in blocking reagent) was used to detect biotin labeled membrane proteins in the Rin cells. Labeling was then visualized using a Zeiss LSM510meta-NLO confocal/multiphoton fluorescence microscope and 514 nm laser wavelength for Cy3 or 633 nm for Cy5 detection.
Co-immunoprecipitation
MDCK43 cells were transiently transfected [using lipofectamine (Invitrogen)] with either mCx37 or mCx37-T154A (using the pcDNA3.1-hygromycin vector). After 24–48 hours, total protein was isolated using non-denaturing buffer [1% Triton X-100, 50 mM Tris-HCl, pH 7.4, 300 mM NaCl, 5 mM EDTA, 0.02% (w/v) NaN3 and protease inhibitors freshly added: 5 mM NaF, 0.025 mM Na3VO4, 2 mM phenylmethyl sulfonyl fluoride (PMSF), and 1 tablet PIC/50 ml (Roche Diagnostics)], DNA sheared and the sample centrifuged for 25 minutes at 16,000 g at 4°C. Supernatant was pre-cleared by incubating with protein-A–Sepharose beads (Pierce Chemical) and either anti-Cx37 or -Cx43 antibody was added to the sample. After 1–2 hours incubation at 4°C, protein-A3 Sepharose beads were added and the mixture mixed overnight at 4°C. The beads were harvested, washed, resuspended in sample buffer and heated, and the supernatant immunoblotted for detection of Cx37 and Cx43. In control experiments, total protein samples isolated (under non-denaturing conditions) from cell lines that expressed only Cx37 or Cx43 were mixed after isolation but before immunoprecipitation. Lack of cross reactivity of both Cx43 and Cx37 antibodies was confirmed by western blotting, method described above, using Cx37-GST and Cx43-GST as the sample protein or protein isolated from cells expressing only one of the full length proteins.
Dye injection studies
MDCK43 cells were plated at low density onto glass coverslips and transfected with GFP (pMAX-GFP, Amaxa) or GFP and either Cx37 (pcDNA3.1-mCx37) or Cx37-T154A (pcDNA3.1-mCx37-T154A). 24 to 48 hours after transfection a coverslip was mounted in a custom-made chamber and cells bathed in external solution. Pairs of GFP expressing cells were identified (Olympus IMT2), and one cell of each pair was injected with a solution containing Alexa Fluor 350 (2 mM) and Rhodamine-labeled dextran (0.4 mM). Each injected cell pair was scored as positive or negative (dye detected or not in non-injected cell) for Alexa Fluor 350 dye coupling 20 minutes after injection. Data from cell pairs wherein Rhodamine-labeled dextran was detected in the non-injected cell (indicative of a cytoplasmic bridge rather than a gap junction connecting the cells) were discarded.
Proliferation
Proliferation assays were performed (with FP supplemented medium) on cells plated at low density in seven six-well plates (~3,125 cells/cm2). At 24 hours after plating (day 0), doxycycline (2 μg/ml) was added to the medium in half of the wells in each plate. Over the ensuing 21 days, medium (with or without doxycycline) was refreshed every 2 days, and cells from one plate were harvested (using trypsin) for counting (hemocytometer) every third day.
Serum deprivation and cell cycle analysis
To assess the effects of serum (FP) deprivation on progression through the cell cycle, Rin 1046, iRin37 and iRin37-T154A 3C3 and 3E4 cells were plated (106 cells per 10-cm plate) and maintained in standard culture medium for 24 hours prior to replacing the medium with serum-free medium (controls continue in serum containing medium). After 48 hour, doxycycline (2 μg/ml) was added to all plates. After an additional 24 hours in the serum-free medium containing doxycycline, cells were either harvested for cell cycle analysis or released from serum deprivation by restoration of medium containing 10% FP and doxycycline. Cells released from serum deprivation were harvested and analyzed for cell cycle position 4, 10, 24, 48, 72 and 96 hours after serum restoration. Results from serum-deprived cells were compared with those from cells never deprived of serum.
Statistics
Statistical comparisons of all data were performed using unpaired, two-tailed Student's t-tests. Significant differences were taken as P<0.05. Error bars represent mean ± s.e.m.
Supplementary Material
Acknowledgments
The authors thank J. F. Ek-Vitorin for his thoughtful suggestions and assistance throughout this project. Cell cycle data were collected at the AZCC/ARL-Division of Biotechnology Cytometry Core Facility, supported by Cancer Center Support Grant (CCSG – CA 023074). Confocal microscopy imaging was done at the Cellular Imaging Facility Core in the Arizona Research Labs Biotechnology center. This work was supported by NIH grants RO1 HL058732 (J.M.B.) and RO1 HL064232 (A.M.S.) and T32 HL07249 (M.E.G.). Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/14/2448/DC1
References
- Alcolea S., Jarry-Guichard T., de Bakker J., Gonzalez D., Lamers W., Coppen S., Barrio L., Jongsma H., Gros D., Van Rijen H. (2004). Replacement of connexin40 by connexin45 in the mouse: impact on cardiac electrical conduction. Circ. Res. 94, 100-109 [DOI] [PubMed] [Google Scholar]
- Ambrosi C., Boassa D., Pranskevich J., Smock A., Oshima A., Xu J., Nicholson B. J., Sosinsky G. E. (2010). Analysis of four connexin26 mutant gap junctions and hemichannels reveals variations in hexamer stability. Biophys. J. 98, 1809-1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X., Lee S. C., Reuss L., Altenberg G. A. (2007). Change in permeant size selectivity by phosphorylation of connexin 43 gap-junctional hemichannels by PKC. Proc. Natl. Acad. Sci. USA 104, 4919-4924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beahm D. L., Oshima A., Gaietta G. M., Hand G. M., Smock A. E., Zucker S. N., Toloue M. M., Chandrasekhar A., Nicholson B. J., Sosinsky G. E. (2006). Mutation of a conserved threonine in the third transmembrane helix of alpha- and beta-connexins creates a dominant-negative closed gap junction channel. J. Biol. Chem. 281, 7994-8009 [DOI] [PubMed] [Google Scholar]
- Brink P. R., Cronin K., Banach K., Peterson E., Westphale E. M., Seul K. H., Ramanan S. V., Beyer E. C. (1997). Evidence for heteromeric gap junction channels formed from rat connexin43 and human connexin37. Am. J. Physiol. 273, C1386-C1396 [DOI] [PubMed] [Google Scholar]
- Burt J. M., Nelson T. K., Simon A. M., Fang J. S. (2008). Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am. J. Physiol. Cell Physiol. 295, C1103-C1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S. A., Burnham B. L., Chick W. L. (1990). Modulation of glucose-induced insulin secretion from a rat clonal beta-cell line. Endocrinology 127, 2779-2788 [DOI] [PubMed] [Google Scholar]
- Dang X., Jeyaraman M., Kardami E. (2006). Regulation of connexin-43-mediated growth inhibition by a phosphorylatable amino-acid is independent of gap junction-forming ability. Mol. Cell. Biochem. 289, 201-207 [DOI] [PubMed] [Google Scholar]
- Doble B. W., Dang X., Ping P., Fandrich R. R., Nickel B. E., Jin Y., Cattini P. A., Kardami E. (2004). Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J. Cell Sci. 117, 507-514 [DOI] [PubMed] [Google Scholar]
- Ek-Vitorin J. F., Burt J. M. (2005). Quantification of gap junction selectivity. Am. J. Physiol. Cell Physiol. 289, C1535-C1546 [DOI] [PubMed] [Google Scholar]
- Fu C. T., Bechberger J. F., Ozog M. A., Perbal B., Naus C. C. (2004). CCN3 (NOV) interacts with connexin43 in C6 glioma cells: possible mechanism of connexin-mediated growth suppression. J. Biol. Chem. 279, 36943-36950 [DOI] [PubMed] [Google Scholar]
- Gazdar A. F., Chick W. L., Oie H. K., Sims H. L., King D. L., Weir G. C., Lauris V. (1980). Continuous, clonal, insulin- and somatostatin-secreting cell lines established from a transplatable rat islet cell tumor. Cell Biol. 77, 3519-3523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellhaus A., Dong X., Propson S., Maass K., Klein-Hitpass L., Kibschull M., Traub O., Willecke K., Perbal B., Lye S. J., et al. (2004). Connexin43 interacts with NOV: a possible mechanism for negative regulation of cell growth in choriocarcinoma cells. J. Biol. Chem. 279, 36931-36942 [DOI] [PubMed] [Google Scholar]
- Goodenough D. A., Paul D. L. (2003). Beyond the gap: functions of unpaired connexon channels. Nat. Rev. Mol. Cell Biol. 4, 285-294 [DOI] [PubMed] [Google Scholar]
- Harris A. L. (2001). Emerging issues of connexin channels: biophysics fills the gap. Q. Rev. Biophys. 34, 325-472 [DOI] [PubMed] [Google Scholar]
- Johnstone S. R., Best A. K., Wright C. S., Isakson B. E., Errington R. J., Martin P. E. (2010). Enhanced connexin 43 expression delays intra-mitotic duration and cell cycle traverse independently of gap junction channel function. J. Cell. Biochem. 110, 772-782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardami E., Dang X., Iacobas D. A., Nickel B. E., Jeyaraman M., Srisakuldee W., Makazan J., Tanguy S., Spray D. C. (2007). The role of connexins in controlling cell growth and gene expression. Prog. Biophys. Mol. Biol. 94, 245-264 [DOI] [PubMed] [Google Scholar]
- King T. J., Bertram J. S. (2005). Connexins as targets for cancer chemoprevention and chemotherapy. Biochim. Biophys. Acta 1719, 146-160 [DOI] [PubMed] [Google Scholar]
- Kruger O., Plum A., Kim J. S., Winterhager E., Maxeiner S., Hallas G., Kirchhoff S., Traub O., Lamers W. H., Willecke K. (2000). Defective vascular development in connexin 45-deficient mice. Development 127, 4179-4193 [DOI] [PubMed] [Google Scholar]
- Kumari S. S., Varadaraj K., Valiunas V., Brink P. R. (2001). Site-directed mutations in the transmembrane domain M3 of human connexin37 alter channel conductance and gating. Biochem. Biophys. Res. Commun. 280, 440-447 [DOI] [PubMed] [Google Scholar]
- Kurjiaka D. T., Steele T. D., Olsen M. V., Burt J. M. (1998). Gap junction permeability is diminished in proliferating vascular smooth muscle cells. Am. J. Physiol. 275, C1674-C1682 [DOI] [PubMed] [Google Scholar]
- Kyle J. W., Minogue P. J., Thomas B. C., Domowicz D. A., Berthoud V. M., Hanck D. A., Beyer E. C. (2008). An intact connexin N-terminus is required for function but not gap junction formation. J. Cell Sci. 121, 2744-2750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird D. W. (2010). The gap junction proteome and its relationship to disease. Trends Cell Biol. 20, 92-101 [DOI] [PubMed] [Google Scholar]
- Larson D. M., Seul K. H., Berthoud V. M., Lau A. F., Sagar G. D., Beyer E. C. (2000). Functional expression and biochemical characterization of an epitope-tagged connexin37. Mol. Cell Biol. Res. Commun. 3, 115-121 [DOI] [PubMed] [Google Scholar]
- Liu S., Taffet S., Stoner L., Delmar M., Vallano M. L., Jalife J. (1993). A structural basis for the unequal sensitivity of the major cardiac and liver gap junctions to intracellular acidification: the carboxyl tail length. Biophys. J. 64, 1422-1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S., Nakagawa S., Suga M., Yamashita E. (2009). Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 458, 597-602 [DOI] [PubMed] [Google Scholar]
- Morel S., Burnier L., Roatti A., Chassot A., Roth I., Sutter E., Galan K., Pfenniger A., Chanson M., Kwak B. R. (2010). Unexpected role for the human Cx37 C1019T polymorphism in tumour cell proliferation. Carcinogenesis 31, 1922-1931 [DOI] [PubMed] [Google Scholar]
- Musil L. S., Goodenough D. A. (1991). Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J. Cell Biol. 115, 1357-1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulauskas N., Pranevicius M., Pranevicius H., Bukauskas F. F. (2009). A stochastic four-state model of contingent gating of gap junction channels containing two “fast” gates sensitive to transjunctional voltage. Biophys. J. 96, 3936-3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenniger A., Derouette J. P., Verma V., Lin X., Foglia B., Coombs W., Roth I., Satta N., Dunoyer-Geindre S., Sorgen P., et al. (2010). Gap junction protein Cx37 interacts with endothelial nitric oxide synthase in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 30, 827-834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum A., Hallas G., Magin T., Dombrowski F., Hagendorff A., Schumacher B., Wolpert C., Kim J., Lamers W. H., Evert M., et al. (2000). Unique and shared functions of different connexins in mice. Curr. Biol. 10, 1083-1091 [DOI] [PubMed] [Google Scholar]
- Reaume A. G., De Sousa P. A., Kulkarni S., Langille B. L., Zhu D., Davies T. C., Juneja S. C., Kidder G. M., Rossant J. (1995). Cardiac malformation in neonatal mice lacking connexin43. Science 267, 1831-1834 [DOI] [PubMed] [Google Scholar]
- Reed K. E., Westphale E. M., Larson D. M., Wang H.-Z., Veenstra R. D., Beyer E. C. (1993). Molecular cloning and functional expression of human connexin37, an endothelial cell gap junction protein. J. Clin. Invest. 91, 997-1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer S. S., Xu Y. T., Nelles E., Fischbeck K., Willecke K., Bone L. J. (1998). Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24, 8-20 [DOI] [PubMed] [Google Scholar]
- Simon A. M., Goodenough D. A., Li E., Paul D. L. (1997). Female infertility in mice lacking connexin 37. Nature 385, 525-529 [DOI] [PubMed] [Google Scholar]
- Simon A. M., Chen H., Jackson C. L. (2006). Cx37 and Cx43 localize to zona pellucida in mouse ovarian follicles. Cell Commun. Adhes. 13, 61-77 [DOI] [PubMed] [Google Scholar]
- Sohl G., Willecke K. (2004). Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228-232 [DOI] [PubMed] [Google Scholar]
- Solan J. L., Lampe P. D. (2007). Key connexin 43 phosphorylation events regulate the gap junction life cycle. J. Membr. Biol. 217, 35-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L., Lampe P. D. (2009). Connexin43 phosphorylation: structural changes and biological effects. Biochem. J. 419, 261-272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorgen P. L., Duffy H. S., Spray D. C., Delmar M. (2004). pH-dependent dimerization of the carboxyl terminal domain of Cx43. Biophys. J. 87, 574-581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergiopoulos K., Alvarado J. L., Mastroianni M., Ek-Vitorin J. F., Taffet S. M., Delmar M. (1999). Hetero-domain interactions as a mechanism for the regulation of connexin channels. Circ. Res. 84, 1144-1155 [DOI] [PubMed] [Google Scholar]
- Traub O., Hertlein B., Kasper M., Eckert R., Krisciukaitis A., Hulser D., Willecke K. (1998). Characterization of the gap junction protein connexin37 in murine endothelium, respiratory epithelium, and after transfection in human HeLa cells. Eur. J. Cell Biol. 77, 313-322 [DOI] [PubMed] [Google Scholar]
- Weber P. A., Chang H. C., Spaeth K. E., Nitsche J. M., Nicholson B. J. (2004). The permeability of gap junction channels to probes of different size is dependent on connexin composition and permeant-pore affinities. Biophys. J. 87, 958-973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T. W. (2002). Unique and redundant connexin contributions to lens development. Science 295, 319-320 [DOI] [PubMed] [Google Scholar]
- Wong C. W., Christen T., Roth I., Chadjichristos C. E., Derouette J. P., Foglia B. F., Chanson M., Goodenough D. A., Kwak B. R. (2006). Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 12, 950-954 [DOI] [PubMed] [Google Scholar]
- Zhang Y. W., Kaneda M., Morita I. (2003a). The gap junction-independent tumor-suppressing effect of connexin 43. J. Biol. Chem. 278, 44852-44856 [DOI] [PubMed] [Google Scholar]
- Zhang Y. W., Nakayama K., Nakayama K., Morita I. (2003b). A novel route for connexin 43 to inhibit cell proliferation: negative regulation of S-phase kinase-associated protein (Skp 2). Cancer Res. 63, 1623-1630 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.