

Abstract
Human APOBEC3G (hA3G), a member of the AID/APOBEC family of deaminases, is a restriction factor for human immunodeficiency virus (HIV). In the absence of the viral Vif protein hA3G is packaged into virions and during reverse transcription in a recipient cell it deaminates cytosines, leading to G --> A hypermutation and inactivation of the viral DNA. Unlike humans, who carry seven APOBEC3 genes, mice only carry one, mA3. Thus the role of mA3 in restriction of retroviral infection could be studied in mA3 −/− knockout mice, where the gene is inactivated. M-MuLV-infected mA3 −/− mice showed substantially higher levels of infection at very early times compared to wild-type mice (ca. 2 logs at 0-10 days), particularly in the bone marrow and spleen. Restriction of M-MuLV infection was studied ex vivo in primary bone marrow-derived dendritic cells (BMDCs) that express or lack mA3, using an M-MuLV-based retroviral vector expressing enhanced jellyfish green fluorscent protein (EGFP). The results indicated that mA3 within the virions as well as mA3 in the recipient cell contribute to resistance to infection in BMDCs. Finally, M-MuLV-infected mA3 +/+ mice developed leukemias more slowly compared to animals lacking one or both copies of mA3 although the resulting disease was similar (T-lymphoma). These studies indicate that mA3 restricts replication and pathogenesis of M-MuLV in vivo.
Keywords: Murine leukemia virus, Moloney murine leukemia virus, APOBEC3, Murine APOBEC3, Leukemia, Lymphoma, Dendritic cell, Bone marrow-derived dendritic cell
INTRODUCTION
The AID/APOBEC family of cytidine deaminases plays diverse roles in cellular physiology. Evolutionarily the original gene is thought to be APOBEC2, from which AID, APOBEC1 and APOBEC3 evolved (Conticello et al., 2005). These proteins have one or two cytidine deaminase domains (CDDs), one of which is catalytically active. APOBEC1 edits single C residues to U on the mRNA encoding apolipoprotein B (ApoB) resulting in translation of a truncated form of ApoB with a different biological function (Teng, Burant, and Davidson, 1993). AID is involved in class switch recombination in B cells by randomly editing dC residues to dU in the immunoglobulin gene locus, causing hypermutation (Muramatsu et al., 2000); the function of APOBEC2 is unknown. APOBEC3 genes are of great interest because they mediate resistance to viral infections, and retroviruses in particular.
Studies on the vif gene of HIV identified human APOBEC3G, 3F and 3H(hA3G, hA3H and hA3F) as host factors that affect viral spread (Mariani et al., 2003; Sheehy et al., 2002; Zhang et al., 2003) (OhAinle et al., 2006). Restriction of HIV infection by hA3G and the counteraction of this restiction by Vif have been extensively studied and reviewed (Cullen, 2006; Holmes, Malim, and Bishop, 2007). In the absence of Vif (i.e. infection with vif-negative HIV mutants), hA3G is packaged into newly forming viral particles. Upon infection of a recipient cell, hA3G deaminates cytosines in the minus strand DNA during reverse transcription, resulting in G to A mutations in the reverse-transcribed plus strand DNA (Harris et al., 2003; Mangeat et al., 2003; Zhang et al., 2003). This change leads to non-functional viral DNA. Vif counteracts hA3G by binding and targeting it for ubiquitination (Mariani et al., 2003); as a result little-or no hA3G is packaged into newly forming virions, and reverse transcription can proceed normally. The importance of hA3 is reflected by extensive genomic duplications resulting in 7 different hA3 genes (hA3A – H), of which hA3B, hA3DE, hA3F and hA3G have activity against HIV (Bishop et al., 2004; Dang et al., 2006; Doehle, Schafer, and Cullen, 2005; Liddament et al., 2004; Rose et al., 2005; Wiegand et al., 2004; Zheng et al., 2004). Additional hA3 family members affect the mobility of ubiquitous human retrotransposons (LTR-retrotransposons such as HERVs and non-LTR elements such as LINEs and SINEs) that make up a substantial fraction of the human genome(Bogerd et al., 2006; Lee, Malim, and Bieniasz, 2008; Muckenfuss et al., 2006; Stenglein and Harris, 2006). Several hA3s also restrict hepatitis B virus by cytidine deamination (Suspene et al., 2005; Turelli et al., 2004).
Unlike humans, mice have only one copy of the APOBEC3 gene (mA3). Since there is only one mA3 gene in mice, any retrovirus that is subject to APOBEC3 restriction would be affected by this one gene. This finding makes possible a genetic study of mA3 since a single genetic inactivation would eliminate its activity from mice. In this report we used mA3 −/− mice to test the hypothesis that mA3 restricts Moloney murine leukemia virus (M-MuLV) replication. M-MuLV is a simple gamma retrovirus and does not encode any known vif analog. mA3 can be incorporated into HIV virions and is not degraded by Vif; moreover mA3 restricts HIV by cytidine deamination. Conversely hA3G can be incorporated into MLV-based vectors and restrict infection in vitro (Abudu et al., 2006; Doehle et al., 2005; Kobayashi et al., 2004). However, previous studies on the effects of mA3 on MuLV infection have been less clear. Some studies indicated that MuLV is resistant to mA3 because it is not efficiently packaged into virions (Doehle et al., 2005; Kobayashi et al., 2004; Zhang et al., 2008), or because the MuLV protease degrades the full-length form of the protein in the virion (Abudu et al., 2006). However, other studies reported efficient packaging of mA3 into MuLV virions and subsequent inhibition of infection (Browne and Littman, 2008; Mariani et al., 2003; Rulli et al., 2008). The emerging consensus is that mA3 can indeed be packaged into MuLV virions, and that mA3 does restrict MuLV infection in vitro, although the degree of restriction is less than for hA3G (or mA3) on HIV (Browne and Littman, 2008; Rulli et al., 2008; Zhang et al., 2008). However, mA3 does not induce cytidine deamination when incorporated into M-MuLV virions (Browne and Littman, 2008; Rulli et al., 2008).
In this study we utilized the mA3 insertional knockout animals to test if mA3 restricts M-MuLV infection in vivo, by measuring rates of initial infection, disease progression and pathogenesis in these animals compared to their wild-type counterparts. We also employed primary bone marrow-derived dendritic cells (BMDCs) from these mice to examine if mA3 inhibits M-MuLV infection in vitro. We found that mA3 plays a role in reducing viral replication and pathogenicity of M-MuLV.
RESULTS
Effects of mA3 on in vivo infection by M-MuLV
Previous studies have indicated that MuLVs are relatively resistant to mA3 in vitro (Abudu et al., 2006; Doehle et al., 2005; Kobayashi et al., 2004), although other recent studies have reported that mA3 does in fact restrict MuLV infection (Browne and Littman, 2008; Rulli et al., 2008). Since these studies were carried out using tissue culture cells transfected with mA3 expression vectors, we wished to determine if endogenous levels of mA3 had any effect on MuLV infection in vivo in mice. To this end we compared in vivo infection by Moloney MuLV (M-MuLV) in mA3 homozygous knock-out (−/−) mice vs. wild-type (+/+) animals. The mA3 −/− mice used in these studies (initially from 129/Ola embryos) were at the ninth back-cross generation to C57Bl6 (B6) mice. Neonatal mA3 and B6 mice were inoculated intraperitoneally with an M-MuLV stock, and levels of infection in bone marrow, spleen and thymus were measured at different times post-infection (6 – 12 days) by an infectious center assay as described previously (Lander, Chesebro, and Fan, 1999). As shown in Fig 1, mA3 −/− animals showed more rapid early infection in all tissues than their wild-type counterparts up to 10-12 days post-infection. Differences in infection levels were substantial at the earlier time points (8-10 days, typically 2-3 logs), and they differed significantly by a two-tailed T-test at 8 (P < 0.04), 10 (P < 0.03) and 12 days (P < 0.01 for spleen and P < 0.0003 for thymus and bone marrow). These results suggested that mA3 does restrict M-MuLV infection in vivo, with the effects being evident by slower establishment of initial infection in mA3 −/− vs. +/+ B6 mice.
Figure 1. M-MuLV infection in C57/Bl6 and mA3−/− mice.

Neonatal mice were inoculated i.p. with M-MuLV. At 6, 8, 10 and 12 days post inoculation, the animals were sacrificed and single cell suspensions were prepared from bone marrow, thymus and spleen. The levels of infection (infectious centers (ICs)/106 cells plated) are shown. Each point represents results from one animal. Circles, C57/Bl6; diamonds, mA3−/−. Mean values are indicated by horizontal bars. A) Bone marrow, B) Spleen, C) Thymus.
Since the mA3 −/− mice used in Fig. 1 might not have been completely isogenic with B6, it was possible that some or all of the effects observed could have reflected other genes besides mA3. To exclude this possibility, the mA3 −/− mice were crossed with B6 mice and the heterozygous mA3 +/− F1 animals were intercrossed to generate F2 animals. These litters were inoculated with M-MuLV and the levels of infection in bone marrow, spleen and thymus at 6 – 14 days were measured as in Fig. 1. Each animal was genotyped for mA3 status, and the results were plotted according to genotype (+/+, +/− or −/−) as shown in Fig. 2. These results again indicated that mA3 +/+ animals established infection more slowly than animals mA3 −/− mice, although in this case differences between the different genotypes were smaller, especially after day 10 post-infection. The bone marrow was of particular interest, since we previously showed that the efficiency of M-MuLV leukemogenesis is dependent on establishing infection in the bone marrow at early times (Belli and Fan, 1994). As shown in Fig 2A, 5/7 mA3 +/+ mice showed no measurable infection at 6 or 8 days post infection, while 6/6 −/− animals showed infection ranging from 1 × 100 to 2 × 103/106 cells. More extensive infection was also observed in the spleen of mA3 −/− compared to +/+ animals at 6 or 8 days; the levels of infection in the thymus at these early times were lower than in the bone marrow and spleen, as we described previously (Davis et al., 1987).
Figure 2. M-MuLV infection in mA3+/− F2 mice.

Neonatal mice from inter-crosses between two mA3 +/− mice were inoculated i.p. with M-MuLV. At 6, 8, 10, 12 and 14 days post-inoculation, the animals were sacrificed and single cell suspensions were prepared from bone marrow, thymus and spleen. Genotypes of the animals were determined. The levels of infection (ICs/106 cells plated) are shown. Each point represents results from one animal. Squares, mA3+/+ mice; triangles, mA3+/− mice; circles, mA3−/− mice. Mean values are indicated by horizontal bars. A) Bone marrow, B) Spleen, C) Thymus.
To ensure that the differences in infection were significant, larger numbers of animals were analyzed at day 10 as shown in Fig 2. In all tissues, mA3 −/− animals showed significantly higher levels of infection than mA3 +/+ mice (Table 1). In the the bone marrow the differences were greater than 2.5 logs. There was a trend towards statistical significance in the differences between +/− and +/+ animals in the bone marrow and spleen, and the levels of infection in the bone marrow of heterozygous +/− and homozygous −/− mice appeared similar. Past 10 days, the levels of infection in +/+ animals increased so that by 12 and 14 days levels of infection in all organs resembled those of their −/− and +/− littermates. Taken together, these results indicated that mA3 has a significant effect in controlling early infection in M-MuLV-inoculated mice, particularly in the bone marrow. However, by two weeks post-inoculation M-MuLV established similar levels of infection in mA3 +/+ and −/− animals, consistent with its ability to induce leukemia.
Table 1.
M-MuLV Infection levels at 10d post-inoculation in mA3 F2 mice.1
| Genotype |
Bone Marrow |
Spleen |
Thymus |
|||
|---|---|---|---|---|---|---|
| +/+ | 7.8 × 101 | 3.7 × 102 | 6.2 × 102 | |||
| +/− | 6.9 × 103 | p ≤ 0.093 | 3.4 × 103 | p ≤ 0.11 | 8.4 × 102 | p ≤ 0.70 |
| −/− | 3.7 × 104 | p ≤ 0.0046 | 1.3 × 104 |
p ≤
0.00001 |
8.0 × 103 | p ≤ 0.053 |
Mean values for infectious centers (per 106 cells) in different tissues for animals shown in Fig 2 at 10d post-infection, according to genotype are shown. Statistical analyses (2-tailed T-test) for +/− and −/− animals in comparison to +/+ animals are shown; comparisons reaching statistical significance are in bold.
Effects of mA3 on in vitro M-MuLV infection of BMDCs
We previously showed that early infection in the bone marrow is important for efficient leukemogenesis by M-MuLV (Belli and Fan, 1994). Moreover, the early difference in infection levels was greatest in this tissue when comparing mA3 −/− and wild-type mice as shown above. Thus we were interested if mA3 could restrict in vitro infection of particular bone marrow cells. Dendritic cells (DCs) from humans and mice have been shown to express human A3s and mA3 respectively (Okeoma et al., 2007; Pion et al., 2006). Therefore we tested whether M-MuLV could infect BMDCs in vitro, and whether the levels of infection were different in BMDCs derived from mA3 +/+ and −/− mice.
For these experiments, an MuLV-based vector expressing enhanced green fluorescent protein (LEGFP-N1) was used. Vector stocks were generated by co-transfecting the 293T-based ecotropic MuLV packing cell line Phoenix-eco with the vector plasmid pLEGFP-N1 along with expression plasmids for HA epitope-tagged mA3 or the backbone plasmid pcDNA3.1. There are actually two isoforms of mA3 that result from the presence or absence of exon 5 in the spliced mA3 mRNA (Abudu et al., 2006); these two isoforms also encode a number polymorphic amino acids (Takeda et al., 2008). cDNAs for epitope-tagged forms of both proteins (mA3HA and mA3Δexon5HA) were used to generate vector stocks containing either mA3 isoform. Tissue culture supernatants were harvested, concentrated and then tested for the presence of vector and mA3 by SDS-PAGE and western blot analyses with antibodies for MuLV CA or HA epitopes respectively as shown in Fig 3A. The amounts of vector particles present in the three stocks were similar, as measured by the presence of CA protein (anti-p30). Moreover, mA3 could be readily detected by the anti-HA antibody, and the relative amounts of mA3 were present regardless of whether exon5 was present, consistent with previous reports (Browne and Littman, 2008; Mariani et al., 2003; Rulli et al., 2008).
Figure 3. M-MuLV infection of BMDCs.

(A) Protein levels in LEGFP-N1 vector stocks Western produced in the absence (pcDNA 3.1) or presences of mA3HA or mA3Δexon5HA. Western blots for mA3 (anti-HA) and MuLV CA protein (anti-p30) are shown. All samples were run on the same gel (B) After standardization of viral stocks by RT-PCR for vector RNA, BMDCs from mA3 +/+ and −/− mice were infected by vector produced in the absence (light grey) or presence (dark grey) of mA3Δexon5HA. FACS analysis for EGFP expression after 24 hr determined infection levels as shown.
The vector pseudovirions with or without mA3 were then tested for in vitro infectivity in primary bone marrow-derived dendritic cells (BMDCs) derived from +/+ or −/− mice. The extents of infection were measured by flow cytometry for the EGFP reporter protein on the vector. Since C57Bl6 mice only express the Δexon5 form of mA3 (Santiago et al., 2008; Takeda et al., 2008), the results shown in Fig 3B are for this form of mA3, although similar results also were obtained for wild-type mA3 (not shown). The results indicated that MuLV restriction resulted both from the presence of mA3 in the virus particles, as well as from the presence of mA3 in the infected cells. The effects of mA3 in the virus particles were evident by comparison of the MuLV vector lacking or containing mA3 in either the +/+ or −/− BMDCs. The presence of Δexon5 mA3 in particles reduced infectivity by 45-50% compared to particles lacking mA3 in both mA3 +/+ and −/− BMDC preparations. The effect of mA3 in the recipient cells was apparent by comparing the efficiencies of infection for the same vector preparation in +/+ vs −/− BMDCs. For both vector preparations there was a ca. 40% reduction in infectivity in +/+ BMDCs compared to −/− cells. In total the effects of mA3 in virions combined with mA3 in the recipient cells resulted in a ca. 3-fold reduction in infectivity (vector lacking mA3 in −/− BMDCs compared to vector containing mA3 in +/+ cells).
Effects of mA3 status on pathogenesis by M-MuLV
In light of the effects of mA3 status on the rate of early M-MuLV infection in the bone marrow and its ability to restrict infection of BMDCs, we next investigated if there was an effect on M-MuLV-induced pathogenesis. M-MuLV induces lymphoma (largely T-lymphoma) in B6 mice, and it seemed possible that knockout or reduction in mA3 copies might accelerate the rate of disease development or cause additional kinds of tumors. Neonatal F2 litters from F1 mA3 +/− intercrosses were inoculated IP with M-MuLV and the animals were observed for development of disease. Moribund animals were sacrificed, subjected to gross pathological analysis, and enlarged or abnormal tissues (thymus, spleen, lymph nodes and in some cases kidneys) were obtained for DNA extraction or paraffin embedding. The animals were also genotyped, and the results were tabulated according to genotype. All but one of the +/+ animals, and all but one of the +/− animals had tumors at the time of sacrifice; all −/− animals had tumors. The time courses for mortality (Kaplan-Meier plots) of the three genotypes are shown in Fig 4. The time courses for the −/− and +/− animals were similar, so the results for these two groups were pooled. The median survival time for M-MuLV-infected mA3 +/+ animals was 269.5 days, compared to 207.5 days for infected animals missing at least one copy of the mA3 gene (−/− or +/−). The survival of the infected mA3 +/+ animals was significantly different from that of the inoculated −/− or +/− animals (log rank test p < 0.023 or by the Wilcoxon test p < 0.021). Secondary analyses showed a statistical difference in time to disease between +/+ and +/− animals (log rank test p < 0.021; Wilcoxon test p < 0.011), but not between +/+ and −/− animals (log rank p<0.078; Wilcoxon p<0.092), due at least in part the lower number of −/− animals. Taken together, these results indicated that mA3 reduces the kinetics of tumor induction by M-MuLV, and that loss of one or both copies of the gene significantly increases the rate of disease development.
Figure 4. Pathogenesis by M-MuLV in mA3 −/− animals.
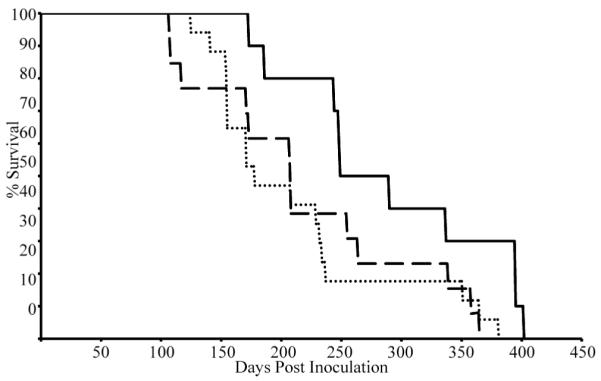
Kaplan-Meier plots of survival of animals from mA3 +/− intercrosses after i.p. inoculation with M-MuLV. Dashed line, mA3−/− animals (n=13; median latency 208 days); dotted line, mA3+/− animals (n=17; median latency 178 days); and the solid line, mA3+/+ animals (n=10, median latency 269.5 days).
The gross pathology of the sacrificed animals indicated enlarged thymus, spleen or lymph nodes for animals of all genotypes, characteristic of the lymphoblastic lymphoma induced by M-MuLV in B6 and other mouse strains (Brightman et al., 1991). Molecular diagnosis of the tumors was performed by testing tumor DNAs for rearrangements of T-cell receptor beta (TCRβ), immunoglobulin Mu heavy chain (IgH) and immunoglobulin kappa light chain (IgK) genes. Tumors with TCRβ gene rearrangements (with or without IgH rearrangements) were classified as T-lymphoid, those with IgK and IgH rearrangements were classified as B-lymphoid, and those with IgH but no IgK or TCRb rearrangements were classified as preB (Hanecak, Pattengale, and Fan, 1988). As shown in Table 2, the great majority of tumors in all three genotypes showed rearrangements in these genes, consistent with T-lymphoma and to a lesser extent B- or preB-lymphomas. If anything the −/− and +/− animals developed a higher percentage of pure T-lymphomas. Thus, while loss of one or both mA3 alleles reduced the time to tumor formation, it did not appear to expand the range of tumor types observed.
Table 2.
Tumor types in M-MuLC-infected mice by mA3 genotype.
Tumors with TCRβ rearrangement with or without IgH rearrangement were classified as T-cell lymphomas.
Tumor tissues with IgK and IgH rearrangement were classified as B-lymphomas; tumors with IgH but not IgK or TCRβ were classified as preB lymphomas.
Tumor tissues that did not show any rearrangement in TCRβ, IgH, or IgK
Interestingly three out of 13 mA3 −/− animals and 2 out of 17 mA3 +/− animals showed evidence of tumor involvement in the kidneys, as evidenced by visible masses on necropsy. In contrast none of the 10 +/+ animals showed this disease pattern. Histopathological analyses of the kidneys from two of these animals are shown in Fig 5. The tumor cells were found to be lymphoid by H&E staining (Fig 5A and C), consistent with metasases from primary lymphoid organs. The tumor infiltrates (localized or extensive) contained small round or oval cells with non-cleaved nuclei that resembled tumor cells in the spleen or thymus from the same animal (not shown). They were positive for immunohistochemical staining for CD3, a marker for T-lymphoid cells and tumors (Fig 5B), indicating that they were of T-cell origin.
Figure 5. Kidney tumors in mA3 −/− and mA3+/− animals.

(A) H&E staining of kidney tissue from an mA3−/− animal showing lymphocyte infiltrates (B) Immunohistochemistry for CD3 staining in the same kidney verified that infiltration was T-lymphoid. (C) H & E staining of a tumor from an mA3+/− animal also shows lymphocyte infiltrates in the kidney.
DISCUSSION
In these experiments, we investigated if mA3 has an effect on M-MuLV infection. Our approach was to employ mA3 knockout mice, and to compare their rates of infection and pathogenesis to animals containing the mA3 gene. Assays of M-MuLV in vivo infection and pathogenesis provided both sensitive and relevant measures of mA3 restriction. The results indicated that mA3 significantly delays establishment of M-MuLV infection at early times, particularly in the bone marrow. At 6 – 10 days post-inoculation, the levels of M-MuLV infection in mA3 −/− mice were 2-3 logs higher than in mA3 +/+ animals in that tissue. Clearly mA3 restriction of M-MuLV is not absolute, and by two weeks, the levels of infection approached equivalency in −/− and +/+ animals. Nevertheless mA3 +/+ animals developed M-MuLV-induced leukemias more slowly (ca. 2 months later on average) than inoculated −/− mice. This was consistent with the less efficient establishment of bone marrow infection in +/+ mice, since we previously found that efficient establishment of early bone marrow infection is correlated with the efficiency of M-MuLV-induced disease (Belli and Fan, 1994).
When we initiated these experiments, it was unclear if there would be a difference between M-MuLV infection in mA3 +/+ and −/− mice. While mA3 efficiently inhibits HIV-1 infection (Bishop et al., 2004; Mariani et al., 2003), early reports indicated that MuLVs are resistant to this factor (Doehle et al., 2005; Kobayashi et al., 2004). This was attributed to exclusion of mA3 from MuLV (but not HIV-1) particles (Doehle et al., 2005; Kobayashi et al., 2004) or degradation of packaged mA3 by viral protease (Abudu et al., 2006). In the latter case, mA3Δexon5 was reported to be relatively resistant to viral PR (Abudu et al., 2006). On the other hand, recent reports have concluded that both mA3 and mA3Δexon5 are efficiently packaged in MuLV virions (Browne and Littman, 2008; Rulli et al., 2008), which are in agreement with other early reports. In fact, the results shown in Fig. 3A are consistent with the conclusion of those investigators favoring packaging of mA3 into MuLV virions. Regardless of whether mA3 (or the Δexon5 version) is packaged into virions or not, our results indicated that mA3 does indeed restrict M-MuLV infection in vivo even when the protein is not over-expressed. Since these experiments were carried out in the B6 genetic background, these conclusions predominantly pertain to the Δexon5 form of mA3.
As shown in Fig. 4, mA3 also affected the kinetics of M-MuLV disease induction. Infected +/+ animals developed disease more slowly than infected −/− and/or +/− mice. This was consistent with the fact that infected +/+ animals show lower levels of infection in the bone marrow (Fig. 2A) (and spleen, Fig. 2B) between days 6 and 10; early bone marrow infection is correlated with efficient disease induction (Belli and Fan, 1994). The early bone marrow infection (perhaps involving BMDCs) could result in defects in bone marrow stroma, leading to extramedullary hematopoiesis (Davis et al., 1987; Li and Fan, 1990) and/or early generation of mink cell focus-inducing (MCF) env recombinants (Brightman, Davis, and Fan, 1990). These preleukemic events have been associated with efficient M-MuLV leukemogenesis. It was also interesting that heterozygous +/− animals showed more rapid disease development than +/+ animals, similar to the faster rate of disease for −/− animals. This might suggest that both mA3 gene copies are required for relative resistance to M-MuLV leukemogenesis. Indeed, at day 10 the levels of M-MuLV infection in the bone marrow of +/− mice more closely resembled those of −/− animals than +/+ animals (Fig. 2A). Pathogenicity in +/− animals was not reported in two recent reports involving different MuLVs ( acute disease induced by the Friend complex) (Santiago et al., 2008; Takeda et al., 2008).
The kinds of disease arising in mA3 −/− animals were of interest. It seemed possible that mA3 might affect the kinds of tumors, if the protein were differentially expressed in different cells. Thus mA3 −/− mice might show novel kinds of tumors compared to +/+ animals. However, the −/− animals also developed T-lymphoma, which indicated that the lack of mA3 did not expand the resulting tumor types. In fact, a higher percentage of the infected −/− and +/− animals developed T-lymphoma than the +/+ mice; the latter also showed development of B- or preB-lymphomas. One possible explanation could be related to the overall time required for disease development. If the target cells for T-lymphoma (likely T-lymphoid progenitors in the thymus) decrease with age while the targets for B-lymphoma do not, then conditions where disease is induced more rapidly could show higher percentages of T-lymphoma. Indeed, an SV40 enhancer-driven variant of M-MuLV that induced disease with extremely long latency (17 months) induced exclusively B-lymphomas (Hanecak, Pattengale, and Fan, 1988). In mouse strains where M-MuLV leukemogenesis is efficient (e.g. ca. 100 days for NIH Swiss mice), the tumors are almost exclusively T-lymphoid (Brightman et al., 1988).
It was interesting that some −/− and +/− animals showed M-MuLV-induced tumors at a novel anatomical site, the kidney. This location has not been observed previously for M-MuLV-induced tumors, and inoculated +/+ animals did not show kidney involvement by gross pathology. These tumors appeared to represent T-lymphoma metastases as shown in Fig.5. In the future it will be interesting to study the basis for the apparently more metastatic T-lymphomas in mA3 −/− mice.
The mechanism(s) by which mA3 restricts MuLVs remain to be determined. Several groups have reported that mA3 does not cause cytidine deamination and DNA hypermutation during MuLV infection (Browne and Littman, 2008; Rulli et al., 2008; Takeda et al., 2008), which suggests that other mechanisms may be involved. Indeed, it has been reported that the cytidine deaminase activity of hA3G is not required for its restriction of HIV (Bishop, Holmes, and Malim, 2006). Chiu et al. (Chiu et al., 2005) have reported that HIV is restricted for infection by a low molecular mass complex of hA3G in recipient resting human T-cells. It is possible that this mechanism may also be applicable to mA3 restriction of MuLV in BMDCs. We therefore also employed the mA3 −/− mice to study M-MuLV infection of BMDCs from mA3 +/+ and −/− animals. We used an M-MuLV-derived vector expressing EGFP, which allowed quantification of infection by flow cytometry. Co-expression of mA3 in the vector packaging cells allowed investigation of the role of packaged mA3 in BMDC cell infection, while comparison of infection in BMDCs from −/− vs +/+ animals allowed investigation of the role of mA3 in the recipient cell. As shown in Fig. 3, the restriction of MuLV by mA3 is influenced by both mA3 brought in by the infecting virion, as well as by mA3 in the infected cell. The presence of mA3 in the virion reduced infectivity by approximately 50%. Similarly, the presence of mA3 in the recipient BMDC also reduced infection by ca. 40%.
We have previously studied restriction by mA3 of another murine retrovirus, mouse mammary tumor virus (MMTV, (Okeoma et al., 2008)). We found that mA3 also restricts infection of MMTV in vivo, and that restriction was mediated by both mA3 packaged in the virions as well as mA3 in the dendritic cells (Okeoma et al., 2008). Thus mA3 restricts infection by two different murine retroviruses, and in both cases the restriction is partial. It will be interesting to compare the mechanisms of restriction for these two viruses. The results obtained so far indicate that mA3 in the virion as well as in the recipient dendritic cell both contribute to the restriction. Dendritic cells are known to be the first targets for MMTV infection. Whether these are also the initial targets for M-MuLV and if DC infection affects M-MuLV- disease are under investigation. In addition ongoing experiments are studying how M-MuLV and MMTV, neither of which apparently encodes a Vif-like function, avoid complete restriction by mA3.
While this manuscript was in preparation, Santiago et al. (Santiago et al., 2008) reported that the mouse resistance gene Rfv3 (recovery from Friend virus gene-3) is in fact associated with mA3 status. A similar conclusion was also reached in a recent report by Takeda et al. (Takeda et al., 2008). Rfv3 was originally defined on the basis of the ability of certain mouse strains to recover from infection and rapid erythroid proliferation induced by the Friend virus complex (consisting of the acute transforming Friend Spleen Focus-forming virus, an ecotropic Friend MuLV [F-MuLV] helper virus, and Lactate dehydrogenase-elevating virus). These recent reports showed that Rfv3 maps to the same chromosomal region as mA3 and that mouse strains harboring resistance alleles of Rfv3 (e.g. B6) encode functional mA3 (predominantly the Δexon5 form) while Rfv3 sensitive strains (e.g. Balb/c) encode defective or less active forms of mA3. Moreover, mA3 −/− mice showed behavior similar to Rfv3 sensitive strains in terms of rapid erythroid disease induced by the Friend virus complex. In agreement with the results reported here for M-MuLV, Takeda et al. (Takeda et al., 2008) also reported that infection with F-MuLV alone showed 2 logs higher infection in mA3 −/− mice compared to +/+ mice at 6 days post-infection, although effects on F-MuLV-induced leukemia were not reported. The results of Santigao et al. and Ikeda et al. also differed from these results in that their infections were into adult mice that can establish effective immune responses. Taken together these two recent reports and our results indicate that mA3 restricts murine leukemia virus replication in vivo. Moreover our studies with M-MuLV indicate that mA3 has long-term effects in modulating M-MuLV-induced leukemogenesis.
EXPERIMENTAL METHODS
Cells
43-D cells are NIH3T3 fibroblasts stably infected with wild type M-MuLV. They were maintained in Dulbecco-modified Eagle’s Medium (DMEM) supplemented with 10% calf serum and antibiotics. The Phoenix-eco cell line is an ecotropic MuLV-based packaging cell line created from Human 293T cells (http://www.stanford.edu/group/nolan/retroviral_systems/phx.html) was obtained from Dr. Garry Nolan and was grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100U/mL penicillin and 100ug/mL streptomycin. Bone marrow-derived dendritic cells (BMDCs) from mA3−/− and mA3+/+ mice were generated according to published procedures (Lutz et al., 1999). Briefly, bone marrow cells from 10 day old mice were cultured for eight days in RPMI medium containing 10% FBS, 100U/mL penicillin, 100ug/mL streptomycin, 0.05mM 2-mercaptoethanol, and 10ng/ml recombinant murine-GM-CSF (Peprotech, Inc. Rocky Hill, NJ). BMDCs were matured by treatment with 100ng/mL lipopolysaccharide (LPS) (Sigma, Inc., St. Louis, MO) for 24 hours. Differentiation into immature or mature DCs was documented by flow cytometry for cell-surface expression of CD40 and anti-CD86. All cells were cultured at 37°C with 5% CO2.
Virus and Vector Production
To prepare M-MuLV viral stocks, 5×105 43-D cells (productively infected NIH-3T3 cells), were seeded on 10cm plates in growth medium, and 24 hours later media was changed. 24 hours after the medium change, supernatants were collected and passed through a .45 micron filter. Supernatants were then divided into aliquots, frozen and stored at −80°.
For generation of vector stocks, Phoenix-eco cells were plated at 2×106 cells per 10 cm plate 24 hours prior to transfection. They were then transiently transfected using the CalPhos mammalian transfection kit (BD Biosciences, Palo Alto, Calif.) with 28 μg of total DNA per plate: 10 μg of pLEGFP-N1 M-MuLV based vector plasmid expressing enhanced green fluorescent protein (EGFP) (Clonetech)) and 18 μg of mA3 expression plasmids (pmA3HA, pmA3D5HA or control pcDNA3.1). pmA3HA and pmA3D5HA were provided by Alan Rein (Rulli et al., 2008). Cells were washed with phosphate-buffered saline (PBS) 12 hours post-transfection and re-fed with fresh media. 24 hours post-transfection, media was replaced with Optimem (Invitrogen, Carlsbad, Calif.) supplemented with 2% FBS (10 ml per dish). 48 hours post-transfection media was collected and filtered through a 0.45 micron filter. Supernantants were then pooled and concentrated 500-fold using a Centricon Plus-70, Ultracel-PL Membrane, 100 kDa (Millipore, USA). Briefly, Centricon units were sterilized with 70% ethanol, and washed with 70 mL of 1x PBS. 70 mL of supernatant was applied to the filter unit followed by centrifugation in a Beckman Allegra 6 centrifuge at 3000 rpm and 4°C for 15 minutes. The flow-through was removed and additional supernatant media was applied to the unit, followed by centrifugation under the same conditions for 30 minutes. To recover the concentrated vector, the unit was inverted followed by centrifugation at 1200 rpm for 1 minute. The concentrated vector was stored in aliquots at −80° C.
Infectivity titrations
M-MuLV titers were determined by a focal immunofluorescence assay (FIA) as described previously (Lander, Chesebro, and Fan, 1999). NIH-3T3 cells were plated at 7×104 cells per 6cm plate 24 hours prior to infection. They were pretreated with 1mL of 2 μg/ml Polybrene in TD (Tris-buffered saline lacking calcium and magnesium) 1 hour prior to infection. The polybrene was then aspirated and 1 mL of diluted virus stock (ten-fold serial dilutions) was applied, followed by incubation at 37° C for 1 hour, after which 3 ml media was added to each plate. Four days post infection plates were stained for 1 hour at 4°C with the 538 env monoclonal antibody. The plates were washed with PBS supplemented with 2% FBS. The plates were then stained for 30 minutes at 4°C with a secondary antibody (fluorescein-conjugated goat IgG fraction to mouse immunoglobulins, MP Bio) which was diluted 1:200 in 2%FBS/PBS. Plates were washed 3 times and then colonies of green cells were counted on a microscope under UV illumination. Viral titers were calculated from the numbers of fluorescent colonies corrected for the dilution factors of the viral stocks in each plate.
Infection levels in primary bone marrow, spleen and thymus were measured in an analogous infectious center assay, as described previously (Lander, Chesebro, and Fan, 1999). Single cells suspensions (106 cells and serial ten-fold dilutions) from animals at different times post-inoculation (6 – 14d) were co-cultivated with NIH-3T3 cells after pretreatment with 1mL of 2 μg/ml Polybrene in TD for 1 hour, and after 24 hours the non-adherent cells were aspirated. After 4 days, the infected NIH-3T3 cell cultures were processed as above, and regions of focal immunofluorescence were counted. The number of infected hematopoietic cells per 106 cells were then calculated from the the number of foci of infection, corrected for the dilution factors of the seeded hematopoietic cells.
Titrations of vector stocks were also performed on NIH-3T3 cells. The cells were plated 24 hours prior to infection at 2×105 cells per 6cm plate. Cells were pretreated and infected as above for viral titrations. 2 days post-infection the plates were observed under UV illumination, and green fluorescent cells (individuals or small groups) were counted. Infectivity titers were calculated from the numbers of fluorescent cells/colonies, corrected for the dilution factors.
Animals
mA3 animals knockout were created by gene trap/ES cell technology (Baygenomics) as described previously (Okeoma et al., 2007). Briefly, the beta-galactosidase (β gal) gene was randomly inserted by way of a retroviral vector into an ES cell line derived from 129/Ola mouse embryos. Expression of the inserted bgal gene was used to identify ES cells with insertions, and also to locate and determine the sites of insertion. The ES cell with mA3 knocked out, were selected and showed disruption by β gal in exon 4, resulting in a fusion product of mA3 and β gal. This ES cell was then used to create the knockout animals; ES cells were cultured and then injected into 129/Ola bastocytes by the Transgenic Knockout Shared Resource at UCDavis. The resulting mA3 knockout mice were backcrossed to C57/Bl6 (B6) mice and used in the ninth backcross generation to B6 for these experiments. We previously showed that cells from these animals lack functional mA3 protein (Okeoma et al., 2007).
Genotyping
Tail samples were obtained from animals and DNA was extracted using the DNeasy Kit (Qiagen). PCR was performed on the samples to identify the wildtype mA3 and the mA3 gene with the insertional knockout cassette. Both PCR reactions utilized the same forward primer in exon 4 )5′-AAT TTA AAA AGT GTT GGA AGA AGT TTG TGG ACA- 3′); PCR which detected wildtype mA3 used a reverse primer specific for exon 5 5′ –TGA AGA GCT GGA AGG GAC CGA GAT GTA GCA AGG – 3′ and the PCR detecting the insertional knockout version of mA3 utilized a reverse primer specific for the β gal cassette used to create the insertional knockout (5′- CCA CAA CGG GTT CTT CTG TT – 3′).
In vivo pathogenesis
Neonatal mice were inoculated intraperitoneally (IP) with 0.2 ml of M-MuLV stock (5 ×105 to 1 ×106 IU/ml), as described previously (Davis, Linney, and Fan, 1985). The animals that were allowed to go to disease were observed until they showed signs of disease, at which point they were sacrificed and subjected to necropsy (Bundy et al., 1995). Hematocrits were measured; spleen, thymi, lymph nodes and other enlarged or abnormal organs were prepared for genomic DNA extraction by phenol/chloroform isolation. Portions of tumors and enlarged or abnormal tissues were also frozen and fixed in formaldehyde and embedded in parrafin.
Tumor diagnosis
Tumor diagnosis was conducted as described previously (Bundy et al., 1995). In light of the fact that M-MuLV induces lymphoblastic lymphomas (predominantly T-lymphoma) in most mice (Brightman et al., 1988), animals with enlarged thymus, spleen and/or lymph nodes and normal hematocrits were considered presumptively to have lymphoblastic lymphoma. To identify the types of lymphoma, cellular DNAs from enlarged organs were analyzed for rearrangements of the T-cell receptor beta (TCRβ), immunoglobulin mu heavy chain (IgM) and kappa light chain (IgK) genes by restriction endonuclease digestion and Southern blot hybridization as described previously (Brightman et al., 1988). Tumor tissues showing gene rearrangements for TCRβ were classified as containing T-lymphoma, while those containing IgK gene rearrangements were classified as containing Blymphoma. Tumors that showed IgM rearrangements (but no TCRβ rearrangements) were classified as containing B lymphoma or pre-B-lymphoma, depending on whether the IgK gene showed rearrangement as well.
Histopathological Analysis
4-6 μm sections were cut from the paraffin blocks and studied by light microscopy after staining with normalized hematoxilin-eosin (H&E) and Congo red for amyloid procedures. In addition, standard immunohistochemistry for CD3 and CD79 lymphocyte antigens were carried out.
In vitro infection of BMDCs
BMDCs were infected with LEGFP-N1 vector stocks (vector alone, vector containing mA3, or vector containing mA3Δ5) for 24 hrs. Cells treated with 3 mg/ml of the reverse transcription inhibitor azidothymidine (AZT; Sigma, Inc.) at 37°C for 2 hrs served as controls. Infection assays were done by the spinoculation method, as previously described (Courreges et al., 2007). Briefly, BMDCs were plated in a 96-well plate at a cell density of 1 × 106 per well and vector stocks were added to cells. The approximate MOIs were 0.03 NIH-3T3 infectious units per cell. The plate was centrifuged at 1,200 × g for 120 minutes at room temperature. After centrifugation, the supernatants were discarded and fresh RPMI complete medium was added to the cells. The cells were cultured for 24 hrs prior to harvesting. Fluorescence-activated cell-sorting (FACS) analysis for EGFP expression was used to detect infection and data are presented as relative infectivity normalized to viral RNA levels. All infectivity assays were done in triplicate; each assay was performed a minimum of 3 times with similar results. The results were normalized for the relative amounts of vector RNA in each of the stocks as determined by quantitative RT-PCR – in general the amounts of vector RNAs in the different stocks were similar.
Reverse transcription-real time quantitative PCR
Total RNA was isolated from vector stocks using the RNeasy Mini Kit (Qiagen, Inc.) according to manufacturer’s instructions. Isolated RNA was treated with DNase I (Qiagen, Inc.) and reverse transcribed (or not) with SuperScript III (Invitrogen, Inc.). For quantification of MLV retroviral vector RNA levels, cDNA was amplified with the following primer pair detecting the EGFP gene: 5′ – ACG TAA ACG GCC ACA AGT TC – 3′ and 5′ – AAG TCG TGC TGC TTC ATG TG – 3′. RT-qPCR was performed using the ABI 7900 real-time PCR machine as previously described (Okeoma et al., 2008).
Western Blots
To prepare vector stocks for western anaylsis, 100ul of vector stock was diluted in 30% sucrose in 1 X PBS, and pellet at 21,000rpm in a SW28 rotor for 1 hour at 4°C. The pellet was then resuspended in virion lysis buffer containing 25mM Tris-HCl (pH8), 150mM NaCl, 2mM EDTA, 0.1% SDS, 0.5% Nonidet P-40, 0.5% deoxycholic acid. Equal portions of the vector stocks were loaded, separated by 12% SDS-PAGE and electrophoretically transferred to a nitrocellulose membrane. For quantification of viral protein in the vector stocks, the membrane was incubated with a primary rabbit polyclonal antibody for M-MuLV p30 (CA) protein (1:3,333 dilution, (Mueller-Lantzsch and Fan, 1976)), followed by washing and incubation with a horseradish peroxidase-conjugated secondary antibody (donkey anti-rabbit IgG, Amersham). The blots were also probed with anti-HA (Invitrogen, Inc., Carlsbad, CA) and species-appropriate HRP-conjugated secondary antibody was used to detect the mA3HA or mA3Δexon5HA proteins. Bands were detected with ECL reagents (Amersham Biosciences, Inc.) and X-ray film.
ACKNOWLEDGEMENTS
This work was supported by an IDEA research grant from the California HIV Research Program and a grant from the Penn Center for AIDS Research (CFAR), an NIH-funded program (P30 AI045008). CMO was supported by training grant PHS T32-CA-009140. We thank the UCI Cancer Research Institute for administrative support, Daniel Talvares for technical support, and the UCI Cancer Center Biostatistcs Shared Resource.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abudu A, Takaori-Kondo A, Izumi T, Shirakawa K, Kobayashi M, Sasada A, Fukunaga K, Uchiyama T. Murine retrovirus escapes from murine APOBEC3 via two distinct novel mechanisms. Curr Biol. 2006;16(15):1565–70. doi: 10.1016/j.cub.2006.06.055. [DOI] [PubMed] [Google Scholar]
- Belli B, Fan H. The leukemogenic potential of an enhancer variant of Moloney murine leukemia virus varies with the route of inoculation. J Virol. 1994;68(11):6883–9. doi: 10.1128/jvi.68.11.6883-6889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Malim MH. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol. 2006;80(17):8450–8. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14(15):1392–6. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O’Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci U S A. 2006;103(23):8780–5. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightman BK, Chandy KG, Spencer RH, Gupta S, Pattengale PK, Fan H. Characterization of lymphoid tumors induced by a recombinant murine retrovirus carrying the avian v-myc oncogene. Identification of novel (B-lymphoid) tumors in the thymus. J Immunol. 1988;141(8):2844–54. [PubMed] [Google Scholar]
- Brightman BK, Davis BR, Fan H. Preleukemic hematopoietic hyperplasia induced by Moloney murine leukemia virus is an indirect consequence of viral infection. J Virol. 1990;64(9):4582–4. doi: 10.1128/jvi.64.9.4582-4584.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightman BK, Rein A, Trepp DJ, Fan H. An enhancer variant of Moloney murine leukemia virus defective in leukemogenesis does not generate detectable mink cell focus-inducing virus in vivo. Proc Natl Acad Sci U S A. 1991;88(6):2264–8. doi: 10.1073/pnas.88.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne EP, Littman DR. Species-specific restriction of apobec3-mediated hypermutation. J Virol. 2008;82(3):1305–13. doi: 10.1128/JVI.01371-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundy LM, Ru M, Zheng BF, Cheng L, Pattengale PK, Portis JL, Fan H. Biological characterization and molecular cloning of murine C-type retroviruses derived from the TSZ complex from mainland China. Virology. 1995;212(2):367–82. doi: 10.1006/viro.1995.1494. [DOI] [PubMed] [Google Scholar]
- Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435(7038):108–14. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol Biol Evol. 2005;22(2):367–77. doi: 10.1093/molbev/msi026. [DOI] [PubMed] [Google Scholar]
- Courreges MC, Burzyn D, Nepomnaschy I, Piazzon I, Ross SR. Critical role of dendritic cells in mouse mammary tumor virus in vivo infection. J Virol. 2007;81(8):3769–77. doi: 10.1128/JVI.02728-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J Virol. 2006;80(3):1067–76. doi: 10.1128/JVI.80.3.1067-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y, Wang X, Esselman WJ, Zheng YH. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol. 2006;80(21):10522–33. doi: 10.1128/JVI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis B, Linney E, Fan H. Suppression of leukaemia virus pathogenicity by polyoma virus enhancers. Nature. 1985;314(6011):550–3. doi: 10.1038/314550a0. [DOI] [PubMed] [Google Scholar]
- Davis BR, Brightman BK, Chandy KG, Fan H. Characterization of a preleukemic state induced by Moloney murine leukemia virus: evidence for two infection events during leukemogenesis. Proc Natl Acad Sci U S A. 1987;84(14):4875–9. doi: 10.1073/pnas.84.14.4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehle BP, Schafer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339(2):281–8. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Doehle BP, Schafer A, Wiegand HL, Bogerd HP, Cullen BR. Differential sensitivity of murine leukemia virus to APOBEC3-mediated inhibition is governed by virion exclusion. J Virol. 2005;79(13):8201–7. doi: 10.1128/JVI.79.13.8201-8207.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanecak R, Pattengale PK, Fan H. Addition of substitution of simian virus 40 enhancer sequences into the Moloney murine leukemia virus (M-MuLV) long terminal repeat yields infectious M-MuLV with altered biological properties. J Virol. 1988;62(7):2427–36. doi: 10.1128/jvi.62.7.2427-2436.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113(6):803–9. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Holmes RK, Malim MH, Bishop KN. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci. 2007;32(3):118–28. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Takaori-Kondo A, Shindo K, Abudu A, Fukunaga K, Uchiyama T. APOBEC3G targets specific virus species. J Virol. 2004;78(15):8238–44. doi: 10.1128/JVI.78.15.8238-8244.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander JK, Chesebro B, Fan H. Appearance of mink cell focus-inducing recombinants during in vivo infection by moloney murine leukemia virus (M-MuLV) or the Mo+PyF101 M-MuLV enhancer variant: implications for sites of generation and roles in leukemogenesis. J Virol. 1999;73(7):5671–80. doi: 10.1128/jvi.73.7.5671-5680.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YN, Malim MH, Bieniasz PD. Hypermutation of an ancient human retrovirus by APOBEC3G. J Virol. 2008;82(17):8762–70. doi: 10.1128/JVI.00751-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QX, Fan H. Combined infection by Moloney murine leukemia virus and a mink cell focus-forming virus recombinant induces cytopathic effects in fibroblasts or in long-term bone marrow cultures from preleukemic mice. J Virol. 1990;64(8):3701–11. doi: 10.1128/jvi.64.8.3701-3711.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddament MT, Brown WL, Schumacher AJ, Harris RS. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol. 2004;14(15):1385–91. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223(1):77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424(6944):99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114(1):21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, Flory E, Schumann GG, Munk C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem. 2006;281(31):22161–72. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- Mueller-Lantzsch N, Fan H. Monospecific immunoprecipitation of murine leukemia virus polyribosomes: identification of p30 protein-specific messenger RNA. Cell. 1976;9(4 Pt 1):579–88. doi: 10.1016/0092-8674(76)90040-4. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–63. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- OhAinle M, Kerns JA, Malik HS, Emerman M. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J Virol. 2006;80(8):3853–62. doi: 10.1128/JVI.80.8.3853-3862.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeoma CM, Lovsin N, Peterlin BM, Ross SR. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature. 2007;445(7130):927–30. doi: 10.1038/nature05540. [DOI] [PubMed] [Google Scholar]
- Okeoma CM, Low A, Bailis W, Hultine S, Fan HY, Peterlin BM, Ross SR. APOBEC3 in viral particles and in target cells cooperatively restricts mouse mammary tumor virus infection in vivo. 2008 [Google Scholar]
- Pion M, Granelli-Piperno A, Mangeat B, Stalder R, Correa R, Steinman RM, Piguet V. APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J Exp Med. 2006;203(13):2887–93. doi: 10.1084/jem.20061519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose KM, Marin M, Kozak SL, Kabat D. Regulated production and anti-HIV type 1 activities of cytidine deaminases APOBEC3B, 3F, and 3G. AIDS Res Hum Retroviruses. 2005;21(7):611–9. doi: 10.1089/aid.2005.21.611. [DOI] [PubMed] [Google Scholar]
- Rulli SJ, Jr., Mirro J, Hill SA, Lloyd P, Gorelick RJ, Coffin JM, Derse D, Rein A. Interactions of murine APOBEC3 and human APOBEC3G with murine leukemia viruses. J Virol. 2008;82(13):6566–75. doi: 10.1128/JVI.01357-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago ML, Montano M, Benitez R, Messer RJ, Yonemoto W, Chesebro B, Hasenkrug KJ, Greene WC. Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science. 2008;321(5894):1343–6. doi: 10.1126/science.1161121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–50. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Stenglein MD, Harris RS. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem. 2006;281(25):16837–41. doi: 10.1074/jbc.M602367200. [DOI] [PubMed] [Google Scholar]
- Suspene R, Guetard D, Henry M, Sommer P, Wain-Hobson S, Vartanian JP. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102(23):8321–6. doi: 10.1073/pnas.0408223102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda E, Tsuji-Kawahara S, Sakamoto M, Langlois MA, Neuberger MS, Rada C, Miyazawa M. Mouse APOBEC3 restricts Friend leukemia virus infection and pathogenesis in vivo. J Virol. 2008 doi: 10.1128/JVI.01311-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260(5115):1816–9. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- Turelli P, Mangeat B, Jost S, Vianin S, Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303(5665):1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. Embo J. 2004;23(12):2451–8. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424(6944):94–8. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li X, Ma J, Yu L, Jiang J, Cen S. The incorporation of APOBEC3 proteins into murine leukemia viruses. Virology. 2008;378(1):69–78. doi: 10.1016/j.virol.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol. 2004;78(11):6073–6. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]