

Abstract
We report the syntheses and activities of a wide range of thiazolides [viz. 2-hydroxyaroyl-N-(thiazol-2-yl)amides] against hepatitis B virus replication, with QSAR analysis of our results. The prototypical thiazolide, nitazoxanide [2-hydroxybenzoyl-N-(5-nitrothiazol-2-yl)amide; NTZ] 1 is a broad spectrum antiinfective agent, effective against anaerobic bacteria, viruses and parasites. By contrast, 2-hydroxybenzoyl-N-(5-chlorothiazol-2-yl)amide 3 is a novel, potent and selective inhibitor of hepatitis B replication (EC50 = 0.33 μm) but is inactive against anaerobes. Several 4′- and 5′-substituted thiazolides show good activity against HBV; by contrast, some related salicyloylanilides show a narrower spectrum of activity. The ADME properties of 3 are similar to 1, viz. the O-acetate is an effective prodrug and the O-aryl glucuronide is a major metabolite. The QSAR study shows a good correlation of observed EC90 s for intracellular virions with thiazolide structural parameters. Finally we discuss the mechanism of action of thiazolides in relation to the present results.
Introduction
Hepatitis B Virus
It is currently estimated that two billion people worldwide have been affected by hepatitis B virus (HBV) and that of these around 360 million are chronically affected.1 The so-called Dane particle, observed in the blood of patients with hepatitis, was identified as the viral agent in 1970.2 The virion is characterized by the surface antigen HBsAg, originally known as the ‘Australia antigen’, and related to HBV by the pioneering research of Blumberg.3 While vaccination is often effective as a preventative measure,4 treatment of patients with chronic HBV requires chemotherapy. Since HBV, like HIV, replicates via reverse transcriptase through an RNA template, and HBV is common in patients who develop AIDS, structurally related nucleoside analogues may have valuable activity against both these viruses: the HBV polymerase and the HIV-1 reverse transcriptase have significant homology.5
Indeed, the earliest chemotherapeutic agents for HBV were viral DNA polymerase inhibitors such as lamivudine, adefovir and entecavir. All these agents were effective, with sub-micromolar anti-HBV IC50 values, but as usual with such agents resistant strains quickly appeared.6-8 HBV displays a particularly fast mutation rate (1010-11 point mutations per day in individuals with active replication)9 and additionally cross-resistance has been noted between chemical agents from different structural groups.
Among other approaches, agents which target the encapsidation step (viz. assembly of viral RNA, polymerase and core into the nucleocapsid prior to replication) have been developed, notably heteroaryldihydropyrimidines10 and phenopropenamides.11 Since the HBsAg is a heavily glycosylated protein, inhibitors of the N-glycosylation pathway have also been studied12 and found effective both in vitro and in an experimental in vivo infection model.13 Finally, other nucleic acid-based approaches have been studied, notably antisense nuceotides14 and gene therapy employing short interfering RNA (siRNA), which has shown promising results in model infection.15 In summarizing new approaches to HBV therapy, including immune modulation, Loomba and Liang16 commented that ‘an improved understanding of virus-host interactions’ was a key need in the development of new therapies. Indeed there is a clear need for effective new small molecule drugs as anti-HBV agents, ideally ones which would act through a mechanism not leading to rapid generation of resistant viral mutants. We now report that a series of thiazolide analogues [2-hydroxyaroyl-N-(thiazol-2-yl)amides] appear to fulfil these requirements.
Thiazolides
The aminothiazole derivative nitazoxanide 1 (Figure 1) was first developed as an antiparasistic agent, particularly indicated against Cryptosporidium parvum17 and has been marketed in the USA since 2002. Indeed 1 is a broad-spectrum antiinfective agent, also active against anaerobic bacteria18 and as an antiprotozoal19 and anthelmintic agent. It was in the late 1990s that the antiviral activity of nitazoxanide was first noted,20 during its use in the treatment of AIDS patients who had developed cryptosporidiosis. In most of its indications, nitazoxanide 1 behaves as a prodrug for the free phenol, tizoxanide 2 which is the effective circulating drug in vivo.
Figure 1.

Basic thiazolide structures 1-3.
There is strong evidence that 1 owes its activity against anaerobic bacteria and parasites to inhibition of the enzyme pyruvate:ferredoxin reductase (PFOR),21 whereby production of acetyl CoA within the anaerobe is prevented. Nevertheless, the nitro group is not directly involved (e. g. by reduction), nor is it essential for broader antiinfective activity. For instance, halothiazolides are also effective against the parasites S. neurona and C. parvum.17(b), 22 We used nitazoxanide as the starting-point for a programme of synthesis and evaluation of a wide range of thiazolide analogues, particularly as selective antiviral agents. We have already presented data showing that nitazoxanide and a few close analogues are effective inhibitors of HBV and in some cases of hepatitis C virus (HCV) replication in cell cultures.23 Further testing has revealed that 1 is active against a range of both DNA and RNA viruses, and it has been evaluated in clinical trials.24
In this paper we present a full account of the synthesis and structure-activity relationships of a wide range of thiazolides, plus some related salicyloyl anilides, against HBV. We also present a quantitative structure-activity relationship (QSAR) study, showing that the activities of thiazolides against HBV demonstrate excellent correlation for intracellular virions. Our results have led to the identification of 2-hydroxybenzoyl-N-(5-chlorothiazol-2-yl)amide 3 as a potent, selective anti-HBV agent. The O-glucuronide of 3, the primary in vivo metabolite of this analogue, has been independently synthesized and evaluated.
Chemistry
Coupling Methods
The synthesis of nitazoxanide 1 and analogues (Tables 1 and 2) without further aryl substitution is generally achieved straightforwardly from commercially available acetylsalicyloyl chloride (Scheme 1, X = H) or a substituted salicyloyl chloride. In the case of nitazoxanide 1 itself,25 anhydrous coupling with 2-amino-5-nitrothiazole (Et3N-THF) is necessary in view of the amine’s low nucleophilicity. More nucleophilic 2-aminothiazoles, e.g. 5-Br and 5-Cl (both commercially available), can be successfully coupled in a two-phase system (CH2Cl2/ aq. NaHCO3); analogues 3-13, Table 1, were made using one or other of these procedures. The fluoro analogue 14 required the prior synthesis of 2-amino-5-fluorothiazole (see below), followed by acylation as above. When using the two-phase acylation method, bis-acylated products of type 15 may also be isolated: their amounts decrease with time, in keeping with their base instability: indeed, the imides have proved more base-labile than the phenolic acetates. The imides are inactive under the assay conditions described.
Table 1.
Activities of 5′-nitro and 5′-halothiazolides against HBV replication.
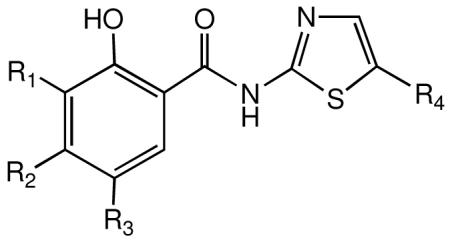
| Compound | R1 | R2 | R3 | R4 | CC50a μM |
EC50b (VIR) μM |
EC50c (RI) μM |
EC90d (VIR) μM |
EC90e (RI)μM |
SIf (VIR) |
SIg (RI) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | H | H | H | No2 | >100 | 0.15 | 0.46 | 0.58 | 1.20 | >172 | >83 |
| 3 | H | H | H | Cl | 38 | 0.33 | 1.00 | 0.89 | 2.90 | 43 | 13 |
| 4 | Me | H | H | No2 | 85 | 0.64 | 2.10 | 3.5 | 12.0 | 24 | 7.1 |
| 5 | H | Me | H | No2 | >100 | 1.30 | 4.20 | 6.90 | 15.0 | >14 | >6.7 |
| 6 | H | H | H | Br | >100 | 1.20 | 2.90 | 4.00 | 8.7 | >25.0 | >12.0 |
| 7 | Me | H | H | Br | >100 | 3.50 | 7.60 | 9.0 | 22.0 | >11 | >4.5 |
| 8 | Cl | H | H | Br | 30.0 | 0.32 | 0.59 | 2.00 | 3.90 | 15 | 7.7 |
| 9 | H | F | H | Br | 12.0 | 2.10 | 5.60 | 2.1 | |||
| 10 | H | H | Cl | Br | 45.0 | 0.21 | 3.50 | 1.10 | 11.0 | 41 | 4.1 |
| 11 | Me | H | H | Cl | 100 | >10.0 | >10.0 | >10.0 | >10.0 | - | - |
| 12 | H | H | Me | Cl | 100 | 0.33 | 0.90 | 0.83 | 2.0 | >120 | >51 |
| 13 | H | Me | H | Cl | 100 | 1.00 | 2.70 | 3.30 | 6.3 | >30 | >16 |
| 14 | H | H | H | F | 51 | 3.1 | - | 12.0 | - | 4.3 | - |
Drug concentration at which a two-fold lower level of neutral red dye uptake is observed relative to average level in untreated cultures.34
Drug concentration required to reduce extracellular (virion) HBV DNA by 50% relative to untreated cells. All EC50 and EC90 figures were determined in triplicate, with standard deviations ±20% of the figure quoted.
Drug concentration required to reduce intracellular HBV DNA by 50%.
Drug concentration required to reduce extracellular (virion) HBV DNA by 90%
Drug concentration required to reduce intracellular HBV DNA by 90%
Selectivity index: CC50/EC90 for extracellular HBV
Selectivity index: CC50/EC90 for intracellular HBV.
Table 2.
Activities of other thiazolides including prodrugs against HBV replication.
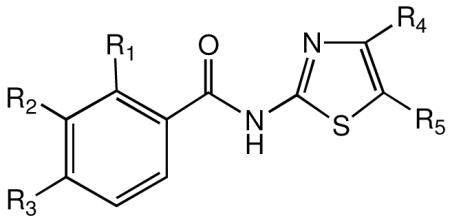
| Compound | R1 | R2 | R3 | R4 | R5 | CC50a μM |
EC50 (VIR) μM |
EC50 (RI) μM |
EC90 (VIR) μM |
EC90 (RI)μM |
SI (VIR) |
SI (RI) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | AcO | H | H | H | NO2 | >100 | 0.12 | 0.59 | 0.83 | 2.10 | >121 | >48 |
| 16 | OH | H | H | H | H | 16.0 | >10.0 | >10.0 | ||||
| 17 | OH | H | H | H | i-Pr | >100 | >10.0 | >10.0 | >10.0 | >10.0 | - | - |
| 18 | OH | H | H | H | C6H4Cl | >100 | >10.0 | >10.0 | >10.0 | >10.0 | - | - |
| 19 | OH | H | H | Ph | H | 22.0 | 5.60 | 21.0 | 1.0 | - | ||
| 20 | OH | Me | H | Ph | H | >100 | 0.22 | 7.80 | 0.73 | 30.0 | >137 | 3.3 |
| 21 | OH | Cl | H | Ph | H | >100 | 0.15 | 0.60 | >166 | |||
| 22 | H | H | OH | H | NO2 | >100 | >10.0 | >10.0 | ||||
| 23 | OH | H | H | H | NHAc | >100 | >10.0 | >10.0 | >10.0 | >10.0 | ||
| 24 | OH | H | H | SO2Me | H | >100 | >10.0 | >10.0 | ||||
| 25 | OH | H | H | H | SO2Me | >100 | >10.0 | >10.0 | >10.0 | |||
| 26 | OH | H | H | H | CN | >100 | 3.2 | 5.5 | 9.4 | 14.0 | >11 | >7.1 |
| 27 | OH | H | H | H | CO2Me | 15.0 | >10.0 | >10.0 | ||||
| 28 | OH | H | H | H | CF3 | >100 | 3.8 | 9.1 | 11.0 | 27.0 | >9.1 | >3.7 |
| 29 | EtO2CO | H | H | H | NO2 | 9.9 | 0.21 | 0.54 | 0.69 | 1.80 | 13 | 5.5 |
Scheme 1.

a General synthetic procedures for thiazolides.
a i) Coupling step: Carboxylic acid reacted with SOCl2/ pyridine or (COCl)2/ DMF, DCM, then either add acid Cl to amine in aq. NaHCO3− organic solvent with stirring, or add to amine in dry THF with Et3N: or BrPyBOP or BOP-Cl, NMM, CH2Cl2; deprotection: aq. HCl, 60°C or aq. NH3.
Similarly compounds 16-22 and 26-28, Table 2, were obtainable by coupling of the appropriate 2-aminothiazole with the appropriate acid chloride; 2-amino-4-phenylthiazole is also commercially available. Analogue 23 (v. i.) was prepared directly from nitazoxanide, and special chemistry was used for the sulfone analogues 24 and 25: see below for these analogues. Finally, the salicyloylanilides 30-37 (Table 3) were made by acylation of the appropriate aniline with acetylsalicyloyl chloride; compounds 35 and 37 were commercially available.
Table 3.
Activities of salicoyl anilides against HBV replication.
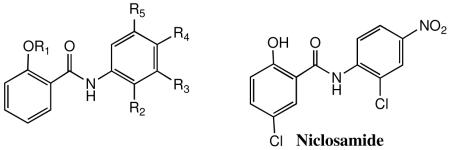
| Compound | R1 | R2 | R3 | R4 | R5 | CC50a μM |
EC50 (VIR) μM |
EC50 (RI) μM |
EC90 (VIR) μM |
EC90 (RI)μM |
SI (VIR) |
SI (RI) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 30 | Ac | H | H | Cl | H | >100 | 3.50 | 9.7 | 11 | |||
| 31 | H | H | H | Cl | H | >100 | >10 | >10 | ||||
| 32 | Ac | H | H | Br | H | >100 | 0.76 | 2.10 | 3.00 | 6.80 | >33 | >15 |
| 33 | Ac | H | H | I | H | >100 | 0.20 | 0.73 | 1.20 | 3.30 | >83 | >30 |
| 34 | H | H | H | NO2 | H | >100 | >10 | >10 | ||||
| 35 | H | Cl | H | H | CF3 | >100 | 3.70 | 13.0 | ||||
| 36 | Ac | H | CF3 | H | CF3 | >100 | 3.80 | 14.0 | >7.1 | |||
| 37 | H | Me | H | NO2 | Cl | 89.0 | 0.28 | 0.66 | 0.73 | 1.7 | 122 | 52 |
| Niclosamide | >100 | >10.0 | >10.0 |
For aryl-substituted analogues where the acid chloride is not commercially available, e. g. 3-methylsalicylic acid, standard acid-chloride forming conditions may be used from the O-acetyl acids [(COCl)2/ cat. DMF or SOCl2/ pyridine]. In general, carbodiimide-based methods lead to very sluggish reactions even when HOBt and/or DMAP are added. However, both BrPyBOP26 and BOP-Cl27 gave reasonable yields with 2-amino-4-phenylthiazole, using NMM as base, though the reactions were slow (3-4 days at 20°C). Both 2-acetoxy-3-methylbenzoic acid (see analogues 19 and 20, Table 2) and 4-acetoxybenzoic acid could be coupled in this way: in the case of 2-acetoxy-3-chlorobenzoic acid, a reasonable yield was obtained using 3eq. of BOP-Cl for an extended period. These conditions are less satisfactory for 2-amino-5-bromo- or 2-amino-5-chlorothiazole, however, with generally slow reactions resulting: in general the rapid acid chloride method is preferred here. Carpino’s reagent HATU28 gives a good yield when coupling 2-amino-5-chlorothiazole with 2-acetoxy-3-methylbenzoic acid, but gives a very sluggish coupling between 2-amino-4-phenylthiazole and the same acid. In the Experimental section we have only described the acid chloride methods in detail.
Other Thiazoles
Earlier results suggested that the nitro group was essential for activity against anaerobes, via inhibition of PFOR (see above), but not for antiviral activity. It was therefore of interest to study the reduction of nitazoxanide 1, Scheme 2. This was achieved using Raney Ni/H2, as reported for 2-acetamido-5-nitrothiazole.29 The free 5-amino compound could not be isolated, but by performing the reaction in Ac2O the diacetate 37 resulted in high yield; mild base-catalysed deprotection afforded the free phenol 23. This compound was antivirally inactive.
Scheme 2.

a Syntheses of 5′-acetamido and 5′-fluorothiazolides
a i) H2-Raney Ni, Ac2O; ii) aq. NH3; iii) SelectFluor, MeCN, heat; iv) aq. NaOH/EtOH.
To prepare the 5′-F analogue (Scheme 2), 2-acetamidothiazole 38 was reacted with excess of SelectFluor30 in MeCN: optimum reaction (ca. 40% conversion to 39) was achieved after about 3h, longer reaction leading to decomposition. Hydrolysis of the acetyl group afforded free amine 40. After we had used this procedure, a report appeared confirming our result.31 These authors, however, found that on a larger scale treatment of Boc-2-aminothiazole with two equivalents of ButLi, followed by addition of N-fluorobenzenesulfonimide, then acidolysis with HCl, was a more reliable route to 40. Coupling of 40 with acetylsalicyloyl chloride followed by standard deprotection gave the 5′-F analogue 14.
Syntheses of the 4′- and 5′-sulfone analogues led to some interesting chemistry, Scheme 3. Heating the 5′-Br intermediate 41 with sodium sulfinate under CuI catalysis in DMF afforded a sulfone product in 37% yield. However, this product proved to be the 4′-sulfone 24 (concomitant deacetylation occurs) and not the 5′- isomer.32 Several variations of this procedure, however, failed to repeat the result: complex mixtures of products resulted. Subsequent rigorous comparison of 24 and the 5′-sulfone 25, prepared unambiguously as described below, using HPLC and 1H/13C NMR studies confirmed that they were indeed isomeric products. Their physical properties are almost identical but there are small characteristic differences in their 13C NMR shifts and HPLC retention times.
Scheme 3.

a Syntheses of 4′-and 5′- methanesulfonylthiazolides
a i) MeSO2Na, CuI, DMF, heat; ii) MeSNa, EtOH; iii) acetylsalicyloyl chloride; iv) 3-ClC6H4CO3H (2 eq.); v) aq. NH3.
A reasonable mechanism for the sulfinate displacement reaction involves addition at C(4′), followed by a hydride shift and loss of Br−. In fact, the 5′-sulfone may be unambiguously prepared by displacement of Br− from 2-amino-5-bromothiazole 42 using MeSNa, Scheme 3, giving 2-amino-5-methylthiothiazole 43. Reaction of 43 with acetylsalicyloyl chloride, followed by peracid oxidation of the resulting 5′-SMe thiazolide 44, then deprotection, afforded 25. Rather surprisingly, both these analogues were inactive against HBV (the 5′-methylthio thiazolide intermediate was also inactive) though as we have reported23 the 4′-sulfone 24 is a potent inhibitor of HCV.
To prepare the 5′-cyano- and 5-methoxycarbonyl analogues 26 and 27, methyl 3-cyano- 45 and methyl 3-methoxyacrylate 46 (Scheme 4) were treated with NBS and the crude bromo products condensed directly with thiourea. After purification, the 2-amino-5- substituted thiazole products 47 and 48 were condensed with acetylsalicyloyl chloride to give 26 and 27 after deacetylation. Recently both 47 and 48 have become commercially available. A similar Hantzsch synthesis via the appropriate α-bromoaldehyde (Scheme 4) was used for the synthesis of the isopropyl and 4-chlorophenyl analogues 17 and 18 via the appropriate thiazoles 49 and 50.
Scheme 4.
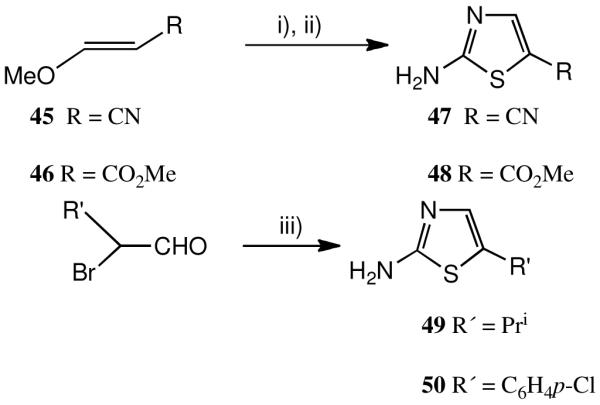
a Syntheses of 5′- aryl, alkyl, cyano and methoxycarbonylthiazolides.
a i) N-bromosuccinimide, MeOH; ii), iii) (H2N)2C=S.
Further prodrugs
Although in general the O-acetyl derivatives serve as efficient prodrugs for the corresponding free phenols, we studied a range of other prodrugs made by acylation of tizoxanide 2. The O-ethyl carbonate 29, Table 2, is given as an example. A variety of other sterically bulkier prodrugs (not shown) were less effective in cell culture; nevertheless their potential utility in animal PK and in vivo studies is being pursued.
Deacetylation
In general, free phenols are used in the biological assays for hepatitis B although the O-acetates are equiactive. The acetates of the precursors may be cleaved using acid or base catalysis: we have generally employed mild heating in aq. HCl, as given in Scheme 1 but aq. ammonia is also effective, as for the sulfone analogue 25. Traditional Zemplen deprotection using catalytic methoxide is rather inefficient, probably because of the highly acidic NH proton.
Finally, the 5-trifluoromethyl analogue 28 was prepared (Scheme 5) from the αβ-unsaturated sulfone 51 which was itself obtained by a known procedure.33 Base-catalysed epoxidation of 51, using MCPBA- K2CO3 in MeCN rather than BunLi-TBHP in THF as described, followed by treatment of the epoxide 52 with thiourea, afforded the desired 2-amino-5-trifluoromethylthiazole 53 in good yield.34 Without further purification, 53 was coupled with acetylsalicyloyl chloride; the intermediate was deacetylated as described above (acid conditions) to afford the desired analogue 28.
Scheme 5.

a Synthesis of 5′-trifluoromethylthiazolide 28.
a) i) MCPBA, K2CO3, CH3CN, RT to 40 0C; ii) NH2C(=S)NH2 (2 equiv.) DMF, 75 0C; iii) Acetylsalicyloyl chloride, Et3N, CH2Cl2; iv) HCl, H2O, THF, 50 0C
Glucuronide Metabolite
Once it was established that the O-aryl glucuronide was a general in vivo metabolite for this class, as already known for tizoxanide itself, it was important to prepare a reference sample of the glucuronide of the lead candidate 3. The protected glucuronide of salicylic acid 54 prepared by us before35 was condensed with 2-amino-5-chlorothiazole using EDCI and DMAP as catalyst, Scheme 6, giving a reasonable yield of the protected glucuronide 55. It is important not to use too strong a base: when Et3N was employed instead of DMAP, significant amounts of the unsaturated glucuronide 56 were obtained. This elimination is well known in the literature, 36 but stronger bases than Et3N have generally been used. Finally, hydrolysis of 55 led to the free glucuronide 57 which was isolated as its sodium salt.
Scheme 6.

a Synthesis of the O-glucuronide of 3.
a i) 2-amino-5-chlorothiazole hydrochloride, NMM, EDCI, DMAP, HOBt, CH2Cl2; ii) NaOH, aq. MeOH, then pH 6.
Results: Antiviral Activity
In general terms, the best activity against HBV was observed for thiazolides with electron-withdrawing groups at C(5′), especially 5′-nitro and 5′-halo, and this data, obtained in Hep G2 (2.2.15 cells),37 is summarized in Table 1. Here the therapeutic (selectivity) index, SI, is shown for each analogue as a ratio of the cytotoxicity (CC50) to efficacy (EC90: the drug concentration at which a 10-fold depression of intracellular HBV DNA was observed relative to the average levels in untreated cultures). Both intracellular HBV replication and extracellular virus production were measured, using the virion (VIR) and replication intermediate (RI) assays, respectively.
Considering first tizoxanide 2 and other nitro analogues, introduction of a methyl group at positions 3, 4 or 5 (not shown) or halogenation at these positions led to 5- to 10-fold loss of activity and selectivity, as typified in compounds 4 and 5. Ortho-substitution as in 4 did not have such a drastic effect as in the antiparasitic screens, where as we have noted38 activity against a Neospora sp. was virtually abolished by a 3-Me group. In the 5′-bromo series, a reasonable level of activity, though at a lower level than the nitro compounds, was observed in compounds 6 to 10. Here again a loss of activity for the 3-Me analogue 7 was noted: the 4- and 5-Me compounds (not shown) were superior but offered no advantage over 6. The 3- and 5-chloro analogues 8 and 10 were rather more potent than 6, but there were concerns over their reduced SI; by contrast, fluorinated analogues typified by 9 were significantly less active and demonstrated low selectivity.
The 5′-chloro analogues 3 and 11-13 showed significantly better activity than the bromo compounds and retained high SI values (as noted above, the direct 5′-Br analogues of 12 and 13 are not shown but were less active). Here the loss of activity in the 3-methyl analogue was more severe: compound 11 was essentially inactive, but both the unsubstituted compound 3 and the 5-methyl analogue 12 exhibited sub-micromolar EC90 values; the 4-methyl compound 13 was less active than 3 or 12. Nominally the activity of 12 was slightly superior to 3, but it later proved to have an unfavourable metabolic/toxicity profile, probably owing to CYP oxidation to a reactive p-quinonemethide: this is discussed in more detail in the next section. The p-cresol structural element is indeed now regarded as a ‘structural alert’.39 The 5′-fluoro analogue 14 was significantly less active than 2, 3 or 6, a trend also noted in the activities of halo-analogues against C. parvum .17(b)
We screened a large number of other thiazolides, and the most interesting of these in structure-activity terms are summarized in Table 2. As stated above, nitazoxanide 1 is a prodrug of tizoxanide 2 and their activities are virtually identical in this assay: this may not be true under the conditions of all antiviral assays, depending on the local availability of esterases in each case. From a range of other possible prodrugs, the ethyl carbonate 29 was the most effective though its SI value appears less than that of 1. The unsubstituted thiazole 1640 showed complete loss of HBV activity, and nonpolar, electronically neutral alkyl and aryl groups at C(5′) were also inactive, compounds 17 and 18.
The 4′-phenyl analogues 19, 20 and 21 were of interest. With no further substitution in ring A, compound 19, a modest level of activity was seen but with a low selectivity index. Introduction of a methyl or chloro group at R2, however, led to a significant improvement in both potency and selectivity as in compounds 20 and 21. It would appear nevertheless that these compounds are significantly less effective against intracellular HBV RNA replication intermediates. Thus the ratio of efficacy EC50(VIR) : EC50(RI) at about 1:35 is much less than that seen with the 5′-chloro analogues, ca. 1:3 (compare compound 3, Table 1, with compound 20, Table 2). Replacement of the 2-hydroxy group by 4-hydroxy as in 22 41 led to complete loss of HBV activity.
A range of other heteroatom and electron-withdrawing C(5′) substituents was also screened, compounds 23 to 28. The cyano and trifluoromethyl analogues 26 and 28 retained slight activity but the 5-methoxycarbonyl analogue 27 was inactive, as were the acetamido analogue 23 and sulfonyl analogues 24 and 25.
Finally, we prepared and screened a set of salicyloyl anilides and the activities of the most interesting, compounds 30-37, are summarized in Table 3. In general the activity profile was more restricted than the thiazolides, and among monosubstituted analogues only the 8-bromo 32 and 8-iodo 33 analogues showed useful activity; here the 8-nitro compound 34 was inactive. Solubility problems are probably responsible for the lack of activity of compound 31 (cf. acetate analog 30, which has low potency) and in these analogues the EC50s of the O-acetates are probably more reliable. Among other analogues, the mono- and bis-trifluoromethyl derivatives 35 and 36 showed moderate activity and the trisubstituted 37 showed good activity. From a practical point of view, however, these molecules are close in structure to niclosamide (Table 3), which is inactive against HBV and has poor aqueous solubility and oral absorption. Additionally the scope for finding new compounds is much less, e. g. the free phenol form of 31 is known.42
In summary, the 5′-chloro analogue 3 was selected for further evaluation in view of its good activity against both extra- and intracellular HBV replication, EC90: EC50 of about 3:1 and its good selectivity index (viz. low cell toxicity).
Drug absorption, distribution, pharmacokinetics and metabolism
Nitazoxanide 1 and the thiazolides generally are rather insoluble compounds and the free phenolic forms are highly protein bound (v. i.). Nevertheless, their log P values (e. g. clogP=1.24 for 1) and other physicochemical parameters, notably H-bond donors (2) and acceptors (7) are favourable for oral absorption according to well-known guidelines.43, 44 Indeed, nitazoxanide is well absorbed from the gastrointestinal tract: 45, 46 when administered after food, the oral bioavailability rises to about 50%. This behaviour contrasts with the poor absorption of the related series of N-salicyloylanilides, typified by the anthelmintic agent niclosamide (Table 3). In clinical trials vs. hepatitis C, nitazoxanide is generally administered as two 500 mg tablets per day: recent clinical trial experience has helped to define the optimum dosing schedule.47 Nitazoxanide, tizoxanide and the newer thiazolides are strongly bound to plasma proteins, typically >95%, but as we have reported23 the consequent raising of EC90 does not prevent an effective therapeutic concentration being achieved.
In ADME/Tox studies, compound 12 was not an inhibitor of 14 different CYP isozymes and showed complete stability in human blood and plasma after a one hour incubation period. However it was found to be >99% protein bound and was extensively metabolized by human liver microsomes and S9 fraction, with only 16% and 6% of the parent compound remaining after a 60 minute incubation period. In rat and dog PK studies, 12 is not orally absorbed. For this reason, compound 3 is the preferred new generation thiazolide as it shows no adverse effects against a panel of CYPs, nor does it show unfavourable drug-drug interactions. Full details of the pharmacokinetic study will be published elsewhere.
Deacetylation of 1 by blood plasma esterases is rapid (t1/2 6 min) and subsequently tizoxanide 2 is extensively metabolized in the liver as its O-arylglucuronide 58,35 Figure 2, which is excreted in both urine and bile. This behaviour is also general for the new thiazolides. We have reported that 58 retains some moderate activity against anaerobic bacteria but it is essentially devoid of antiviral activity: 35 as noted above, the glucuronide 57 of compound 3 has also been independently synthesized as a standard. Pharmacokinetic studies for the new generation thiazolides, including prodrugs, are under way and the results will be reported elsewhere.
Figure 2.
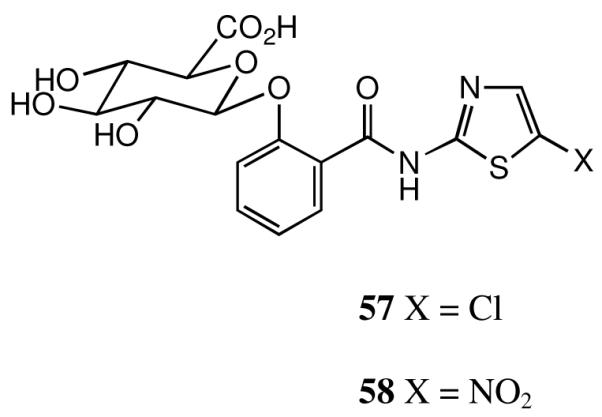
Thiazolide O-glucuronides.
Quantitative Structure Activity Relationships
Correlation of EC90 RI values (viz. inhibition of replication within cells)
Using the data in Tables 1 to 3, quantitative structure activity relationships for the drug concentration required to reduce intracellular HBV DNA by 90%, pEC90 RI, were constructed with the GA-MLR method using autoscaled and filtered subset of the descriptor set generated by the Pipeline Pilot, 48 DRAGON 49 and CDK 50 descriptor generation software as described in the Experimental Methods section.
Recent guidelines recommend that a training set for QSAR model development must contain at least 10 data points/compounds51 and other sources strongly recommend that an external test set should contain a minimum of 5 data points/compounds.52 As the EC90 RI dataset contained 11 compounds there were not a sufficient number data points to split into a training and test set and still have an adequate number for either, therefore internal leave-one-out, leave-many-out and bootstrap internal validations were performed on the constructed models.
The quality of each model was evaluated by calculation of the coefficient of determination (r2), the adjusted coefficient of determination (r2adj), mean absolute error (MAE), standard deviation of regression, F-value, average fold error, leave-one-out r2 (r2LOO), 10-fold leave-many-out r2 (r2LMO) averaged over 1000 runs and bootstrap r2 (r2BS) averaged over 5000 runs. Table 5 shows the contributing terms and performance statistics for pEC90 RI QSAR models generated with the three different fitness functions.
Table 5.
QSAR models and their performance statistics for the pEC90 VIR (M) end point, as determined by GA-MLR using different fitness functions for subjective descriptor selection.
| Subjective Descriptor Selection Fitness Function | |||
|---|---|---|---|
| Model02A r2adj |
Model02B r2adj in LOO validation |
Model02C r2adj in bootstrap validation |
|
| Model activity (EC90 RI) = |
15.27-1.53(BEHe8) −70.97(BEHp7) +0.50(khs.sCl) |
4.37+1.85(X5sol) −15.27(VC-5) |
74.81− 63.96(MATS6m) +31.83(ATSc2) |
|
| |||
| r2 | 0.721 | 0.415 | 0.551 |
| r2adj | 0.666 | 0.361 | 0.511 |
| MAE | 0.185 | 0.288 | 0.231 |
| S.D of regression | 0.273 | 0.376 | 0.329 |
| F-value | 7.762 | 3.540 | 6.153 |
| Av. Fold error | 1.035 | 1.053 | 1.043 |
|
| |||
| r2LOO | 0.443 | 0.125 | 0.281 |
| r2LMO | 0.436 | 0.109 | 0.270 |
| r2BS | −1.200 | −1516.856 | 0.041 |
The QSAR models using r2adj and r2adj in leave-one-out validation as the fitness function yielded the same model as did the best model using average r2 for bootstrap validation as the fitness function. This convergence of the descriptors that were selected by different fitness functions empirically indicates that this model is likely to be the best available with respect to this dataset and set of descriptors. The descriptors within this model are IDDE, the mean information content on the distance degree equality,53 BEHp7, the highest Eigen-value n. 7 of the Burden matrix weighted by atomic polarisabilities BCUT descriptors54 and ATSc3, the Moreau-Broto autocorrelation descriptors using partial charges. 55 Figure 3 shows a graph for predicted pEC90 RI against experimentally determined pEC90 RI for model01A: it can be seen that there are no outliers as the residuals are of the same magnitude for all predicted data points.
Figure 3.

Predicted vs Experimental pEC90 RI as predicted by the QSAR model01A for pEC90 RI.
The performance measures (Table 4) indicate a robust and predictive model with most measures falling within the recommended guidelines.56 In particular r2 is greater than 0.7, mean absolute error is less than 0.1, r2LOO is greater than 0.5 and the r2BS internal validation performance parameter is > 0.5 which indicates a robust model.
Table 4.
QSAR models and their performance statistics for the pEC90 RI (M) end point, as determined by GA-MLR using different fitness functions for subjective descriptor selection.
| Subjective Descriptor Selection Fitness Function | |||
|---|---|---|---|
| Model01A r2adj |
Model01B r2adj in LOO validation |
Model01C r2adj in bootstrap validation |
|
| Model activity (EC90 RI) = |
21.31-1.85(IDDE)− 3.30(BEHp7) −23.63(ATSc3) |
21.31-1.85(IDDE)− 3.30(BEHp7) −23.63(ATSc3) |
21.31-1.85(IDDE)− 3.30(BEHp7) −23.63(ATSc3) |
|
| |||
| r2 | 0.923 | 0.923 | 0.923 |
| r2adj | 0.904 | 0.904 | 0.904 |
| MAE | 0.088 | 0.088 | 0.088 |
| S.D of regression | 0.131 | 0.131 | 0.131 |
| F-value | 28.046 | 28.046 | 28.046 |
| Av. Fold error | 1.013 | 1.013 | 1.013 |
|
| |||
| r2LOO | 0.767 | 0.767 | 0.767 |
| r2LMO | 0.763 | 0.763 | 0.763 |
| r2BS | 0.528 | 0.528 | 0.528 |
Aside from giving an empirical model of pEC90 RI activity these descriptors are not readily physically interpretable, therefore a correlation was sought with other descriptors that are more physically interpretable. Descriptors with > 0.75 correlation with the above three descriptors were identified (Table 2, supplementary information), showing the identity of the descriptors as well as their correlation coefficient. For the BEHp7 descriptor which is negatively correlated with the activity, there are a number of physically interpretable descriptors that are highly positively correlated to it; these are number of atoms, number of bonds, number of non-hydrogen atoms, number of non-hydrogen bonds, number of rotatable bonds, rotatable bond fraction. This indicates a small, rigid molecule is more likely to have activity.
Correlation of EC90 VIR values (viz. inhibition of virions released from the cell)
Quantitative structure activity relationships for the drug concentration required to reduce extracellular HBV DNA by 90%, pEC90 VIR, were constructed with the GA-MLR method using autoscaled and filtered subset of the descriptor set generated by the Pipeline Pilot, DRAGON and CDK descriptor generation software as defined in the Methods section.
In analogy to the EC90 RI dataset the EC90 VIR dataset contained 13 compounds: therefore there were insufficient data points to split into training and test sets and still have an adequate number for either. Hence internal leave-one-out, leave-many-out and bootstrap internal validations was performed on the constructed models. Table 5 shows the contributing terms and performance statistics for pEC90 VIR QSAR models generated with the three different subjective fitness functions. All QSAR models constructed for the pEC90 VIR end point fall short of thresholds recommended for the mean absolute error and internal validation r2 statistics. The performance of these models indicates that a robust model could not be constructed for the pEC90 VIR end point.
In summary, our SAR data were substantiated by a QSAR study based on the activities against intracellular virions which showed an excellent correlation. In the future we will aim to employ the QSAR to inform the molecular design of novel derivatives.
Discussion and mechanism of action
In summary, we have synthesized a range of thiazolides [2-hydroxyaroyl-N-(thiazol-2-yl)amides] together with some related salicyloyl anilides and screened them for inhibition of HBV replication. A number of the thiazolides exhibited sub micromolar EC50 values against both extracellular HBV virions and intracellular replication intermediates; the salicyloyl anilides were generally less potent. In general thiazolides with an electron-withdrawing substituent at C(5′), especially nitro and chloro, were most potent in this assay; substitution in the phenyl ring had less effect, with a 3-methyl group generally causing loss of potency. The introduction of a 3-chloro substituent improved potency at the cost of selectivity. Other electron-withdrawing 5′-substituents were significantly less effective, as were 5′-alkyl or aryl groups. Our results were substantiated by a QSAR study based on the activities against intracellular virions which showed an excellent correlation.
Analogues with a 4′-phenyl group were of interest. Although the activity of the unsubstituted aryl analogue was modest, here the introduction of a 3-methyl or 3-chloro substituent dramatically improved activity, the likely drawback being a significant increase in log P value for this series. Finally a number of salicyloyl anilides, analogues of niclosamide, were screened. In general these compounds were less active, though it is interesting that here again analogues with electron-withdrawing substituents were most effective, particularly the p-iodo compound. The known poor oral absorption of this class of drug, in addition to the reduced scope for novelty, makes them less attractive for development than the thiazolides.
From the analogues described, 2-hydroxybenzoyl-N-(5-chlorothiazol-2-yl)amide 3 was selected for further development: in activity terms, this was very similar to the 5-methyl analogue 12, but the latter proved to have an unfavourable metabolic profile. After scale-up, the pharmacokinetic behaviour of the two chloro analogues, as their acetate pro-drugs, will be determined in both rats and dogs and compared to that of nitazoxanide 1. The results of these studies will be reported in due course.
The mechanism of action of the thiazolides is still under active investigation, but over the last few years some important findings have appeared. The inhibition of PFOR as a mechanism of action against anaerobes alluded to above21 is postulated to be due to mimicry of TPP anion by the anion of nitazoxanide, without involving direct redox reactions of the nitro group. This does not explain, however, why other thiazolides with an acidic NH [viz. in particular, those with strong electron-withdrawing groups at C(5)] apparently lack significant anaerobic activity. A very recent publication57 reported the activity of a number of analogues of 1, including some other heterocycles, against anaerobic bacteria: here again activity was essentially confined to nitro analogues, including a dinitrothiophene amide. Also, it has been shown by affinity chromatography that 1 inhibits the action of a nitroreductase in Giardia lamblia.58
Turning to antiviral activity, studies in rotavirus have shown that nitazoxanide is cytoprotective,24(a) that is, it acts at a post-entry level, as shown specifically in MDCK cells infected with influenza A virus.59 Thus when infected cells were treated with nitazoxanide between 0 and 6 h post-infection, viral replication was effectively inhibited and a single administration remained effective for up to 48h. Pretreatment of cells with 1 prior to infection, however, was not cytoprotective. In the same study, it was shown that 1 effects post-translational modification of haemagglutinin (HA), specifically by inhibition of the maturation of HA glycoprotein at a stage before resistance to endoglycosidase H digestion. However, it must be noted that, in the same studies 1 was not found to be an inhibitor of cellular glycosidation pathways.59
In the case of HBV, it is known that 1 induces reductions in key HBV proteins, especially the surface antigen HBsAg, also HBeAg and HBcAg,23 and in view of the observation with HA it is probably significant that all these proteins are heavily glycosylated. However, there is no effect of 1 on levels of HBV RNA transcription, consistent with a post-translational mechanism. The difference in mechanism is valuable in that 1 has been shown to be synergistic with lamivudine and adefovir against HBV, and indeed it maintains equivalent activity against HBV strains resistant to those agents.23
Recent studies including clinical trial results have shown that 1 is active against different genotypes of the Hepatitis C virus (HCV), and is also synergistic with interferon (with and without ribavirin), telaprevir (an HCV protease inhibitor) and 2′C-methylcytidine (an HCV RNA polymerase inhibitor).47, 60 In the most recent trials, Phase II clinical studies carried out in 511 experienced and naïve patients with chronic hepatitis C Genotype 1 (240 subjects) and 4 (271 subjects) combined with pegylated interferon-alpha 2a (Pegasys ®) with or without ribavirin showed that NTZ 1 added a 40-50% increase in the SVR rate of the standard of care combining pegylated interferon-alpha 2a and ribavirin.61
Here too, equivalent activity is maintained by 1 against representative drug-resistant strains of HCV.60 HCV replicon-containing cell lines resistant to 1 have been isolated, but the resistance phenotype was not found to be transferred to naive cells by HCV replicons isolated from these cell lines, indicating that a cellular mechanism is most likely responsible for this property. 62 In other studies in HCV containing cell lines, 1 was observed to enhance the phosphorylation of eukaryotic initiation factor-2 α (eIF2α).63 In the same report, 1 was found capable of enhancing the activity of PKR in enzymatic assays, a protein kinase activated by double-stranded RNA that is part of innate cell defense mechanisms and activates eIF2α via phosphorylation.63 The weight of evidence therefore supports a mechanism involving stimulation of the host cell immune system. Most recently, it was discovered that in peripheral blood mononuclear cells from human volunteers thiazolides showed potent immuno-modulating effect of both the innate and adaptive immune systems.64
In summary, it may be said that the mechanism of antiinfective action of nitazoxanide and other thiazolides is complex, and most likely involves more than one pathway: the complete elucidation of the mechanism is a work in progress. In the case of antiviral activity, an increasing body of evidence is consistent with a post-translational, host cell-mediated effect which may either stimulate innate cell defence processes or inhibit maturation of key viral proteins. What is beyond doubt is that the thiazolides are highly effective antiviral agents both in vitro and in vivo, as we have demonstrated here for HBV, and in further publications we shall report on their structure-activity properties against other important viruses.
Experimental Section
Chemical Procedures
Organic extracts were washed finally with satd. aq. NaCl and dried over anhydrous Na2SO4 prior to rotary evaporation at <30 °C. Analytical thin-layer chromatography was performed using Merck Kieselgel 60 F 254 silica plates. Preparative column chromatography was performed on Merck 938S silica gel. Unless otherwise stated, 1H and 13C NMR spectra were recorded on CDCl3 solutions using either Bruker 250 or 400 MHz (100 MHz for 13C) instruments with tetramethylsilane as internal standard. Both low- and high-resolution mass spectra were obtained by direct injection of sample solutions into a Micromass LCT mass spectrometer operated in the electrospray mode, +ve or −ve ion as indicated. CI mass spectra (NH3) were obtained on a Fisons Instruments Trio 1000. All compounds tested were analysed by HPLC using an Agilent 1100 system, eluting with a variable percentage of MeCN in water containing 0.1% CF3CO2H and were of at least 97% peak area purity.
Antiviral assays were performed as described previously:37, 65 in summary, confluent cultures of the chronically HBV-producing cell line, 2.2.15, were maintained on 96-well flat-bottomed tissue culture plates (confluence in this culture system is required for active, high levels of HBV replication equivalent to that observed in chronically-infected individuals37, 65). Cultures were treated with nine consecutive daily doses of the test compounds. HBV DNA levels were assessed by quantitative blot hybridization 24 hr. after the last treatment. Cytotoxicity was assessed by uptake of neutral red dye 24 hr. following the last treatment.
General procedures for acid chloride couplings and O-acetate deprotections
Acetylsalicyloyl chloride was used directly; substituted versions were made from the O- acetates as indicated below.
1. Two-phase method
To a stirred solution of a protected salicylic acid (1 mmol) in dry Et2O (10 mL) at 0° C was added pyridine (1.2 mmol) followed by dropwise addition of thionyl chloride (1.2 mmol). Stirring was continued at 0° C for 4 hrs, then the white precipitate formed was filtered off and the filtrate was concentrated under vacuum to give the acid chloride as an oil which was used without further purification. The appropriate 2-aminothiazole (1 mmol) was added to a vigorously stirred two-phase mixture of NaHCO3 (3 mmol for a free base, 4 mmol for an HCl or HBr salt) in H2O (3 mL per mmol) and EtOAc (3 mL per mmol). A solution of the above acid chloride in EtOAc (2 ml per mmol) was then added with vigorous stirring. The reaction mixture was stirred at 20°C for 12 h. The layers were separated and the aqueous layer was extracted once with EtOAc. The combined organic extracts were washed with 0.5 N HCl (2x), followed by brine. The organic layer was dried and concentrated under vacuum to give a pale yellow solid which was chromatographed on silica, eluting with EtOAc-hexane mixtures; in favourable cases trituration of crude product with Et2O followed by drying the resulting solid gave the O-acetyl intermediate directly. This material in conc. was heated in conc. aq. HCl (3ml per mmol) at 50° C for 24 hrs. The reaction mixture was cooled to ambient temp., then filtered and the solid washed with H2O (dist.) until the washings were at neutral pH. The solid was dried under vacuum to give the product. On a small scale it was more efficient to extract the final product into EtOAc, followed by washing with H2O (3x) and brine, then drying and evaporation. Chromatography of the final product, if necessary, was again carried out using EtOAc-hexane mixtures.
2-Hydroxy-3-methylbenzoyl-N-(5-bromothiazol-2-yl) amide (7) (75 mg, 66%)
Mp 187-188°C; Anal. (C11H9BrN2O2S) C, H, N; 1H NMR [400 MHz, (CD3)2SO] 2.21 (3H, s, CH3Ar), 6.86 (1H, d, J = 7.7 Hz, ArH), 7.39 (1 H, d, J = 7.2 Hz, ArH), 7.73 (1 H, s, 4′-H) and 7.95 (1 H, d, J = 7.8 Hz, ArH); 13C NMR [100 MHz(CD3)2SO] 15.6, 101.9, 114.3, 118.8, 126.2, 126.6, 135.7, 136.3, 158.4, 159.9 and 168.8; MS (CI) m/z 313 and 315 (M+ for 79Br, 81Br respectively); HRMS, found, m/z 312.9641, C11H10BrN2O2S (MH+ for 79Br) requires m/z, 312.9646.
2. Anhydrous conditions
Either the free amine was used directly, or the HBr or HCl salt of the appropriate 2-aminothiazole was partitioned between dil. aq. NaOH and EtOAc, then the organic layer was separated, dried and evaporated to dryness. A solution of the thiazole (1 mmol) in THF (3 mL) was added to a stirred solution of salicyloyl chloride (1 eq.) in THF (3 mL per mmol) at 0°C before the addition of Et3N (1 eq.). The solution was stirred at room temperature until reaction was complete, then the reaction mixture was poured into water and extracted with ethyl acetate (2 x). The organic layer was washed with 1M HCl, water, dried and evaporated. The product was purified by column chromatography to give the intermediate O-acetate, which was hydrolysed as above to deliver the product.
2-Hydroxybenzoyl-N-(5-chlorothiazol-2-yl) amide (3)
Mp 227-228°C (dec.); 1H NMR [500 MHz, (CD3)2SO] 7.00 (1 H, t, ArH), 7.04 (1 H, d, ArH), 7.48 (1 H, t, 4-H), 7.60 (1 H, s, 4′-H) and 7.96 (1 H, d, 6-H); 13C NMR [125 MHz, (CD3)2SO] 116.5, 117.1, 118.5, 119.7, 130.3, 134.6, 135.4, 155.8, 157.3 and 164.7; m/z (ES +ve ion mode) 277 (MNa+, 100%); Found: m/z, 276.9806; C10H735ClN2O2SNa requires m/z, 276.9809.
3-Chloro-2-hydroxybenzoyl-N-(5-bromothiazol-2-yl)amide (8) (0.116g, 48%)
Mp 200°C. Found: m/z, 332.90930. C10H7BrClN2O2S (MH+) requires m/z, 332.91003. 1H NMR [400 MHz, (CD3)2SO] 7.02 (1 H, t, J = 7.9 Hz, ArH), 7.69 (1 H, dd, J = 7.9 and 1.4 Hz, ArH), 7.84 (1 H, s, 4′-H) and 8.02 (1 H, dd, J = 7.9 and 1.4 Hz, ArH); m/z (CI, NH3) 333 (MH+, 35%).
2-Hydroxy-5-methylbenzoyl-N-(5-chlorothiazol-2-yl) amide (12)
Mp 212-213°C; Anal. (C11H9ClN2O2S) C, H, N; 1H NMR [(CD3)2SO] 2.27 (3 H, s, CH3Ar), 6.94 (1 H, d, J = 8.3 Hz, ArH), 7.29 (1 H, d, J = 8.1 Hz, ArH), 7.59 (1 H, s, 4′-H) and 7.78 (1 H, s, ArH); 13C NMR [(CD3)2SO] 19.9, 99.1, 115.7, 117.0, 118.5, 128.5, 130.1, 135.3, 135.7, 155.0 and 164.1; MS (CI) m/z 269, 271 (MH+ for 35Cl, 37Cl respectively); HRMS, found, m/z 269.0149, C11H10ClN2O2S (MH+ for 35Cl) requires m/z, 269.0151.
2-Hydroxy-3-methylbenzoyl-N-(4-phenylthiazol-2-yl)amide (20)
Mp 180°C; 1H NMR (400 MHz, CDCl3) 7.94 (1H, d, J = 8.1, ArH), 7.83 (2H, d, J = 8.5, 2 × ArH), 7.52-7.41 (4H, m, ArH), 7.2 (1H, s, ArH), 6.69 (1H, t, J = 7.7, ArH), 2.31 (3H, s, CH3); 13C NMR [125 MHz, (CDCl3] 15.8, 108.4, 112.0, 118.7, 123.2, 126.0, 128.2, 128.8, 134.1, 136.4, 150.2, 157.2, 160.4 and 167.7; m/z (CI) 311 (35%, [M+H]+); (ES +ve ion mode) 333 (MNa+, 100%); Found: m/z, 333.0659; C17H14N2O2SNa requires m/z, 333.0668.
See Supporting Information for the characterisation of all other analogues made using either of the above methods. The thiazole precursors of compounds 17 and 18 are known.66, 67 Acetate deprotection, basic conditions. Mild base hydrolysis using aq. ammonia is also used for deacetylation, as in the following example.
2-Hydroxybenzoyl-N-(5-acetamidothiazol-2-yl)amide (23)
To a solution of nitazoxanide 1 (615 mg, 2.0 mmol) in acetic anhydride (~70 cm3), Raney Ni was added with vigorous stirring. The reaction mixture was evacuated and twice refilled with hydrogen, then allowed to stir at room temperature under the atmosphere of hydrogen until the theoretical volume (~135 cm3, 6.0 mmol) was consumed after 30 minutes. The reaction mixture was stirred for a few minutes under atmosphere to allow the escape of hydrogen present in flask. The solution was filtered through a sinter followed by the evaporation of solvent in vacuo. The crude product was re-dissolved in EtOAc (50 cm3) and washed with excess of satd. aq. NaHCO3. The organic fraction was again evaporated and the crude product was dissolved in acetone (10 cm3) and stirred for two hours with 1N HCl (5 cm3) to hydrolyse over-acetylated material. The reaction was neutralized with satd. aq. NaHCO3 and extracted with EtOAc (4 × 20 cm3). The combined organic fractions were evaporated and the O-acetate of 23 was purified through flash column chromatography as white amorphous solid (548 mg, 86%), mp 176.5°C, 1H NMR (400 MHz, CDCl3) 2.08 (3H, s, CH3), 2.25 (3H, s, CH3), 7.16 (1H, s, CH), 7.26 (1H, dd, J 0.6, 8.1 Hz, ArH), 7.39 (1H, td, J 1.0, 7.6 Hz, ArH), 7.60 (1H, td, J 1.6, 8.0 Hz, ArH), 7.75 (1H, dd, J 1.6, 7.6 Hz, ArH), 11.0 (1H, s, NH); 13C NMR (100 MHz, CDCl3) 21.1, 22.6, 123.7, 126.1, 127.3, 130.0, 131.0, 132.7, 148.8, 166.8, 169.1, 169.4, 184.5, 201.8; m/z EI [M + Na]+ 342 [(M + Na)+, 100%]; Found: m/z, 342.0524; C14H13N3O4NaS requires 342.0538. To a solution of this O-acetate (319 mg, 1.0 mmol) in acetone (5 cm3), 20 cm3 of aq. NH3 was added, then the mixture was stirred overnight at room temperature, followed by evaporation. The crude mixture was dissolved in EtOAc (20 mL) and was washed with 1M HCl. The desired product 23 was purified through flash column chromatography as a white solid (0.25g, 90%); 1H NMR (400 MHz, CDCl3) 2.10 (3H, s, CH3), 6.98 (1H, t, J 7.0 Hz, ArH), 7.03 (1H, d, J 8.0 Hz, ArH), 7.18 (1H, s, ArH), 7.45 (1H, td, J 1.75, 7.0 Hz, ArH), 8.01 (1H, dd, J 1.75, 8.0 Hz, ArH); 13C NMR (100 MHz, CDCl3) 22.7, 117.3, 117.5, 119.8, 130.5, 130.7, 134.5, 158.2, 167.0; m/z CI [M + H]+ 278, [278, 100%]; Found: m/z, 278.05983; C12H12N3O3S requires 278.05994.
Ethyl 2-[(5-nitro-1,3-thiazol-2-yl)carbamoyl]phenyl carbonate (29)
A solution of ethyl chloroformate (0.114 g, 1.11 mmol) in dry THF (3 mL) was added to a bright yellow solution of tizoxanide 2 (0.250 g, 0.942 mmol) and Et3N (0.118 g, 1.16 mmol) in dry THF (6 mL), yielding a white precipitate. The reaction was stirred at 20°C for 6 hours, then concentrated in vacuo and the residue was partitioned between water and dichloromethane. The organic layer was washed with water (2x), then with brine, dried and concentrated to give crude 14 (0.343 g, >100%) as a yellow solid. The crude product was purified by chromatography using a gradient of 0- 50% ethyl acetate in hexane. Appropriate fractions were combined and evaporated to give 29 (0.254 g, 80%) as a tan solid, mp 159-161 °C; HPLC area purity 100% (conditions C). 1H NMR [400 MHz, (CD3)2SO] δ 1.25 (t, J = 7 Hz, 3H), 4.22 (q, J = 7 Hz, 2H), 7.43 (dd, J = 1, 8 Hz, 1H), 7.48 (td, J = 1, 8 Hz, 1H), 7.71 (td, J = 2, 8 Hz, 1H), 7.88 (dd, J = 2, 8 Hz, 1H), 8.71 (s, 1H) and 13.69 (br s, 1H); MS (ES +ve mode) m/z 360.0 (M+Na)+, 338.0 (M+H)+ and (ES −ve mode) m/z 336.1 (M-H)−.
Methyl 1-[2-N-(5-Chlorothiazol-2-yl)carboxamido]phenyl-2,3,4-tri-O-acetyl-β-D-glucopyranuronate (55)
Methyl 1-(2-carboxyphenyl)-2,3,4-tri-O-acetyl-β-D-glucopyranuronate 5435 (0.45 g, 1 mmol) was dissolved with 2-amino-5-chlorothiazole, hydrochloride (0.17 g, 1 mmol), DMAP (0.12 g, 1 mmol) and anhydrous 1-hydroxybenzotriazole (0.14 g, 1 mmol) in anhydrous CH2Cl2 (5 mL). The clear solution was stirred and cooled to 0°C, then N-methylmorpholine (0.11 mL, 1 mmol) and EDCI (0.19 g, 1 mmol) were added. The pale yellow solution was stored at 0°C for 72 h, then diluted with EtOAc (30 mL) and washed with 5% aq. citric acid (25 mL), backwashing with EtOAc, then the combined organic phases were washed with satd. aq. NaHCO3, water and brine, dried and evaporated to crude product which was purified by chromatography, eluting first with a gradient of 30-100% EtOAc in hexane, then with 10% MeOH-CHCl3. Appropriate fractions were combined and evaporated to give reasonably pure product as a pale yellow solid (0.31g, 54%) which was recrystallised from CH2Cl2-EtOH-hexane to give 55 as off-white crystals (0.169 g, homogeneous by TLC); concentration gave a second crop (0.032g) also of good purity; mp 245-246.5°C dec.; 1H NMR [500 MHz, (CD3)2SO] 1.91, 1.95, 2.03 (9 H, 3s, 3×CH3CO), 3.66 (3 H, s, CH3O), 4.77 (1 H, d, J = 10.0 Hz, 5′-H), 5.00-5.10 (2 H, m) and 5.48 (1 H, m, 2′-H + 3′-H + 4′-H), 5.66 (1 H, d, J = 7.9 Hz, 1′-H), 7.18-7.25 (2 H, m, ArH), 7.50-7.60 (2 H, m, ArH), 7.58 (1 H, s, thiazole 4-H) and 12.6 (1 H, br s, NH); 13C NMR [125 MHz, (CD3)2SO] 20.2, 20.3 (x2), 52.6, 68.9, 69.9, 70.9 (x2), 97.7, 115.4, 118.2, 122.9, 124.8, 128.9, 132.3, 135.7, 153.4, 155.7, 164.8, 167.0, 168.6 and 169.3(x2); m/z (ES+ve mode) 593 (MNa+, 100%); Found: m/z, 593.0603. C23H23ClN2O11S Na requires m/z, 593.0603.
Any traces of elimination by-product 56 were distinguished by: 1H NMR [400 MHz, CDCl3] 2.02, 2.13 (6 H, 2s, 2×CH3CO), 3.76 (3 H, s, CH3O), 5.40 (1 H, m), 5.44 (1 H, m), 6.10 (1 H, brs), 6.42 (1 H, m), 7.26 (1 H, s, thiazole 4-H), 7.32, 7.46, 7.62 and 8.13 (4 H, 4m, ArH). See also reference 33.
1-[2-N-(5-Chlorothiazol-2-yl )carboxamido]phenyl-β-D-glucopyranosiduronic Acid (57)
The ester 55 (0.159 g, 0.28 mmol) suspended in MeOH (1.5 mL) was stirred at 0°C with 2.5 M NaOH (0.56 mL, added dropwise). After 2 h, allowing the reaction to regain ambient temperature, reaction appeared complete by TLC and glacial AcOH was added dropwise to achieve a pH of 5.6, then EtOH (4 mL) was added and the mixture cooled to 0°C. The resulting light beige solid was filtered, washed with ether and dried to give substantially pure product (0.113 g, 90%); analytical material was obtained by chromatography on Lichroprep (Merck), eluting with a gradient of 0-60% MeCN in H2O. Appropriate fractions were combined and evaporated to give off-white solid which was triturated with a few drops of moist EtOH and excess ether to afford the product, which was filtered, washed with ether and dried to give highly pure 57 (0.066g) as Na salt; 1H NMR [400 MHz, (CD3)2SO + D2O] 3.25-3.40 (3 H, m, 2′-H + 3′-H + 4′-H), 3.65 (1 H, m, 5′-H), 5.15 (1 H, m, 1′-H), 7.22 (1 H, m, ArH), 7.41 (1 h, m, ArH), 7.59 (1 H, s, thiazole 4′-H), 7.61 (1 H, m, Ar H) and 7.84 (1 H, m, ArH); 13C NMR[125 MHz, (CD3)2SO + D2O] 71.7, 73.3, 74.4, 76.1, 101.4, 116.6, 118.5, 121.6, 122.7, 130.8, 134.0, 135.9, 155.3, 155.6, 163.7 and 171.1; m/z (ES +ve mode) 452 (100%, M+ for Na salt); HPLC analytical purity 98.4% at 280 nm (C18 reverse-phase column, MeCN-H2O); m/z (ES +ve mode) 475 (100%); Found: m/z, 474.9945. C16H14ClN2O8SNa2 requires m/z, 474.9949.
Quantitative Structure Activity Relationship Methods
In order to assist in analysing and interpreting the structure-activity relationships (SAR) associated with the 5′-nitro and 5′-halothiazolide compounds quantitative structure activity relationship (QSAR) models were developed for the some of the biological endpoints/activities against HBV replication.
QSAR models were developed and validated for data concerning the drug concentration required to reduce intracellular HBV DNA by 90% (EC90 RI) and the drug concentration required to reduce extracellular HBV DNA by 90% (EC90 VIR). These data were chosen for their amenability towards modelling in terms of range of values and distribution of data (vide infra). The EC90 RI dataset contains 11 compounds and the EC90 VIR dataset contains 13 compounds (see Tables 4 and 5). The range for the range the EC90 RI is from 1.20 to 22.00 M whilst the EC90 VIR data spans from 0.58 to 12.0 M; both datasets are spread relatively evenly. The biological end points for these assays are reported in M; for the development of QSAR models, however, the activities were converted to pEC90 values in M (pEC90 = −log10(EC90/106). These end-points offer the greatest range of activity of the biological end points for datasets containing low-micromolar or sub-micromolar activities. Given the above characteristics these datasets provide were subject to QSAR analysis.
In total 685 0, 1 and 2-dimensional molecular descriptors/properties were calculated for the set of compounds using Pipeline Pilot Student Edition,48 DRAGON Web Version 3.0 49 and CDK.50
The combined descriptor set of 685 variables was autoscaled and filtered using two objective selection methods. Firstly descriptors that had the same value for 80% of the dataset were removed as these contained minimal information: this left 554 and 557 descriptors for the EC90 RI and EC90 VIR datasets respectively. Secondly the CHORCHOP procedure68 (a), (b) was used to eliminate one of a pair of descriptors that exhibited very high inter-correlation (r > 0.99). The procedure removed the descriptor whose distribution deviated the most from normal (as defined by maximum kurtosis 100). This left 37 descriptors for the EC90 RI dataset and 46 descriptors for the EC90 VIR dataset.
The multiple linear regression machine learning method coupled with genetic algorithm subjective descriptor selection (GA-MLR) as implemented in the PHAKISO program was used to relate the activities (Y) of a set compounds to their molecular descriptors (X) using a linear equation.69, 70
The genetic algorithm was set to have population size 50, replacement rate 0.6, cross-over rate 1.0 and maximum number of generations 100. The maximum number of descriptors allowed was set to 3, in order to follow the recommended 5:1 to ratio of number of descriptors to molecule so minimise the occurrence of chance correlations. The subjective fitness function for descriptor selection in this case was chosen to be either the highest adjusted r2 for the training set, leave-one-out validation or bootstrap validation (averaged of 1000 repeats each validation). For the bootstrap method a number of descriptor selection runs were performed and the best model of the multiple runs selected. QSAR models were developed using all three adjusted R2 criteria as a subjective fitness function with the model that gave the best performance for training set and internal validation tests was selected for further analysis if minimum performance threshold are met.
Supplementary Material
Acknowledgments
We are grateful to Romark Laboratories for funding this work, at the University of Liverpool, UK (2003-2007) and the University of Oxford, UK (from 2009). HBV analyses were supported by NIAID contract NO1-AI-30046 to GUMC. We also thank K. Gaye (GUMC) for technical assistance with the HBV analyses and Kalexsyn Inc. Kalamazoo, Michigan, for the synthesis of certain analogues.
Abbreviations used
- BOP-Cl
bis(2-oxo-3-oxazolidinyl)phosphonic chloride
- GA-MLR
genetic algorithm and multiple linear regression
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate
- HBsAg
hepatitis B surface antigen
- NMM
N-methylmorpholine
- NTZ
nitazoxanide [2-hydroxybenzoyl-N-(5-nitrothiazol-2-yl)amide]
- PFOR
pyruvate ferredoxin reductase
- PyBroP®
bromotripyrrolidinophosphonium hexafluorophosphate
- QSAR
quantitative structure-activity relationship(s)
- SelectFluor®
1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis (tetrafluoroborate)
Footnotes
Supporting Information Available: Spectroscopic data for compounds 3-6, 9-13, 18-21, 26-27, 31, 32, 33 and 35-37 along with C, H and N analyses for selected examples. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- (1).Chen DS. Hepatitis B vaccination: the key towards elimination and eradication of hepatitis B. J. Hepatol. 2009;50:805–816. doi: 10.1016/j.jhep.2009.01.002. [DOI] [PubMed] [Google Scholar]
- (2) (a).Dane DS, Cameron CH, Briggs M. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet. 1970:695–698. doi: 10.1016/s0140-6736(70)90926-8. [DOI] [PubMed] [Google Scholar]; (b) Summers J, O’Connell A, Millman I. Genome of hepatitis B virus: restriction enzyme cleavage and structure of DNA extracted from Dane particles. Proc. Nat. Acad. Sci. USA. 1975;72:4597–4601. doi: 10.1073/pnas.72.11.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Blumberg BS. Australia Antigen and the Biology of Hepatitis B. Science. 1977;197:17–25. doi: 10.1126/science.325649. [DOI] [PubMed] [Google Scholar]
- (4) (a).Poland GA. Evaluating existing recommendations for hepatitis A and B vaccination. Am. J. Med. 2005;118(10A):16S–20S. doi: 10.1016/j.amjmed.2005.07.029. [DOI] [PubMed] [Google Scholar]; (b) Ni YH, Chang MH, Huang LM, Chen HL, Hsu HY, Chiu TY, Tsai KS, Chen DS. Hepatitis B virus infection in children and adolescents in a hyperendemic area: 15 years after mass hepatitis B vaccination. Ann. Intern. Med. 2001;135:796–800. doi: 10.7326/0003-4819-135-9-200111060-00009. [DOI] [PubMed] [Google Scholar]
- (5).Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5Å resolution of HIV reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- (6) (a).Doong S-L, Tsai C-H, Schinazi R-F, Liotta DC, Cheng Y-C. Inhibition of the replication of hepatitis B virus in vitro by 2′, 3′-dideoxy-3′-thiacytidine and related analogues. Proc. Nat. Acad. Sci. USA. 1991;88:8495–8499. doi: 10.1073/pnas.88.19.8495. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lok AS, Lai CL, Leung N, Yao GB, Cui ZY, Schiff ER, Fienstag JL, Heathcote EJ, Little NR, Griffiths DA, Gardner SD, Castiglia M. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology. 2003;125:1714–1722. doi: 10.1053/j.gastro.2003.09.033. [DOI] [PubMed] [Google Scholar]
- (7).Yang H, Westland CE, Delaney WET, Heathcote EJ, Ho V, Fry J, Brosgart C, Gibbs CS, Miller MD, Xiong S. Resistance surveillance in chronic hepatitis B patients treated with adefovir dipivoxil for up to 60 weeks. Hepatology. 2002;36:464–473. doi: 10.1053/jhep.2002.34740. [DOI] [PubMed] [Google Scholar]
- (8).Warner N, Locarnini SA, Colledge D, Edwards R, Angus P, et al. Molecular modeling of entecavir resistant mutations in the hepatitis B virus polymerase selected during therapy. Hepatology. 2003;40:245A. [Google Scholar]
- (9).Ghany M, Liang TJ. Drug targets and molecular mechanisms of drug resistance in chronic hepatitis B. Gastroenterology. 2007;132:1574–1585. doi: 10.1053/j.gastro.2007.02.039. [DOI] [PubMed] [Google Scholar]
- (10).Weber O, Schlemmer KH, Hartmann E, Hagelschuer I, Paessens A, Graef E, Deres K, Goldman S, Niewoehner U, Stoltefuss J, Haebich D, Ruebsamen-Waigmann H, Wohlfeil S. Inhibition of human hepatitis B virus (HBV) by a novel non-nucleosidic compoundin a transgenic mouse model. Antiviral Res. 2002;54:69–78. doi: 10.1016/s0166-3542(01)00216-9. [DOI] [PubMed] [Google Scholar]
- (11).King RW, Ladner SK, Miler TJ, Zaifert K, Perni RB, Conway SC, Otto MJ. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (−)β-L-2′,3′-dideoxy-3′-thiacytidine. Antimicrob. Agents Chemother. 1998;42:3179–3186. doi: 10.1128/aac.42.12.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12) (a).Dwek RA, Butters TD, Platt FM, Zitzmann N. Targeting glycosidation as a therapeutic approach. Nature Rev. Drug Discovery. 2002;1:65–75. doi: 10.1038/nrd708. [DOI] [PubMed] [Google Scholar]; (b) Mehta A, Zitzmann N, Rudd PM, Block TM, Dwek RA. Alpha-glucosidase inhibitors as potential broad based anti-viral agents. FEBS Lett. 1998;430:17–22. doi: 10.1016/s0014-5793(98)00525-0. [DOI] [PubMed] [Google Scholar]
- (13).Block TM, Lu X, Mehta AS, Blumberg BS, Tennant B, Ebling M, Korba BE, Lansky DM, Jacob GS, Dwek RA. Treatment of chronic hepadnavirus infection in a woodchuck animal model with an inhibitor of protein folding and trafficking. Nature Med. 1998;4:610–614. doi: 10.1038/nm0598-610. [DOI] [PubMed] [Google Scholar]
- (14).Korba BE, Gerin JL. Antisense oligonucleotides are effective inhibitors of hepatitis b virus replication in vitro. Antivir. Res. 1995;28:225–242. doi: 10.1016/0166-3542(95)00050-v. [DOI] [PubMed] [Google Scholar]
- (15).Wu HL, Huang LR, Huang CC, Lai HL, Liu CJ, Huang YT, Hsu YW, Lu CY, Chen DS, Chen PJ. RNA interference-mediated control of hepatitis B virus and emergence of resistant mutant. Gastroenterology. 2005;128:708–716. doi: 10.1053/j.gastro.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Loomba R, Liang TJ. Novel approaches to new therapies for hepatitis B viral infection. Antiviral Therapy. 2006;11:1–15. [PubMed] [Google Scholar]
- (17) (a).Fox LM, Saravolatz LD. Nitazoxanide: a new thiazolide antiparasitic agent. Clin. Infect. Dis. 2005;40:1173–1180. doi: 10.1086/428839. [DOI] [PubMed] [Google Scholar]; (b) Gargala G, Le Goff L, Ballet JJ, Favennec L, Rossignol J-F, Stachulski AV. Evaluation of new thiazolide/thiadiazolide derivatives reveals nitro group-independent efficacy against in vitro development of Cryptosporidium parvum. Antimicrob. Agents Chemother. 2010;54:1315–1318. doi: 10.1128/AAC.00614-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dubrueil L, Houcke I, Mouton Y, Rossignol J-F. In vitro evaluation of activities of nitazoxanide and tizoxanide against anaerobes and aerobic organisms. Antimicrob. Agents Chemother. 1996;40:2266–2270. doi: 10.1128/aac.40.10.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19) (a).Rossignol J-F, Maisonneuve H. Nitazoxanide in the treatment of Taenia saginata and Hymenolepis nana. Am. J. Trop. Med. Hyg. 1984;33:511–512. doi: 10.4269/ajtmh.1984.33.511. [DOI] [PubMed] [Google Scholar]; (b) Doumbo O, Rossignol J-F, Pichard E, Traore H, Dembele M, Diakite M, Traore F, Diallo DA. Nitazoxanide in the treatment of cryptosporidial diahorrea and other intestinal parasitic infections associated with acquired immunodeficiency syndrome in tropical Africa. Am. J. Trop. Med. Hyg. 1997;56:637–639. doi: 10.4269/ajtmh.1997.56.637. [DOI] [PubMed] [Google Scholar]
- (20).Rossignol J-F. Nitazoxanide in the treatment of acquired immune deficiency syndrome-related cryptosporidiosis: results of the United States compassionate program in 365 patients. Alimentary Pharmacology and Therapeutics. 2006;24:887–894. doi: 10.1111/j.1365-2036.2006.03033.x. [DOI] [PubMed] [Google Scholar]
- (21).Hoffmann PS, Sisson G, Croxen MA, Welch K, Harman WD, Cremades N, Morash MG. Antiparasitic drug nitazoxanide inhibits the pyruvate oxidoreductase of Helicobacter pylori and selected anaerobic bacteria and parasites, and Campylobacter jejuni. Antimicrob. Agents Chemother. 2007;51:868–876. doi: 10.1128/AAC.01159-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gargala G, Le Goff L, Ballet JJ, Favennec L, Stachulski AV, Rossignol J-F. In vitro efficacy of nitro-and halogeno-thiazolide/thiadiazolide derivatives against Sarcocystis neurona. Vet. Parasitol. 2009;162:230–235. doi: 10.1016/j.vetpar.2009.03.022. [DOI] [PubMed] [Google Scholar]
- (23).Korba BE, Montero AB, Farrar K, Gaye K, Mukerjee S, Ayers MS, Rossignol J-F. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antivir. Res. 2008;77:56–63. doi: 10.1016/j.antiviral.2007.08.005. [DOI] [PubMed] [Google Scholar]
- (24) (a).Rossignol J-F, Abu-Zekry M, Hussein A, Santoro MG. Effect of nitazoxanide for treatment of severe rotavirus diahorrea: randomized double-blind placebo-controlled trial. Lancet. 2006;368:124–129. doi: 10.1016/S0140-6736(06)68852-1. [DOI] [PubMed] [Google Scholar]; (b) Rossignol J-F, El-Gohary Y. Nitazoxanide in the treatment of viral gastroenteritis: a randomized double-blind, placebo-controlled trial. Aliment. Pharmacol. Ther. 2006;24:1423–1430. doi: 10.1111/j.1365-2036.2006.03128.x. [DOI] [PubMed] [Google Scholar]
- (25).Rossignol J-F, Cavier R. Chem. Abs. 1975;83:28216. [Google Scholar]
- (26).Coste J, Frérot E, Jouin P. Coupling N-methylated amino acids using PyBroP and PyCloP halogenophosphonium salts: mechanism and fields of application. J. Org. Chem. 1994;59:2437–2446. [Google Scholar]
- (27).Tung RD, Rich DH. Bis(2-oxo-3-oxazolidinyl)phosphinic chloride as a coupling reagent for N-alkylamino acids. J. Am. Chem. Soc. 1985;107:4342–4343. [Google Scholar]
- (28).Carpino LA. 1-Hydroxy-7-azabenzotriazole-an effective peptide coupling additive. J. Am. Chem. Soc. 1993;115:4397–4398. [Google Scholar]
- (29).Parent RA. 2,4-Dinitrothiazole.The boron trifluoride-nitrogen tetroxide nitration of 2-nitrothiazole. J. Org. Chem. 1962;27:2282–2283. [Google Scholar]
- (30).Banks RE, Besheesh MK, Mohialdin-Khaffaf SN, Sharif I. N-Halogeno compounds. Part 18. 1-Alkyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane salts: user-friendly site-selective electrophilic fluorinating agents of the N-fluoroammonium class. J. Chem. Soc., Perkin Trans. 1. 1996:2069–2076. [Google Scholar]
- (31).Briner PH, Fyfe MCT, Martin P, Murray PJ, Naud F, Procter MJ. Practical synthesis of 2-amino-5-fluorothiazole hydrochloride. Org. Process Res. Dev. 2006;10:346–348. [Google Scholar]
- (32).Our results should be compared with: Dahlbom R, Ekstrand T. Sulfides and sulfones containing an aminothiazole group. Svensk Kemisk Tidskrift. 1945;57:229–234., where it was claimed that a similar displacement of an aminothiazole-5-halide by an arylsulfinate Na salt gave the 5-sulfone.
- (33).Yamazaki T, Ishikawa N. Building-blocks for trifluoromethylated organic molecules. 2. (E)-3,3,3-Trifluoro-1-propenyl phenyl sulfoxide- a useful building block for trifluoromethylated organic molecules. Chem. Lett. 1985:889. [Google Scholar]
- (34).Laduron F, Janousek Z, Viehe HG. α- or β-Trifluoromethyl epoxysulfones: new C3 reagents for heterocyclisation. J. Fluorine Chem. 1995;73:83–86. [Google Scholar]
- (35).Stachulski AV, Rossignol J-F. Syntheses and Antibacterial Activities of Tizoxanide, an N-(Nitrothiazolyl)salicylamide, and its O-Aryl Glucuronide. J. Chem. Res. 1999:44–45. [Google Scholar]
- (36).For instance, Stanford D, Stachulski AV. Convenient syntheses of deoxypyranose sugars from glucuronolactone. Tetrahedron Lett. 2007;48:2361–2364. and refs. therein.
- (37).Korba BE, Gerin JL. Use of a standardized cell culture assay to determine activities of nucleoside analogues against hepatitis B virus replication. Antivir. Res. 1992;19:55–70. doi: 10.1016/0166-3542(92)90056-b. [DOI] [PubMed] [Google Scholar]
- (38).Esposito M, Stettler R, Moores SL, Pidathala C, Muller N, Berry NG, Stachulski AV, Rossignol J-F, Hemphill A. In vitro efficacy of nitazoxanide and other thiazolides against Neospora caninum tachyzoites reveals anti-parasitic activity independent of the nitro group. Antimicrob. Agents Chemother. 2005;49:3715. doi: 10.1128/AAC.49.9.3715-3723.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Baillie TA. Metabolism and toxicity of drugs. Two decades of progress in industrial drug metabolism. Chem. Res. Toxicol. 2008;21:129–137. doi: 10.1021/tx7002273. [DOI] [PubMed] [Google Scholar]
- (40) (a).Matyk J, Waisser K, Drazkova K, Kunes J, Klimesova V, Palat K, jr., Kaustova J. Heterocyclic isosteres of antimycobacterial salicylamides. Farmaco. 2005;60:399–408. doi: 10.1016/j.farmac.2005.02.002. [DOI] [PubMed] [Google Scholar]; (b) Luo Q-L, Li J-Y, Liu Z-Y, Chen L-L, Li J, Ye Q-Z, Nan F-J. Inhibitors of type I MetAPs containing pyridine-2-carboxylic acid thiazol-2-yl amide. Part I: SAR studies on the determination of the key scaffold. Bioorg. Med. Chem. Lett. 2005;15:635–638. doi: 10.1016/j.bmcl.2004.11.034. [DOI] [PubMed] [Google Scholar]
- (41).Dymicky M, Huhtanen CN, Wassreman AE. Inhibition of clostridium botulinum by 5-nitrothiazoles. Antimicrob. Agents Chemother. 1977;12:353–356. doi: 10.1128/aac.12.3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Waisser K, Hladuvkova J, Kunes J, Kubicova L, Klimesova V, Karajannis P, Kaustova J. Synthesis and antimycobacterial activity of salicylanilides substituted in position 5. Chem. Papers. 2001;55:121–129. [Google Scholar]
- (43).Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- (44).Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- (45).Rossignol J-F. Thiazolides: a new class of antiviral drugs. Expert Opin. Drug Metab. Toxicol. 2009;5(6):667–674. doi: 10.1517/17425250902988487. [DOI] [PubMed] [Google Scholar]
- (46) (a).Stockis A, De Bruyn S, Gengler C, Rosillon D. Nitazoxanide pharmacokinetics and tolerability in man during 7 days dosing with 0.5 g and 1 g b.i.d. Int. J. Clin. Pharmacol. Ther. 2002;40:221–227. doi: 10.5414/cpp40221. [DOI] [PubMed] [Google Scholar]; (b) Broekhuysen J, Stockis A, Lins RL, De Graeve J, Rossignol J-F. Nitazoxanide: pharmacokinetics and metabolism in man. Int. J. Clin. Pharmacol. Ther. 2000;38:387–94. doi: 10.5414/cpp38387. [DOI] [PubMed] [Google Scholar]
- (47).Rossignol J-F, Elfert A, El-Gohary Y, Keeffe EB. Improved virologic response in chronic hepatitis C genotype 4 treated with nitazoxanide, peginterferon, and ribavirin. Gastroenterology. 2009;136:856–862. doi: 10.1053/j.gastro.2008.11.037. [DOI] [PubMed] [Google Scholar]
- (48). http://accelerys.com/products/scitegic/
- (49).Todeschini R, Mauri A, Pavan M. DRAGON (Web Version 3.0) 2003.
- (50).Steinbeck C, Hoppe C, Kuhn S, Floris M, Guha R, Willighagen EL. Recent Developments of the Chemistry Development Kit (CDK, version 0.94) - An Open-Source Java Library for Chemo- and Bioinformatics. Curr. Pharm. Design. 2006;12:2111–2120. doi: 10.2174/138161206777585274. [DOI] [PubMed] [Google Scholar]
- (51).Durham SK, Pearl GM. Computational methods to predict drug safety liabilities. Curr. Opin. Drug Discovery Dev. 2001;4:102–109. [PubMed] [Google Scholar]
- (52).Eriksson L, Jaworska J, Worth AP, Cronin MTD, McDowell RM. Methods for reliability and uncertainty assessment and for applicability evaluation of classification and regression-based QSARs. Environment Health Perspectives. 2003;111:1361–1375. doi: 10.1289/ehp.5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Bonchev D. Information Theoretic Indices for Characterization of Chemical Structures. RSP-Wiley; Chichester (UK): 1983. [Google Scholar]
- (54).Benigni R, Passerini L, Pino A, Giuliani A. The information content of the eigenvalues from modified adjacency matrices: large scale and small-scale correlations. Quant. Struct. -Act. Relat. 1999;15:449–455. [Google Scholar]
- (55).Todeschini R, Consonni V. Handbook of Molecular Descriptors. Wiley-VCH; Weinheim: 2000. [Google Scholar]
- (56).Guidance document on the validation of (quantitative) structure-activity relationships [(Q)SAR] models, qecd. Volume 69. Paris: 2007. [Google Scholar]
- (57).Ballard TE, Wang X, Olekhnovich I, Koerner T, Seymour C, Hoffman PS, Macdonald TL. Biological activity of modified and exchanged 2-amino-5-nitrothiaole amide analogues of nitazoxanide. Bioorg. Med. Chem. Lett. 2010;20:3537–3539. doi: 10.1016/j.bmcl.2010.04.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Muller J, Wastling J, Sanderson S, Muller N, Hemphill A. A novel Giardia lamblia nitroreductase, G1NR1, interacts with nitazoxanide and other thiazolides. Antimicrob. Agents Chemother. 2007;51:1979–1986. doi: 10.1128/AAC.01548-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Rossignol J-F, La Frazia S, Chiappa L, Ciucci A, Santoro MG. Thiazolides, a new class of anti-influenza molecules targeting viral haemagglutinin at the post-translational level. J. Biol. Chem. 2009;284:29798–29808. doi: 10.1074/jbc.M109.029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Korba BE, Elazar M, Lui P, Glenn JS, Rossignol J-F. Potential role for nitazoxanide in combination with STAT-C agents for the inhibition of HCV replication without the development of resistance. Hepatology. 2008;48(suppl):356A. [Google Scholar]
- (61).Latest results, to be published elsewhere; cf. Rossignol J-F, Elfert A, Keeffe EB. Treatment of chronic hepatitis C using a 4-week lead-in with nitazoxanide before peginterferon plus nitazoxanide. J. Clin. Gastroenerol. 2010;44:504–509. doi: 10.1097/MCG.0b013e3181bf9b15.
- (62).Korba BE, Elazar M, Lui P, Rossignol J-F, Glenn JS. Studies of the Potential for Nitazoxanide or Tizoxanide Resistance in Hepatitis C Virus Replicon-Containing Cell Lines. Antimicrob. Agents Chemother. 2008;52:4069–4071. doi: 10.1128/AAC.00078-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Elazar M, Liu M, McKenna SA, Liu P, Gehrig EA, Puglisi JD, Rossignol J-F, Glenn JS. The anti-hepatitis C agent nitazoxanide induces phosphorylation of eukaryotic initiation factor 2 alpha via protein kinase activated by double-stranded RNA activation. Gastroenterology. 2009;137:1827–1835. doi: 10.1053/j.gastro.2009.07.056. [DOI] [PubMed] [Google Scholar]
- (64).Trabattoni D, Pacei M, Biasin M, De Luca C, Clerici M, Rossignol J-F. Antimicrobial nitazoxanide shows strong immunomodulating effects. Abstracts of the 98th. Annual Meeting of the American Association of Immunologists; San Francisco, California. May 13th-17th, 2011. [Google Scholar]
- (65).Sells MA, Zelent AZ, Shvartsman M, Acs G. Replicative intermediates of hepatitis B virus in Hep G2 cells that produce infectious virions. J Virol. 1988;62:2836–2844. doi: 10.1128/jvi.62.8.2836-2844.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Helal CJ, Sanner MA, Cooper CB, Gant T, Adam M, Lucas JC, Kang Z, Kupchinsky S, Ahlijanian MK, Tate B, Menniti FS, Kelly K, Peterson M. Discovery and SAR of 2-aminothiazole inhibitors of cyclin-dependent kinase 5/p25 as a potential treatment for Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2004;14:5521–5525. doi: 10.1016/j.bmcl.2004.09.006. [DOI] [PubMed] [Google Scholar]
- (67).Bal’on Ya. G., Shul’man MD, Kuznetsov NV. New reactions of aryldichloromethylcarbinols. Zhur. Org. Chem. 1979;15:2351–2356. [Google Scholar]
- (68) (a).Livingstone DJ, Rahr E. Chorchop – an Interactive Routine for the Dimension Reduction of Large QSAR Data Sets. Quant. Struct-Act. Relat. 1989;8:103–108. [Google Scholar]; (b) Leach AR, Gillet VJ. Introduction to Chemoinformatics. Springer; Netherlands: 2007. [Google Scholar]
- (69).Draper NR, Smith H. Applied Regression Analysis. John Wiley and Sons; NY, USA: 1991. http://www.phakiso.com. [Google Scholar]
- (70).Eriksson L, Jaworska J, Worth AP, Cronin MTD, McDowell RM. Methods for reliability and uncertainty assessment and for applicability evaluation of classification and regression-based QSARs. Environment Health Perspectives. 2003;111:1361–1375. doi: 10.1289/ehp.5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.