

Abstract
Protein amyloid oligomers have been strongly linked to amyloid diseases and can be intermediates to amyloid fibers. β-Sheets have been identified in amyloid oligomers. However, because of their transient and highly polymorphic properties, the details of their self-association remain elusive. Here we explore oligomer structure using a model system—macrocyclic peptides. Key amyloidogenic sequences from Aβ and tau were incorporated into macrocycles, thereby restraining them to β-strands, but limiting the growth of the oligomers so they may crystallize and cannot fibrillate. We determined the atomic structures for four such oligomers, and all four reveal tetrameric interfaces in which β-sheet dimers pair together by highly complementary, dry interfaces, analogous to steric zippers found in fibers, suggesting a common structure for amyloid oligomers and fibers. In amyloid fibers, the axes of the paired sheets are either parallel or antiparallel, whereas the oligomeric interfaces display a variety of sheet-to-sheet pairing angles, offering a structural explanation for the heterogeneity of amyloid oligomers.
Introduction
A wide range of human pathologies, including Alzheimer’s disease, dialysis-related amyloidosis and Parkinson disease, are associated with amyloid fiber formation from diverse proteins1. Despite the enormous variation in sequences and structures of the amyloidogenic proteins, the interaction of β-sheets is central to the assembly of soluble oligomers and mature amyloid fibers2–7. Crystallographic studies have now revealed the fiber-like atomic structures of numerous amyloidogenic segments from fiber-forming proteins8. The formation of parallel or anti-parallel β-sheets and the assembly of pairs of β-sheets into a steric-zipper are two key steps of fiber formation4.
Evidence has recently accumulated suggesting that instead of amyloid fibers, soluble oligomers are the more pathogenic species in several types of protein deposition diseases9–11. Studied by NMR, FTIR, EPR spectroscopy and other methods, amyloid oligomers have been found to exhibit several common biochemical and biophysical properties: 1) amyloid oligomers contain β-sheet rich structures2,3,5,6; 2) different sizes of the oligomer species co-exist in solution and contribute to the heterogeneity of the oligomer mixtures12; 3) as an intermediate state, most of the oligomeric species are transient13; 4) mediated by different protein segments, different types of oligomers can form, indicating polymorphism of amyloid oligomers14–17, with some of the species showing strong structural resemblance to fibers18; 5) oligomers formed from different amyloidogenic proteins seem to share common structural features because they are recognized by the same antibody A1119; 6) some of the oligomeric species show higher cytotoxicity than fibers20. Despite this knowledge, the dynamic, polymorphic, and noncrystalline behavior of the oligomeric species hinder structural studies at atomic resolution. Learning the structures of amyloid oligomers seems necessary for understanding of their cellular toxicity and fiber formation, and for chemical interventions against amyloid disease.
Here we adopt macrocyclic peptides to explore the nature of amyloid oligomers. Because of the prevalence of β-sheets in biological processes such as protein-protein interactions, protein self-association and protein aggregation, peptidic model systems which mimic β-sheets have been established21–24. Nowick and co-workers recently developed macrocyclic peptides as a constrained chemical model to investigate interactions within and between β-sheets25–27. The macrocyclic peptide is a 42-membered ring consisting of a pentapeptide β-strand (recognition strand in Fig. 1. The term “recognition” is used since it has been established that the sequence of this strand confers the ability to recognize and bind like-sequence segments in the context of larger proteins28), two δ-linked ornithines mimicking β-turns, and an antiparallel β-strand (blocking strand in Fig. 1) composed of two amino acids and a “Hao” unit. The Hao unit mimics a tripeptide β-strand and is conformationally restricted to an extended β-sheet geometry by an aromatic group fused to its backbone. By forming hydrogen bonds with the recognition strand, Hao supports the β-strand conformation of the pentapeptide recognition strand. Therefore, the recognition strand is open to form edge-to-edge β-sheets with the recognition strand from a second macrocyclic peptide, whereas Hao prevents the blocking strand from further aggregation.
Figure 1.
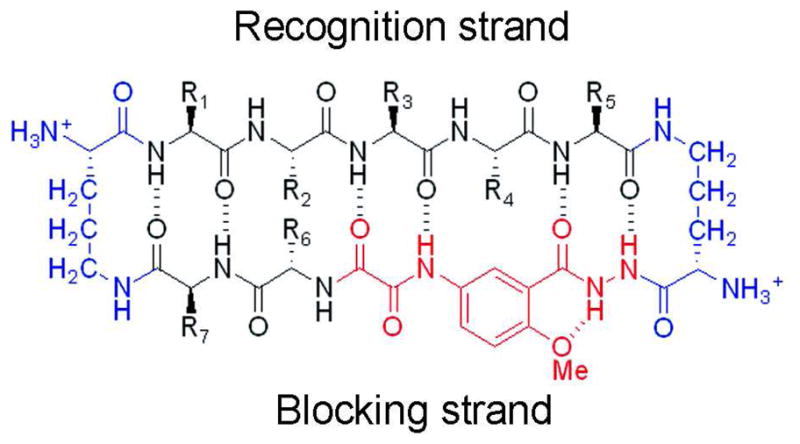
The 42-membered macrocyclic framework used in this study. Two δ ornithine turn units are in blue. The Hao unit (red) blocks the lower edge of the recognition strand. The pentapeptide of the recognition strand (positions R1–R5) accommodates the amyloidogenic sequence of interest (Table 2). Residues in the blocking strand (positions R6 and R7) can be varied for better folding and solubility.25
The 42-membered ring macrocyclic peptide mimics the β-strand conformation of polypeptides in oligomeric states. By displaying a pentapeptide sequence from the amyloidogenic polypeptide in the recognition strand, the conformation of the pentapeptide is restrained to be a β-strand, whereas the stacking of β-strands into infinite β-sheets is prevented by Hao in the blocking strand. Therefore macrocyclic rings can freeze and homogenize the transient amyloid oligomers, and make atomic structure determination possible. Specifically, sheets would be restricted to dimers in the hydrogen bonding direction, but sheet-to-sheet interactions can expand the oligomer to tetramers.
In this work, we set out to mimic with such macrocyclic peptides the assembly and structures of amyloid oligomers associated with Alzheimer’s disease. Three amyloidogenic peptides from Aβ and tau known to account for the aggregation of these proteins in amyloid assemblies were displayed on macrocycles in β-strand conformation. We determined the crystal structures of these macrocycles and analyzed their structural characteristics at an atomic-level. The crystal structures show β-sheet dimers assemble into tetramers through dry, complementary interfaces between sheets. These observations, common to all four oligomeric interfaces, suggest dry, complementary interfaces are characteristic features of amyloid oligomer assembly as they are of fiber assembly. Unlike amyloid fiber structures, we observe the sheet-to-sheet pairing geometries in tetrameric oligomers to deviate from cross-β geometry by the angles of intersection of the axes of the two β-sheets of the steric zipper motifs. These variations help to explain the diversity of previously observed amyloid oligomeric polymorphs. The resulting understanding of amyloidogenic oligomer assembly at the atomic level offer clues to design of structure-based therapeutics. In addition, macrocyclic peptides have the potential to inhibit the growth of amyloid oligomers and fibers.
Experimental Section
1. Crystallization
The four macrocyclic peptides were prepared as described previously25. The mcLVFBrFA, mcLVFFA, mcAIIFL and mcVQIVFBr were dissolved in 20 mM sodium phosphate buffer pH 7.0 containing 100 mM sodium chloride to 20 mg ml−1, 20 mg ml−1, 25 mg ml−1 and 15 mg ml−1, respectively. Crystals of macrocyclic peptides were grown by mixing the peptides with an equal amount of well solution by the hanging drop vapor diffusion method. Peptide mcVQIVFBr was crystallized under the condition containing Na/K phosphate pH 6.2, 35% (v/v) (+/−)-2-methyl-2, 4-pentanediol, at 18°C. The crystallization condition which gave crystals of peptide mcLVFBrFA contains 0.1 M sodium acetate pH 4.6, 0.17 mM calcium chloride dehydrate, 20% (v/v) 2-propanol. Crystals of peptide mcLVFFA were obtained in 0.1 M MES pH 6.0, 200 mM Li2SO4, 20% (v/v) 1, 4-butanediol. Crystals of peptide mcAIIFL were obtained in 0.1 M MES pH 6.2, 0.15 mM Zn(OAc)2, 12.5% (w/v) PEG 8,000. Crystals of all four macrocyclic peptides were soaked in the cryoprotectant buffer containing the reservoir solution plus 20% (v/v) glycerol. Crystals were frozen in Hampton loops in liquid nitrogen before data collection.
2. Data collection
X-ray diffraction data of peptide mcVQIVFBr were collected at 100 K with a Rigaku FR-D X-ray generator equipped with an Raxis4++ imaging plate detector. Data of peptides mcAIIFL, mcLVFBrFA and mcLVFFA were collected at 100 K at beamline 24-ID-C, Advanced Photo Source, Argonne National Laboratory. Denzo and XSCALE29 were used for data integration and scaling. Statistics of data collection are listed in Table 1.
Table 1.
Statistics of crystallographic data collection and atomic refinement for the four structures reported in this paper.
| Macrocycle | mcVQIVFBr | mcAIIFL | mcLVFBrFA | mcLVFFA | |||
|---|---|---|---|---|---|---|---|
| Data collection | |||||||
| Space group | I4122 | Refinement | Peak | Remote | Refinement | Phasing | P43212 |
| P31 | P6422 | P6422 | P43212 | P43212 | |||
| Cell dimensions | |||||||
| a, b, c (Å) | 32.9, 32.9, 55.4 | 41.8, 41.8, 63.1 | 41.8, 41.8, 63.1 | 41.5, 41.5, 62.6 | 59.0, 59.0, 128.5 | 59.2, 59.2, 128.7 | 57.9, 57.9, 129.7 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Wavelength (Å) | 1.5418 | 0.9197 | 0.9197 | 0.9129 | 0.9794 | 0.9197 | 0.9791 |
| Molecules per asymmetric unit | 1 | 8 | 2 | 2 | 8 | 8 | |
| Resolution (Å) | 20–2.01 (2.05–2.10) | 18–2.55 (2.62–2.55) | 20–2.6 (2.72–2.60) | 90–2.8 (2.95–2.80) | 54–1.99 (2.04–1.99) | 20–2.3 (2.37–2.30) | 100–2.25 (2.33–2.25) |
| Rmerge (%) | 5.0 (10.9) | 3.9 (50.9) | 4.6 (58.7) | 8.4 (50.4) | 6.5 (47.8) | 6.2 (51.7) | 7.3 (47.0) |
| I/σ I | 42.1 (22.9) | 15.6 (1.7) | 29.5 (3.7) | 16.1 (4.2) | 26.1 (6.5) | 14.6 (2.6) | 15.7 (2.9) |
| Completeness (%) | 98.2 (97.1) | 98.1 (100.0) | 98.8 (100.0) | 99.5 (100.0) | 100.0(100.0) | 99.0 (98.3) | 98.8 (99.6) |
| Redundancy | 12.0 (12.2) | 2.2 (2.3) | 8.6 (8.8) | 8.0 (8.2) | 13.6 (14.0) | 4.4 (4.2) | 3.3 (3.4) |
| Refinement | |||||||
| Resolution (Å) | 2.05 | 2.55 | 1.99 | 2.25 | |||
| No. reflections | 1007 | 3980 | 15269 | 10451 | |||
| Rwork/Rfree (%) | 17.9/20.3 | 17.8/22.4 | 19.6/21.8 | 20.5/22.2 | |||
| No. atoms | |||||||
| Macrocycle | 93 | 816 (includes alternate conformations) | 744 | 736 | |||
| Ligand/ion | glycerol (6), acetate acid (1) | glycerol (18), zinc (8) | glycerol (18), chloride (7), isopropanol (8) | glycerol (5), sulfate (6), 1,4-butanediol (11) | |||
| Water | 14 | 19 | 48 | 31 | |||
| B-factors (Å2) | |||||||
| Peptide | 13.5 | 74.0 | 26.8 | 41.5 | |||
| Water | 27.1 | 73.8 | 40.7 | 45.8 | |||
| RMS deviations | |||||||
| Bond lengths (Å) | 0.017 | 0.010 | 0.016 | 0.010 | |||
| Bond angles (°) | 1.3 | 1.2 | 1.2 | 1.3 | |||
3. Structure determination and refinement
The crystal structure of mcVQIVFBr was determined to 2.0 Å resolution using phases determined from a single anomalous dispersion (SAD) data set. One bromine site was located using SHELXD30 and phases were calculated with SHELXE31. Model building was performed with Coot32 and illustrated with PyMOL from Delano Scientific. Crystallographic refinement was performed with program REFMAC33. The model was finally refined with a TLS model using REFMAC to an Rwork = 17.9%, and Rfree = 20.3%. Coordinates have been deposited with PDB accession code 3Q9G.
The crystal structure of mcAIIFL was determined to 2.55 Å resolution using phases determined from a two-wavelength anomalous dispersion data set. Three zinc sites were located in space group P6422 using SHELXD and phases were calculated with SHELXE. Model building was performed with Coot and illustrated with PyMOL. Examination of side-chain packing patterns indicated the crystallographic 2-fold axes parallel and perpendicular to the 64 screw axis were broken, so the space group symmetry was expanded to P31. Crystallographic refinement was performed with program REFMAC and BUSTER-TNT34. The model was finally refined to an Rwork = 17.6%, and Rfree = 22.3%. Coordinates have been deposited with PDB accession code 3Q9J.
The crystal structure of mcLVFBrFA was determined to 2.0 Å resolution using phases determined from a single-wavelength anomalous dispersion data set. Eight bromine sites were located in space group P43212 using SHELXD and phases were calculated with SHELXE. Model building was performed with Coot and illustrated with PyMOL. Crystallographic refinement was performed with program REFMAC. The model was finally refined to an Rwork = 19.6%, and Rfree = 21.8%. Coordinates have been deposited with PDB accession code 3Q9I. The native mcLVFFA crystal was isomorphous with the bromo derivative. Phases were obtained by difference Fourier methods. The mode was refined with REFMAC, then Buster/TNT to an Rwork =20.5, Rfree = 22.2%. Coordinates have been deposited with PDB accession code 3Q9H.
4. Oligomer modeling
Models of extended oligomers were built from crystal structures of the tetrameric oligomers by repeated application of 9.6 Å translations in the hydrogen-bonding direction. The models were energy minimized using conjugate gradient and simulated annealing algorithms available with the program CNS35 with hydrogen bonding restraints36.
Results
1. Macrocyclic peptide design
The abnormal aggregation of Aβ into amyloid plaques and tau into paired helical filaments (PHFs) are the hallmarks of Alzheimer’s disease and related tauopathies37–39. Oligomers formed by Aβ have been claimed to be the causative agents of Alzheimer’s disease40. Even in the absence of Aβ, tau oligomers can cause memory impairment and neurodegeneration41. Based on the participation of Aβ and tau in neurodegenerative disease, key amyloidogenic segments from Aβ and tau were selected and incorporated into macrocycles for structural study.
In tau oligomerization and fibrillation, segment 306VQIVYK311 located at the third microtubule-binding domain was revealed to be essential in mediating molecular assembly42. It has the highest predicted β-sheet potential and shows a high tendency to self associate6. Proline-scanning mutations show that conformational changes in this segment from random coil to β-strand drive tau molecular assembly and aggregation42. Because of the importance of 306VQIVYK311 in tau aggregation, the pentapeptide VQIVY was incorporated into the macrocyclic peptide mcVQIVFBr (Table 2). The tyrosine hydroxyl group was replaced by a bromine atom so that crystallographic phases could be obtained by anomalous scattering methods.
Table 2.
Sequences of the macrocyclic peptides.
| Macrocycle | Amyloid sequence | R1 | R2 | R3 | R4 | R5 | R6 | R7 |
|---|---|---|---|---|---|---|---|---|
| mcLVFFA | Aβ17–21 | Leu | Val | Phe | Phe | Ala | Leu | Lys |
| mcLVFBrFA | Aβ17–21F19FBr | Leu | Val | Phe(Br) | Phe | Ala | Leu | Lys |
| mcAIIFL | Aβ30–34G33F | Ala | Ile | Ile | Phe | Leu | Tyr | Lys |
| mcVQIVFBr | Tau306–310Y310FBr | Val | Gln | Ile | Val | Phe(Br) | Lys | Leu |
Column 1 gives the abbreviation used in the text. Column 2 gives the protein from which an amyloidogenic segment between the numbered positions has been selected. Columns 3–7 give the residues inserted into the framework shown in Fig. 1.
Solid-state NMR studies on Aβ fibers have revealed a U-shaped structure—two β-strand segments (residues 10–24 and 30–40) joined by a U-turn43,44. These two segments were also observed to adopt β-strand conformations in various soluble oligomeric species and in a monomeric state stabilized by a binding partner2,5,45. An observed shift from random coil to β-strand triggers the assembly of Aβ molecules2. We inserted Aβ residues 17–21, LVFFA, from the first amyloidogenic segment into the recognition strand of the macrocyclic peptide mcLVFFA. In order to obtain the crystallographic phases for structure determination, we also prepared mcLVFBrFA, in which the phenylalanine at position R3 is replaced by 4-bromo-phenylalanine (FBr). To mimic the second amyloidogenic segment, we prepared the macrocyclic peptide mcAIIFL, a derivative of Aβ residues 30–34, AIIGL, substituting Gly with Phe. This replacement improved the entire folding of the macrocycle and made it possible for crystallization and structure determination26.
2. Structures of the monomeric macrocyclic peptides
Extensive solution-phase studies on 42-membered macrocyclic peptides have indicated that the recognition strands tend to adopt a β-strand conformation in solution25. Here we present the first crystal structures of macrocyclic peptides in this family. Statistics for crystallographic data collection and structure refinement of macrocyclic peptides mcLVFFA, mcAIIFL and mcVQIVFBr are listed in Table 1. Fig. 2a shows the crystal structures of the monomeric macrocycles. As designed, each macrocycle displays a pair of hydrogen-bonded β-strands locked into an antiparallel topology by two peptidomimetic δ-linked ornithine residues. Despite the diverse peptide sequences in the three macrocycles, the RMSD values for backbone atoms between any pair of the three structures do not exceed 1.3 Å (Table S2). The ability to accommodate a variety of sequences indicates the stability of the conserved framework (Fig. S2a).
Figure 2.

Crystal structures of macrocyclic peptides mcVQIVFBr, mcAIIFL and mcLVFFA. Within the monomeric structures (a), hydrogen bonds link the blocking and recognition strands and are shown as dotted lines. β-Sheet formation by dimeric assemblies are either parallel or antiparallel as shown in (b). (c) Molecular packing of four different tetramers. The tetrameric macrocyclic molecules form a variety of β-sheet-to-β-sheet packing geometries, deviating by 0° to 45° from cross-β geometry as shown in (d).
The designed incorporation of the Hao residue in the blocking strand also appears to be effective in promoting oligomeric β-sheet assembly while blocking run-away fiber formation. In all three structures, the Hao residue in the blocking strand acts as an essential building block constraining the recognition strand to an extended β-strand conformation by a network of backbone hydrogen bonds with the blocking strand’s Hao, R6, and R7 residues. The constrained conformation of the recognition strand facilitates formation of an intermolecular β-sheet with other β-strand molecules at its exposed edge. But, the exposed edge of the blocking strand is prohibited from interacting with other β-strand molecules by the aromatic ring and an intramolecular hydrogen bond in the Hao residue. Indeed, hydrogen-bonded self-association of macrocycles is observed in our crystal structures only in the β-sheet formed between recognition strands containing the amyloidogenic segment. This designed feature makes the macrocycle system suitable for study of the interaction patterns of amyloidogenic segments in the context of oligomers.
In addition to these designed features, an unexpected feature, a bend, was observed in the blocking strands of mcVQIVFBr, mcLVFFA, and mcAIIFL (Fig. S2b). Because the bend is located at the same residue in each macrocycle (at the α-carbon of R6), a likely explanation is that it arises from an interruption of the natural pleat of the β-sheet by the Hao residue. The natural pleat of β-sheets arises from the 109° bond angle of sp3 hybridized α-carbons that connect the 3.3 Å long planar peptide linkages. The α-carbon of each successive amino acid creates a pleat opposite in direction and equal in depth to the preceding amino acid (the zig-zag pattern is obvious when a sheet is viewed down the hydrogen bonding direction). The alternating directions of the pleats with equal depth give the sheet an overall flat geometry. However, in the macrocycle, the pleats are not all of equal depth. The Hao residue enforces planarity over the distance of three amino acids (about 12 Å), so the depth of the pleats (distance between tetrahedral α-carbons) varies within the blocking strand: 12 Å on the N-terminal side of R6 (where the Hao moiety is located) and 3.3 Å on the C-terminal side (where the standard peptide is located). Because the pleats are of unequal depth in the blocking strand, the appearance of an overall bent topology is conferred where the natural peptide joins the Hao plane at R6 (Fig. S2b). The bend is propagated to the recognition strand (at the α-carbon of R2) by the hydrogen bonds that link it to the blocking strand. However, the bend in the recognition strand is less pronounced than the blocking strand. The degree of curvature is well within the range observed in natural β-sheets (Fig. S2c), especially in those with fewer strands46.
Previous studies have revealed that macrocycle solubility and folding are influenced by the choice of residues incorporated at the R6 and R7 positions25; our crystal structures offer some explanation. As might be expected, macrocycle solubility improves by incorporating a charged residue such as lysine at R6 or R7, as has been done in all three macrocycles presented here (Table 2). In all three structures, the lysine side chain extends into bulk solvent, improving the macrocycle interaction with solvent. Less obvious is the effect of the R6 and R7 side chains on folding. The interactions between the neighboring strands are limited to backbone hydrogen bonding (Fig. 2a) as is typical in conventional β-sheets. The R6 and R7 side chains of the blocking strand have no van der Waals contacts with R2 and R1 of the recognition strand. In thips respect, macrocycle folding should be relatively insensitive to the specific residues at R6 or R7. The reported improvement in macrocycle folding with the incorporation of an aromatic residue at R6 compared to Leu and Ala25 might be attributed to a van der Waals contact between the R6 aromatic ring and the nearby ornithine hairpin turn (Fig. 2a mcAIIFL, R6=PheBr). Such stabilization is not possible with Leu or Ala side chains because they are too short to form a contact with the ornithine turn.
3. Assembly of dimers through β-sheet hydrogen-bonding interactions
In all three crystal structures, the macrocycles assemble into dimers via standard backbone hydrogen bonds between recognition β-strands, forming intermolecular β-sheets (Fig. 2b). One particular pattern of hydrogen bonding predominates: a two-fold symmetric antiparallel arrangement of strands where the symmetry axis (marked by ellipses in Fig. 2b) is normal to the plane of the sheet. The position of the two-fold differs among the three structures and alters the registration between recognition strands. In mcLVFFA, the two-fold operator passes between R3 residues, whereas in mcVQIVFBr and mcAIIFL the two-fold operator passes between R4 residues (Fig 2b) leaving some backbone amides at the N-termini solvent exposed. In these arrangements, the intermolecular sheet has a sidedness; the side-chains displayed on the face have different identities than those on the back. An additional parallel, out-of-register sheet is observed in the mcLVFFA crystal structure (Fig. 2b); the topology of the macrocyclic framework precludes homodimeric parallel in-register sheets47.
Other types of intermolecular hydrogen-bonded arrangements are possible, but not observed. For example, one can imagine a different type of antiparallel arrangement in which the face and back of the sheets are related by two-fold symmetry. The particular hydrogen-bonding geometry adopted by a pair of macrocycles, is specified by the sequence of the recognition strand (the amyloidogenic insert). That is, the identity of side chains that come in contact across the dimer interface—their complementarity in shape and physical properties—depends on the geometry of the dimer. Side-chain packing interactions are also observed between pairs of dimers as described below, and likely play a role in specifying hydrogen-bonding geometry.
Lastly, it should be noted that there are some hydrogen-bonded arrangements which are common among globular and fibrillar proteins, but cannot be formed by macrocycle homodimers. These include parallel in-register dimers and some anti-parallel pairing arrangements. Whereas the recognition strand has two non-equivalent hydrogen bonding edges, only one edge is available to form the dimer interface (formed by backbone N and O of R2 and R4); the other edge is hydrogen-bonded to the blocking strand (formed by backbone N and O of R1, R3, and R5). Hydrogen bonding with the blocking strand prohibits the two edges from interchanging. Thus, antiparallel sheet formation is limited to dimer interfaces involving backbone atoms of even numbered residues.
4. Assembly of tetramers through complementary side-chain interactions
In all three macrocycles studied here, dimers assemble into tetramers through the interdigitation of side-chains protruding from the surfaces of the dimeric β-sheets (Fig. 2c). The different tetrameric interfaces formed by the three macrocycles in crystal structures are listed in Table S3. Four of these interfaces (including two in the mcLVFFA crystal) are large, burying surface areas ranging from 894 Å2 to 1089 Å2 (Table S1), and mainly formed by inserted amyloidogenic segments. Their shape complementarities (0.60 to 0.77; Table S1) are comparable to those observed in steric zippers (0.57–0.92)4 (Fig. S3) and typical oligomeric interfaces between globular proteins (from 0.70–0.74)48. The side chains in all four interfaces are mainly hydrophobic (both aromatic and aliphatic), exclude water and create a stable nucleation site in the tetramer. The majority of these dry interfaces are packed face-to-face (approximate D2 symmetry), using side-chains from positions R1, R3, and R5. For example, the nucleation site of mcVQIVFBr is formed mainly by π–π stacking between two pairs of Tyr side chains at the R5 positions (Fig. 3a) and is supported by hydrophobic contacts between pairs of Ile side chains at position R3. In mcAIIFL, the hydrophobic nucleation site is formed by alanine, isoleucine and leucine side chains at positions R1, R3, and R5 (Fig. 3b). Note that the lack of participation of R4 in this tetramer interface alleviates concern about the biological relevance raised by the mutation of R4 from Gly in Aβ30–34 to Phe. In mcLVFFA there are two types of interfaces. In the first, the hydrophobic nucleation site is formed by leucine, phenylalanine, and alanine side chains at positions R1, R3, and R5 (Fig. 3c). But, the second interface lacks symmetry (an interface between a parallel and an antiparallel dimer), so different strands contribute different side chains (Fig. 3d). Three of the strands contribute side chains at positions R2 and R4, while one of the strands from the parallel sheet contributes side chains at positions R1, R3, and R5. Although residues in the blocking strand also have some contribution to the dry interface, the majority of the interface derives from the amyloidogenic insert in the recognition strand (Table S1). The predominant role played by the amyloidogenic insert suggests that the dry interfaces observed here present a very likely possibility for amyloidogenic oligomer assembly at the atomic level and may even be representative of the oligomeric packing patterns in solution. Furthermore, the tight oligomeric packing of these inserts illustrates that these segments have a strong intrinsic tendency of self-association, which is in agreement with previous studies5,42,45.
Figure 3.

Side-chain interactions of the amyloidogenic segments in the macrocyclic tetramers. The designed amyloidogenic segments mediate tight and highly complementary hydrophobic interactions between macrocycle molecules. The side chains of the segments are shown as sticks and spheres.
Another key feature of the oligomeric interface is the variety of β-sheet-to-β-sheet packing geometries observed. In all four interfaces, the pairs of sheets interact through their flat surfaces (Fig. 3). The recognition strands of the monomers can assemble as antiparallel (mcLVFFA, mcAIIFL and mcVQIVFBr) or parallel (mcLVFFA) (Fig. 2b). Variations in dimer interfaces lead to variations in tetramer interfaces; interfaces were observed between pairs of antiparallel sheets (mcLVFFA interface 1, mcAIIFL and mcVQIVFBr) and between a parallel and antiparallel sheet (mcLVFFA interface 2). Further variations in tetramer interfaces involve differences in orientation between the faces of opposed β-sheets (Fig. 2c); the crossing angle between β-strands of opposing sheets can be orthogonal (mcVQIVFBr), anti-parallel (mcAIIFL and mcLVFFA interface 1), or somewhere in-between (mcLVFFA interface 2) (Table S1; Fig. 2d).
5. Structural features that distinguish amyloid-like oligomers from fibers
A close structural relationship exists between our tetrameric amyloid-like oligomers and fibers. The formation of a dry, highly complementary interface between pairs of β-sheets, is not only a characteristic feature of these amyloid oligomers, but of amyloid fibers as well. Recently, numerous crystal structures have been reported for various amyloidogenic peptides in the fibrillar form4,49. The steric zipper motif was observed in all these fiber structures. A typical steric zipper is formed by a pair of interdigitated β-sheets with no water in the interface. Figure 4 compares various amyloidogenic segments in oligomeric and fibrillar states. The structures in both states share two features: 1) architecture: the segments form β-strands, β-strands stack to β-sheets, and β-sheets pair with one another via side chains; 2) dry interface: the interactions between β-sheet layers are all dry interfaces with high shape complementarity. The area buried and shape complementarity have similar values for both oligomeric and fibrillar structures (Fig. S3; Table S4). These two shared characteristics suggest that both oligomerization and fibrillation are driven by the formation of highly complementary, dry interfaces. The common observation of dry, complementary interfaces in both oligomers and fibers demonstrates the structural similarity of the two states, which is indicated also by NMR, FTIR spectrometry and conformation-dependent antibodies3,14,16,18.
Figure 4.

Comparison of the packing of amyloidogenic segments in oligomeric states (this paper) and in fibrillar states (PDB codes: 2ON9, 3FQP, 2OMQ, 3FVA). In the structures of both oligomeric states and fibrillar states, water molecules are entirely excluded from the interfaces. Amyloidogenic segments are assembled through a highly complementary, dry interface between pairs of β-sheets. The difference in packing between these two states is that in the oligomeric state, the orientation between opposing β-sheets ranges from orthogonal to parallel; whereas, in the fibrillar state, the strands are constrained to either parallel or antiparallel orientations (cross-β). Water is shown in cyan spheres, and zinc is in green cyan spheres. For AIIGL from Aβ, Gly is replaced to Phe in the macrocycle. Phe adopts two conformations in the crystal structure and does not contribute to the tetramer packing.
The primary structural difference between our macrocyclic oligomers and peptide fibers appears to be an additional degree of freedom in sheet-to-sheet packing observed in the oligomers. In the fibrillar state, the strands in opposing sheets are constrained to either parallel or antiparallel orientations (definition of cross-β architecture)4. But, in the oligomeric state, the orientation between opposing β-sheets ranges from orthogonal to parallel (Fig. 2c–d). Notably, the orientations between strands of opposing sheets observed in mcVQIVY (90°) and mcLVFFA (30°) are similar to values commonly reported for β-sandwiches in small globular proteins and first noted in the earlier years of protein crystallography50,51. Orientations of 0° or 180° (as in mcAIIFL and amyloid fibers) were not observed until structures with significantly larger sheets, such as GNNQQNY8 and β-helix proteins52, were revealed. Indeed, it seems likely that the greater geometric constraints imposed in the fibers compared to oligomers arise from the larger number of molecules in each β-sheet; there might be thousands of β-strands in a sheet within a fiber, but only a few dozen in an oligomer. Consequently, there are additional degrees of freedom in the side-chain rotamers of oligomers compared to fibers. The additional degrees of freedom can accommodate additional (non cross-β) sheet-to-sheet packing geometries. In short, the most pronounced difference in the geometries of some of these amyloid oligomers from amyloid fibers is the deviation of the axes of the interacting sheets of the oligomers from 0° or 180°.
Discussion
A variety of morphologies have been described for amyloidogenic oligomers. Considering Aβ alone, nine types of oligomers have been identified including, prefibrillar oligomers, fibrillar oligomers, annular protofibers and others2,12,15,16,18. Molecular weights of these oligomers range from 10 kDa dimers to 700 kDa amylospheroids. Models proposed for these β-rich solution oligomers can be roughly divided into two groups depending on the way the β-sheets self-assemble. In one group, the β-sheets wrap around to form a topologically closed cylinder or β-barrel so that all main chain hydrogen bond donors and acceptors of the β-strands are satisfied. Models of β-barrels include an antiparallel single β-sheet cylinder3 and a parallel double layer β-barrel53. In the other group the β-sheets are open, leaving exposed main chain hydrogen bond donors and acceptors on the β-sheet edges. Models of open sheet oligomers include one constructed from a U-shaped building block54, and one constructed from a β-hairpin building block45,55,56.
Our atomic oligomer models more closely resemble the second group: open sheet oligomers. Indeed, a cylindrical topology is prohibited by the blocking strand used in the macrocyclic design. We propose that the tetrameric oligomers of the type described here could exist in solution in the absence of the blocking strand. Since these tetramers are smaller than most reported oligomers of Aβ, it is tempting to speculate that the tetramers could be building blocks for larger molecular weight oligomers. In the absence of a blocking strand, expansion of the oligomer size is most likely to occur through addition of β-strands at the exposed edges of the β-sheets. Growth of the oligomer is likely to stop more quickly if the crossing angle between the two sheets is large, since the addition of strands would not increase the size of the dry interface, and the growing single sheets would be solvent exposed and labile. But, if the axes of the two sheets are nearly parallel, that is closer to cross-β geometry, the addition of each strand would proportionally increase the area of the dry interface, providing the driving force that could lead to the formation of amyloid fibers (Fig. 5).
Figure 5.

Models of extended amyloid-like oligomers (right column), extrapolated from crystallographic tetramers (left column). Extended oligomers were modeled by continuing the hydrogen-bonding pattern in the tetramer with the addition of β-strands at the exposed edges of the tetramer until a 20mer is achieved (middle column). Side chains are shown for those residues that face the opposing sheet. The side chain atoms are colored red if they are solvent exposed and blue if they are buried by the opposing sheet. The maximum stable size of the oligomer was estimated by removing those strands in which over 50% of the side chain atoms are exposed (right column).
The particular sheet-to-sheet packing adopted by an oligomer may distinguish it as being either on-pathway or off-pathway to fiber formation. Only the oligomeric species with approximate cross-β geometry can further associate into higher oligomers and eventually form fibers. Other oligomers may be trapped in various off-pathway oligomeric species (Fig. 5). Some of these oligomers are toxic with characteristically high β-sheet content2,10,14–16,18. One might imagine that if cell toxicity requires the β-sheet aggregate to have a particular structure, an oligomer might be able to achieve that structure more readily than a fiber, because oligomers can sample more conformations than fibers. This might explain the phenomenon that some amyloid oligomeric species are pathogenic or more toxic than the fibers.
Our structures help to explain the great polymorphism of amyloid oligomers. The observed variation in oligomeric geometry among the three macrocycles presumably arises from differences in the sequence of the amyloidogenic insert, but also polymorphic variations occur within the same crystal (compare mcLVFFA interface 1 and 2, Fig. 3c–d). The polymorphism observed with the mcLVFFA crystal suggests that polymorphism observed in Aβ and other amyloid oligomers may result in part from analogous differences in hydrogen bonding patterns and sheet-to-sheet packing geometries. Furthermore, many amyloidogenic proteins have more than one amyloidogenic segment and may encounter different environments during molecular assembly49. The local environment may influence the type of interface formed during self-assembly. Thus, under different conditions, different segments, or different residues in the same segment may be involved in different types of oligomer formation. Therefore, the number of different potential interactions may account for one of the most distinct features of amyloid oligomers—polymorphism.
Despite the diversity of oligomer assembly, our crystal structures also show that soluble proteins with entirely different sequences fold into the β-sheet-rich structures with the common dry steric complimentary interfaces. Polyphenols and small aromatic peptides such as (−)-epigallocatechin gallate (EGCG) and resveratrol which were shown to disrupt the amyloid oligomers and fibers formed by different proteins57–60 may target the dry interface. By disrupting the dry interface, polyphenols may dissolve amyloid oligomers, incorporate into the amyloidogenic polypeptides and form off-pathway oligomers with diminished cytotoxicity and reduced strand content58,59. This suggests that compounds which can disrupt the dry interface of the amyloid oligomers could be potential drug candidates for broad-spectrum therapeutic treatments against amyloidogenic diseases.
In addition to facilitating structural research on amyloid oligomers, macrocycles with amyloidogenic segments inserted are potential inhibitors against amyloid oligomerization and fibrillation, as summarized in Figure 6. The amyloidogenic proteins are intrinsically disordered. They expose their amyloidogenic segments, which can self-associate into self-complementary complexes. The initial aggregation process leads to the formation of oligomeric intermediates. By mimicking the conformation of amyloid oligomeric state, the macrocycle with the same amyloidogenic segment inserted could interact with the exposed segment of the protein. With Hao molecules preventing further assembly, the macrocycle molecules trap the amyloidogenic protein in a low molecular weight oligomer. Furthermore the β-sheet conformation of the amyloidogenic segments of the macrocycles is compatible with the conformation of β-strands in steric zipper structures (Fig. S4). The macrocycle could bind stably to the growing end of the protofilament and prevent additional molecules from binding and elongating the fibers. Indeed, the macrocycle which contains VQIVY in the recognition strand shows strong inhibition of AcPHF6 fibrillation28.
Figure 6.
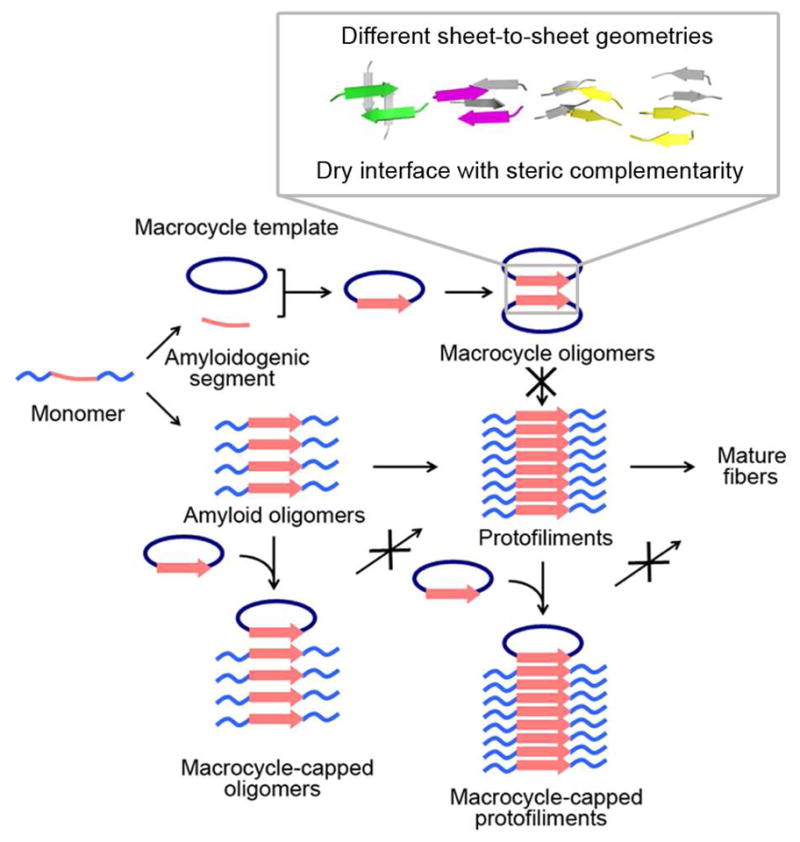
Schematic diagram of macrocyclic peptides mimicking amyloidogenic protein self-association. Amyloid proteins are shown in light blue with amyloidogenic segments in pink. Mediated by amyloidogenic segments, an amyloidogenic protein forms transient and highly polymorphic oligomers, protofilaments and eventually mature fibers. By displaying the amyloidogenic peptides in the recognition strand of a macrocycle as a β-strand, the conformation of the peptides during self-assembly is mimicked. The Hao residue in the macrocyclic ring blocks the infinite molecular assembly and captures a single oligomeric state for X-ray crystallographic studies. The structures of the macrocycles reflect one very likely possibility for amyloid oligomer assembly. By mixing a macrocycle with an amyloidogenic protein, the β-strand mimics can interact with the same segment in the native protein. The Hao residue as a blocker may stop oligomers from further association into fibers and also cap protofilaments from elongation and maturation by binding at the growing edge of the sheets.
Supplementary Material
Acknowledgments
We thank the NE-CAT beamline, the Advanced Photon Source for beam time and collection assistance. We thank NIH (GM-49076 and AG-029430), NSF, and HHMI for grant support.
Footnotes
Supporting Information Available
Complete Reference 14, figures including electron density maps and superposition of the macrocyclic structures, further analysis of the amyloidogenic segments in oligomeric and fibrillar forms, overlapping the amyloidogenic segments of macrocycles with steric zipper fibril structures; tables including sequences and structural statistics of the macrocyclic peptides. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Buxbaum JN. Curr Opin Rheumatol. 2004;16:67–75. doi: 10.1097/00002281-200401000-00013. [DOI] [PubMed] [Google Scholar]
- 2.Ono K, Condron MM, Teplow DB. Proc Natl Acad Sci U S A. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cerf E, Sarroukh R, Tamamizu-Kato S, Breydo L, Derclaye S, Dufrene YF, Narayanaswami V, Goormaghtigh E, Ruysschaert JM, Raussens V. Biochem J. 2009;421:415–423. doi: 10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]
- 4.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 5.Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y. Nat Struct Mol Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 6.von Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E. Biochim Biophys Acta. 2005;1739:158–166. doi: 10.1016/j.bbadis.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 7.Makin OS, Serpell LC. FEBS J. 2005;272:5950–5961. doi: 10.1111/j.1742-4658.2005.05025.x. [DOI] [PubMed] [Google Scholar]
- 8.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakono M, Zako T. Febs J. 2010;277:1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- 11.Meraz-Rios MA, Lira-De Leon KI, Campos-Pena V, De Anda-Hernandez MA, Mena-Lopez R. J Neurochem. 2010;112:1353–1367. doi: 10.1111/j.1471-4159.2009.06511.x. [DOI] [PubMed] [Google Scholar]
- 12.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, Bowers MT. Nat Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith DP, Radford SE, Ashcroft AE. Proc Natl Acad Sci U S A. 2010;107:6794–6798. doi: 10.1073/pnas.0913046107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu LP, et al. Biochemistry. 2009;48:1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- 15.Glabe CG. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO. Nat Struct Mol Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kayed R, Pensalfini A, Margol L, Sokolov Y, Sarsoza F, Head E, Hall J, Glabe C. J Biol Chem. 2009;284:4230–4237. doi: 10.1074/jbc.M808591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu JW, Breydo L, Isas JM, Lee J, Kuznetsov YG, Langen R, Glabe C. J Biol Chem. 2010;285:6071–6079. doi: 10.1074/jbc.M109.069542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 20.Caughey B, Lansbury PT. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 21.Hughes RM, Waters ML. Curr Opin Struct Biol. 2006;16:514–524. doi: 10.1016/j.sbi.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 22.Gellman SH. Curr Opin Chem Biol. 1998;2:717–725. doi: 10.1016/s1367-5931(98)80109-9. [DOI] [PubMed] [Google Scholar]
- 23.Nowick JS. Acc Chem Res. 2008;41:1319–1330. doi: 10.1021/ar800064f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khakshoor O, Nowick JS. Curr Opin Chem Biol. 2008;12:722–729. doi: 10.1016/j.cbpa.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woods RJ, Brower JO, Castellanos E, Hashemzadeh M, Khakshoor O, Russu WA, Nowick JS. J Am Chem Soc. 2007;129:2548–2558. doi: 10.1021/ja0667965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khakshoor O, Demeler B, Nowick JS. J Am Chem Soc. 2007;129:5558–5569. doi: 10.1021/ja068511u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khakshoor O, Lin AJ, Korman TP, Sawaya MR, Tsai SC, Eisenberg D, Nowick JS. J Am Chem Soc. 2010;132:11622–11628. doi: 10.1021/ja103438w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng J, Liu C, Sawaya MR, Vadla B, Khan S, Woods RJ, Eisenberg D, Goux WJ, Nowick JS. J Am Chem Soc. 2011 doi: 10.1021/ja110545h. Article ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabsch W. Journal of Applied Crystallography. 1993;26:795–800. [Google Scholar]
- 30.Schneider TR, Sheldrick GM. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 31.Sheldrick GM. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 34.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Smart OS, Vonrhein C, Womack TO. United Kingdom, Global Phasing Ltd. Cambridge: 2009. [Google Scholar]
- 35.Korostelev A, Bertram R, Chapman MS. Acta Crystallogr D Biol Crystallogr. 2002;58:761–767. doi: 10.1107/s0907444902003402. [DOI] [PubMed] [Google Scholar]
- 36.Fabiola F, Bertram R, Korostelev A, Chapman MS. Protein Sci. 2002;11:1415–1423. doi: 10.1110/ps.4890102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.LaFerla FM, Oddo S. Trends Mol Med. 2005;11:170–176. doi: 10.1016/j.molmed.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 38.Hardy JA, Higgins GA. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 39.Ballatore C, Lee VM, Trojanowski JQ. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 40.Walsh DM, Selkoe DJ. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 41.Kayed R, Jackson GR. Curr Opin Immunol. 2009;21:359–363. doi: 10.1016/j.coi.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 42.von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Proc Natl Acad Sci U S A. 2000;97:5129–5134. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paravastu AK, Leapman RD, Yau WM, Tycko R. Proc Natl Acad Sci U S A. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. Proc Natl Acad Sci U S A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoyer W, Gronwall C, Jonsson A, Stahl S, Hard T. Proc Natl Acad Sci U S A. 2008;105:5099–5104. doi: 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koh E, Kim T, Cho H. Bioinformatics. 2006;22:297–302. doi: 10.1093/bioinformatics/bti775. [DOI] [PubMed] [Google Scholar]
- 47.Levin S, Nowick JS. J Am Chem Soc. 2007;129:13043–13048. doi: 10.1021/ja073391r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawrence MC, Colman PM. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 49.Wiltzius JJ, Landau M, Nelson R, Sawaya MR, Apostol MI, Goldschmidt L, Soriaga AB, Cascio D, Rajashankar K, Eisenberg D. Nat Struct Mol Biol. 2009;16:973–978. doi: 10.1038/nsmb.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chothia C, Janin J. Proc Natl Acad Sci U S A. 1981;78:4146–4150. doi: 10.1073/pnas.78.7.4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chothia C, Janin J. Biochemistry. 1982;21:3955–3965. doi: 10.1021/bi00260a009. [DOI] [PubMed] [Google Scholar]
- 52.Jenkins J, Pickersgill R. Prog Biophys Mol Biol. 2001;77:111–175. doi: 10.1016/s0079-6107(01)00013-x. [DOI] [PubMed] [Google Scholar]
- 53.Jang H, Zheng J, Nussinov R. Biophys J. 2007;93:1938–1949. doi: 10.1529/biophysj.107.110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng J, Yu X, Wang J, Yang JC, Wang Q. J Phys Chem B. 2010;114:463–470. doi: 10.1021/jp907608s. [DOI] [PubMed] [Google Scholar]
- 55.Sandberg A, Luheshi LM, Sollvander S, Pereira de Barros T, Macao B, Knowles TP, Biverstal H, Lendel C, Ekholm-Petterson F, Dubnovitsky A, Lannfelt L, Dobson CM, Hard T. Proc Natl Acad Sci U S A. 2010;107:15595–15600. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strodel B, Lee JW, Whittleston CS, Wales DJ. J Am Chem Soc. 2010;132:13300–13312. doi: 10.1021/ja103725c. [DOI] [PubMed] [Google Scholar]
- 57.Scherzer-Attali R, Pellarin R, Convertino M, Frydman-Marom A, Egoz-Matia N, Peled S, Levy-Sakin M, Shalev DE, Caflisch A, Gazit E, Segal D. PLoS One. 2010;5:e11101. doi: 10.1371/journal.pone.0011101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE. Nat Struct Mol Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 59.Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE. Proc Natl Acad Sci U S A. 2010;107:7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ladiwala AR, Lin JC, Bale SS, Marcelino-Cruz AM, Bhattacharya M, Dordick JS, Tessier PM. J Biol Chem. 2010;285:24228–24237. doi: 10.1074/jbc.M110.133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.