

Abstract
Protein 4.1R is a membrane-cytoskeleton adaptor protein that has diverse roles in controlling the cell surface expression and/or function of transmembrane proteins, and in organizing F-actin. 4.1R is expressed in keratinocytes, but its role in these cells has not been explored. Here, we have investigated the role of 4.1R in skin using 4.1R−/− mice. Cell adhesion, spreading, migration and motility were significantly impaired in 4.1R−/− keratinocytes, and 4.1R−/− mice exhibited defective epidermal wound healing. Cultured 4.1R−/− keratinocytes on fibronectin failed to form actin stress fibres and focal adhesions. Furthermore, in the absence of 4.1R, the surface expression, and consequently the activity of β1 integrin were reduced. These data enabled the identification of a functional role for protein 4.1R in keratinocytes by modulating the surface expression of β1 integrin, possibly through a direct association between 4.1R and β1 integrin.
Key words: 4.1R, β1 integrin, Cell motility, Keratinocyte
Introduction
The processes of maintaining and repairing the skin throughout the lifetime of a mammalian organism are of fundamental importance (Blanpain and Fuchs, 2009; Fuchs and Raghavan, 2002). In the skin, a layer of squamous stratified epithelium (the epidermis) is separated from an underlying layer of connective tissue by a basement membrane. The only cells capable of division in the epidermis are keratinocytes of the innermost layer attached to the basement membrane (Fuchs, 1990; Fuchs, 1994). Terminal differentiation of these keratinocytes provides cells of the suprabasal layers. In response to a wound, keratinocytes from the wound margin migrate to the wound bed and proliferate to form the new epithelium (Martin, 1997). The processes of keratinocyte proliferation, differentiation and migration therefore govern skin morphology and homeostasis: loss of control of these processes can lead to defective healing and tumorigenesis (Raja et al., 2007).
Similarly to most motile cells, the cell movement of keratinocytes is controlled by the dynamic rearrangements of the actin cytoskeleton (Kirfel and Herzog, 2004). In cultured motile cells, polymerization of actin forms actin stress fibers, filopodia and lamellipodia (Pollard and Borisy, 2003). At the tips of actin stress fibers, integrins cluster with a group of integrin-interacting proteins (such as talin, vinculin and paxillin) to form focal adhesions. The formation of actin stress fibers and focal adhesions allows cells to adhere, spread and migrate efficiently (Calderwood et al., 1999; Danen et al., 2005; Galbraith et al., 2007; Geiger and Bershadsky, 2001; Kaverina et al., 2002).
4.1R (also known as protein 4.1R) is the prototypical member of the FERM domain-containing superfamily of proteins along with ezrin, radixin and moesin (Chishti et al., 1998; Diakowski et al., 2006). First discovered in erythrocytes as a major cytoskeleton protein, 4.1R is now viewed as an adaptor protein that links a diverse range of transmembrane proteins (among others cell adhesion molecules, ion channels and receptors) and the cytoskeleton. The functions of 4.1R have been revealed from studies of mutations in the human EPB41 gene, by knockout of its mouse ortholog, and by overexpression or knockdown experiments in cells expressing the protein. The first function to be revealed was a requirement for 4.1R in erythrocyte membranes: in the absence of 4.1R they are mechanically unstable because of altered actin skeleton organization (Shi et al., 1999), as well as the loss or reduction in number of several transmembrane proteins (Salomao et al., 2008). Subsequently, it became clear that 4.1R is required for such diverse functions as control of ion channel activities in the heart (Stagg et al., 2008; Taylor-Harris et al., 2005), involvement in the organization of stomach epithelia adherens junctions (Yang et al., 2009) and modulation of T-cell antigen-receptor-mediated signal transduction in CD4+ T cells (Kang et al., 2009b).
4.1R was detected in skin keratinocytes by immunological methods and molecular cloning nearly two decades ago (Nunomura et al., 1997), but its function has not been further explored. In the present study, using 4.1R−/− mice, we investigate the function of 4.1R in keratinocytes, both in vitro and in vivo. Our results demonstrate a crucial role for 4.1R in the processes of keratinocyte motility in vitro, and in wound healing in vivo, and show that it does so, in part, by modulating the surface expression of β1 integrin.
Results
Expression of 4.1R in keratinocytes
As the first step to explore the role of 4.1R in keratinocytes, we examined the expression of 4.1R by RT-PCR and western blotting analysis. RT-PCR analysis revealed four isoforms: ATG1 4.1R Δexons14,15; ATG1 4.1R Δexons14,15,17B; ATG2 4.1R Δexons14,15; and ATG2 4.1R Δexons14,15,17B. The exon composition of these isoforms is depicted in Fig. 1A. Consistent with RT-PCR results, western blots probed with anti-4.1R-exon18 antibody revealed four bands (Fig. 1Ba): two highly expressed proteins migrating at ~80 kDa and ~115 kDa and two low abundance proteins at ~135 kDa and ~170 kDa. Western blot using an antibody against the head-piece region of 4.1R that only recognizes proteins initiating at ATG1 revealed the two upper bands (Fig. 1Bb), implying that these two high molecular mass polypetides (~135 kDa and ~170 kDa) are isoforms initiating at ATG1, whereas the two smaller polypeptides (~80 kDa and ~115 kDa) are isoforms initiating at ATG2. The specificity of these bands was supported by the absence of all four bands in the 4.1R−/− samples (Fig. 1Ba,Bb). We surmise the difference between the two high and the low molecular mass bands is the inclusion or exclusion of sequences encoded by exon 17B. Additionally, 4.1G, 4.1N and 4.1B, three other members of the protein 4.1 family, are also expressed in keratinocytes. Interestingly, both 4.1G and 4.1N are upregulated in 4.1R−/− cells (Fig. 1Bc,Bd), whereas 4.1B is unchanged (Fig. 1Be). Quantitative analysis from three independent experiments revealed a ~1.5-fold and ~2.5-fold increase for 4.1G and 4.1N, respectively (Fig. 1Bf). GAPDH was used as a control in all western blot analysis.
Fig. 1.

Expression and localization of 4.1R in keratinocytes. (A) Schematic view of 4.1R isoforms expressed in keratinocytes. The top diagram shows a general view of 4.1R exon organization, with alternatively spliced (black), constitutive (grey) and non-coding exons (open box) shown. Start codons are indicated. The lower diagrams show the exon composition of four isoforms revealed by sequencing of RT-PCR amplicons. (B) Immunoblot analysis of protein 4.1 members in keratinocytes. Total lysates (30 μg protein) were probed with polyclonal rabbit antibodies against 4.1R exon18 (a), 4.1R head piece (b), 4.1G head piece (c), 4.1N head piece (d) and 4.1B head piece (e). Quantitative analysis of immunoblot results from three independent experiments is shown in (f). GAPDH was used as loading control. (C) Confocal images of transfected GFP–4.1R. GFP–ATG2-4.1RΔexons14,15,17B was transiently expressed in live keratinocytes. A representative example of four images from a stack of 60 taken over a period of 10 hours, with a 10 minute interval between each panel is shown. Scale bar: 20 μm. (D) Immunofluorescence staining of endogenous 4.1R. Keratinocytes were cultured on fibronectin-coated surface for 36 hours, the cells were then fixed and stained using anti-4.1R-exon16 antibody.
Localization of 4.1R in keratinocytes
The localization of the two major 4.1R proteins (~80 kDa and ~115 kDa) was investigated by transiently expressing GFP fusion proteins and filming live cells. The ~80 kDa isoform was localized in the cytoplasm, as well as at the leading edge of a moving cell (Fig. 1C). By contrast, the ~115 kDa isoform was primarily retained in the nucleus (data not shown). The location of endogenous 4.1R was examined by immunofluorescence staining using anti-4.1R-exon16 antibody. Fig. 1D shows that the endogenous 4.1R was seen in cytoplasm and leading edge (probably the ~80 kDa isoform) as well as the nucleus (probably the ~115 kDa isoform). No staining was seen with secondary antibody only (data not shown).
Decreased adhesion and restricted spreading of 4.1R−/− keratinocytes
The finding that the ~80 kDa 4.1R isoform is localized at the leading edge of the moving cells suggests a possible role of 4.1R in cell motility and movement. To test this, we first examined the adhesion of the cells to the fibronectin-coated surface. After 4 hours, the adhesion of 4.1R−/− cells to the surface was ~70% less than that of 4.1R+/+ cells (Fig. 2A). We also examined the spreading of cells on a fibronectin-coated surface. After 12 hours of incubation, the extent of spreading of 4.1R−/− cells was significantly less than that of the 4.1R+/+ cells (Fig. 2B). Quantitative analysis revealed a ~50% reduction in the spreading area of the 4.1R−/− cells compared with that of 4.1R+/+ cells (Fig. 2C).
Fig. 2.
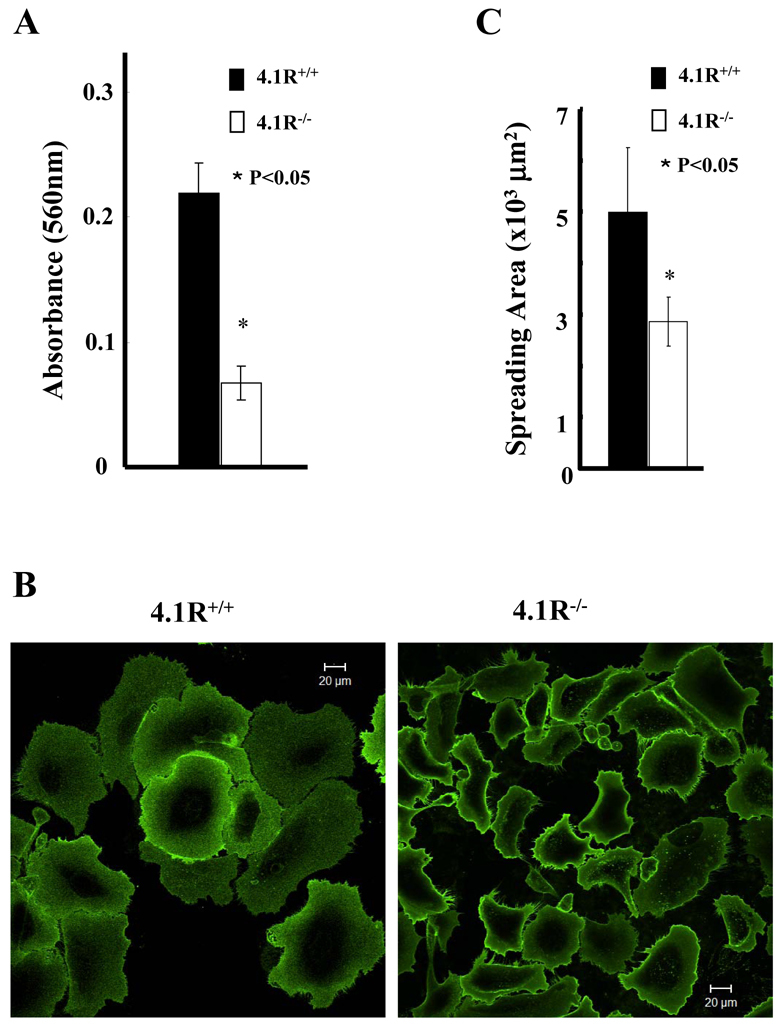
Impaired adhesion and spreading of 4.1R−/− keratinocytes on fibronectin. (A) Cells were plated on fibronectin-coated 96-well plates and incubated for 4 hours. The adherent cells were stained with Crystal Violet and the staining intensity was quantified by spectrophotometry at 560 nm. The results are mean ± s.e.m. of three independent experiments. (B) Cells were plated on fibronectin-coated four-well chambers and allowed to spread for 12 hours. The cells were labeled with Alexa-Fluor-488-conjugated WGA (Invitrogen) and the images were collected using a Zeiss Axiophot wide-field epifluorescence microscope. Scale bars: 20 μm. (C) The mean surface area from 35 individual cells was calculated using LSM 5 Pascal software. The data shown are mean ± s.e.m. of three experiments. One-tailed Student's t-tests were applied to test the statistical significance of the data.
Impaired migration of 4.1R−/− keratinocytes
We next examined the directional migration of keratinocytes by in vitro wound-healing assay and by Transwell migration assay. Fig. 3A,B shows the results of the migration of keratinocytes toward a pseudo wound inflicted on a monolayer of cells at 12 hours and beyond. These results reveal that the closure of the wounded area of 4.1R−/− cells occurred at a much-reduced rate. At around 12 hours post wounding, although the closure of the wound area of 4.1R+/+ cells was nearly complete, >50% of the wounded area of 4.1R−/− cells was still not closed. Representative video clips showing the in vitro wound-healing process of 4.1R+/+ or 4.1R−/− cells are shown in supplementary material Movies 1 and 2, respectively. 4.1R−/− keratinocytes migrated through a fibronectin matrix in 8-μm-diameter pore Transwell cell culture inserts approximately six times slower than was observed with 4.1R+/+ cells (Fig. 3C).
Fig. 3.

Impaired directional migration and motility of 4.1R−/− keratinocytes. (A) A pseudo-wound was inflicted in a >90% confluent monolayer of keratinocytes. ‘Wound areas’ were filmed every 20 minutes for 16 hours. The representative images at 12 hours are shown. Scale bar: 50 μm. (B) The wounded area was measured using ImageJ for each representative time point. The data shown are from six experiments. Representative video clips showing the in vitro wound healing process of 4.1R+/+ or 4.1R−/− cells are shown in supplementary material Movie 1 and Movie 2, respectively. (C) 8-μm-diameter pore Transwell cell culture inserts were placed in six-well plates, the bottom of which was coated with fibronectin. Cells were seeded on top of the insert and incubated for 12 hours. The cells migrated to the bottom of the well were fixed and stained with Crystal Violet. Absorbance was read using a multi-well plate reader at 560 nm wavelength. The averages from three experiments are shown ± s.e.m. (D) Live-cell images were obtained every 5 minutes over a period of 3 hours (60 images in total). Each track represents an individual cell; red tracks are cells migrating negatively along the y-axis, whereas black tracks indicate cells migrating positively. The representative video clips showing the motility of an individual 4.1R+/+ or 4.1R−/− cell are shown in supplementary material Movie 3 and Movie 4, respectively. (E) The distance migrated was measured using the cell migration and chemotaxis plug-in (Ibidi) for ImageJ. 4.1R−/− cells (white bars) show a significant increase in overall distance migrated. n=30 (4.1R+/+) and n=41 (4.1R−/−).
Impaired motility of 4.1R−/− keratinocytes
To investigate the cell motility in greater detail, we used live-cell videomicroscopy to monitor the random motility of keratinocytes on a fibronectin substrate. Track plots of randomly migrating keratinocytes revealed that 4.1R+/+ cells migrated in a highly direction manner (Fig. 3D). Once a cell was committed to a given route or path it rarely deviated or changed direction, and continued on that path for the entire period of observation. By contrast, the majority of 4.1R−/− cells migrated around a central point, frequently changing direction and going back on itself, but rarely continuing in one direction for more than several frames. The distance, and hence velocity, migrated by 4.1R−/− cells were also significantly reduced (Fig. 3E). Representative video clips showing the motility of individual 4.1R+/+ or 4.1R−/− cells are shown in supplementary material Movies 3 and 4, respectively.
Impaired epidermal repair in 4.1R−/− mice
To validate our in vitro findings, we performed an in vivo wound-healing assay. After dermal biopsy, wound areas were measured every day for a week and expressed as a percentage of unhealed wound as a function of time. As shown in Fig. 4A, at day 1, there was no significant difference between the two experimental groups, which both had ~50% of the original wound area remaining unhealed. At day 2, wounds from the 4.1R−/− group appeared to be healing slower than the 4.1R+/+ controls, with 4.1R−/− wounds remaining 45% of their original size, whereas the 4.1R+/+ wounds had reduced to around 20% of the original size. Statistically significant differences were observed for the remaining time points, with 4.1R−/− wounds healing much slower than 4.1R+/+ wounds. Fig. 4B shows the representative images of haematoxylin and eosin staining of sections 5 days after wounding and it reveals that 4.1R+/+ mice had re-epithelialized the wound, as shown by a thick epithelium covering completely the wound bed; however, 4.1R−/− mice failed to do so. It should be mentioned that although 4.1R−/− mice were born in sub-mendelian ratios (Kang et al., 2009b), those that were born survived normally. Moreover, no differences in the quality, quantity or distribution of hair or skin from birth until 1 year of age were observed between 4.1R+/+ and 4.1R−/− mice.
Fig. 4.

Impaired re-epithelialization in 4.1R−/− mice. (A) Quantitative assay for wound healing. Wound sizes at any given time point after wounding are expressed as percentage ± s.e.m. of the initial (day 0) wound area. Blue line, 4.1R+/+; red line, 4.1R−/−. n=5. (B) Histological examination of wound. Haemtoxylin and eosin staining of sections 5 days post-wounding. Magnification ×20.
Altered actin stress fiber formation of 4.1R−/− keratinocytes
Our findings thus far clearly identified an important role of 4.1R in cell motility. The motility of the cell is tightly controlled by the actin dynamics. To explore the molecular mechanisms by which lack of 4.1R led to impaired cell motility, we next examined whether the loss of 4.1R has an effect on actin structure using two approaches: staining endogenous actin filaments with Texas-Red–phalloidin, and transfecting cells with exogenous GFP-actin. Fig. 5A shows representative images of endogenous actin filaments. Quantitative analysis reveals that more than 90% of 4.1R+/+ keratinocytes exhibited robust actin stress fibers, which formed a fiber network across the cell body (n=150). In marked contrast, more than 70% of 4.1R−/− cells failed to form the well-defined actin stress fiber network seen in 4.1R+/+ cells (n=150). Actin filaments in 4.1R−/− cells were restricted to the cell periphery, and often ran parallel to the plasma membrane. Similar results were obtained when GFP–actin was transfected into 4.1R+/+ or 4.1R−/− keratinocytes (Fig. 5B).
Fig. 5.

4.1R is required for actin stress fiber and focal adhesion formation in keratinocytes. (A) Endogenous actin filament staining. Keratinocytes were cultured on fibronectin-coated surface for 36 hours, the cells were then fixed and stained for actin using Rhodamine–phalloidin. (B) Exogenously transfected GFP–actin. Keratinocytes were transiently transfected with GFP–actin. 24 hours after transformation, samples were fixed and viewed. All images shown were collected by wide-field epifluorescence microscopy, and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). (C) Immunostaining of focal adhesion proteins. Cells were stained for β1 integrin, talin, paxillin and vinculin. All samples were counterstained for actin with Rhodamine–phalloidin (red) and DAPI (blue). Scale bars: 20 μm. (D) Localization of transiently transfected GFP–vinculin in keratinocytes. EGFP–vinculin was transiently transfected into 4.1R+/+ or 4.1R−/− primary keratinocytes using Fugene6. GFP-positive cells were sorted by FACS. Cells were viewed for fluorescence using a Zeiss Axiophot wide-field epifluorescence microscope. (E) Western blot analysis of actin and focal adhesion proteins. Total lysates (20 μg protein) were probed with indicated antibodies. GAPDH immunoblot is shown as a loading control. (F) Quantitative analysis from three independent experiments.
Focal adhesion proteins fail to localize in the absence of 4.1R
Next, we investigated whether the formation of focal adhesions (FAs) in 4.1R−/− keratinocytes was affected. To do this, we immunostained keratinocytes for three well-characterized structural and signaling components of FAs: talin, paxillin, and vinculin (Fig. 5C). As expected, in 4.1R+/+ cells, all three molecules localized robustly to sharp, distinct complexes at the tips of actin stress fibers. In marked contrast, in 4.1R−/− cells, all these proteins were diffusely stained in the cell body and perinuclear region. The severely compromised ability of 4.1R−/− cells to form focal adhesions was further confirmed by the finding that transfected EGFP-tagged vinculin localized to form focal adhesions in 4.1R+/+ keratinocytes, but failed to do so in 4.1R−/− keratinocytes (Fig. 5D). By scoring cells for proper focal adhesion localization, we observed that approximately 70% of 4.1R−/− keratinocytes (n=85) displayed the abnormal staining patterns described above. Western blot analysis revealed no discernible difference in endogenous expression levels of these proteins between 4.1R+/+ and 4.1R−/− cells (Fig. 5E,F). These findings demonstrate that lack of 4.1R affects assembly, but not the expression levels of these focal adhesion proteins.
Decreased surface expression and activity of β1 integrin in 4.1R−/− keratinocytes
Cell adhesion, spreading, migration and actin dynamic are triggered by the attachment of cells to extracellular matrix through interaction between integrins and fibronectin. To explore the mechanisms responsible for the above-described defects, we examined the surface expression and activity of integrins by flow cytometry. Representative flow cytometry results (Fig. 6A) and quantitative analysis from three independent experiments (Fig. 6B) revealed that whereas the surface expression of β1 integrin was decreased by ~50%, the surface expression levels of α5 and α6 integrins increased by approximately 100% and 30%, respectively, in 4.1R−/− cells. The surface expression of β4 integrin was unchanged. It is well documented that in red blood cells, 4.1R directly associates with several transmembrane proteins and that deficiency of 4.1R results in the decreased expression of its binding partners (Salomao et al., 2008). The above findings suggest a possible direct association between 4.1R and β1 integrin. To further examine the effect of 4.1R deficiency of β1 integrin, we compared the activities of β1 integrin in 4.1R+/+ and 4.1R−/− keratinocytes. We found a ~43% reduction in the activated form of β1 integrin, both in the absence and presence of MnCl2 (Fig. 6D). As the extent of decreased surface expression and activity of β1 integrin was similar, the decreased activity was probably due to the decreased surface expression. Surprisingly, this decreased surface expression was accompanied by a more than fourfold increase in total β1 integrin expression in cells as measured by western blot analysis (Fig. 6E,F).
Fig. 6.

Decreased expression and activity of β1 integrin in 4.1R−/− keratinocytes. (A,B) Expression of integrins in keratinocytes. The surface expression of β1, α5, α6 and β4 integrins was measured by flow cytometry. The representative profiles and quantitative analysis (the mean fluorescence intensity ± s.e.m from three independent experiments) are shown in A and B, respectively. For simplicity, autofluorescence control from only wild-type cells is shown. (C,D) Activity of β1 integrin in keratinocytes. The active form of β1 integrin in the absence or presence of Mn2+ was stained by anti-mouse CD29 and measured by flow cytometry. The representative profiles are shown in C and quantitative data from three experiments is shown in D. (E) Western blot analysis of β1 integrin in keratinocytes. 20 μg protein of total lysates was probed with indicated antibodies. GAPDH immunoblot is shown as a loading control. (F) Quantitative analysis from three independent experiments.
Direct association of β1 integrin and 4.1R
Members of the protein 4.1 superfamily have previously been shown to bind to integrins (Calderwood et al., 1999; McCarty et al., 2005; Tang et al., 2007). To examine whether the effect of 4.1R on β1 integrin in keratinocytes was due to direct association between 4.1R and β1 integrin, we first performed co-immunoprecipitation experiments. Both ATG1 4.1R isoforms were pulled down by β1 integrin antibody (Fig. 7A). Similarly, β1 integrin was pulled down with 4.1R by anti-4.1R antibody (Fig. 7B). In addition, actin was also pulled down with 4.1R. As a negative control, α6 integrin did not co-immunoprecipitate with 4.1R. Furthermore, an in vitro GST pull-down assay showed that both ATG1 4.1R and ATG2 4.1R bound to the cytoplasmic domain of β1 integrin (Fig. 7E) and that β1 integrin bound to the 30 kDa membrane-binding domain of 4.1R (Fig. 7F). These results demonstrate a direct interaction between 4.1R and β1 integrin.
Fig. 7.

Direct association of β1 integrin with 4.1R. (A,B) 4.1R and β1 integrin associate in situ. (A) Immunoprecipitation of β1 integrin. β1 integrin was immunoprecipitated from keratinocytes using anti-β1-integrin. β1 integrin or 4.1R in the immunoprecipitate was detected using anti-β1-integrin antibody or anti-4.1R-exon18 antibody. (B) Immunoprecipitation of 4.1R. 4.1R was immunoprecipitated from keratinocytes using anti-4.1R-exon18 antibody. 4.1R, β1 integrin, α6 integrin or actin in the immunoprecipitate was detected using corresponding antibodies. (C) Schematic presentation of 4.1R domain structure. HP, head piece; MBD, membrane binding domain; SABD, spectrin–actin binding domain; CTD, C-terminal domain. (D) Proteins used in the binding assays. 2 μg of each affinity-purified recombinant protein was separated by 18% SDS-PAGE and stained with Gel Blue. (E) Binding of 4.1R to the cytoplasmic domain of β1 integrin. His-tagged ATG-1 4.1R or ATG-2 4.1R was incubated for 30 minutes at room temperature with GST-tagged cytoplasmic domain of β1 integrin, and binding was assessed by pull-down assay. 4.1R binding was detected by blotting with anti-His antibody. (F) Binding of cytoplasmic domain of β1 integrin to recombinant 4.1R fragments. GST-tagged cytoplasmic domain of β1 integrin was incubated with His-tagged 4.1R fragments. Binding was assayed as above, using anti-His antibody for detection.
Discussion
Protein 4.1R was first identified as a major cytoskeleton component of the erythrocyte membrane, where it serves as a core for both the organization of actin skeleton as well as the assembly of transmembrane proteins (Marfatia et al., 1995; Salomao et al., 2008). A diverse function for 4.1R in non-erythroid cells has also been demonstrated from studies using cell lines (Huang et al., 2004; Krauss et al., 2008; Perez-Ferreiro et al., 2001). We recently started to explore the function of 4.1R using 4.1R−/− mice. We have demonstrated that 4.1R negatively regulates T cell activation by modulating intracellular signal transduction (Kang et al., 2009b). We also have shown that lack of 4.1R resulted in impaired integrity of stomach epithelia adherens junctions (Yang et al., 2009). In the present study, we have defined a previously unrecognized role for 4.1R in the motile behavior of keratinocytes, both in vitro and in vivo.
One striking finding of the present study is the selective reduction in surface expression of β1 integrin in the context of increased expression levels of β1 integrin and its heterodimer partner α5 integrin (probably to compensate for the decreased surface expression of β1 integrin) in the cells lacking 4.1R. These findings imply a crucial role of 4.1R in the surface expression of β1 integrin in keratinocytes. Given the importance of 4.1R in the organization of transmembrane proteins, it is reasonable to speculate that the assembly of β1 integrin into the membrane or its retention in the membrane might have been altered in the absence of 4.1R. It is also interesting to note that although α5 integrin forms a heterodimer with β1 integrin (Watt, 2002), our finding that the surface expression of β1 integrin is decreased, whereas that of α5 integrin is increased, suggests that these two molecules are assembled into the membrane independently.
FERM domains represent genetically versatile protein modules that have adapted for interactions with many membrane proteins and/or lipids in vertebrates: 48 different human genes are listed in Ensembl (Hubbard et al., 2002) that encode FERM domains. These proteins represent a variety of functional categories, among which are the 4.1 proteins (4.1R, 4.1G, 4.1N and 4.1B), the ERM family (ezrin, radixin and moesin), talin-related molecules, PTPH (protein tyrosine phosphatase) proteins and NBL4 (a novel band-4.1-like protein) (Sun et al., 2002). It is interesting to note that several of these are also known to interact directly with integrin subunits. The FERM domain of talin contains a major binding site for several integrins, and is crucial for regulating integrin activation (Calderwood et al., 1999). Radixin binds to αMβ2 integrin and enhances its adhesive properties (Tang et al., 2007), and protein 4.1B is reported to bind to αvβ8 integrin via its highly conserved C-terminal domain (McCarty et al., 2005). These findings together highlight the important roles played by proteins containing FERM domains in integrin signalling.
In keratinocytes, it has been shown that Kindlin-1, another FERM-containing protein, binds to β1 integrin (Herz et al., 2006) and is required for the β1-integrin-mediated signal pathway (Has et al., 2009). We now show that the other three members of protein 4.1 family, 4.1N, 4.1G and 4.1B, are also expressed in keratinocytes. Whereas the expression levels of 4.1B is unaltered in 4.1R−/− keratinocytes, both 4.1N and 4.1G are significantly upregulated, suggesting that 4.1N or/and 4.1G partially compensate for the function of 4.1R. However, it should be noted that in spite of the expression of these 4.1 paralogs, 4.1R-deficient keratinocytes exhibit a well-defined phenotype. Although it is not clear whether these protein 4.1 members have redundant or distinct function in keratinocytes, given the upregulation of 4.1N and 4.1G in the absence of 4.1R, one could speculate that depletion of the multiple members in keratinocytes would lead to more severe impairment in keratinocyte function.
It has been documented that members of protein 4.1 family associate with a variety of transmembrane proteins and regulate the expression and/or the function of these proteins. For example, in red blood cells, 4.1R binds to transmembrane proteins GPC, band 3, XK and Duffy and deficiency of 4.1R in erythrocytes leads to the altered expression of these proteins (Salomao et al., 2008). In CD4+ T cells, 4.1R binds to the adaptor protein LAT (linker of activation of T cells) and inhibits the phosphorylation of LAT (Kang et al., 2009b). The association of other members of protein 4.1 family with various membrane proteins has also been documented, such as the binding of 4.1G to metabotropic glutamate receptors (Lu et al., 2004a), adenosine receptors (Lu et al., 2004b) and parathyroid hormone receptors (Saito et al., 2005), as well as the binding of 4.1N to IP3 receptors (Maximov et al., 2003) and dopamine receptors (Binda et al., 2002). We now show that in keratinocytes, 4.1R binds to β1 integrin and is required for the surface expression of β1 integrin. These findings imply a unifying function of the protein 4.1 family in promoting the plasma membrane retention and cell surface exposure of a variety of transmembrane proteins.
Materials and Methods
Mice
The generation of 4.1R knockout (4.1R−/−) mice has been described previously (Shi et al., 1999). The mice were backcrossed onto C57BL/6 background. The wild-type C57BL/6 mice were purchased from Jackson Laboratory. All the mice were maintained at the animal facility of New York Blood Center under pathogen-free conditions according to institutional guidelines. Animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee.
Antibodies and reagents
Monoclonal antibodies used were as follows. Mouse anti-paxillin (Chemicon), vinculin (Sigma), anti-integrin β1 (clone MB1.2, Millipore), rat anti-mouse CD29 (clone 9EG7; BD Bioscience), anti-integrin α5 (Abcam), anti-integrin-α6 (BD Bioscience), anti-integrin β4 (BD Bioscience). Rabbit polyclonal antibodies used were: anti-GAPDH (Sigma) and anti-talin (Santa Cruz). All antibodies against members of the protein 4.1 family were generated by our laboratory (Kang et al., 2009a). Alexa-Fluor-conjugated secondary antibodies and Texas-Red–phalloidin were from Invitrogen. HRP-conjugated secondary antibodies were from Amersham Biosciences or Jackson ImmunoResearch. Fibronectin was purchased from BD Biocoat.
Culture of primary mouse keratinocytes and immortalization of keratinocytes
Primary mouse keratinocytes were cultured according to the method first outlined by Rheinwald and Green (Rheinwald and Green, 1975). Cells were isolated from 4.1R+/+ or 4.1R−/− C57Bl/6 mouse pups (P0–P3) by floating the skins on Dispase (Roche) overnight at 4°C. The following morning, the dermis was carefully separated from the epidermis and discarded. The epidermis was incubated in 0.25% trypsin–EDTA (ATCC) for 15 minutes at room temperature. Basal keratinocytes were separated from the cornified sheets by filtration through a 70 μM cell strainer (BD). Basal keratinocytes were cultured in E medium (Rheinwald and Green, 1977): calcium-free DMEM (US Biologicals) was combined with calcium-free F12 nutrient mix at a ratio of 3:1, and supplemented with 15% chelexed fetal bovine serum (Hyclone), 0.05 mM CaCl2, 0.4 μg/ml hydrocortisone (Sigma), 6.225×10−6 M insulin (Sigma), 2×10−9 M 3,3′,5-triiodo-L-thyronine (Sigma), 5 μg/ml transferrin (Sigma), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen), 7.5×10−9 M cholera toxin (Calbiochem) and L-Glutamine (Sigma). Cells were cultured at 37°C in a humidified environment with 5% CO2 on mitotically-inactivated mouse 3T3 feeder layer cells for the initial passages, and after 2–3 generations, the feeder cells could be removed and the cells propagated on normal uncoated tissue culture plastic dishes. Cells were immortalized by infecting with virus containing SV40 Large-T antigen. Immortalized keratinocytes were used for western blotting analysis of 4.1N, 4.1G and 4.1R as well as co-immunoprecipitation assay, all other experiments were performed using primary cells.
Cloning of 4.1R cDNA isoforms from keratinocyte RNA
Total RNA was isolated from 4.1R+/+ or 4.1R−/− keratinocytes by RNeasy mini kit (Qiagen, Valencia, Ca). RNA (1 μg) was reverse-transcribed into cDNA using random nonamers and M-MuLV reverse transcriptase (New England Biolabs, Ipswich, MA) for 50 minutes at 42°C. An equivalent of 5 ng of cDNA was used for PCR. PCR was performed using Accuprime Platinum Pfx DNA polymerase (Invitrogen, Carlsbad, CA). Transcripts of 4.1R can initiate at two distinct start sites, therefore PCR primers used were: AUG1F, 5′-ATGACAACAGAGAAGAGTTTAGTGGCTGAAGC-3′; AUG2F, 5′-ATGCACTGTAAGGTCTCCTTGTTGGATGACACG-3′; epb41R, 5′-CTCCTCAGAGATCTCTGTCTCCTGGTGGA-3′. Primers were designed to incorporate recognition sequences for the restriction enzymes XhoI and XmaI at the 5′ and 3′ ends of the PCR product, respectively. N-terminal GFP-fusion constructs were created by ligating 4.1R digested with XhoI and XmaI cDNAs downstream of the GFP coding sequence in pEGFP-C3 vector. The fidelity of the constructs was confirmed by sequencing and the expression of the GFP fusion proteins was validated by expression in 293T cells followed by western blotting of 293T lysates with anti-GFP and anti-4.1R antibodies (data not shown).
Transfection of primary mouse keratinocytes
An EGFP–β-actin expression construct and an EGFP–vinculin expression construct were provided by Daniel Soong and Daniel Worth (Kings College London, UK), respectively. Primary keratinocytes were transiently transfected with Fugene6 (Roche) according to the manufacturer's recommendations in serum-free E medium. Efficiency of transfection was generally around 1–2%. Recombinant 4.1R-expressing retrovirus particles were generated by cloning 4.1R into the SnaBI site of pBabe-GFP (Addgene plasmid 10668). Virus particles were produced by co-transfecting pBabe constructs into 293Ebna cells together with the retroviral packaging plasmid pCL-Eco and harvested according to previously published methods (Morgenstern and Land, 1990). Keratinocytes were infected with recombinant virus particles and incubated overnight before GFP-positive cells were sorted by FACS (MoFlo, Becton Dickinson).
Immunofluorescence
Cells were seeded on 13-mm-diameter 1.5 German glass coverslips (BD) in a 24-well format at approximately 30 % confluency in E medium. Cells were left to adhere and spread completely for 36–48 hours before processing. Samples were fixed in 4% paraformaldehyde (PFA) in PBS (Electron Microscopy Services) for 10 minutes at room temperature. After three washes in 100 mM glycine-PBS, samples were permeabilized in 0.1% Triton X-100 in PBS for 4 minutes. Samples were blocked in Abdil, 1% BSA in PBS. Primary antibodies were diluted in Abdil and generally incubated with the samples for 1–2 hours. Alexa-Fluor-conjugated secondary antibodies were purchased from Molecular Probes and diluted 1:700 in Abdil. For analysis of the actin cytoskeleton, cells were stained with either Alexa-Fluor-488 or Texas-Red–phalloidin, diluted 1:40 in PBS for 45 minutes at room temperature followed by three washes in PBS. Coverslips were mounted in Vectashild mounting medium (Vector Laboratories) supplemented with DAPI for nuclear staining. Cells were viewed for fluorescence using a Zeiss Axiophot wide-field epifluorescence microscope.
Adhesion assay
2×104 cells were plated on 96-well polystyrene assay plates coated with 10 μg/ml fibronectin. After 4 hours, the cells were washed with PBS and fixed with 4% formaldehyde for 20 minutes before being stained with 0.2% Crystal Violet for 15 minutes. The plates were then washed extensively, dried. The crystal violet was then released using 2% SDS in PBS and the absorbance was read by spectrophotometry at 560 nm.
Cell spreading assay
1×105 cells were plated on four-well chambers (Nunc) pre-coated with 10 μg/ml of fibronectin, and allowed to spread for 12 hours at 37°C in the presence of complete medium. Cells were then fixed and labeled with Alexa-Fluor-488-conjugated WGA (Invitrogen) for 30 minutes to visualize the cell outlines. Representative digital images were collected using a Zeiss Axiophot wide-field epifluorescence microscope. Cell-surface boundaries were outlined for 35 individual cells chosen randomly, and LSM 5 Pascal software was used to calculate the mean surface area and standard deviation of each population. One-tailed Student's t-tests were applied to test the statistical significance of the data.
Transwell migration assay
For migration assays, 8-μm-diameter pore Transwell cell culture inserts (BD Biocoat) were placed in six-well plates. The underside of the insert and the bottom of the well were coated with 10 μg/ml of fibronectin overnight. Keratinocytes were seeded on top of the insert (5×105/well) in E medium with 50 μM calcium. Cells were incubated for 12 hours during which cells migrated through the pores in the insert onto the bottom of the well. After incubation, the inserts were discarded, and the migratory cells in the bottom of the well were fixed with 1% glutaraldehyde (EMS) in PBS. Cells were stained with crystal violet (0.1% in ddH2O, freshly prepared and filtered through a 0.45 μm syringe filter) for 30 minutes at room temperature. Wells were extensively washed with ddH2O, and the dye was liberated with 0.5% Triton X-100 in PBS. Absorbance was read using a multiwell plate reader at 560 nm wavelength.
In vitro wound-healing assay
An in vitro wound-healing assay was carried out with cells that had been mitotically arrested by incubation with 8 μg/ml mitomycin C (Roche) in E medium for 2 hours at normal culture conditions. Mitomycin C was removed by three washes in PBS. A pseudo-wound was inflicted in a >90% confluent monolayer of keratinocytes by lightly scratching with a 10 μl pipette tip across the cell layer. Cell debris was removed by two washes with E medium. A minimum of six ‘wounded areas’ was filmed for each sample by obtaining images every 20 minutes for 16 hours. The wounded area was measured using ImageJ for each representative time point.
Cell motility assay
Motility assays were performed in E medium with 50 μM calcium using glass-bottomed 30 mm cell culture dishes (Matek). For generating movies of randomly migrating live cells, repetitive images were collected every 5 minutes for a total of 3 hours. DIC Image stacks were compressed into .mov format using ImageJ (NIH). A minimum of eight individual fields was filmed for each sample. All live cell microscopy was carried out using a Zeiss LSM 510 Meta microscope fitted with a 25× objective with samples maintained at 37°C in a 5% CO2 environment. The velocity and distance travelled for >30 cells were quantified using ImageJ with the cell migration and chemotaxis plug-in (Ibidi). For live cell fluorescence microscopy, keratinocytes were transfected with GFP fusion constructs with Fugene as above and incubated for 24 hours before microscopy. Cells were filmed over a 16 hour period throughout which samples were maintained. Images were collected every 10 minutes for a 10 hour period. The resulting microscopy data was analysed using the Cell Migration and Chemotaxis plug-in for ImageJ.
In vivo wound-healing assay and histology
Mice were anaesthetised by intraperitoneal injection of ketamine (10 mg/ml) and xylazine (1 mg/ml) solution at 0.01 ml per gram body weight. The backs of the mice were shaved and sterilized with alcohol followed by povidone-iodine solution. For histological analysis, two full-thickness excisional wounds (down to and including the panniculus carnosus) were made on either side of the dorsal midline using a 3-mm-diameter punch biopsy (Miltex). Wounds were left uncovered and harvested 2 or 5 days after injury. The complete excisional wound area was removed, fixed overnight in 10% formalin in PBS, and embedded in paraffin wax. Microtomy was performed at 4 μm. Sections from the center of the wound were stained with haematoxylin and eosin to examine general tissue and cellular morphology. To quantify the rate of wound healing, a single 8-mm-diameter wound was made through the dorsal skin of mice from each experimental group (n=5). A digital image of each wound was captured (day 0), and mice were caged singly without bedding for the remainder of the experiment. At the indicated time point, further digital images were captured and the size of the wound area was measured using ImageJ (version 1.37; NIH).
Flow cytometry
4.1R+/+ and 4.1R−/− keratinocytes were serum starved for 24 hours. The cells were trypsinized and washed twice with 0.5% BSA in PBS solution. The cells were stained respectively with monoclonal anti-integrin-β1 antibody (clone MB1.2, which recognizes total surface β1 integrin), rat anti-mouse CD29 antibody (Clone 9EG7, which recognizes active form of β1 integrin) or rat anti-mouse CD104 (which recognizes β4 integrin), in 0.5% BSA in PBS solution for 30 minutes on ice. The cells were washed twice and incubated with PE-conjugated anti-rat secondary antibody for a further 30 minutes on ice. To detect the surface expression of α5 and α6 integrin, the cells were stained with PE-conjugated rat anti-mouse CD49e or integrin α6 antibody directly in 0.5% BSA in PBS solution for 30 minutes on ice. To stain the active form of β1 integrin in the presence of MnCl2, cells were suspended in Tris buffer (25 mM Tris-HCl, pH 7.5, 137 mM NaCl, 2.7 mM KCl, 5% BSA) containing 2 mM EDTA and incubated at 37°C for 10 minutes. Subsequently, the cells were washed twice with Tris buffer and incubated in the same buffer containing 5 mM MnCl2 at 37°C for 10 minutes, followed by incubation with rat anti-mouse CD29 as described above. Flow cytometric analysis was performed on a FACSCanto flow cytometer (Becton Dickinson) and flow data overlay plots were produced using the CellQuest Pro software (BD).
Co-immunoprecipitation assay
Immortalized 4.1R+/+ and 4.1R−/− keratinocytes were lysed with ice-cold lysis buffer (50 mM HEPES, pH 8.3, 420 mM KCl, 0.1% NP-40, 1 mM EDTA) for 30 minutes on ice. Supernatant was collected after centrifugation at 14,000 r.p.m. at 4°C for 10 minutes and the concentration of protein in the supernatant was determined by the Bradford method using BSA as standard (Bio-Rad). 500 μg extract was incubated with 5 μg anti-β1-integrin antibody or 10 μg anti-4.1R-exon18 antibody or preimmune IgG in 500 μl of Co-IP buffer (Active motif) at 4°C overnight with rotation. The immunoprecipitates were isolated on magnetic Protein-G beads and separated by 10% SDS-PAGE followed by transfer to nitrocellulose membrane. The membrane was probed with antibodies against β1 integrin, anti-4.1R-exon18, anti-actin or anti-α6-integrin.
Construction, expression and purification of 4.1R, domains of 4.1R and cytoplasmic domain of β1 integrin
Construction, expression and purification of 4.1R were described previously (An et al., 2001). His-tagged 4.1R headpiece (HP) and the 30 kDa domain (MBD) was cloned into pET-31b(+) with NsiI and XhoI upstream and downstream, respectively. His-tagged 4.1R 16 kDa, 10 kDa (SABD) and C-terminal domains (CTD) were cloned into pET-28b(+) with NcoI and XhoI upstream and downstream, respectively. The cytoplasmic domain of β1 integrin was cloned into pGEX 4T-2 vector using restriction enzymes SmaI and XhoI. The template used for amplifying the cytoplasmic domain of β1 integrin was reversely transcribed from keratinocyte total mRNA. The fidelity of all constructs was confirmed by sequencing.
GST pull-down assay
GST-tagged recombinant cytoplasmic domain of β1 integrin was coupled to glutathione–Sepharose-4B beads at room temperature for 30 minutes. Beads were pelleted and washed. His-tagged 4.1R or various functional domains of 4.1R were added to the coupled beads in a final volume of 100 μl. The final concentration of the coupled protein was 2 μM. The mixture was incubated for 1 hour at room temperature, pelleted, washed and eluted with 10% SDS. The pellet was analyzed by SDS-PAGE. The binding of 4.1R or His-tagged 4.1R domains to the cytoplasmic domain of β1 integrin fragments was detected by western blot using anti-His antibody. GST was used as negative control in all experiments.
Western blotting
Cultured keratinocyte lysates were prepared by scraping the cells into ice-cold RIPA buffer (150 mM NaCl, 25 mM Tris-HCl, pH 7.4, 0.1% SDS, 1% Triton X-100, 1% deoxycholate, 2 mM EDTA, supplemented with protease and phosphatase inhibitor cocktails (Sigma). Protein concentrations were assayed using the Bradford Assay Reagent (Bio-Rad). Approximately 20–30 μg of total protein was fractionated by SDS-PAGE. Proteins were transferred electrophoretically onto nitrocellulose membrane (Bio-Rad) and the membrane was blocked in TBS with 1% BSA and 5% non-fat milk. Membranes were probed overnight with primary antibodies, and detected with the appropriate horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) followed by exposure to SuperSignal West Pico Chemiluminescent substrate (Pierce). For quantification, the intensity of the band was evaluated by densitometry using ImageJ software.
Supplementary Material
Acknowledgments
We thank Wu He for flow cytometry analysis and Lyudmil Angelov for confocal images. This work was supported in part by NIH grants DK 26263, DK 32094, HL31579. The authors declare that they have no conflicts of interest. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/14/2478/DC1
References
- An X. L., Takakuwa Y., Manno S., Han B. G., Gascard P., Mohandas N. (2001). Structural and functional characterization of protein 4.1R-phosphatidylserine interaction: potential role in 4.1R sorting within cells. J. Biol. Chem. 276, 35778-35785 [DOI] [PubMed] [Google Scholar]
- Binda A. V., Kabbani N., Lin R., Levenson R. (2002). D2 and D3 dopamine receptor cell surface localization mediated by interaction with protein 4.1N. Mol. Pharmacol. 62, 507-513 [DOI] [PubMed] [Google Scholar]
- Blanpain C., Fuchs E. (2009). Epidermal homeostasis: a balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 10, 207-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood D. A., Zent R., Grant R., Rees D. J., Hynes R. O., Ginsberg M. H. (1999). The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274, 28071-28074 [DOI] [PubMed] [Google Scholar]
- Chishti A. H., Kim A. C., Marfatia S. M., Lutchman M., Hanspal M., Jindal H., Liu S. C., Low P. S., Rouleau G. A., Mohandas N., et al. (1998). The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem. Sci. 23, 281-282 [DOI] [PubMed] [Google Scholar]
- Danen E. H., van Rheenen J., Franken W., Huveneers S., Sonneveld P., Jalink K., Sonnenberg A. (2005). Integrins control motile strategy through a Rho-cofilin pathway. J. Cell Biol. 169, 515-526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diakowski W., Grzybek M., Sikorski A. F. (2006). Protein 4.1, a component of the erythrocyte membrane skeleton and its related homologue proteins forming the protein 4.1/FERM superfamily. Folia Histochem. Cytobiol. 44, 231-248 [PubMed] [Google Scholar]
- Fuchs E. (1990). Epidermal differentiation. Curr. Opin. Cell Biol. 2, 1028-1035 [DOI] [PubMed] [Google Scholar]
- Fuchs E. (1994). Epidermal differentiation and keratin gene expression. Princess Takamatsu Symp. 24, 290-302 [PubMed] [Google Scholar]
- Fuchs E., Raghavan S. (2002). Getting under the skin of epidermal morphogenesis. Nat. Rev. Genet. 3, 199-209 [DOI] [PubMed] [Google Scholar]
- Galbraith C. G., Yamada K. M., Galbraith J. A. (2007). Polymerizing actin fibers position integrins primed to probe for adhesion sites. Science 315, 992-995 [DOI] [PubMed] [Google Scholar]
- Geiger B., Bershadsky A. (2001). Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 13, 584-592 [DOI] [PubMed] [Google Scholar]
- Has C., Herz C., Zimina E., Qu H. Y., He Y., Zhang Z. G., Wen T. T., Gache Y., Aumailley M., Bruckner-Tuderman L. (2009). Kindlin-1 Is required for RhoGTPase-mediated lamellipodia formation in keratinocytes. Am. J. Pathol. 175, 1442-1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz C., Aumailley M., Schulte C., Schlotzer-Schrehardt U., Bruckner-Tuderman L., Has C. (2006). Kindlin-1 is a phosphoprotein involved in regulation of polarity, proliferation, and motility of epidermal keratinocytes. J. Biol. Chem. 281, 36082-36090 [DOI] [PubMed] [Google Scholar]
- Huang S. C., Jagadeeswaran R., Liu E. S., Benz E. J., Jr (2004). Protein 4.1R, a microtubule-associated protein involved in microtubule aster assembly in mammalian mitotic extract. J. Biol. Chem. 279, 34595-34602 [DOI] [PubMed] [Google Scholar]
- Hubbard T., Barker D., Birney E., Cameron G., Chen Y., Clark L., Cox T., Cuff J., Curwen V., Down T., et al. (2002). The Ensembl genome database project. Nucleic Acids Res. 30, 38-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Q., Wang T., Zhang H., Mohandas N., An X. (2009a). A Golgi-associated protein 4.1B variant is required for assimilation of proteins in the membrane. J. Cell Sci. 122, 1091-1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Q., Yu Y., Pei X., Hughes R., Heck S., Zhang X., Guo X., Halverson G., Mohandas N., An X. (2009b). Cytoskeletal protein 4.1R negatively regulates T-cell activation by inhibiting the phosphorylation of LAT. Blood 113, 6128-6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaverina I., Krylyshkina O., Small J. V. (2002). Regulation of substrate adhesion dynamics during cell motility. Int. J. Biochem. Cell Biol. 34, 746-761 [DOI] [PubMed] [Google Scholar]
- Kirfel G., Herzog V. (2004). Migration of epidermal keratinocytes: mechanisms, regulation, and biological significance. Protoplasma 223, 67-78 [DOI] [PubMed] [Google Scholar]
- Krauss S. W., Spence J. R., Bahmanyar S., Barth A. I., Go M. M., Czerwinski D., Meyer A. J. (2008). Downregulation of protein 4.1R, a mature centriole protein, disrupts centrosomes, alters cell cycle progression, and perturbs mitotic spindles and anaphase. Mol. Cell. Biol. 28, 2283-2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D., Yan H., Othman T., Rivkees S. A. (2004a). Cytoskeletal protein 4.1G is a binding partner of the metabotropic glutamate receptor subtype 1 alpha. J. Neurosci. Res. 78, 49-55 [DOI] [PubMed] [Google Scholar]
- Lu D., Yan H., Othman T., Turner C. P., Woolf T., Rivkees S. A. (2004b). Cytoskeletal protein 4.1G binds to the third intracellular loop of the A1 adenosine receptor and inhibits receptor action. Biochem. J. 377, 51-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfatia S. M., Leu R. A., Branton D., Chishti A. H. (1995). Identification of the protein 4.1 binding interface on glycophorin C and p55, a homologue of the Drosophila discs-large tumor suppressor protein. J. Biol. Chem. 270, 715-719 [DOI] [PubMed] [Google Scholar]
- Martin P. (1997). Wound healing-aiming for perfect skin regeneration. Science 276, 75-81 [DOI] [PubMed] [Google Scholar]
- Maximov A., Tang T. S., Bezprozvanny I. (2003). Association of the type 1 inositol (1,4,5)-trisphosphate receptor with 4.1N protein in neurons. Mol. Cell. Neurosci. 22, 271-283 [DOI] [PubMed] [Google Scholar]
- McCarty J. H., Cook A. A., Hynes R. O. (2005). An interaction between {alpha}v{beta}8 integrin and Band 4.1B via a highly conserved region of the Band 4.1 C-terminal domain. Proc. Natl. Acad. Sci. USA 102, 13479-13483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern J. P., Land H. (1990). Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587-3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura W., Takakuwa Y., Tokimitsu R., Krauss S. W., Kawashima M., Mohandas N. (1997). Regulation of CD44-protein 4.1 interaction by Ca2+ and calmodulin. Implications for modulation of CD44-ankyrin interaction. J. Biol. Chem. 272, 30322-30328 [DOI] [PubMed] [Google Scholar]
- Perez-Ferreiro C. M., Luque C. M., Correas I. (2001). 4.1R proteins associate with interphase microtubules in human T cells: a 4.1R constitutive region is involved in tubulin binding. J. Biol. Chem. 276, 44785-44791 [DOI] [PubMed] [Google Scholar]
- Pollard T. D., Borisy G. G. (2003). Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453-465 [DOI] [PubMed] [Google Scholar]
- Raja S. K., Garcia M. S., Isseroff R. R. (2007). Wound re-epithelialization: modulating keratinocyte migration in wound healing. Front. Biosci. 12, 2849-2868 [DOI] [PubMed] [Google Scholar]
- Rheinwald J. G., Green H. (1975). Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell 6, 331-343 [DOI] [PubMed] [Google Scholar]
- Rheinwald J. G., Green H. (1977). Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature 265, 421-424 [DOI] [PubMed] [Google Scholar]
- Saito M., Sugai M., Katsushima Y., Yanagisawa T., Sukegawa J., Nakahata N. (2005). Increase in cell-surface localization of parathyroid hormone receptor by cytoskeletal protein 4.1G. Biochem. J. 392, 75-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomao M., Zhang X., Yang Y., Lee S., Hartwig J. H., Chasis J. A., Mohandas N., An X. (2008). Protein 4.1R-dependent multiprotein complex: new insights into the structural organization of the red blood cell membrane. Proc. Natl. Acad. Sci. USA 105, 8026-8031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z. T., Afzal V., Coller B., Patel D., Chasis J. A., Parra M., Lee G., Paszty C., Stevens M., Walensky L., et al. (1999). Protein 4.1R-deficient mice are viable but have erythroid membrane skeleton abnormalities. J. Clin. Invest. 103, 331-340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg M. A., Carter E., Sohrabi N., Siedlecka U., Soppa G. K., Mead F., Mohandas N., Taylor-Harris P., Baines A., Bennett P., et al. (2008). Cytoskeletal protein 4.1R affects repolarization and regulates calcium handling in the heart. Circ. Res. 103, 855-863 [DOI] [PubMed] [Google Scholar]
- Sun C. X., Robb V. A., Gutmann D. H. (2002). Protein 4.1 tumor suppressors: getting a FERM grip on growth regulation. J. Cell Sci. 115, 3991-4000 [DOI] [PubMed] [Google Scholar]
- Tang P., Cao C., Xu M., Zhang L. (2007). Cytoskeletal protein radixin activates integrin alpha(M)beta(2) by binding to its cytoplasmic tail. FEBS Lett. 581, 1103-1108 [DOI] [PubMed] [Google Scholar]
- Taylor-Harris P. M., Keating L. A., Maggs A. M., Phillips G. W., Birks E. J., Franklin R. C., Yacoub M. H., Baines A. J., Pinder J. C. (2005). Cardiac muscle cell cytoskeletal protein 4.1: analysis of transcripts and subcellular location-relevance to membrane integrity, microstructure, and possible role in heart failure. Mamm. Genome 16, 137-151 [DOI] [PubMed] [Google Scholar]
- Watt F. M. (2002). Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 21, 3919-3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Guo X., Debnath G., Mohandas N., An X. (2009). Protein 4.1R links E-cadherin/beta-catenin complex to the cytoskeleton through its direct interaction with beta-catenin and modulates adherens junction integrity. Biochim. Biophys. Acta 1788, 1458-1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.