

Abstract
Autoimmunity leads to the activation of innate effector pathways, pro-inflammatory cytokine production, and end-organ injury. Macrophage migration inhibitory factor (MIF) is an upstream activator of the innate response that mediates the recruitment and retention of monocytes via CD74 and associated chemokine receptors, and it has a role in the maintenance of B lymphocytes. High-expression MIF alleles also are associated with end-organ damage in different autoimmune diseases. We assessed the therapeutic efficacy of ISO-1, an orally bioavailable, MIF antagonist, in two distinct models of systemic lupus erythematosus (SLE): the NZB/NZW F1 and the MRL/lpr mouse strains. ISO-1, like anti-MIF, inhibited the interaction between MIF and its receptor, CD74, and in each model of disease, it reduced functional and histological indices of glomerulonephritis, CD74+ and CXCR4+ leukocyte recruitment, and pro-inflammatory cytokine and chemokine expression. Neither autoantibody production nor T and B cell activation were significantly affected, pointing to the specificity of MIF antagonism in reducing excessive pro-inflammatory responses. These data highlight the feasibility of targeting the MIF–MIF receptor interaction by small molecule antagonism and support the therapeutic value of downregulating MIF-dependent pathways of tissue damage in SLE.
Keywords: Autoimmunity, Cytokine, Innate Immunity
INTRODUCTION
Systemic lupus erythematosus (SLE) is a multi-system autoimmune disease that is characterized by the loss of immune tolerance and the production of autoantibodies to nucleic acids and nucleoproteins (1). Immunopathology results primarily from immune complex deposition in the small vessels of the skin, kidney and other organs; this leads to the activation of complement and immunoglobulin Fc receptors, and the recruitment of neutrophils and monocytes. Monocytes/macrophages are retained and persist within inflammatory sites, producing cytokines that propagate inflammatory tissue damage. In the kidney, for instance, infiltrating monocytes/macrophages are major constituents of the crescentic lesions that develop in rapidly progressive lupus nephritis and their presence signifies severe glomerular injury (2).
Macrophage migration inhibitory factor (MIF) inhibits the movement or egress of macrophages and it exerts an upstream role in regulating the innate immune response (3,4). MIF is present pre-formed within monocytes/macrophages and its rapid release results in the autocrine/paracrine activation of both immune and non-immune cell types (5,6). MIF counter-regulates the immunosuppressive actions of glucocorticoids and it promotes TNFα and IL-1β production, leading to further MIF release and a re-entrant activation response that supports the maximal expression of cytokines, matrix-degrading enzymes, and cyclooxygenases (3,7,8). Genetic knockout studies additionally have established an important role for MIF in inhibiting activation-induced apoptosis (9), which sustains monocyte/macrophage activation within inflammatory sites and contributes to the maintenance of mature immune cell populations (10–12). MIF signal transduction is initiated by high affinity binding to CD74 13. Recent studies indicate that MIF also may act as a non-cognate ligand for the chemokine receptors CXCR2 and CXCR4; these proteins form complexes with CD74 and are necessary for MIF-driven atherogenic leukocyte recruitment (4,14).
Evidence for a role for MIF in autoimmunity has been provided by studies showing that immunoneutralization or genetic deletion of MIF confers protection from pathologic progression in different experimental models of disease (15–17). MIF is known to be expressed in increased levels in the SLE-prone, MRL/MpJ-Faslpr mouse, and an intercross between this strain and mif−/− mice reduces glomerular injury and lethality (18). Both the circulating level and the tissue expression of MIF are elevated in patients with autoimmune inflammatory disorders, and high-expression MIF alleles have been associated with more severe end-organ damage in rheumatoid arthritis (19,20), asthma (21), scleroderma (22), and with disease risk in SLE (23). Circulating levels of MIF are increased in patients with SLE and may correlate with indices of disease severity, renal dysfunction, and steroid resistance (24).
MIF is encoded by a unique gene and crystallographic studies have revealed the protein to share structural homology with a class of prokaryotic tautomerases (25). While in vitro studies have shown that MIF also tautomerizes model substrates (26), a physiologic role for this tautomerization activity has not been established. Indeed, genetic knockin studies with a catalytically inactive MIF have led to the conclusion that enzymatic activity is a vestigial property of the protein that may have originated from the gene's ancestral role in invertebrate immunity (27). The MIF tautomerase site nevertheless has been proposed to be an attractive entry point for the design of small molecules that might be targeted to the protein surface to inhibit receptor interaction, and proof-of-concept for this approach has been provided by the observation that covalent modification of MIF's catalytic, N-terminal proline, reduces both MIF bioactivity and its binding to target cell receptors (28,29).
The investigation of new treatments for SLE remains challenging and several recently developed biologic agents that are effective in other autoimmune disorders have not shown benefit in lupus (30). Given the unmet need for new therapeutic approaches in SLE, we tested the efficacy of a small molecule MIF antagonist, ISO-1, which binds to the MIF tautomerase site (31) in two different experimental models of SLE: the NZB/NZW F1 and the MRl/lpr mouse strains. We report herein that in each model of spontaneous lupus, treatment with ISO-1 reduced MIF-dependent pro-inflammatory cytokine production and leukocyte recruitment, and ameliorated immune-mediated renal injury.
MATERIALS and METHODS
Reagents
ISO-1 ((S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester) was synthesized in three steps from 4-hydroxy-benzaldehyde by minor modifications of a previously reported procedure (32). The structure and purity of the synthetic product was verified by 1H-NMR and electrospray mass spectrometry (M+=236.1). NAPQI (N-acetyl-p-benzoquinone imine) was prepared as previously described (28). A neutralizing murine monoclonal anti-MIF IgG1 (NIHIIID.9) (15,33) was produced from ascites, and an IgG1 isotypic control antibody (clone HB9) was obtained from ATCC (Manassas, VA). Recombinant human and mouse MIF were prepared as described by Bernhagen et al. (34), and the soluble MIF receptor ectodomain (CD7473–232 = sCD74) was purified from an E. coli expression system as previously reported (13).
MIF Binding Studies
For epitope mapping, individual human MIF 10-mer peptides were synthesized on polyethylene rods compatible with 96-well ELISA assays (35). The rod-coupled peptides were incubated in 96-well plates for 1 hour with 1% BSA, 1% ovalbumin, 0.1% Tween-20 in PBS, pH 7.4. Diluted anti-MIF or control antibody was incubated overnight with peptides in the 96-well plates at 4°C and washed four times for 10 min in PBS with 0.05% Tween-20. Antibodies bound to peptide were detected with a peroxidase-coupled goat-anti-rabbit IgG, the addition of substrate solution, and measurement of absorption at 405 nm (OD405).
The binding of MIF to the MIF receptor (CD74) was quantified by an in vitro competition assay employing immobilized MIF receptor ectodomain (CD7473–232) and biotinylated human MIF (13). The OD405 was measured after addition of test inhibitors and the values plotted as percent OD405 relative to wells containing biotinylated human MIF alone.
Mice and Study Design
Female NZB/NZW F1 and MRL-Faslpr (MRL/lpr) mice were obtained from Charles River and acclimated for two weeks prior to study. All mice were maintained under specific pathogen-free conditions and studies were performed in accordance with an approved IACUC protocol. Blood was obtained for baseline studies, following which the mice were divided into groups of 10–11 individuals. The NZB/NZW F1 mice were treated for 12 weeks beginning at 22 weeks of age, and the MRL/lpr mice were treated for 10 weeks beginning at 9 weeks of age. ISO-1 was administered in sterile 10% DMSO/H2O at a dose of 40 mg/kg by daily intraperitoneal (ip) injection. Control mice received vehicle alone. Anti-MIF mAb or control IgG1 was administered ip in sterile saline at a dose of 20 mg/kg twice weekly. All mice were observed daily and weighed weekly for evidence of drug toxicity. Midway through the treatment protocol, blood was sampled from the retro-orbital plexus for measurement of blood urea nitrogen, cytokines, and autoantibodies. At the completion of the studies, mice were euthanized by CO2 asphyxiation, blood sampled by cardiac puncture, and tissues removed and processed for flow cytometric, histologic, and mRNA and protein analysis.
Analyses for Autoantibodies, Cytokines, and Urea Nitrogen
Serum anti-dsDNA IgG antibodies were measured by ELISA using S1 nuclease-treated DNA as described previously (36). A positive serum sample from a 20 wk old MRL/lpr mouse was used as an internal control. MIF was measured using a murine-specific ELISA and native-sequence, recombinant mouse MIF as a standard (21). The IFNα ELISA kit was from PBL laboratories. The remaining cytokines were measured using a multi-cytokine beadmaster kit (Luminex, Upstate, NY). Blood urea nitrogen levels were quantified by the Clinical Chemistry Laboratory of Yale-New Haven Hospital.
Renal Histopathology and Immunohistochemistry
To assess pathologic changes, kidney tissues were stained with hematoxylin and eosin and with periodic acid Schiff's reagent, and numbered slides were evaluated by a pathologist (MK) blinded to the treatment protocol. Scoring was on a scale of 0 – 4+ and included the assessment of endocapillary proliferation, capillary loop thickening, leukocyte exudation, and glomerular necrosis (karyorrhexis, fibrinoid changes, cellular crescents and hyaline deposits) (36,37). Sections were examined in 8–10 individual kidneys from each treatment group. Immunoglobulin deposition was assessed by immunofluorescense staining with anti-mouse IgG (Invitrogen A11001). Slides were analyzed at the lowest positive dilution (1:25,000), and the fluorescence intensity within glomeruli evaluated with ImageJ software and expressed on a scale of 1–4 (38). Kidney tissue additionally was processed (n=4 per group) and each section stained individually for MIF+-cells (anti-MIF R102) (39), F4/80+-macrophages (clone BM8), CD3+T cells (anti-CD3, Abcam) (40), CD74+ cells (clone sc-5438) and CXCR4+ cells (clone 247506, R&D Systems) (41). An avidin-biotin-HRP system or secondary antibodies conjugated with fluorescent dyes were employed, and non-immune IgG was used as a specificity control. Immunoreactive cells were enumerated in approximately 50 glomeruli within at least 4 sections per experimental condition (33). The presence of interstitial nephritis was assessed by enumerating F4/80+macrophages in at least twenty grid defined (100×) fields of renal interstitium.
Flow Cytometry Analysis
Spleen and cervical lymph nodes were harvested, weighed, cleared of erythrocytes, and the cells pooled from individual mice for phenotypic analysis using four color flow cytometry (FACSCalibur, BD Biosciences, San Jose, CAS), commercially available antibodies, and FLOWJO software (Tree Star, Ashland, OR) as previously described (36).
Quantitative PCR Analysis
RNA was extracted from frozen tissue samples using the RNeasy extraction kit (Qiagen, Valencia, CA) and cDNA was synthesized from 1 μg of RNA using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Real-time PCR was carried out with the iQ SYBR Green system (Bio-Rad) using previously published primers (21,42). The emitted fluorescence for each reaction was measured during the annealing/extension phase and relative quantity values were calculated by the standard curve method. The quantity value of GAPDH in each sample was used as a normalizing control. Differences were evaluated by non-parametric testing using the Mann-Whitney test.
Transcriptome Analysis
Total RNA from kidneys (3 samples per experimental group) was isolated using RNeasy miniprep columns (Qiagen), and labeling and hybridization were performed with the Genisphere Array900 Expression Array Detection kit (http://www.genisphere.com/array_detection_900.html) according to the manufacturer's protocol. The OMM25K oligonucleotide gene array set (18,000 genes) from Yale University Keck Facility was utilized (http://keck.med.yale.edu/dnaarrays/genelists), and the cDNA probe and the fluorescent 3DNA reagent were hybridized to the microarray in succession. Hybridization was performed with a Advalytix Slide Booster hybridization station. The hybridized slides were scanned with a GenePix 4000 scanner (Axon Instrument, Union City, CA) and raw data analyzed using GenePix 5.0 analysis software. Hierarchical clustering analysis was performed using GeneSpring GX 7.3 (Agilent Technologies) to show the relationships between the expression levels of the four experimental conditions: MRL ISO-1 / MRL vehicle control, MRL anti-MIF / IgG control, NZB ISO-1/control, NZB anti-MIF/control. Pearson correlation was used to measure the similarity of the expression levels. Selected immune response genes were extracted from the full transcriptional profile analysis by a combination of statistical testing of absolute and relative changes in expression across the different experimental conditions and a permutation-based test to estimate false discovery (43). Statistical analysis between experimental groups was performed using the Student's T-test with genes with a FDR of <0.05 and a fold change of >1.5 being considered differentially expressed. Gene expression data is available upon publication on http://www.biochemmcb.rwth-aachen.de/mif_consortium.
RESULTS
The MIF Tautomerase Site Mediates Binding to the MIF Receptor, CD74
To better validate the pharmacologic targeting of the MIF tautomerase site, we quantified the ability of selected MIF antagonists to interfere with MIF binding to an immobilized, recombinant MIF receptor ectodomain (CD7473–232) (Fig. 1A). NAPQI, which covalently and irreversibly modifies the catalytic Pro1 within the MIF tautomerase site (28), showed potent, dose-dependent inhibition of MIF binding to its receptor (IC50=90 nM). The small molecule pharmacophore, ISO-1, binds reversibly to the MIF tautomerase site and inhibits MIF-dependent MAPK activation in target cells (31). ISO-1 also reduced MIF interaction with its receptor, albeit with a more modest dose-dependent effect (estimated IC50=50 μM) than the irreversible inhibitor, NAPQI. We additionally tested the inhibitory activity of a biologically neutralizing anti-MIF IgG1 (15). This antibody showed significant, dose-dependent inhibition of MIF binding to the MIF receptor (IC50 = 400 nM), with the steep slope of inhibition likely due to the high avidity of bivalent antibody. Notably, an epitope scan of MIF using a neutralizing anti-MIF polyclonal Ab (15) also showed recognition of a predominantly single epitope (Fig. 1B) that borders the tautomerase substrate binding pocket (Fig. 1C). These data support a role for the MIF tautomerase site in receptor engagement and the notion that small molecules that target this site may be useful pharmacologically.
Figure 1. Pharmacologic Targeting of the MIF N-terminal Region.

(A) Competition studies of MIF interaction with its receptor in the presence of NAPQI, ISO-1, or anti-MIF. The MIF receptor ectodomain (CD7473–232) was immobilized in 96 wells and recombinant MIF added together with increasing concentrations of antagonists as described in the Materials and Methods. Data are shown for a biologically neutralizing, anti-MIF IgG1 (15). Symbols depict means of quadruplicate measurements and lines show log regression analyses. No influence of vehicle or control IgG1 was observed (data not shown). (B) Amino acid epitope scan of human MIF performed by reacting a neutralizing anti-MIF polyclonal antibody (15) with sequential peptide 10-mers (each offset by two residues). Peptide 4 shows the highest reactivity and corresponds to MIF6–15 (NTNVPRASVP). The inset shows the MIF primary sequence. (C) Simulated view of the immunoreactive epitopes superimposed on the MIF homotrimer. Axial and lateral views of MIF are shown with the model dopachrome tautomerase substrate, D-dopachrome methyl ester (blue space filling model). The epitopes defined by peptides 2–6 and 23–26 are in red and green in one subunit, revealing their adjacent position to the tautomerase site.
MIF is Expressed in Elevated Levels in Lupus-prone Mice
In preparation for studying the potential therapeutic effect of MIF inhibition, we examined MIF expression in two experimental models of lupus, the NZB/NZW F1 and the MRL/lpr mouse strains. The NZB/NZW F1 mouse strain is a useful model for autoimmune B cell and T cell interactions, and for the time-dependent diversification of the autoimmune response. A progressive serum autoantibody response results in the development of severe nephritis at 24–48 weeks of age. The MRL/lpr mouse develops a lymphoproliferative autoimmune syndrome that mimics several features of SLE; these include a similar spectrum of autoantibodies and an immune complex glomerulonephritis that develops over 12–24 weeks of age 44.
Both the NZB/NZW F1 and the MLR/lpr mice manifest a time-dependent elevation in circulating MIF at ages that correspond with disease progression and the development of glomerulonephritis (Fig. 2). MIF mRNA and protein expression in kidneys also increased significantly with inflammatory progression in the two lupus-prone mouse strains, and this effect was associated with an increase in the number of MIF+ mononuclear cells within glomeruli. These data are consistent with the notion that the development of autoimmune pathology in lupus-prone mice is associated with increased MIF production, both systemically and within inflammatory renal lesions.
Figure 2. Plasma and Intrarenal MIF Expression Increases During Disease Progression in Lupus-prone Mice.

Plasma and renal MIF was assessed in NZB/NZW F1 (upper panels) and MRl/lpr mice (lower panels). MIF levels in plasma and renal tissue lysates were measured by specific ELISA, and renal MIF mRNA by qPCR (n=4 per experimental group, see Methods). Plasma samples were obtained at the ages shown and compared to plasma from healthy, C57BL/6 mice. Renal histologic sections were stained with anti-MIF (R102) and the MIF-positive cells enumerated within 50 individual glomeruli of representative kidney sections (n=4 kidneys per group). Inset images show MIF immunostaining within a representative glomerulus of an NZB/NZW F1 mouse at 22 and 34 weeks. Diffuse staining for MIF within proximal tubular epithelial cells also is evident, as previously described (66). MIF protein and cell count data are expressed as Mean ± SD, with P values calculated by Student's T-test, two-tailed. For the qPCR data, the upper and lower edges of the boxes represent the 75th and 25th percentiles, respectively, the error bars denote the range of observations, and the horizontal line shows the median, with P values calculated by the Mann-Whitney test. *P<0.005, **P<0.001.
Pharmacologic Inhibition of MIF Attenuates Renal Dysfunction and Glomerulonephritis in Lupus-prone Mice
We initiated a therapeutic trial of ISO-1 and anti-MIF mAb in the NZB/NZW F1 and the MLR/lpr mouse strains. Mice were divided into groups (n=10–11 per group) and treated daily with ISO-1 or its vehicle, or twice weekly with anti-MIF or an isotypic control (IgG1) antibody. In NZB/NZW F1 mice, anti-dsDNA autoantibodies become detectable in the circulation at ~24 weeks of age and renal disease may be detected at 28 weeks. Treatment was begun at 22 weeks, which is prior to the onset of nephritis, and continued until 34 weeks of age in order to encompass the period of autoimmunity. ISO-1 or anti-MIF mAb were well-tolerated and treatment for up to 12 weeks was not associated with any evident toxicity or change in body weight when compared with vehicle-treated controls (data not shown). NZB/NZW F1 mice treated with ISO-1 or anti-MIF showed a significant reduction in the progressive rise in serum blood urea nitrogen (BUN), which is a sensitive indicator of renal function (Fig. 3A). Renal tissue that was examined at the end of the treatment protocol and scored histologically for glomerular damage confirmed that ISO-1 or anti-MIF ameliorated the development of glomerulonephritis (Fig. 3B,C). The kidneys from treated mice showed a decrease in glomerular crescents and karyorrhexis. Neither of the MIF antagonists reduced the glomerular deposition of circulating immunoglobulin (Fig. 3D). MIF inhibition did reduce interstitial inflammation, as assessed by the enumeration of F4/80+ macrophages (Fig. 3E,F), and it reduced the intraglomerular content of infiltrating F4/80+ macrophages, CD3+ T cells, and cells expressing MIF (Fig. 3G). A corresponding decrease in the glomerular content of infiltrating MIF receptor positive (CD74+, CXCR4+) cells also was observed.
Figure 3. ISO-1 or Anti-MIF Ameliorates Glomerulonephritis in NZB/NZW F1 Mice.

(A) Serum BUN levels in mice treated with ISO-1, anti-MIF mAb, or controls (vehicle or IgG1). Blood was sampled in 4 mice per group before treatment (22 weeks), midway through treatment (28 weeks), and at the end of the treatment protocol (34 weeks). (B) Glomerulonephritis scores (0 – 4+) reflecting indices of cellularity, focal and segmental lesions, and sclerosis in histologic sections after 12 weeks of treatment (n=8 kidneys per group). (C) Representative, periodic acid Schiff's stained kidney sections obtained at 34 weeks of age showing the typical inflammatory lesions that develop in NZB/NZW F1 and the less intense glomerulonephritis observed in mice treated with ISO-1 (200×). (D) Glomerular immunoglobulin (Ig) deposition detected using fluorescein-conjugated, anti-mouse IgG. Fluorescence intensity was examined microscopically and scored on a scale of intensity of 0–4. P=NS among groups. (E) Assessment of interstitial inflammation by enumeration of F4/80+ macrophages in ≥20 interstitial fields selected from 4 representative kidneys per group. (F) Reduced inflammatory infiltration is evident in immunoperoxidase stained images from mice treated with ISO-1, or anti-MIF. (G) Enumeration of intra-glomerular F4/80+ macrophages, CD3+ T cells, MIF+ cells, and MIF receptor positive (CD74+, CXCR4+) cells. Data are summed from approximately 50 glomeruli visualized in 4 kidneys per experimental group. Values shown are Mean ± SD and P values are by Student's T test, two-tailed; *P<0.05, **P<0.01.
For the study of the MRL/lpr mice, treatment was initiated at 9 weeks of age, continued for 10 weeks, and terminated when mice reached 19 weeks of age. Circulating anti-DNA antibodies become evident in MRL/lpr mice at 9 weeks and their appearance precedes the development of glomerulonephritis (44). ISO-1 reduced the time-dependent increase in renal insufficiency and histologic indices of renal damage (Fig. 4A–C). While anti-MIF also ameliorated the development of glomerular disease, its effect was not significantly different from that observed by treatment with control IgG1. A protective action of non-specific IgG on renal immunopathology was observed, which may reflect an immunosuppressive action by IgG on FcR ITIM signaling, or by depletion of serum complement (45,46). The not-quite significant effect of anti-MIF mAb in MRL/lpr mice also may have resulted from an insufficient dose of anti-MIF in this model of renal immunopathology. Representative, periodic acid Schiff's stained sections of kidneys nevertheless illustrate the reduction in glomerular cellularity, focal and segmental lesions, and glomerulosclerosis that was evident in the ISO-1 treated MRL/lpr mice when compared to vehicle controls. As in the case of NZB/NZW F1 mice, ISO-1 treatment of MRL/lpr mice was unaccompanied by a reduction in immune complex deposition (Fig. 4D), but it significantly reduced the content of infiltrating inflammatory cells (F4/80+ macrophages) in renal interstitium (Fig. 4E–F) as well as the number of intraglomerular F4/80+ macrophages, CD3+ T cells, MIF+ cells, and MIF receptor positive (CD74+, CXCR4+) cells (Fig. 4G).
Figure 4. ISO-1 Ameliorates Glomerulonephritis in MRL/lpr Mice.

(A) Serum BUN levels in mice treated with ISO-1, anti-MIF mAb, or controls (vehicle or IgG1). Blood was sampled in 4 mice per group before treatment (9 weeks), midway through treatment (14 weeks), and at the end of the treatment (19 weeks). (B) Glomerulonephritis scores (0 – 4+) reflecting indices of cellularity, focal and segmental lesions, and sclerosis were obtained in kidney sections (n=8 kidneys per group) after 10 weeks of treatment. P values are by Student's T test, two-tailed. (C) Representative, periodic acid Schiff's stained kidney sections obtained at 19 weeks of age showing inflammatory renal damage in MRL/lpr mice and the less intense glomerulonephritis observed in mice treated with ISO-1 (200×). (D) Glomerular IgG deposition detected using fluoresceinconjugated, anti-mouse IgG. Fluorescence intensity was examined microscopically and scored as described 38. P=NS among groups. (E) Assessment of interstitial inflammation by enumeration of F4/80+ macrophages in ≥20 interstitial fields selected from 4 representative kidneys per group. (F) Reduced inflammatory infiltration is evident in a representative immunoperoxidase-stained renal section from an ISO-1 treated mouse, which shows a few periglomerular F4/80+ cells (200×). (G) Enumeration of intra-glomerular F4/80+ macrophages, CD3+ T cells, MIF+ cells, and MIF receptor (CD74+, CXCR4+) positive cells. Data are summed from approximately 50 glomeruli visualized in 4 kidneys per experimental group. Values shown are Mean ± SD and P values are by Student's T test, two-tailed; *P<0.05, **P<0.01.
Influence of MIF Inhibition on Serum Autoantibody and Splenic Lymphocyte Populations in Lupus-prone Mice
Serum anti-dsDNA autoantibody is a serologic hallmark of SLE. Circulating levels of anti-dsDNA in NZB/NZW F1 mice were unaffected by ISO-1 treatment, although a modest decrease in anti-dsDNA antibody titer was discernable at the 28 day time point in the anti-MIF treated group (Fig. 5A). An analysis of pooled splenic and lymph node cells in NZB/NZW F1 mice did not show any effect of MIF inhibition on B cell (CD3−B220+) or T cell populations (CD3+CD4+, CD3+CD8+), and this included a specific analysis of double-negative (CD3+B220−), naïve (CD3+CD4+CD44loCD62hi, CD3+CD8+CD44loCD62hi), and mature (CD3+CD4+CD44hiCD62hi, CD3+CD8+CD44hiCD62hi) T cells (Fig. 5B). In the case of the MRL/lpr lupus-prone mice, circulating concentrations of anti-dsDNA autoantibody were unchanged (Fig. 5C), however a modest increase in the percentage of total CD3+ T cells and a decrease in the percentage of B cell (CD3−B220+) and naïve T cell (CD3+CD4+/CD3+CD8+Cd44loCD62hi) populations was noted after ISO-1 treatment (Fig. 5D). MIF has been described to provide survival signals to murine B cells via a Syk-Akt dependent pathway (11,12) and it is plausible that pharmacologic MIF antagonism may influence the composition of the secondary lymphoid organs in the lymphoproliferative MLR/lpr mice. Nevertheless, this modest difference in lymphoid subpopulations after ISO-1 treatment was not associated with a significant change in the circulating level of anti-dsDNA autoantibody (Fig. 5C).
Figure 5. Influence of ISO-1 and Anti-MIF on Circulating anti-dsDNA Autoantibody Levels and Secondary Lymphoid Organ Subpopulations.

Serum anti-dsDNA antibody titers were measured by specific ELISA and lymphocyte cell surface markers by flow cytometry in NZB/NZW F1 (A, B) and MRL/lpr (C,D) mice. Autoantibody titers were measured on 7–10 individuals per treatment group. Lymphocytes were pooled from the spleen and cervical lymph nodes of each individual mouse (n=4–7 mice per group). No treatment-specific effects in absolute lymphocyte numbers were observed (data not shown). Flow cytometry analyses show the Mean ± SD and P values are by T test (two-tailed).
Influence of MIF Inhibition on Plasma Cytokine Expression
We next measured the circulating levels of selected cytokines during disease development. Serum levels of the inflammatory effector, TNFα, and the chemokine, MCP-1 (CCL2), increased significantly in both the NZB/NZW F1 and MRL/lpr lupus strains, with the highest levels of these mediators observed in the lymphoproliferative MRL/lpr mice (Fig. 6). MIF is known to upregulate innate cytokine production by suppressing activation induced apoptosis (9,10) and ISO-1 or anti-MIF treatment resulted in a significant reduction in plasma TNFα and CCL2 in the MRL/lpr and NZB/NZW F1 strains respectively. The production of Type I interferons (IFNα/β) is associated with the development of lupus immunopathology 1. Our measurements showed the highest circulating levels of IFNα in the lymphoproliferative MRL/lpr strain at 9 weeks, however plasma IFNα concentrations decreased during disease course irrespective of treatment. These data indicate that one impact of MIF inhibition is to reduce the systemic production of effector cytokines such as TNFα, which initiates endothelial and end-organ damage by several mechanisms, and CCL2, which mediates monocyte/macrophage and T cell trafficking into inflammatory sites (1).
Figure 6. Serum Cytokine Levels in NZB/NZW F1 and MRL/lpr Mice Before and After Treatment with MIF Inhibitors.

Sera were isolated at the indicated times in the two mouse models and analyzed for cytokine content by Luminex beadlyte methodology (TNFα, IL-1β, IL-12, CCL2) or specific ELISA (IFNα). Values are the Mean±SD of 4–5 mice measured per group. Only significant differences are marked, and the 22 and 9 week “before treatment” groups were compared to vehicle and IgG controls (Mean ± SD, P values by two-tailed T test); *P<0.05, **P<0.005, ***P<0.001.
Influence of MIF Inhibition on Renal Cytokine and Inflammatory Gene Expression
An examination of kidney tissue from the two lupus-prone mouse strains by quantitative PCR (qPCR) showed a disease associated increase in the expression of mRNA for several effector cytokines, including TNFα, IL-1β, and CCL2, as well as an elevation in IL-12 and IFNα (in the NZB/NZW F1 and MLR/lpr strains respectively) (Fig. 7). After administration of MIF inhibitors, a statistically significant reduction was observed in the NZB/NZW F1 strain for TNFα, IL-1β, and CCL2, and in the MRL/lpr strain for TNFα, CCL2, and IL-1β (after anti-MIF). Of note, while control IgG1 treatment was noted to confer some protection on glomerular disease in the MRL/lpr mouse (Fig. 4B), both ISO-1 and anti-MIF were associated with a significant reduction in the expression of renal TNFα and CCL2 in this mouse strain. Overall, these data suggest a specific action for MIF antagonism in reducing pro-inflammatory cytokine expression in the NZB/NZW F1 and MRL/lpr models of spontaneous SLE.
Figure 7. Quantitative PCR Analyses of mRNA Expression in Renal Tissue in NZB/NZW F1 and MRL/lpr and Mice Before and After Treatment with MIF Inhibitors.

The mRNA values for each treatment group (n=4 kidneys per group) are expressed in units relative to mRNA for GAPDH. The values for wild-type mice are from aged-matched, disease-free (C57BL/6) controls. The upper and lower edges of the boxes represent the 75th and 25th percentiles, respectively, the error bars denote the range of observations, and the horizontal line shows the median, with P values calculated by the Mann-Whitney test. Only significant differences are marked (P values by Mann-Whitney test); *P<0.05.
To gain a more comprehensive assessment of the influence of MIF neutralization on the nephritic phenotype of the NZB/NZW F1 and MRL/lpr lupus-prone mice, we performed a comparative analysis of mRNA isolated from the kidney tissue of treated and untreated mice using genome-scale DNA microarrays. Using statistical and fold-change filtering procedures, a significant change in the expression of 50 genes was observed after ISO-1 or anti-MIF treatment in both the NZB/NZW F1 and MRl/lpr strains (Fig. 8). These genes could be grouped into 3 functional networks: cytokine/chemokine/receptor, T cell, and MIF receptor. Forty of these genes encoded pro-inflammatory cytokines, chemokines, and their receptors. In addition to the decrease in the expression of TNFα, IL-1β and CCL2 noted by qPCR and described above, reduced levels of IL-12, IL-17, MIF and a number of additional pro-inflammatory cytokines as well as chemokine receptors (CCR1,4,5,6,8; CXCR1,4,6) was apparent. The treatment induced reduction of this gene cluster appeared to be more marked in the MRL/lpr than in the NZB/NZW F1 strain, and there appeared to be a greater effect with ISO-1 than for anti-MIF in both strains, although exceptions are evident for particular genes. The presence of the CD2, CD4, and TCRα gene transcripts also was reduced MIF inhibition, which most likely reflects the reduced infiltration of kidneys by CD3+ T cells (Figs. 3E, 4E). Among genes known to be regulated by activation of the MIF receptor, MIF antagonism reduced the expression of the ERK effector, RhoGTPase (47), and the two tyrosine kinases, SRC and LCK (48,49). MIF-dependent, Src family kinase activation is known to inhibit p53-mediated apoptosis (9,50) and a corresponding decrease in the downstream expression of cyclin-dependent kinases also was observed (47,51).
Figure 8. Two-way Coupled Cluster Analysis of Gene Expression in Lupus-prone Mice Treated with ISO-1 or Anti-MIF.
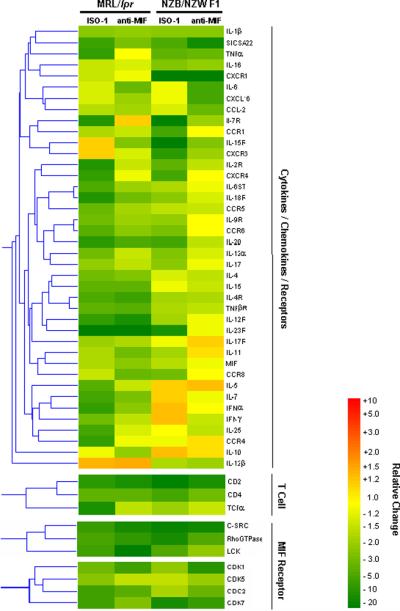
Each column represents an experimental condition (ISO-1 or anti-MIF) relative to control (vehicle or IgG1). The genes selected for display showed significant differences (P<0.05) in ≥2 of the experimental conditions shown. N=3 samples analyzed per experimental group.
In summary, the amelioration in renal injury observed in the NZB/NZW F1 and the MRL/lpr strains, whether mediated by anti-MIF or by the small-molecule, pharmacologic antagonist ISO-1, was associated with a broad downregulation in the expression of inflammatory cytokines and a reduction in the recruitment and retention of infiltrating immune cells.
DISCUSSION
MIF is an innate mediator that exists pre-formed in monocytes/macrophages and is released rapidly upon pro-inflammatory activation (3). Historically, MIF's main role has been considered to be in the retention of infiltrating mononuclear cells within inflammatory lesions. More recent investigations have emphasized MIF's ability to sustain cellular responses by inhibiting activation-induced apoptosis (10,12) and to regulate leukocyte trafficking via non-cognate interactions with CXCR2 and CXCR4, which are expressed in association with the MIF receptor, CD74 (4,14). MIF also influences the differentiation of the adaptive T and B cell response (6,21). While MIFKO mice are developmentally normal, inflammatory or infectious provocation results not only in deficiencies in innate cytokine production (10,52) but also in lymphocyte survival, T cell polarization, and antibody production (6,12,21).
Emerging clinical (24), genetic (23), and murine experimental data (18,33) support an important role for MIF in the immunopathology of SLE. Serum MIF concentrations are elevated in lupus patients and are positively associated with end-organ damage (SLICC/ACR index) (24). MIF is encoded within a polymorphic genetic locus and high-expression MIF alleles may be associated with susceptibility to SLE (23). MIF also has been shown to be a critical mediator of inflammatory renal damage in the anti-glomerular basement membrane model of glomerulonephritis, which mimics many of the immunopathologic features of lupus nephritis (33).
The pharmacologic targeting of MIF by humanized monoclonal antibodies or by small molecule antagonists has attracted considerable interest. A small molecule approach has been facilitated by the presence of an intrinsic tautomerase activity that, while not physiologically relevant, resides in a domain that interacts with the MIF receptor, CD74 (27,28). ISO-1 is an orally active MIF tautomerase inhibitor that has been localized crystallographically to the protein's N-terminal, substrate binding site (31). We verified a role for the tautomerase site in MIF receptor interaction by observing an inhibitory effect of ISO-1 or NAPQI, which covalently modifies the catalytic N-terminal proline (28), on MIF binding to its receptor ectodomain. A biologically neutralizing anti-MIF polyclonal antibody (15) also was found to bind pre-dominantly to a single epitope at the protein's N-terminus, further supporting the importance of this region for inflammatory function.
Several studies have shown a beneficial action of ISO-1 in models of inflammatory tissue damage (42,53) however it should be noted that ISO-1 inhibits MIF binding to its receptor with an IC50 of only 10 μM. Moreover, ISO-1 was developed from a class of platelet glycoprotein IIb/IIIa inhibitors (54) and its selectivity for MIF remains to be established; this may account for certain of the divergent effects of anti-MIF versus ISO-1 in the microarray analysis. In contrast to anti-MIF, ISO-1 also penetrates cells (55), which may elicit additional actions. Indeed, the recently reported MIF inhibitor, 4-iodopyrimidine, influences the interaction between MIF and its intracellular chaperone, p115, which is necessary for MIF secretion (56).
We hypothesized that immunoneutralization or pharmacologic inhibition of MIF may be beneficial in lupus, particularly with respect to protection from inflammatory end-organ damage. In an evaluation of two genetically distinct murine models of spontaneous SLE, MIF was expressed in increased levels, both systemically in the serum and locally within the infiltrating mononuclear cells of the kidney. Treatment with ISO-1 during the time of disease progression ameliorated the decline in renal function and reduced histologic parameters of glomerular injury and interstitial inflammation. Neither glomerular IgG deposition, circulating anti-dsDNA autoantibody levels, or major indices of splenic T or B cell activation were markedly affected by MIF inhibition, although in the case of the MRL/lpr mice, a modest reduction in the secondary lymphoid tissue content of B cells and naïve CD4+, CD8+ T cells was observed. Whether there is a greater reliance on MIF-dependent pathways for lymphocyte survival and the development of autoimmunity in the lymphoproliferative MRL/lpr mice remains to be more closely evaluated (11,12,44). An alteration in B and T cell populations was not observed in the NZB/NZW F1 lupus prone mice, which harbor distinct abnormalities in these lymphocytes (57).
Among the circulating cytokines measured, lupus development was associated with a significant increase in TNFα and in the monocyte chemoattractant, CCL2. MIF inhibition in turn led to a significant reduction in circulating plasma levels of CCL2 (NZB/NZW F1 mice) and TNFα (NZB/NZW F1 and MRL/lpr mice). Intrarenal mRNA levels of TNFα, IL-1β, and CCL2 also were reduced in response to anti-MIF or ISO-1 treatment. A reduction in the expression of both tissue damaging cytokines such as TNFα or IL-1β, and in the chemokine CCL2 are consistent with MIF's upstream role in the expression of these mediators (3,33) and may explain in large part the protective action of MIF inhibition in these models of lupus nephritis (58,59). CCL2 induces the transendothelial migration of monocytes, thereby facilitating tissue injury (60), and MIF itself directly activates the chemokine receptor CXCR4, which is expressed in association with CD74 (4,14). Leukocyte recruitment is an important early event in autoimmune kidney injury (61) and the persistence of macrophages is a consistent feature of rapidly progressive lupus nephritis (2). CXCR4 is known to be upregulated in different mouse models of lupus and treatment with a CXCR4 peptide antagonist has been shown recently to reduce intrarenal leukocyte trafficking and prolong survival in the B6. Sle1Yaa mouse model of SLE (62). The lower indices of inflammatory cytokine activation and intrarenal leukocyte content that were observed after anti-MIF and ISO-1 treatment was supported by the microarray-based survey of gene expression, which showed that in both lupus prone mouse strains, there was a generalized downregulation in the expression of numerous pro-inflammatory cytokines, chemokines, and MIF-dependent signaling intermediates. These conclusions also appear in agreement with the the protective effect of MIF deficiency on lethal renal injury that was reported in MRL/MpJ-Faslpr mice backcrossed onto a mif−/− background (18). In that study, mif deletion reduced renal macrophage recruitment and intrarenal TNFα and IL-1β expression, and urinary CCL2 excretion.
Experimental studies of murine lymphoid development and human lymphoproliferative disorders also support a functional role for MIF in B cell survival signaling (11,12). While neutralization of MIF in the MRl/lpr lupus-prone mice appeared to influence immune cell subpopulations in the spleen, it is unlikely that this effect was therapeutically beneficial because circulating anti-dsDNA levels and renal immunoglobulin deposition were not significantly affected. B cells are presently being targeted in the clinical application of anti-CD20 and soluble human B lymphocyte stimulator (BLyS) therapies, and it is conceivable that more potent MIF antagonists may exert a similar action in downregulating B cell responses (11).
In summary, the present data support the therapeutic value of reducing MIF-dependent effector responses in SLE and they highlight the feasibility of targeting the MIF‐MIF receptor interaction by a small molecule approach. Recent disappointments in the application of biologically-based therapies such as anti-TNF or anti-CD20 to SLE underscore the importance of evaluating new therapeutic targets (30). The small molecule approach represented by the orally-active MIF antagonist ISO-1 or more recent pharmacophores (29) is especially attractive given the high cost of production and parenteral administration of antibody-based therapies, and by the loss of efficacy that may arise from anti-idiotype responses. Patients with SLE also suffer from significant atherosclerosis and cardiovascular mortality (63), and MIF's role in the inflammatory pathogenesis of insulin resistance and atherosclerosis (4,64) would further support its therapeutic targeting in this disease. Finally, the possibility that some SLE patients demonstrate a MIF-dependent form of disease based on their MIF allele (65) suggests that a pharmacogenomic approach may be applied to the clinical evaluation and application of new therapies.
ACKNOWLEDGEMENTS
We are grateful to Drs. Joe Craft, Mark Mamula, and Idit Shachar for their suggestions, and to Anne Davidson for her recommendations about the intervention studies.
Funding: Supported by grants from the Alliance for Lupus Research and the NIH.
Abbreviations
- anti-dsDNA
anti-double-stranded DNA
- ISO-1
(S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5 isoxazole acetic acid methyl ester
- mAb
monoclonal antibody
- MIF
macrophage migration inhibitory factor
- NAPQI
N-acetyl-p-benzoquinone imine
- SLE
systemic lupus erythematosus
- SLICC/ACR
Systemic Lupus International Collaborating Clinics/American College of Rheumatology
REFERENCES
- 1.Rahman A, Isenberg DA. Mechanisms of disease: Systemic lupus erythematosus. New England Journal of Medicine. 2008;358:929–939. doi: 10.1056/NEJMra071297. [DOI] [PubMed] [Google Scholar]
- 2.Peterson KS, Winchester R. Systemic Lupus Erythematosus: Pathogenesis. In: Koopman WJ, Moreland LW, editors. Arthritis and Allied Conditions. Lippincott Williams & Wilkins; Philadelphia: 2005. pp. 1523–1574. [Google Scholar]
- 3.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, Mccoll SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nature Medicine. 2007;13:587–596. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 5.Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179:1895–902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bacher M, Metz CN, Calandra T, Mayer K, Chesney J, Lohoff M, Gemsa D, Donnelly T, Bucala R. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci USA. 1996;93:7849–7854. doi: 10.1073/pnas.93.15.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274:18100–6. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 8.Onodera S, Kaneda K, Mizue Y, Koyama Y, Fujinaga M, Nishihira J. Macrophage migration inhibitory factor up-regulates expression of matrix metalloproteinases in synovial fibroblasts of rheumatoid arthritis. J Biol Chem. 2000;275:444–50. doi: 10.1074/jbc.275.1.444. [DOI] [PubMed] [Google Scholar]
- 9.Fingerle-Rowson G, Petrenko O, Netz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Müller W, Bucala R. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USA. 2003;100:9354–9359. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci USA. 2002;99:345–350. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gore Y, Starlets D, Maharshak N, Becker-Herman S, Kaneyuki U, Leng L, Bucala R, Shachar I. Macrophage migration inhibitory factor (MIF) induces B cell survival by activation of a CD74/CD44 receptor complex. J Biol Chem. 2008;283:2784–2792. doi: 10.1074/jbc.M703265200. [DOI] [PubMed] [Google Scholar]
- 12.Sapoznikov A, Pewzner-Jung Y, Kalchenko V, Krauthgamer R, Shachar I, Jung S. Perivascular clusters of dendritic cells provide critical survival signals to B cells in bone marrow niches. Nature Immunology. 2008;9:388–395. doi: 10.1038/ni1571. [DOI] [PubMed] [Google Scholar]
- 13.Leng L, Metz C, Fang Y, Xu J, Donnelly S, Baugh J, Delonery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–1476. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz V, Lue H, Kraemer S, Korbiel J, Krohn R, Ohl K, Bucala R, Weber C, Bernhagen J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009;583:2749–2757. doi: 10.1016/j.febslet.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mikulowska A, Metz CN, Bucala R, Holmdahl R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J Immunol. 1997;158:5514–5517. [PubMed] [Google Scholar]
- 16.de Jong YP, Abadia-Molina AC, Satoskar AR, Clarke K, Rietdijk ST, Faubion WA, Mizoguchi E, Metz CN, Sahli MA, ten Hove T, Keates AC, Lubetsky JB, Farrell RJ, Michetti P, van Deventer SJ, Lolis E, David JR, Bhan AK, Terhorst C. Development of chronic colitis is dependent on the cytokine MIF. Nat. Immunol. 2001;2:1061–1066. doi: 10.1038/ni720. [DOI] [PubMed] [Google Scholar]
- 17.Denkinger CM, Denkinger M, Kort JJ, Metz C, Forsthuber TG. In vivo blockade of macrophage migration inhibitory factor ameliorates acute experimental autoimmune encephalomyelitis by impairing the homing of encephalitogenic T cells to the central nervous system. J Immunol. 2003;170:1274–1282. doi: 10.4049/jimmunol.170.3.1274. [DOI] [PubMed] [Google Scholar]
- 18.Hoi AY, Hickey MJ, Hall P, Yamana J, O'Sullivan KM, Santos LL, James WG, Kitching AR, Morand EF. Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. Journal of Immunology. 2006;177:5687–5696. doi: 10.4049/jimmunol.177.8.5687. [DOI] [PubMed] [Google Scholar]
- 19.Baugh JA, Chitnis S, Donnelly SC, Monteiro J, Lin X, Plant BJ, Wolfe F, Gregersen PK, Bucala R. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002;3:170–176. doi: 10.1038/sj.gene.6363867. [DOI] [PubMed] [Google Scholar]
- 20.Radstake TRDJ, Sweep FCGJ, Welsing P, Franke B, Vermeulen SHHM, Geurts-Moespot A, Calandra T, Donn R, van Riel PLCM. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis and Rheumatism. 2005;52:3020–3029. doi: 10.1002/art.21285. [DOI] [PubMed] [Google Scholar]
- 21.Mizue Y, Ghani S, Leng L, McDonald C, Kong P, Baugh J, Lane SJ, Craft J, Nishihira J, Donnelly SC, Zhu Z, Bucala R. Role for Macrophage Migration Inhibitory Factor (MIF) in Asthma. Proc Natl Acad Sci USA. 2005;102:14410–14415. doi: 10.1073/pnas.0507189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu S-P, Leng L, Feng Z, Liu N, Zhao H, McDonald C, Lee A, Arnett FC, Gregersen PK, Mayes MD, Bucala R. MIF Promoter Polymorphisms Influence the Clinical Expression of Scleroderma. Arthritis & Rheum. 2006;54:3661–3669. doi: 10.1002/art.22179. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez E, Gomez LM, Lopez-Nevot MA, Gonzalez-Gay MA, Sabio JM, Ortego-Centeno N, de Ramon E, Anaya JM, Gonzalez-Escribano MF, Koeleman BP, Martin J. Evidence of association of macrophage migration inhibitory factor gene polymorphisms with systemic lupus erythematosus. Genes and Immunity. 2006;7:433–436. doi: 10.1038/sj.gene.6364310. [DOI] [PubMed] [Google Scholar]
- 24.Foote A, Briganti EM, Kipen Y, Santos L, Leech M, Morand EF. Macrophage migration inhibitory factor in Systemic Lupus Erythematosus. J Rheumatol. 2004;31:268–273. [PubMed] [Google Scholar]
- 25.Suzuki M, Sugimoto H, Nakagawa A, Tenaka I, Nishihira J, Sakai M. Crystal structure of the macrophage migration inhibitory factor from rat liver. Nature Structural Biology. 1996;3:259–66. doi: 10.1038/nsb0396-259. [DOI] [PubMed] [Google Scholar]
- 26.Rosengren E, Bucala R, Aman P, Jacobsson L, Odh G, Metz CN, Rorsman H. The immunoregulatory mediator macrophage migration inhibitory factor (MIF) catalyzes a tautomerization reaction. Molecular Medicine. 1996;2:143–149. [PMC free article] [PubMed] [Google Scholar]
- 27.Fingerle-Rowson G, Kaleswarapu DR, Schlander C, Kabgani N, Brocks T, Reinart N, Busch R, Schutz A, Lue HQ, Du X, Liu AH, Xiong HB, Chen YB, Nemajerova A, Hallek M, Bernhagen J, Leng L, Bucala R. A Tautomerase-Null Macrophage Migration-Inhibitory Factor (MIF) Gene Knock-In Mouse Model Reveals That Protein Interactions and Not Enzymatic Activity Mediate MIF-Dependent Growth Regulation. Molecular and Cellular Biology. 2009;29:1922–1932. doi: 10.1128/MCB.01907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senter PD, Al-Abed Y, Metz CN, Benigni F, Mitchell RA, Chesney J, Han J, Gartner CG, Nelson SD, Todaro GJ, Bucala R. Inhibition of macrophage migration inhibitory factor (MIF) tautomerase and biological activities by acetaminophen metabolites. Proc Natl Acad Sci USA. 2002;99:144–9. doi: 10.1073/pnas.011569399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cournia Z, Leng L, Gandavadi S, Du X, Bucala R, Jorgensen WL. Discovery of Human Macrophage Migration Inhibitory Factor (MIF)-CD74 Antagonists via Virtual Screening. J Med Chem. 2009;52:416–424. doi: 10.1021/jm801100v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schröder JO, Zeuner RA. Biologics as treatment for systemic lupus: Great efforts, sobering results, new challenges. Curr Drug Discov Technol. 2009;6:252–255. doi: 10.2174/157016309789869010. [DOI] [PubMed] [Google Scholar]
- 31.Lubetsky JB, Dios A, Han J, Aljabari B, Ruzsicska B, Mitchell R, Lolis E, Al Abed Y. The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J.Biol.Chem. 2002;277:24976–24982. doi: 10.1074/jbc.M203220200. [DOI] [PubMed] [Google Scholar]
- 32.Lubetsky JB, Swope M, Dealwis C, Blake P, Lolis E. Pro-1 of macrophage migration inhibitory factor functions as a catalytic base in the phenylpyruvate tautomerase activity. Biochemistry. 1999;38:7346–54. doi: 10.1021/bi990306m. [DOI] [PubMed] [Google Scholar]
- 33.Lan HY, Bacher M, Yang N, Mu W, Nikolic-Paterson DJ, Metz C, Meinhardt A, Bucala R, Atkins RC. The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J Exp Med. 1997;185:1455–65. doi: 10.1084/jem.185.8.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF) Biochemistry. 1994;33:14144–14155. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- 35.Schwab C, Twardek A, Lo TP, Brayer GD, Bosshard HR. Mapping antibody binding sites on cytochrome c with synthetic peptides: Are results representative of the antigenic structure of proteins? Protein Science. 1993;2:175–182. doi: 10.1002/pro.5560020206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin ZN, Bahtiyar G, Zhang N, Liu LZ, Zhu P, Robert ME, McNiff J, Madaio MP, Craft J. IL-10 regulates murine lupus. Journal of Immunology. 2002;169:2148–2155. doi: 10.4049/jimmunol.169.4.2148. [DOI] [PubMed] [Google Scholar]
- 37.Austin HA, Muenz LR, Joyce KM, Antonovych TT, Balow JE. Diffuse Proliferative Lupus Nephritis - Identification of Specific Pathologic Features Affecting Renal Outcome. Kidney International. 1984;25:689–695. doi: 10.1038/ki.1984.75. [DOI] [PubMed] [Google Scholar]
- 38.Kinoshita K, Tesch G, Schwarting A, Maron R, Sharpe AH, Kelley VR. Costimulation by B7-1 and B7-2 is required for autoimmune disease in MRL-Fas(lpr) mice. Journal of Immunology. 2000;164:6046–6056. doi: 10.4049/jimmunol.164.11.6046. [DOI] [PubMed] [Google Scholar]
- 39.Bacher M, Meinhardt A, Lan HY, Mu W, Metz CN, Chesney JA, Calandra T, Gemsa D, Donnelly T, Atkins RC, Bucala R. Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol. 1997;150:235–246. [PMC free article] [PubMed] [Google Scholar]
- 40.Vera PL, Wang XH, Meyer-Siegler KL. Upregulation of macrophage migration inhibitory factor (MIF) and CD74, receptor for MIF, in rat bladder during persistent cyclophosphamide-induced inflammation. Experimental Biology and Medicine. 2008;233:620–626. doi: 10.3181/0709-RM-240. [DOI] [PubMed] [Google Scholar]
- 41.Togel F, Isaac J, Hu ZM, Weiss K, Westenfelder C. Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney International. 2005;67:1772–1784. doi: 10.1111/j.1523-1755.2005.00275.x. [DOI] [PubMed] [Google Scholar]
- 42.Arjona A, Foellmer H, Town T, Leng L, McDonald C, Wang T, Wong S, Montgomery RR, Fikrig E, Bucala R. Abrogation of Macrophage Migration Inhibitory Factor Decreases West Nile Virus Lethality by Limiting Viral Neuroinvasion. J Clin Invest. 2007;117:3059–3066. doi: 10.1172/JCI32218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300. [Google Scholar]
- 44.Crofford LJ, Wilder RL. Arthritis and Autoimmunity in Animals. In: Koopman WJ, editor. Arthritis and Allied Conditions. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 607–634. [Google Scholar]
- 45.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001;291:484–486. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 46.Basta M, Van Goor F, Luccioli S, Billings EM, Vortmeyer AO, Baranyi L, Szebeni J, Alving CR, Carroll MC, Berkower I, Stojilkovic SS, Metcalfe DD. F(ab)'(2)-mediated neutralization of C3a and C5a anaphylatoxins: a novel effector function of immunoglobulins. Nature Medicine. 2003;9:431–438. doi: 10.1038/nm836. [DOI] [PubMed] [Google Scholar]
- 47.Swant JD, Rendon BE, Symons M, Mitchell RA. Rho GTPase-dependent signaling is required for macrophage migration inhibitory factor-mediated expression of cyclin D1. J Biol Chem. 2005;280:23066–23072. doi: 10.1074/jbc.M500636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi X, Leng L, Wang T, Wang W, Du X, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. CD44 is the Signaling Component of the Macrophage Migration Inhibitory Factor-CD74 Receptor Complex. Immunity. 2006;25:595–606. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lue HQ, Kapurniotu A, Fingerle-Rowson G, Roger T, Leng L, Thiele M, Calandra T, Bucala R, Bernhagen J. Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cellular Signalling. 2006;18:688–703. doi: 10.1016/j.cellsig.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 50.Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190:1375–1382. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fingerle-Rowson G, Petrenko O. MIF coordinates the cell cycle with DNA damage checkpoints. Lessons from knockout mouse models. Cell Division. 2007;2:22. doi: 10.1186/1747-1028-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, David JR. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med. 1999;189:341–346. doi: 10.1084/jem.189.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stojanovic I, Maksimovic-Ivanic D, Al Abed Y, Nicoletti F, Stosic-Grujicic S. Control of the of the final stage of immune-mediated diabetes by Iso-1, an antagonist of macrophage migration inhibitory factor. Archives of Biological Sciences. 2008;60:389–401. [Google Scholar]
- 54.Xue CB, Wityak J, Sielecki TM, Pinto DJ, Batt DG, Cain GA, Sworin M, Rockwell AL, Roderick JJ, Wang SG, Orwat MJ, Frietze WE, Bostrom LL, Liu J, Higley CA, Rankin FW, Tobin AE, Emmett G, Lalka GK, Sze JY, Dimeo SV, Mousa SA, Thoolen MJ, Racanelli AL, Hausner EA, Reilly TM, DeGrado WF, Wexler RR, Olson RE. Discovery of an orally active series of isoxazoline glycoprotein IIb/IIIa antagonists. Journal of Medicinal Chemistry. 1997;40:2064–2084. doi: 10.1021/jm960799i. [DOI] [PubMed] [Google Scholar]
- 55.Al Abed Y, Dabideen D, Aljabari B, Valster A, Messmer D, Ochani M, Tanovic M, Ochani K, Bacher M, Nicoletti F, Metz C, Pavlov VA, Miller EJ, Tracey KJ. ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. Journal of Biological Chemistry. 2005;280:36541–36544. doi: 10.1074/jbc.C500243200. [DOI] [PubMed] [Google Scholar]
- 56.Merk M, Baugh J, Zierow S, Leng L, Pal U, Lee S, Ebert A, Mizue Y, Trent J, Mitchell R, Nickel W, Kavathas P, Bernhagen J, Bucala R. The Golgi-Associated Protein p115 Mediates the Secretion of Macrophage Migration Inhibitory Factor. J Immunol. 2009;182:6896–6906. doi: 10.4049/jimmunol.0803710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sabzevari H, Propp S, Kono DH, Theofilopoulos AN. G1 arrest and high expression of cyclin kinase and apoptosis inhibitors in accumulated activated memory phenotype CD4(+) cells of older lupus mice. European Journal of Immunology. 1997;27:1901–1910. doi: 10.1002/eji.1830270813. [DOI] [PubMed] [Google Scholar]
- 58.Boswell JM, Yui MA, Burt DW, Kelley VE. Increased Tumor Necrosis Factor and IL-1β-Gene Expression in the Kidneys of Mice with Lupus Nephritis. Journal of Immunology. 1988;141:3050–3054. [PubMed] [Google Scholar]
- 59.Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR. Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas(lpr) mice. Journal of Experimental Medicine. 1999;190:1813–1824. doi: 10.1084/jem.190.12.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Randolph GJ, Furie MB. A Soluble Gradient of Endogenous Monocyte Chemoattractant Protein-1 Promotes the Transendothelial Migration of Monocytes In-Vitro. Journal of Immunology. 1995;155:3610–3618. [PubMed] [Google Scholar]
- 61.de Lema GP, Maier H, Franz TJ, Escribese M, Chilla S, Segerer S, Camarasa N, Schmid H, Banas B, Kalaydjiev S, Busch DH, Pfeffer K, Mampaso F, Schlondorff D, Luckow B. Chemokine receptor CCR2 deficiency reduces renal disease and prolongs survival in MRL/1pr lupus-prone mice. Journal of the American Society of Nephrology. 2005;16:3592–3601. doi: 10.1681/ASN.2005040426. [DOI] [PubMed] [Google Scholar]
- 62.Wang A, Fairhurst AM, Tus K, Subramanian S, Liu Y, Lin FM, Igarashi P, Zhou XJ, Batteux F, Wong D, Wakeland EK, Mohan C. CXCR4/CXCL12 Hyperexpression Plays a Pivotal Role in the Pathogenesis of Lupus. Journal of Immunology. 2009;182:4448–4458. doi: 10.4049/jimmunol.0801920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salmon JE, Roman MJ. Subclinical atherosclerosis in rheumatoid arthritis and systemic lupus erythematosus. American Journal of Medicine. 2008;121:3–8. doi: 10.1016/j.amjmed.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verschuren L, Kooistra T, Bernhagen J, Voshol PJ, Ouwens DM, van Erk M, de Vries-van der Weij, Leng L, van Bockel JH, van Dijk KW, Fingerle-Rowson G, Bucala R, Kleemann R. MIF Deficiency Reduces Chronic Inflammation in White Adipose Tissue and Impairs the Development of Insulin Resistance, Glucose Intolerance, and Associated Atherosclerotic Disease. Circulation Research. 2009;105:99–U275. doi: 10.1161/CIRCRESAHA.109.199166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sreih AG, Ezzeddine R, Leng L, LaChance A, Yu G, Svenungsson E, Gunnarsson I, Mizue Y, Cavett J, Glenn S, Pons-Estel B, Perl A, Salmon J, Alacon-Riquelme M, Harley J, Bucala R. Dual Effect of MIF Gene Polymorphisms On the Development and the Severity of SLE. Arthritis and Rheumatism. 2009;60 doi: 10.1002/art.30624. Abstract 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lan HY, Mu W, Yang N, Meinhardt A, Nikolic-Paterson DJ, Ng YY, Bacher M, Atkins RC, Bucala R. De novo renal expression of macrophage migration inhibitory factor during the development of rat crescentic glomerulonephritis. Am J Pathol. 1996;149:1119–1127. [PMC free article] [PubMed] [Google Scholar]