

Abstract
Diaminopimelate decarboxylase (DAPDC) and ornithine decarboxylase (ODC) are pyridoxal 5′-phosphate dependent enzymes that are critical to microbial growth and pathogenicity. The latter is the target of drugs that cure African sleeping sickness, while the former is an attractive target for antibacterials. These two enzymes share the (β/α)8 (i.e., TIM barrel) fold with alanine racemase, another pyridoxal 5′-phosphate dependent enzyme critical to bacterial survival. The active site structural homology between DAPDC and ODC is striking even though DAPDC catalyzes the decarboxylation of a D stereocenter with inversion of configuration and ODC catalyzes the decarboxylation of an L stereocenter with retention of configuration. Here, the structural and mechanistic bases of these interesting properties are explored using reactions of alternate substrates with both enzymes. It is concluded that simple binding determinants do not control the observed stereochemical specificities for decarboxylation, and a concerted decarboxylation/proton transfer at Cα of the D stereocenter of diaminopimelate is a possible mechanism for the observed specificity with DAPDC.
Introduction
Diaminopimelate decarboxylase (DAPDC) is a pyridoxal phosphate (PLP) dependent enzyme that catalyzes the decarboxylation of D,L-diaminopimelate (D,L-DAP) to form L-lysine in the last step in bacterial L-lysine biosynthesis. The presence of multiple pathways to synthesize D,L-DAP in some bacteria suggests that this pathway is highly important for bacterial survival [1]. Indeed, a functional DAPDC gene is required for Mycobacterium tuberculosis survival in immunocompromised mice, demonstrating that de novo lysine biosynthesis is essential for in vivo viability [2]. Combined with the absence of analogous pathways in humans, this makes DAPDC an attractive antibacterial drug target.
Mechanistically, DAPDC has unique features and challenges. First, it must differentiate between the D- and L-stereocenters of D,L-DAP, selectively decarboxylating only the D-stereocenter to generate L-lysine. X-ray crystallographic studies of DAPDC from M. tuberculosis (mtDAPDC), as well as other bacteria, suggest a structural basis for this specificity, especially through specific binding of the L-stereocenter distal to the reaction center [2, 3]. Second, DAPDC is unique among known PLP dependent decarboxylases in decarboxylating a D-stereocenter; all other PLP dependent decarboxylases act on an L-stereocenter. Third, stereochemical studies show that DAPDC catalyzes decarboxylation with inversion of configuration at the reaction center [4, 5], in contrast to all other PLP dependent decarboxylases studied to date [6–11].
Despite these differences, DAPDC shares both sequence and structural similarity to eukaryotic ornithine decarboxylase (ODC), a typical PLP dependent decarboxylase. They have 25% sequence identity [3], and structural overlays of mtDAPDC and Trypanosoma brucei ODC have an rmsd of ~2.2 Å [12]. Furthermore, the active sites of mtDAPDC and ODC have many structural equivalences, including the KAFL motif that contains the lysine that forms the internal aldimine with PLP and a glutamate residue that interacts with the pyridine nitrogen of PLP. In addition, the His that p stacks with the PLP ring is conserved and similarities in the side chain binding pocket including a conserved arginine and serine. The active site structural similarity between mtDAPDC and ODC implies a similar chemical mechanism despite the stereochemical differences between the enzymes. Here, kinetic characterization of mtDAPDC and yeast ODC using alternate substrates is used to explore the strictness of DAPDC specificity for decarboxylating a D stereocenter, as well as the origin of this stereospecificity.
Experimental Procedures
Materials
All materials were purchased from Sigma unless otherwise noted. D,L-DAP, L,L-DAP and D,D-DAP were a gift from Professor John Blanchard (Albert Einstein College of Medicine).
Cloning of Yeast ODC gene
The yeast ODC gene was amplified from genomic DNA from the WDHT668 strain of Saccharomyces cerevisiae using PCR and the following primer pair: 5′-GTG GTG CTC GAG TCA ATC GAG TTC AGA GTC TAT GTA TAC TAT ATC C-3′ (XhoI restriction site is underlined) and 5′-GGA TCC GAA TTC ATG TCT AGT ACT CAA GTA GGA AAT GCT CTA TCT AG-3′ (EcoRI restriction site is underlined). The ~1.4kb PCR product was cleaned using a commercially available kit (Qiagen) and ligated into pET28a (Novagen) digested with XhoI and EcoRI using standard procedures [13]. The construct was sequenced to ensure no mutations had been incorporated during PCR and yielded the final product, an N-terminal His6-tagged yeast ODC fusion protein.
Expression and Purification of Yeast ODC
For overexpression, 1 L of LB growth medium was innoculated with a 5 mL overnight culture and grown at 37 °C until OD600 = 0.5. Expression was induced with IPTG at a final concentration of 500 μM, and growth at 37 °C was continued for 6–8 hours. The cells were pelleted by centrifugation, resuspended in lysis buffer (20 mM TEA-HCl pH 7.8, 10 mM imidazole, 50 μM PLP and 1 mM β-mercaptoethanol) and stored overnight at −80°C. The thawed cell suspension was incubated with 1 mg/mL lysozyme for 1 hour, sonicated and the cell debris removed by centrifugation. The cell free extract was added to 8 mL Ni-NTA Superflow resin (Qiagen) and mixed by end-to-end rotation for 1 hour at 4 °C. After packing, the column was washed extensively (>20 column volumes) with 20 mM TEA-HCl pH 7.8, 20 mM imidazole, 50 μM PLP, 1 mM β-mercaptoethanol and 300 mM KCl. The protein was eluted by running a linear gradient from 20 mM imidazole to 300 mM imidazole in 20 mM TEA-HCl pH 7.8, 50 μM PLP, 1 mM β-mercaptoethanol and 300 mM KCl at 1 mL/min. The fractions containing ODC, as judged by SDS-PAGE, were pooled, concentrated and dialyzed into 50 mM TEA-HCl pH 7.8, 50 mM KCl, 20 μM PLP and 1 mM DTT. The protein was aliquoted, flash frozen and stored at −80°C. The enzyme concentration was determined using the Lowry Assay (BioRad) with IgG as a standard.
Expression and Purification of M. tuberculosis DAPDC
For overexpression, 1 L LB growth medium was innoculated with 3 mL overnight culture and grown at 37°C until OD600 = 0.5. The cells were placed on ice for 30 minutes, induced with IPTG at a final concentration of 500 μM, and grown at 20 °C for 24 hours. The cells were pelleted by centrifugation, resuspended in lysis buffer (20 mM TEA-HCl pH 7.8, 10 mM imidazole, 50 μM PLP and 1 mM β-mercaptoethanol) and stored overnight at −80°C. The thawed cell suspension was incubated with 0.5 mg/mL lysozyme for 1 hour, sonicated and the cell debris removed by centrifugation. The cell free extract was added to 8 mL Ni-NTA Superflow resin (Qiagen), and purified as described above for yeast ODC. The protein was aliquoted, flash frozen and stored at −80°C. The enzyme concentration was determined using the Bradford and Lowry assays (BioRad) with IgG as a standard.
Steady-state Assay
Yeast ODC and mtDAPDC were assayed as described previously [14, 15] by coupling the carbon dioxide produced from decarboxylation to phosphoenol pyruvate (PEP) carboxylase to give oxalacetate, followed by malate dehydrogenase (MDH) catalyzed reduction. Loss of NADH absorbance at 340 nm was followed on a Kontron UVIKON 9420. Reaction mixtures contained 100 mM TEA-HCl pH 7.8, 20 μM PLP, 300 μM NADH, 10 mM MgCl2, 2 mM PEP, 8 units/mL freshly prepared PEP carboxylase and 33 units/mL MDH. All reagents were prepared in water that had been boiled extensively to remove carbon dioxide. Stock solutions were lyophilized and redissolved in water from which the carbon dioxide had been removed.
Inhibition Assays
Inhibition studies for ODC were performed with D-ornithine and putrescine, using the coupled carbon dioxide assay. The concentration of L-ornithine was held at Km (130 μM) and Ki was determined assuming competitive inhibition (eq 1).
| (1) |
Inhibition studies for mtDAPDC used L- and D-lysine, L,L- and D,D-DAP using the coupled carbon dioxide assay. The concentration of D,L-DAP was held at Km (400 μM) and Ki was determined assuming competitive inhibition.
Circular Dichroism-Based Assay
For both ODC and DAPDC, the substrate and product differ in their optical activity, allowing direct detection of the reaction using circular dichroism (CD). A Jasco J-600 CD spectrometer was used to assay ODC activity, as previously reported for T. brucei ODC [16]. The loss of CD signal at 210 nm was monitored over time. The wavelength was chosen to maximize signal while maintaining a linear relationship between CD signal and amino acid substrate concentration. Reaction mixtures contained 20 mM potassium phosphate pH 7.5, 20 μM PLP, and 100 mM KCl.
HPLC Assay for Reaction with Alternate Substrates
Assays to identify the amine or amino acid product of decarboxylation were performed using a previously reported HPLC-based method [17]. Reactions with 100 mM TEA-HCl pH 7.8, 100 mM KCl, 20 μM PLP, and the appropriate alternate substrate and enzyme were allowed to react at 25 °C for 12–16 hours. The protein was denatured by adding 2 μL glacial acetic acid to the 200 μL reaction and the precipitated protein was removed by centrifugation. The reaction mixture was then derivatized with o-phthaldialdehyde (OPA). Briefly, a 100 μL sample was mixed with 100 μL freshly prepared OPA reagent (4 mg solid OPA, mixed with 4.5 mL 0.1 M borate pH 10.4, 15 μL 30% (w/v) Brij detergent and 10 μL β-mercaptoethanol), allowed to react for 1 minute at room temperature and acidified with 2 M acetic acid to pH 4.5. The derivatized sample was immediately run on a Hibar LiChrosorb C18 column using an Agilent 1100 series chromatography system attached to a Perkin-Elmer 650-15 fluorescence spectrophotometer. The HPLC method [17] used 50 mM sodium acetate pH 5.7, 5% (v/v) THF (Solvent A); methanol (Solvent B); acetonitrile (Solvent C). The elution gradient was as follows: 90% A, 10% B to 35% A, 65% B from 0–15 minutes; 35% A, 65% B from 15–20 minutes; 35% A, 65% B to 50% B, 50% C from 20–25 minutes; 50% B, 50% C from 25–30 minutes. The flow rate was 1 mL/min and the products were monitored using fluorescence (ex = 340 nm, em = 455 nm).
HPLC analysis of the reactions was complicated due to the fact that diamines such as putrescine, cadaverine, ornithine or lysine can react with multiple equivalents of the OPA reagent [18]. Although a large excess of OPA was used in an attempt to fully derivatize the species, thereby simplifying the chromatograms, the relatively high concentration of the diamine species did not allow complete derivatization. To circumvent this problem, negative controls in which enzyme was not added but were otherwise identical were always run and compared to the enzymatic samples.
Molecular Dynamics
The structure of M. tuberculosis DAPDC with L-lysine bound (PDB entry 1HKV) was used as the template structure. The external aldimine intermediates for both D,L- and L,L-DAP were constructed by adding a carboxylate group of the appropriate stereochemistry on the bound lysine, then replacing the Schiff base between Lys72 and PLP with one between DAP and PLP and energy minimizing the ligand. The program BALLView was used to perform simulated annealing molecular dynamics using the MMFF94 force field. Residues within 4 Å of any atom of the PLP-DAP external aldimine were included in the calculations, while the rest of the protein remained unaltered. The structures were first energy minimized to a gradient of less than 0.1 kJ/Å, then heated to 300K over 10 ps (1 fs timesteps), equilibrated for 20 ps, then slowly cooled to 10K over 20 ps. The final structures from cooling were again energy minimized.
Results
Yeast ODC
Steady-state kinetic parameters for ODC are given in Table 1. The kinetic parameters for L-ornithine varied with ODC concentration, presumably due to the formation of higher order oligomers (data not shown). This was not studied in detail. Instead, all kinetic studies were performed at the same relatively high enzyme concentration (50 nM) to minimize differences in kcat, Km and Ki due to oligomerization. Since all inhibitor studies were performed at the same substrate concentration and the inhibition data collected was well fitted by a simple competitive inhibition model, it is unlikely that enzyme oligomerization complicated the results significantly. The values obtained here are in agreement with previously reported values for the yeast ODC enzyme [19]. The kinetic parameters from the coupled-enzyme assay agree with those from the direct CD assay, although the errors are larger in the latter.
Table 1.
Steady-state Kinetic Parameters for S. cerevisiae ODCa
| Substrate | kcat (s−1) | Km (μM) | kcat/Km (M−1s−1) | Ki (μM) | |
|---|---|---|---|---|---|
| L-ornithine | CO2 Assay | 3.3 (0.2) | 122 (20) | 2.7 (0.4) × 104 | |
| CD Assay | 4.2 (0.7) | 214 (134) | 2 (1) × 104 | ||
| D-ornithine | ~0.002 | 380 ± 67 | |||
| putrescine | 250 ± 45 |
Errors given in parentheses.
Inhibition studies with ODC show that the enzyme binds D-ornithine (Ki = 380 ± 67 μM). The enzyme also shows significant product inhibition (putrescine Ki = 250 ± 45 μM), in agreement with previous studies that found the concentration of putrescine necessary for 50% inhibition was 100 μM [19].
HPLC analysis of the reaction of ODC with D-ornithine shows that a small amount of putrescine is formed (Figure 1), indicating that ODC can slowly decarboxylate a D-stereocenter. The rate constant for D-ornithine decarboxylation was estimated to be ~0.002 s−1 from these data.
Figure 1.

HPLC fractionation of ODC reaction with D-ornithine. (A) Control reaction without enzyme added. Diamines including putrescine and ornithine can react with multiple equivalents of the OPA reagent [18]. Although a large excess of OPA was used in an attempt simplify the chromatograms, the relatively high concentration of the diamines prevented complete derivatization. (B) Reaction with ODC and D-ornithine.
mtDAPDC
The steady-state kinetic parameters for mtDAPDC are given in Table 2. D,L-DAP and commercial meso-DAP give identical results, when it is taken into account that commercial meso-DAP is a mixture of 50% D,L-, 25% D,D- and 25% L,L-DAP and the Km value is calculated by ignoring the 50% of the meso-DAP that is not D,L-DAP (as reported here). Studies with D,D-DAP showed no detectable inhibition of the D,L-DAP reaction at concentrations of 6 mM (data not shown). In fact, D,D-DAP is a reasonable substrate for mtDAPDC (Table 2) but with a ~100-fold lower kcat/KM value than D,L-DAP. Thus, its contribution to the activity measured with meso-DAP can be ignored.
Table 2.
Steady-state Kinetic Parameters for mtDAPDCa
| Substrate | kcat (s−1) | Km (mM) | kcat/Km (M−1s−1) | Ki (mM)b |
|---|---|---|---|---|
| D,L-DAP | 7.1 (0.1) | 0.43 (0.03) | 1.6 (0.1) × 104 | |
| meso-DAPc | 7.1 (0.1) | 0.40 (0.01) | 1.8 (0.1) × 104 | |
| D,D-DAP | 1.8 (0.3) | 17 (4) | 100 (30) | |
| D-lysined | 26 (5) | |||
| L,L-DAP | 1.6 (0.4) | |||
| L-lysine | 1.8 (0.2) |
Errors are given in parentheses.
Ki was obtained by assuming competitive inhibition and using eq 1.
meso-DAP is a mixture of D,L-, D,D-, and L,L-DAP. It is assumed that L,L- and D,D-DAP do not significantly contribute to the observed rate, thus the Km value given has been corrected for the 50% D,L-DAP present.
Although a rate could not be estimated for D-lysine, HPLC analysis showed a small amount of decarboxlation occurs with this substrate.
mtDAPDC is inhibited by L,L-DAP (Ki = 1.6 ± 0.4 mM), D-lysine (Ki = 26 ± 5 mM), and the product of D,L-DAP decarboxylation, L-lysine (Ki = 1.8 ± 0.2 mM). Coenzyme spectral changes on addition of these inhibitors to mtDAPDC are similar to those observed when D,L-DAP is added to the enzyme, with a red shift in the peak centered around 420 nm characteristic of the ketoenamine tautomer and an increase in absorbance of 330 nm characteristic of the enolimine tautomer. The similarity in the spectral changes observed for the inhibitors to those seen for D,L-DAP suggest that the inhibitors bind covalently (Figure 2).
Figure 2.

Spectra of mtDAPDC with DAP substrates and inhibitors. Reaction conditions: 100 mM TEA-HCl, pH 8.0, 18 μM enzyme, 5 mM substrate/inhibitor, 20 μM PLP. (A) mtDAPDC without substrate or inhibitor. (B) mtDAPDC with D,L-DAP. (C) mtDAPDC with D,D-DAP. (D) mtDAPDC with L,L-DAP.
HPLC analyses of reactions of mtDAPDC with L-lysine and L,L-DAP found no detectable amounts of their decarboxylation products, cadaverine or L-lysine, respectively (data not shown). Both D,D-DAP (Figure 3) and D-lysine (Figure 4) are decarboxylated by mtDAPDC.
Figure 3.
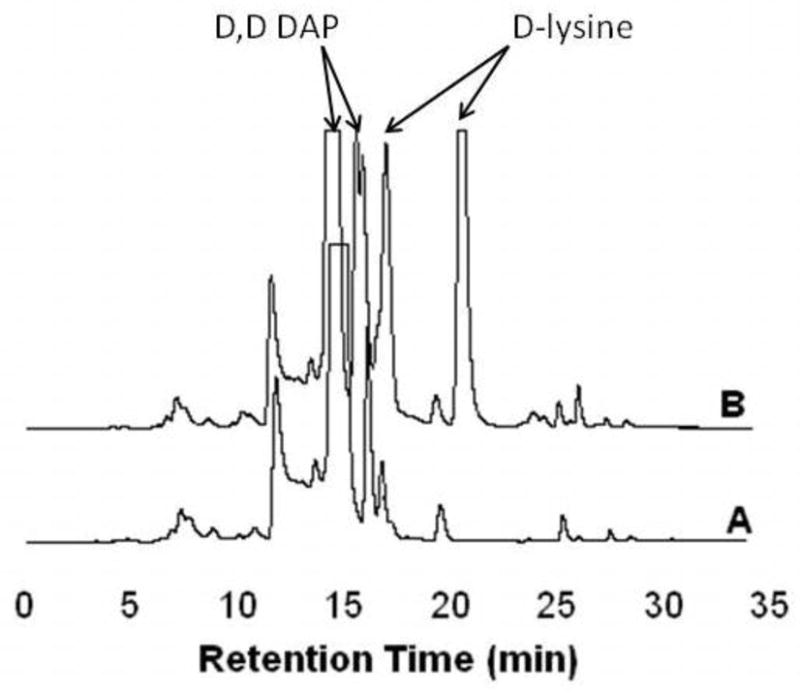
HPLC fractionation of mtDAPDC reaction with D,D-DAP. (A) Control reaction without enzyme added. Diamines including lysine and D,D-DAP can react with multiple equivalents of the OPA reagent [18]. Although a large excess of OPA was used in an attempt simplify the chromatograms, the relatively high concentration of the diamines prevented complete derivatization. (B) Reaction with mtDAPDC and D,D-DAP.
Figure 4.

HPLC fractionation of mtDAPDC reaction with D-lysine. (A) Control reaction without enzyme added. Diamines like cadaverine and lysine react with multiple equivalents of the OPA reagent [18]. Although a large excess of OPA was used in an attempt simplify the chromatograms, the relatively high concentration of the diamines prevented complete derivatization. (B) Reaction with mtDAPDC and D-lysine.
Molecular Dynamics
The results of slow-cooling molecular dynamics calculations on DAPDC with both L,L- and D,L-DAP external aldmines are presented in Figure 5 along with the experimental X-ray structure of ODC with D-ornithine bound. The L,L-DAP structure in Figure 5A shows that the active site readily accommodates the carboxylate in the L configuration at the reaction center. The carboxylate accepts four hydrogen bonds: S377 side chain, E376 backbone amide, Lys72 side chain, and the distal ammonium group of the substrate. The carboxylate group is nearly perpendicular (88°) to the plane of the Schiff base.
Figure 5.

A) Active site of M. tuberculosis DAPDC after slow-cooling molecular dynamics with L,L-DAP present as an external aldimine intermediate. B) Same A except D,L-DAP was present as the external aldimine intermediate. C) Overlay of the structures in parts A and B. D) Active site of ODC bound to D-ornithine determined by X-ray crystallography.
The D,L-DAP structure in Figure 5B shows the carboxylate in the D configuration is also well accommodated by the active site. It accepts three hydrogen bonds: S377 side chain, E376 backbone amide, and H213. The carboxylate group is not perpendicular to the plane of the Schiff base. The dihedral angle for C4′-N-Cα-CO2− is 157° while that for C4-C4′-N-Cα is 22°. The L,L- and D,L-DAP external aldimine structures are similar in energy with the D,L-DAP structure being somewhat lower.
Discussion
ODC
Unlike other known PLP dependent decarboxylases, DAPDC acts on a D stereocenter and decarboxylation occurs with inversion of configuration [4, 5]. Nevertheless, it shows a high degree of sequence and structural homology to eukaryotic ODCs, which are typical PLP dependent decarboxylases that catalyze decarboxylation at L stereocenters with retention of configuration [8, 20]. ODC from S. cerevisiae was cloned, expressed, and purified, and its kinetic characterization is presented in Table 1. The kcat and Km values are in good agreement with previously reported values [19].
For simplicity, here we assume the experimental Km values to be binding constants (i.e. formation of the external aldimine intermediate from free enzyme and substrate is at equilibrium compared to decarboxylation), as is the case for homologous T. brucei ODC [21–23]. Compared to the natural substrate L-ornithine, both D-ornithine and putrescine (the product of decarboxylation of either isomer) bind well to ODC (Table 1). The contribution of a substrate carboxylate group in the L stereoconfiguration is small (−0.2 kcal/mol), as gleaned from comparing the Km value for L-ornithine and the Ki value for putrescine. One can interpret these results as ODC providing only small stabilizing interactions to the bound substrate carboxylate, roughly equivalent to the energy required for desolvating the carboxylate as it binds. Such weak interactions are consistent with previous proposals that the carboxylate is bound in a hydrophobic pocket that enforces ground state destabilization of the negative charge [24].
Similarly, the presence of a carboxylate in the D stereoconfiguration has little effect on binding affinity, as observed previously [20, 25, 26]. Nevertheless, ODC decarboxylates D-ornithine slowly (Figure 1). Given that D-ornithine binds well to ODC, there must be a specific mechanistic reason for its slow CO2 loss since all subsequent steps are identical to those with L-ornithine. The structure of T. brucei ODC with D-ornithine bound was solved by X-ray crystallography, and the active site is shown in Figure 5D. One might speculate that the solvent exposed si face of PLP allows water to hydrate the carboxylate, thereby providing ground state stabilization of the D isomer. Then again, the active site on the si face of PLP in DAPDC, which only decarboxylates a D stereocenter, is similarly solvent exposed.
DAPDC
While ODC binds L-ornithine with the carboxylate on the re face of the PLP ring, X-ray crystallographic structures of DAPDC suggest that D,L-DAP binds with the carboxylate of the D-stereocenter on the si face of the PLP ring [2, 3, 20]. The basis of the specificity for decarboxylation of the D-stereocenter in DAPDC is not easily explained by the available structures, since there are few differences between ODC and DAPDC on the si face of the PLP ring, and none that clearly account for preferential binding of the carboxylate on the si face in DAPDC (Figure 5). For example, Ser377 is the only obvious, conserved difference and it seems unlikely that this residue alone accounts for the observed specificity.
One means for achieving D stereocenter decarboxylation selectivity is simply having a weak affinity for an L stereocenter at the reaction center. To address this possibility, L,L-DAP was tested as a substrate. L,L-DAP binds well (~4-fold weaker than D,L-DAP) to DAPDC although it is not decarboxylated (Table 2). This agrees with molecular dynamics calculations discussed below, and suggests that preferential D stereocenter binding at the reaction center is not the determinant of DAPDC decarboxylation specificity.
The basis for the specificity for decarboxylation of the D stereocenter in DAPDC could be the result of stereoelectronic effects. If DAPDC were to allow only the carboxylate of the D stereocenter to be aligned perpendicular to the p orbitals of the conjugated Schiff base/pyridine ring, then stabilization of negative charge on Cα will selectively occur in decarboxylation of a D stereocenter. This possibility was explored by performing slow-cooling molecular dynamics calculations. The results of these calculations show that the lowest energy structure of the L,L-DAP external aldmine (Figure 5A) places the carboxylate at the reaction center nearly perpendicular to the plane of the Schiff base/pyridine ring, even though no decarboxylation with L,L-DAP is observed. On the other hand, the carboxylate of the D stereocenter of the D,L-DAP external aldmine is far from being perpendicular to either the Schiff base or the pyridine ring (Figure 5B). Rather it binds in an orientation very similar to that found for the carboxylate of D-ornithine in the X-ray structure of ODC [20]. Thus, the determinants of stereospecificity in DAPDC are apparently not simply stereoelectronic effects enforced in the ground state by substrate carboxylate orientation with respect to the plane of the Schiff base/pyridine ring. Rather, they probably reside in another source.
Some other means for selective catalysis of CO2 loss from the D-stereocenter of the external aldimine formed with D,L-DAP, which places the carboxylate on the si face of PLP, must be the major determinant of stereospecificity in DAPDC. This is supported by the results from D,D-DAP and D-lysine reactions with DAPDC. Although these alternate substrates bind relatively weakly to DAPDC, both are decarboxylated by DAPDC. The poor binding observed for D,D-DAP and D-lysine is not unexpected considering the structure of the substrate binding pocket (Figure 5). With the natural substrate, the carboxylate of the L stereocenter interacts with two conserved arginine residues while the amino group of the L stereocenter is anchored by a conserved aspartate. Conceivably, D-lysine could form a Schiff base with PLP through either of its amino groups although this is unlikely. One expects a ~40-fold preference for carboxylate binding at the catalytic site compared to the side-chain site given the higher binding affinity for D,L-DAP compared to D,D-DAP (Table 2). Additionally, the decarboxylation observed with D-lysine requires it to bind to PLP through its α-amino group.
When D,D-DAP binds, a D stereocenter is forced into the substrate binding pocket that is structurally complementary to an L stereocenter, explaining the high Km value observed. Despite weaker binding, D,D-DAP is decarboxylated rapidly with a kcat only 4-fold less than with the natural substrate. This indicates that poor distal interactions at the side-chain site are not transmitted to the active site resulting in poor decarboxylation catalysis, and that placement of the carboxylate on the si face of the PLP is sufficient for efficient catalysis.
It has been suggested that decarboxylation may be facilitated in part by an induced-fit mechanism in DAPDC from Helicobacter pylori (hpDAPDC) on the basis of X-ray crystallographic structures and mutational analysis [27]. In hpDAPDC a mobile active site α-helical loop was observed to be “open” in the absence of a ligand and “closed” in the presence of L-lysine. It was proposed that this loop closes down over the substrate, shielding it from bulk solvent and stabilizing catalytic intermediates. This loop is one of the most divergent elements of DAPDC structure, however, and mtDAPDC differs significantly from other DAPDCs in this regard. mtDAPDC contains a β-sheet structure that is unlikely to undergo the dynamic changes observed in the hpDAPDC structures. Indeed, X-ray crystallographic structures of mtDAPDC with and without L-lysine show no large structural changes [2], making it unlikely that catalysis is facilitated by an induced fit mechanism in mtDAPDC.
A major difference between the structures of the D,L- and L,L-DAP external aldimine structures shown in Figure 5 is the number of hydrogen bonds from positively charged groups the carboxylate at the reaction center accepts: in the L,L-DAP structure, the carboxylate at the reaction center accepts hydrogen bonds from Lys72 and from the distal substrate ammonium group, while in the D,L-DAP structure the carboxylate at the reaction center accepts a hydrogen bond only from His213, which was presumed to be charged for the calculations. The interaction with two positive charges for the L stereocenter vs. one for the D stereocenter could be a major contributor to selectivity for decarboxylation at the D stereocenter by stabilizing the negatively charged ground state of the L stereocenter. Additionally, there are other minor differences in the interactions between the active site and the two external aldmine intermediates (Figure 5C) that may sum to a significant catalytic advantage for decarboxylation at the D stereocenter.
Another possibility for the observed specificity for decarboxylation of the D-stereocenter in DAPDC is that decarboxylation is an obligatorily concerted process, in which loss of CO2 and protonation from the re face at Cα occur in the same step. If the substrate carboxylate binds on the re face, as expected with L,L-DAP or L-lysine, then it is likely that the general acid involved in a concerted decarboxylation/protonation transition state is not able to interact with Cα because of steric interference from the substrate carboxylate (i.e. a front-side electrophilic substitution reaction seems unlikely). Such a concerted decarboxylation/protonation transition state would explain both the stereoselective decarboxylation by DAPDC as well as the unusual inversion of configuration observed.
Given the structural and mechanistic similarity between ODC and DAPDC, one expects that the general acid that protonates Cα would be conserved. It has been suggested that Cys360 acts as the general acid in T. brucei ODC [28]. However, it is highly unlikely that in mtDAPDC the corresponding residue, Cys375, acts as the general acid since it is involved in an intermolecular disulfide bond (Figure 5) [2]. Although other DAPDCs do not contain this disulfide bond, it seems unlikely from a parsimony point of view that DAPDCs from other microorganisms are mechanistically distinct from their mycobacterial counterparts and use the corresponding cysteine residue as the general acid. Other potential general acids, such as His213, are not consistent with the observed stereochemistry.
Thus, the identity of the general acid that protonates Cα after CO2 loss in DAPDC is not clear. The most obvious possibility for the general acid in DAPDC is Lys72 since it is appropriately positioned, and in many PLP dependent enzymes the Schiff base-forming lysine also acts as an acid/base catalyst. Therefore, we propose that the reaction catalyzed by DAPDC on D stereocenters could be a concerted, backside electrophilic substitution reaction with simultaneous CO2 loss from Cα and proton donation to the backside of Cα from Lys72. Although there is a mechanistic preference for frontside electrophilic attack, this is not a strict mechanistic requirement as it is for nucleophilic substitution. This proposal is consistent with all of the available stereochemical, kinetic, and structural data, and provides an elegant means for enforcing stereochemistry where both stereoisomeric carboxylate group bind in similar environments.
Scheme 1.

Highlights.
The stereospecificity of DAPDC and ODC, two structuirally similar PLP dependent enzymes, have been analyzed using alternate substrates
Simple binding determinants do not control reactivity
A concerted transition state in which CO2 is lost and a proton is added to Cα is proposed for DAPDC to account for its stereospecificity
Acknowledgments
Supported by grant GM54779 from the National Institutes of Health
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Born TL, Blanchard JS. Structure/function studies on enzymes in the diaminopimelate pathway of bacterial cell wall biosynthesis. Curr Opin Chem Biol. 1999;3:607–613. doi: 10.1016/s1367-5931(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 2.Gokulan K, Rupp B, Pavelka MS, Jr, Jacobs WR, Jr, Sacchettini JC. Crystal structure of Mycobacterium tuberculosis diaminopimelate decarboxylase, an essential enzyme in bacterial lysine biosynthesis. J Biol Chem. 2003;278:18588–18596. doi: 10.1074/jbc.M301549200. [DOI] [PubMed] [Google Scholar]
- 3.Ray SS, Bonanno JB, Rajashankar KR, Pinho MG, He G, De Lencastre H, Tomasz A, Burley SK. Cocrystal structures of diaminopimelate decarboxylase: mechanism, evolution, and inhibition of an antibiotic resistance accessory factor. Structure (Camb) 2002;10:1499–1508. doi: 10.1016/s0969-2126(02)00880-8. [DOI] [PubMed] [Google Scholar]
- 4.Asada Y, Tanizawa K, Sawada S, Suzuki T, Misono H, Soda K. Stereochemistry of meso-alpha, epsilon-diaminopimelate decarboxylase reaction: the first evidence for pyridoxal 5′-phosphate dependent decarboxylation with inversion of configuration. Biochemistry. 1981;20:6881–6886. doi: 10.1021/bi00527a022. [DOI] [PubMed] [Google Scholar]
- 5.Kelland JG, Palcic MM, Pickard MA, Vederas JC. Stereochemistry of lysine formation by meso-diaminopimelate decarboxylase from wheat germ: use of 1H-13C NMR shift correlation to detect stereospecific deuterium labeling. Biochemistry. 1985;24:3263–3267. doi: 10.1021/bi00334a028. [DOI] [PubMed] [Google Scholar]
- 6.Asada Y, Tanizawa K, Nakamura K, Moriguchi M, Soda K. Stereochemistry of ornithine decarboxylase reaction. J Biochem (Tokyo) 1984;95:277–282. doi: 10.1093/oxfordjournals.jbchem.a134594. [DOI] [PubMed] [Google Scholar]
- 7.Gerdes HJ, Leistner E. Stereochemistry of reactions catalysed by L-lysine decarboxylase and diamine oxidase. Phytochemistry. 1979;18:771–775. [Google Scholar]
- 8.Orr GR, Gould SJ, Pegg AE, Seely JE, Coward JK. Stereochemistry of the decarboxylation of L-ornithine with ornithine decarboxylase from mouse kidney. Bioorganic chemistry. 1984;12:252–258. [Google Scholar]
- 9.Stevenson DE, Akhtar M, Gani D. Streptomyces L-methionine decarboxylase: purification and properties of the enzyme and stereochemical course of substrate decarboxylation. Biochemistry. 1990;29:7660–7666. doi: 10.1021/bi00485a015. [DOI] [PubMed] [Google Scholar]
- 10.Yamada H, O’Leary MH. Stereochemistry of reactions catalyzed by glutamate decarboxylase. Biochemistry. 1978;17:669–672. doi: 10.1021/bi00597a017. [DOI] [PubMed] [Google Scholar]
- 11.Belleau B, Burba J. The stereochemistry of the enzymic decarboxylation of amino acids. J Am Chem Soc. 1960;82:5751–5752. [Google Scholar]
- 12.Sharma V, Arockiasamy A, Ronning DR, Savva CG, Holzenburg A, Braunstein M, Jacobs WR, Jr, Sacchettini JC. Crystal structure of Mycobacterium tuberculosis SecA, a preprotein translocating ATPase. Proc Natl Acad Sci U S A. 2003;100:2243–2248. doi: 10.1073/pnas.0538077100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sambrook J, Russell DW. Molecular cloning : a laboratory manual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 2001. [Google Scholar]
- 14.Scriven F, Wlasichuk KB, Palcic MM. A continual spectrophotometric assay for amino acid decarboxylases. Anal Biochem. 1988;170:367–371. doi: 10.1016/0003-2697(88)90644-6. [DOI] [PubMed] [Google Scholar]
- 15.Osterman A, Grishin NV, Kinch LN, Phillips MA. Formation of functional cross-species heterodimers of ornithine decarboxylase. Biochemistry. 1994;33:13662–13667. doi: 10.1021/bi00250a016. [DOI] [PubMed] [Google Scholar]
- 16.Brooks HB, Phillips MA. Circular dichroism assay for decarboxylation of optically pure amino acids: application to ornithine decarboxylase. Anal Biochem. 1996;238:191–194. doi: 10.1006/abio.1996.0274. [DOI] [PubMed] [Google Scholar]
- 17.Christenson SD, Wu W, Spies MA, Shen B, Toney MD. Kinetic analysis of the 4-methylideneimidazole-5-one-containing tyrosine aminomutase in enediyne antitumor antibiotic C-1027 biosynthesis. Biochemistry. 2003;42:12708–12718. doi: 10.1021/bi035223r. [DOI] [PubMed] [Google Scholar]
- 18.Molnar-Perl I. Derivatization and chromatographic behavior of the o-phthaldialdehyde amino acid derivatives obtained with various SH-group-containing additives. J Chromatogr A. 2001;913:283–302. doi: 10.1016/s0021-9673(00)01200-0. [DOI] [PubMed] [Google Scholar]
- 19.Tyagi AK, Tabor CW, Tabor H. Ornithine decarboxylase from Saccharomyces cerevisiae. Purification, properties, and regulation of activity. J Biol Chem. 1981;256:12156–12163. [PubMed] [Google Scholar]
- 20.Jackson LK, Goldsmith EJ, Phillips MA. X-ray structure determination of Trypanosoma brucei ornithine decarboxylase bound to D-ornithine and to G418: insights into substrate binding and ODC conformational flexibility. J Biol Chem. 2003;278:22037–22043. doi: 10.1074/jbc.M300188200. [DOI] [PubMed] [Google Scholar]
- 21.Brooks HB, Phillips MA. Characterization of the reaction mechanism for Trypanosoma brucei ornithine decarboxylase by multiwavelength stopped-flow spectroscopy. Biochemistry. 1997;36:15147–15155. doi: 10.1021/bi971652b. [DOI] [PubMed] [Google Scholar]
- 22.Osterman AL, Brooks HB, Jackson L, Abbott JJ, Phillips MA. Lysine-69 plays a key role in catalysis by ornithine decarboxylase through acceleration of the Schiff base formation, decarboxylation, and product release steps. Biochemistry. 1999;38:11814–11826. doi: 10.1021/bi9906221. [DOI] [PubMed] [Google Scholar]
- 23.Swanson T, Brooks HB, Osterman AL, O’Leary MH, Phillips MA. Carbon-13 isotope effect studies of Trypanosoma brucei ornithine decarboxylase. Biochemistry. 1998;37:14943–14947. doi: 10.1021/bi981154i. [DOI] [PubMed] [Google Scholar]
- 24.Jackson LK, Brooks HB, Myers DP, Phillips MA. Ornithine decarboxylase promotes catalysis by binding the carboxylate in a buried pocket containing phenylalanine 397. Biochemistry. 2003;42:2933–2940. doi: 10.1021/bi026795z. [DOI] [PubMed] [Google Scholar]
- 25.Eichler W. Properties of purified L-ornithine decarboxylase (EC 4.1.1.17) from Tetrahymena thermophila. J Protozool. 1989;36:577–582. doi: 10.1111/j.1550-7408.1989.tb01100.x. [DOI] [PubMed] [Google Scholar]
- 26.Kitani T, Fujisawa H. Molecular properties of ornithine decarboxylase from mouse kidney: detailed comparison with those of the enzyme from rat liver. J Biochem. 1988;103:547–553. doi: 10.1093/oxfordjournals.jbchem.a122306. [DOI] [PubMed] [Google Scholar]
- 27.Hu T, Wu D, Chen J, Ding J, Jiang H, Shen X. The catalytic intermediate stabilized by a “down” active site loop for diaminopimelate decarboxylase from Helicobacter pylori. Enzymatic characterization with crystal structure analysis. J Biol Chem. 2008;283:21284–21293. doi: 10.1074/jbc.M801823200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson LK, Brooks HB, Osterman AL, Goldsmith EJ, Phillips MA. Altering the reaction specificity of eukaryotic ornithine decarboxylase. Biochemistry. 2000;39:11247–11257. doi: 10.1021/bi001209s. [DOI] [PubMed] [Google Scholar]