

SUMMARY
Autophagy, a catabolic pathway that delivers cellular components to lysosomes for degradation, can be activated by stressful conditions such as nutrient starvation and endoplasmic reticulum (ER) stress. We report that thapsigargin, an ER stressor widely used to induce autophagy, in fact blocks autophagy. Thapsigargin does not affect autophagosome formation but leads to accumulation of mature autophagosomes by blocking autophagosome fusion with the endocytic system. Strikingly, thapsigargin has no effect on endocytosis-mediated degradation of epidermal growth factor receptor. Molecularly, while both Rab7 and Vps16 are essential regulatory components for endocytic fusion with lysosomes, we found that Rab7 but not Vps16 is required for complete autophagy flux, and that thapsigargin blocks recruitment of Rab7 to autophagosomes. Therefore, autophagosomal-lysosomal fusion must be governed by a distinct molecular mechanism compared to general endocytic fusion.
INTRODUCTION
Macroautophagy, herein referred to as autophagy, is a conserved catabolic process in eukaryotic cells. Autophagy is critical for physiological processes such as embryonic development and establishment of self tolerance in the immune system (Nedjic et al., 2008; Tsukamoto et al., 2008). Autophagy is impaired in many human diseases including cancer, Parkinson’s and Crohn’s (Mizushima et al., 2008). Autophagy occurs when a small membrane cistern, called the isolation membrane or phagophore, grows and surrounds a portion of the cytosol. The isolation membrane eventually seals itself to form a double-membrane vesicular structure termed an autophagosome. The autophagosome then fuses with the endocytic system to deliver its contents to the lysosome for degradation (Mizushima, 2007; Yang and Klionsky, 2009).
Although membrane fusion is required at multiple stages within the autophagic pathway, the underlying mechanisms are not well defined. Work in yeast suggests that the initial stages of isolation membrane growth require a novel mechanism to allow membranes to fuse, independent of SNARE proteins that drive conventional membrane fusion (Ishihara et al., 2001; Nakatogawa et al., 2007). Once the yeast autophagosome has formed, fusion with the vacuole is thought to proceed in an essentially fashion to that of endocytic fusion; requiring SNARE proteins, the Rab GTPase Ypt7 and the class C/HOPS tethering complex, all of which have known roles in the endocytic pathway (Darsow et al., 1997; Fischer von Mollard and Stevens, 1999; Harding et al., 1995; Ishihara et al., 2001; Kirisako et al., 1999; Sato et al., 2000). Likewise, SNARE proteins, Rab7 and the HOPS complex have been implicated in mammalian autophagy (Fader et al., 2009; Furuta et al., 2010; Gutierrez et al., 2004; Jager et al., 2004; Kimura et al., 2007; Liang et al., 2008; Pankiv et al., 2010).
Conditions of stress strongly stimulate autophagy, with nutrient starvation the most understood form of autophagy induction. Nutrient starvation leads to mTOR kinase inactivation and concomitant activation of the autophagy-essential Atg1/ULK kinase complex (Chan, 2009; Chang and Neufeld, 2009; Ganley et al., 2009; Hosokawa et al., 2009; Kamada et al., 2000). Under normal growth conditions, when autophagic flux is low, active mTOR negatively regulates autophagy by direct, inhibitory phosphorylation of the ULK kinase complex. Endoplasmic reticulum (ER) stress has also been shown to be a strong inducer of autophagy, potentially by a mechanism independent of mTOR (Bernales et al., 2006; Ding et al., 2007; Hoyer-Hansen et al., 2007; Kawakami et al., 2009; Kim et al., 2010; Kouroku et al., 2007; Ogata et al., 2006; Sakaki et al., 2008; Yorimitsu et al., 2006). One method of inducing ER stress is to use thapsigargin, an inhibitor of SERCA (the sarco/endoplasmic reticulum Ca2+ ATPase) (Thastrup et al., 1990). Here we describe the effect of thapsigargin on autophagy. We found that thapsigargin specifically blocked fusion of autophagosomes with lysosomes, while leaving the endocytic system itself functional. We found that while both Rab7 and the HOPS complex component Vps16 are essential for endocytic fusion with lysosomes, only Rab7 is required for complete autophagic flux. Further, recruitment of Rab7 to autophagosomes can be blocked by thapsigargin. Therefore, autophagosomes employ a distinct molecular mechanism of fusion on their route to lysosomes when compared to other endocytic compartments.
RESULTS
Thapsigargin blocks autophagy
Recent reports indicated that thapsigargin, an inhibitor of the ER SERCA calcium pump, and tunicamycin, an inhibitor of N-acytlglucosamine phosphotransferase, induce the ER stress response and autophagy (Grotemeier et al., 2010; Ogata et al., 2006; Sakaki et al., 2008). To confirm this result we analyzed autophagosome formation in mouse embryonic fibroblasts (MEFs) treated with thapsigargin, tunicamycin, or amino acid starvation (Figure 1). In MEFs stably expressing GFP-LC3, all treatments resulted in an increase in GFP-LC3 puncta formation (Figure 1A, top panels), suggesting induction of autophagy. Thapsigargin treatment produced GFP-LC3 structures larger in size, yet fewer in number, compared with those induced by amino acid starvation (Figures 1E, 2C and 2D). Autophagy is a dynamic process, with autophagosome formation being balanced by turnover upon delivery to lysosomes. Inhibition of lysosome-mediated degradation, for example by addition of bafilomycin A1 to inhibit the vacuolar H+-ATPase, should block LC3 degradation and hence increase GFP-LC3 staining. This effect is evident in starvation-treated and tunicamycin-treated cells, confirming that both treatments can induce a complete autophagic flux (Figure 1A). Bafilomycin also causes a small but significant increase in GFP-LC3 puncta in cells incubated in complete growth media, reflecting the basal level of autophagy. Surprisingly, we did not see any increase in GFP-LC3 staining upon addition of bafilomycin to thapsigargin-treated cells. This raised the possibility of thapsigargin blocking basal autophagy after autophagosome formation but before lysosomal degradation, thus leading to GFP-LC3 puncta accumulation.
Figure 1. Thapsigargin induces autophagosome accumulation in an ATG5 and phosphatidylinositol 3-kinase activity dependent manner.

(A) MEF cells stably expressing GFP-LC3 (green) were incubated in complete media (control), amino acid-free media for 2 h (starvation), complete media with 3 μM thapsigargin for 5 h, or complete media with 20 μg/ml tunicamycin for 5 h; each in the presence or absence of 10 nM bafilomycin A1 for the final two hours of treatment. Cells were stained with DAPI (blue). Bar, 10 μm. Quantitation shown on the right represents mean GFP puncta per cell (n=20) from two independent experiments ± SD. (B) Immunoblots of MEF lysates treated as in (A). (C) Immunoblots of wild type (WT) and ATG5 null MEF lysates, treated as indicated. (D) ATG5 null MEFs, transduced with GFP-ATG5 retrovirus, were treated as indicated, each in the presence or absence of 50 nM wortmannin. Cells were stained with DAPI (blue), anti-GFP (green) and endogenous LC3 (red). Asterisks represent cells that do not express GFP-ATG5, note the lack of LC3 puncta in these cells. Bar, 10 μm. (E) GFP-LC3 expressing MEFs were incubated in amino acid-free media for 1 h (starve), amino acid-free media for 1 h followed by addition of 50 nM wortmannin for 1 h (starve → wort), complete media with 3 μM thapsigargin for 4 h (TG), complete media with 3 μM thapsigargin for 4 h followed by addition of 50 nM wortmannin for 1 h (TG → wort), or complete media with 3 μM thapsigargin and 50 nM wortmannin together for 4 h (TG + wort). Bar, 10 μm. See also Figure S1.
Figure 2. Thapsigargin blocks both basal and starvation-induced autophagy.

(A) MEF cells were incubated in complete media (control), amino acid-free media for 1 h (starve), complete media with 3 μM thapsigargin for 5 h (TG), or complete media with 3 μM thapsigargin for 4 h followed by washing and incubation in amino acid-free media with 3 μM thapsigargin for 1 h (TG + starve); each in the presence or absence of 10 nM bafilomycin A1 for the final hour. (B) Quantitation of immunoblot data in (A) showing autophagic flux (change in LC3-II levels upon bafilomycin treatment) and relative bafilomycin-induced LC3-II levels. Values represent means normalized to tubulin from 5 independent experiments ± SEM. (C) GFP-LC3 expressing MEFs were treated as in (A). For rescue samples, cells were cultured in fresh complete media for an additional hour before fixation. Bar, 10 μm. (D) Quantitation of data presented in (C) shows mean number of GFP-LC3 puncta per cell ± SEM (n=50 cells per condition). (E) MEF cells were treated with 3 μM thapsigargin (TG) and/or amino acid-free media (starve) and the increase in degradation of long-lived proteins was measured. Values represent the mean percentage of protein loss per hour ± SD as compared to bafilomycin treated samples from 3 independent experiments.
We next analyzed levels of endogenous LC3 by western blot (Figure 1B). Consistently, amino acid starvation and tunicamycin treatment caused an increase in LC3-II levels that was further enhanced by bafilomycin. In contrast, even though thapsigargin alone caused a strong increase in LC3-II levels, no further enhancement was observed upon addition of bafilomycin, confirming that thapsigargin is indeed blocking autophagic flux. This block is not due to ER stress per se, because tunicamycin, which does increase autophagic flux, induced a similar level of the ER stress marker CHOP (C/EBP homologous protein) as thapsigargin (Figure 1B, middle panel). Indeed, MEF cells deficient in ER stress response, due to genetic deletion of both isoforms of inositol requiring enzyme 1 (IRE-1α and β) (Wiseman et al., 2010), still show a thapsigargin-induced block in autophagic flux (Figure S1). Importantly, we confirmed the activity of thapsigargin in blocking autophagic flux on several different cell types in addition to MEFs (Figure 4 and Figure S3).
Figure 4. Thapsigargin does not block initial stages of autophagosome formation.

(A) ATG5 null MEFs stably expressing GFP-ATG5K130R were incubated in complete media (control), amino acid-free media for 1 h (starve), or 3 μM thapsigargin for 4 h in the presence of DMSO or 50 nM wortmannin. Samples were stained with anti-GFP (green) and DAPI (blue). Bar, 10 μm. (B) U2OS cells expressing GFP-LC3 were incubated in complete media (control), amino acid-free media for 2 h (starve), or 3 μM thapsigargin for 5 h, followed by staining with DAPI (blue), anti-p62 (red) and GFP (green). Bar, 10 μm. Panels on the right are high magnifications of the boxed regions; arrows indicate co-localization between LC3 and p62, arrowheads indicate p62-only puncta and double arrowhead indicates LC3-only puncta. (C) Immunoblots of U2OS cell lysates, treated as above. (D) Quantitation of p62 levels shown in (C). Values represent relative levels ± SD, normalized to actin from three independent experiments. See also Figure S3.
To test whether thapsigargin-induced LC3 conversion and puncta accumulation require the conventional autophagy machinery, we examined ATG5 null MEFs (Figure 1C). In the absence of ATG5, an essential component for autophagy (Mizushima et al., 2001), neither starvation nor thapsigargin enhanced lipidation of LC3 (Figure 1C). We next transduced ATG5 null MEFs with GFP-ATG5 to see if thapsigargin treatment resulted in GFP-ATG5 puncta formation, a marker for the isolation membrane (Mizushima et al., 2001). Figure 1D shows that upon amino acid starvation, GFP-ATG5 translocated from cytosol to distinct foci that partially co-localized with an increase in LC3 puncta, indicating isolation membrane formation and maturation into autophagosomes. Consistently, this whole process was drastically reduced by wortmannin, an inhibitor of Vps34, a lipid kinase essential for autophagy induction (Blommaart et al., 1997; see Figure 1D). However, upon thapsigargin treatment, although LC3 was strongly relocalized, there was no corresponding increase in GFP-ATG5 foci, similar to control cells with only basal levels of autophagy. Again, wortmannin treatment drastically reduced LC3 puncta caused by thapsigargin treatment; and in cells lacking ATG5 expression (marked by asterisks in Figure 1D), LC3 puncta failed to form. Thus, thapsigargin induced LC3-II accumulation requires a functional autophagic pathway. Wortmannin is sufficient to block thapsigargin induced LC3 puncta accumulation if added at the start of treatment; however, once LC3 puncta have formed, wortmannin cannot reduce them in the presence of thapsigargin (Figure 1E). This is different from starvation induced LC3 puncta, for which later addition of wortmannin results in an overall reduction of LC3 puncta, because wortmannin blocks new LC3 puncta generation while preexisting ones undergo lysosomal degradation (Figure 1E). This reinforced the possibility that thapsigargin does not induce autophagy, but blocks completion of the basal autophagic flux. If thapsigargin can block basal autophagic flux and thus cause LC3-II accumulation, then it is possible that it might also block the flux of induced autophagy. Indeed, when cells were subjected to starvation in combination with thapsigargin, addition of bafilomycin caused no further increase in LC3-II levels, indicating that thapsigargin blocks autophagic flux even upon starvation-induced autophagy (Figure 2A and B).
Blockage of autophagic flux by thapsigargin appears to be irreversible (Figure 2C and D). In MEFs expressing GFP-LC3, both amino acid starvation and thapsigargin treatment led to an increase in GFP-LC3 puncta, with thapsigargin-induced puncta larger in size and fewer in number. Starvation with thapsigargin also lead to an increase in GFP-LC3 puncta with approximately twice as many as with thapsigargin alone, but only half that of starvation alone. Following treatment, these cells were washed and re-incubated in normal growth media for one hour (see “rescue” samples in Figure 2C and D). For starvation, this was sufficient to stop autophagy induction and allow clearance of the autophagosomes already produced. However, a rescue following thapsigargin treatment had little effect on GFP-LC3 puncta, suggesting an irreversible blockage by thapsigargin. This irreversible blockage might be due to thapsigargin irreversibly inhibiting the SERCA pump.
As an alternative, non-LC3 based autophagy assay, we examined the effect of thapsigargin on degradation of long-lived proteins in untreated or amino acid starved cells (Figure 2E). An enhanced rate of autophagy should result in increased protein degradation, even of proteins that are normally long-lived and stable. This is clearly seen under starvation-induced autophagy, which results in a two-fold increase in long-lived protein degradation. However, although thapsigargin induced LC3-II accumulation, it did not stimulate long-lived protein degradation as starvation did. Further, thapsigargin inhibited starvation-stimulated degradation of long-lived proteins. Taken together, these results demonstrate that thapsigargin blocks the flux of both basal and stimulated autophagy.
Thapsigargin blocks fusion of autophagosomes with lysosomes
LC3 positive structure accumulation upon thapsigargin treatment suggests that fusion of autophagosomes with lysosomes might be blocked. To determine whether thapsigargin prevented autophagosomes from reaching lysosomes, we used a tandem tagged GFP-mCherry-LC3 probe. This probe can dissect whether an autophagosome has fused with a lysosome, based on the distinct chemical properties of GFP and mCherry fluorophores (Kimura et al., 2007). Under non-lysosomal and near-neutral pH conditions, both GFP and mCherry fluoresce. However, the low pH in the lumen of the lysosome quenches the GFP signal but not the mCherry. Using this approach, we found that upon amino acid starvation GFP and mCherry positive autophagosomes rapidly formed after 40 minutes of treatment. Figure 3A and Movie S1 shows cells that have been under starvation for 120 minutes and multiple autophagosomes are already visible. Once puncta form, it takes approximately 30 minutes for them to lose the GFP signal, while retaining the mCherry signal, indicating fusion with lysosomes. This agrees with previously published lifetimes of autophagosomes (Jahreiss et al., 2008; Kochl et al., 2006). With thapsigargin, GFP and mCherry positive structures start to appear and accumulate after approximately two hours of treatment (Movie S2 and Figure 3B). Unlike starvation, these structures do not lose the GFP signal, even two hours after their formation, indicating no lysosomal fusion (it was not possible to image and follow individual structures longer than two hours due to photo-bleaching and phototoxicity). Importantly, thapsigargin treatment also blocked the starvation-induced loss of the autophagosomal GFP signal (Movie S3 and Figure 3C). The live-cell microscopy data is supported by immunoblot experiments using different doses of thapsigargin to treat MEF cells for different times, showing a bafilomycin-insensitive accumulation of LC3-II after as little as two hours of 0.3 μM thapsigargin treatment (Figure S2).
Figure 3. Thapsigargin blocks fusion of autophagosomes with lysosomes.
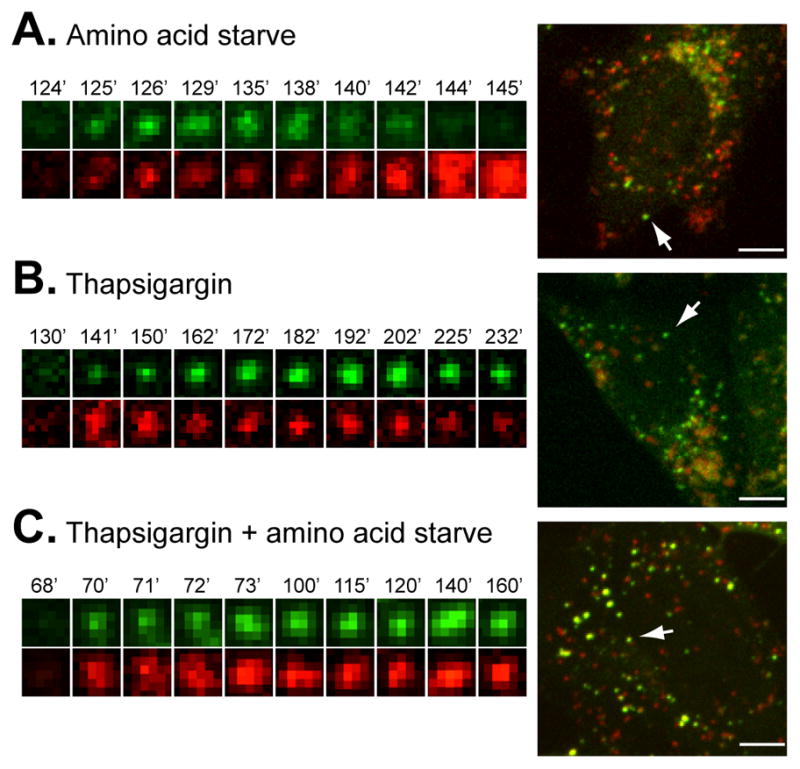
MEF cells stably expressing GFP-mCherry-LC3 were subject to live cell microscopy. Small panels on the left show the life span of an individual LC3 structure, as manifested by its GFP (green, top) and mCherry (red, bottom) fluorescence. The same structure is indicated by an arrow in the whole cell pictured on the right. In (A) times represent minutes post amino acid starvation (see also Movie S1); in (B) times represent minutes post addition of thapsigargin to 3 μM (see also Movie S2); and in (C) times show minutes post amino acid starvation in combination with thapsigargin treatment (see also Movie S3). Bar, 10 μm. See also Figure S2.
Thapsigargin does not affect autophagosome maturation
Since thapsigargin ultimately blocks autophagosome-lysosome fusion, the next question is at what stage does the blockage occur? There are three obvious steps where a block might occur: 1) at the induction stage, prior to isolation membrane formation; 2) at the maturation stage, when the isolation membrane converts to autophagosome; or 3) when mature autophagosomes fuse with the endocytic system and lysosomes. Given that LC3 is conjugated and forms visible punctate structures within the cell, it is unlikely that thapsigargin blocks at a stage prior to autophagy induction.
We further confirmed that thapsigargin does not block initiation of autophagy. Under basal autophagy conditions, the isolation membrane is difficult to detect, perhaps due to small size, low number and/or rapid conversion into mature autophagosomes. The isolation membrane can however be forced to persist by replacing ATG5 with a mutant (ATG5K130R) that is incapable of conjugation to ATG12 (Mizushima et al., 2001). We stably expressed GFP-ATG5K130R in ATG5 null MEFs. Under normal growth conditions isolation membranes were visible (Figure 4A), and thapsigargin did not block their formation. However, addition of wortmannin did reduce GFP-ATG5K130R puncta formation under all conditions. This suggests that thapsigargin does not block isolation membrane formation and that the isolation membranes formed in GFP-ATG5K130R-expressing cells are dynamic and dependent on Vps34 kinase activity, as are their wild-type counterparts.
Concurrent with autophagosome maturation, cargo incorporation occurs. Incorporation of one specific cargo, polyubiquitinated protein aggregates, is mediated by the adaptor protein p62 (Kirkin et al., 2009). This process can be monitored by analyzing p62 and LC3 co-localization. In U2OS cells, which show a similar thapsigargin-induced block of autophagy to MEF cells (Figure 4B and C), p62 and GFP-LC3 are predominantly diffuse under normal growth conditions (Figure 4B, left panels). GFP-LC3 punctate structures are occasionally observed, which co-localize with p62 (See Figure 4B, enlarged panels). This likely represents the basal level of autophagy. Upon starvation, there is a large increase in p62 and GFP-LC3 puncta, many of which co-localize, indicating incorporation of specific p62-labeled cargo into autophagosomes (Figure 4B, middle panels). Addition of thapsigargin also leads to p62 and GFP-LC3 puncta accumulation with extensive co-localization (Figure 4B, right panels). These data indicate that thapsigargin does not block the cargo incorporation process associated with autophagosomal maturation. Incorporation of p62 into autophagosomes results in its degradation upon autophagosome-lysosome fusion, demonstrated by a bafilomycin-sensitive reduction in p62 levels upon starvation-induced autophagy (Figure 4C and D). In contrast, even though thapsigargin treatment results in accumulation of p62 on autophagosomes, there is no bafilomycin-sensitive reduction in p62 levels (Figure 4C and D), again implying a block in the late stage of autophagy.
Subsequently, we examined the morphology of the arrested LC3 structures. High resolution time-lapse confocal microscopy in MEF cells expressing GFP-LC3 and an ER-marker, dsRedER (Figure 5A, B and Movies S4 and S5) showed that upon starvation, GFP-LC3 puncta form and expand to form ring-like structures. Following this, they lose signal intensity until they are no longer visible, probably due to fusion with lysosomes. This process takes just under 60 minutes. The underlying ER structure changes and closely matches the forming autophagosome, in a fashion very similar to that reported previously (Axe et al., 2008), indicating that the ER is involved in autophagosome formation. When samples were treated with thapsigargin and then starved, we noticed that the forming autophagosomes also closely associated with the ER and expanded into ring-like structures (Figure 5B). However, treatment with thapsigargin blocked their disappearance, again highlighting the block in autophagic flux. Three-dimensional analysis of these ring-like structures revealed them to be spherical in shape, suggesting that thapsigargin does not affect closure of the isolation membrane and maturation of the autophagosome (Figure 5B, iii).
Figure 5. Thapsigargin arrests autophagy after autophagosome formation.

(A) MEFs expressing GFP-LC3 and dsRed-ER were incubated in amino acid-free media and subjected to time-lapse confocal microscopy. (i) An example cell with the arrow marking a single GFP-LC3 puncta whose lifetime is represented in the panels of (ii). Time represents minutes and seconds of amino acid starvation. Bar, 10 μm. See also Movie S4. (B) MEFs expressing GFP-LC3 and dsRed-ER were treated with 3 μM thapsigargin for 4 h followed by amino acid-free media, also containing 3 μM thapsigargin. (i) An example cell with the arrow marking a single GFP-LC3 puncta whose lifetime is represented in the panels of (ii). Time shows minutes of amino acid starvation. (iii) Magnification of the GFP-LC3 puncta from the last panel of (B. ii.) in x, y and z dimensions, plus a 3D reconstruction in the bottom right panel. See also Movie S5. (C) Immuno-EM analysis of GFP-LC3 labeled structures, which were classified into four different groups: non-circular, circular, internal label only, or circular clusters. Bars represent mean number of structures per cell section ± SEM for control treated cells (n = 73 cell sections), 1 h 30 min amino acid-free media treated cells (starve; n = 84 cell sections), 4 h 30 min thapsigargin (3 μM) treated cells (TG; n = 103 cell sections), and 4 h 30 min thapsigargin (3 μM) plus amino acid starvation for the final 1 h 30 min (TG + starve; n = 89 cell sections). Panels below the chart represent examples of the indicated structures. Bar, 500 nm. See also Movie S6.
Further evidence that the thapsigargin block occurs after autophagosome formation came from electron microscopy (EM) coupled with gold-enhanced immuno-labeling of GFP-LC3 (Figure 5C). Gold label was identified throughout the cell sections at low density but was greatly concentrated on what appeared to be membranous structures. These structures were classified into four groups: non-circular (concentrated areas of gold, often linear and 200–500 nm in length and may represent isolation membranes); circular (a circular pattern of gold labeling, often containing internal cellular material, ranging from 200–600 nm and might indicate mature autophagosomes); circular clusters (groups of two or more circular labeled structures); and internal only (membranous structures that contained gold label on the inside of the limiting membrane, which are likely autolysosomes) (Figure 5C). Untreated cells contained very few labeled structures, as expected for low level basal autophagy. Starvation resulted in a large increase in isolation membrane-, autophagosome- and autolysosome-type structures. Compared to starvation alone, thapsigargin treatment did not show as large an increase in isolation membrane structures, however, thapsigargin did not block the increase induced by starvation. There was a large increase in autophagosome-like structures upon thapsigargin treatment, either alone or in combination with starvation. Interestingly, the structures resulting from thapsigargin-treatment tended to have denser GFP-LC3 labeling, when compared to starvation alone. Thapsigargin treatment resulted in very few autolysosome-like structures and also blocked formation of such structures by starvation. Taken together, the EM data supports the conclusion that thapsigargin blocks autophagy by preventing fusion of mature autophagosomes with the endocytic system and lysosomes, but has no effect on autophagy initiation or autophagosome formation. One other major difference upon thapsigargin treatment was a three-fold increase in the appearance of clusters of circular GFP-LC3 labeled structures, implying that the accumulated autophagosomes have a tendency to aggregate or cluster. Therefore, some of the larger GFP-LC3 puncta observed by fluorescence microscopy (see Figure 1A and 2C) might represent more than one autophagosome. These large GFP-LC3 puncta form and then disperse as observed by live cell microscopy (Movie S6), consistent with them forming a cluster of autophagosomes.
Thapsigargin does not inhibit endocytosis
Thapsigargin exerts a major effect on the ER and cellular calcium levels, which are important in supplying membrane for intracellular trafficking and vesicular fusion, respectively. It is possible that thapsigargin has a more global effect and can block membrane fusion in multiple transport pathways. To test this, we measured epidermal growth factor receptor (EGFR) turnover. Upon binding extracellular EGF, EGFR travels from the plasma membrane to the lysosome where it is degraded. This endocytic process requires multiple membrane fusion events, including endosomal-lysosomal fusion. Strikingly, although thapsigargin can block autophagy-induced LC3 turnover, endocytosis-mediated EGFR degradation proceeded normally in the presence of thapsigargin (Figure 6A and B). As a control, lysosomal inhibitor bafilomycin blocked EGFR turnover. Therefore, thapsigargin blocks autophagosome fusion with lysosomes but not general endocytic trafficking and membrane fusion, or lysosomal hydrolytic function per se. We also monitored fluid phase endocytosis by fluorescent dextran uptake (Figure 6C and D). Treatment of cells with thapsigargin did not significantly alter LysoTracker staining, suggesting there was no modification in lysosome acidity. In addition, the endocytosis of FITC-labeled dextran was comparable under control or thapsigargin-treated conditions, with approximately 80% of dextran positive structures reaching lysosomes, as assessed by LysoTracker co-localization (Figure 6C and 6D). Taken together, the data strongly suggest that under these conditions thapsigargin does not alter endocytosis, fusion within the endocytic pathway, or lysosomal function, but specifically blocks fusion of autophagosomes with the endocytic system.
Figure 6. Thapsigargin does not block endocytosis-mediated EGFR degradation.

(A) MEFs were treated as described in the methods section for the EGF receptor degradation assay. Post EGF addition, cells were chased in cycloheximide, with/without 3 μM thapsigargin, or 10 nM bafilomycin A1. (B) Quantitation of EGFR degradation. Values represent mean percentage decrease in EGF receptor levels ± SD from three independent experiments. (C) MEFs were incubated in complete media (control) or complete media with 3 μM thapsigargin for 5 h (thapsigargin). Following 2 h of thapsigargin treatment, 5 mg/ml FITC-dextran (green) was added for a further 2.5 h and then cells were washed and incubated in complete media containing 100 nM Lysotracker (red) with/without thapsigargin for the final 30 min. Cells were stained with DAPI (blue). Bar, 10 μm. (D) Quantitation of data represented in (C). Values represent mean number of dextran structures per cell that were positive for lysotracker stain ± SD, from two independent experiments and a total of 20 cells. See also Figure S4.
Rab7, but not Vps16, is required for autophagosomes to fuse with lysosomes
We next studied how thapsigargin blocks fusion of mature autophagosomes with the endocytic system and how autophagosomal-lysosomal fusion differs from general endocytic-lysosomal fusion. We first checked the potential effect of thapsigargin on several specific molecular events relevant to fusion of autophagosomes with the endocytic system, such as interaction of Beclin 1 with ATG14L, Rubicon, or UVRAG (Itakura et al., 2008; Liang et al., 2008; Matsunaga et al., 2009; Sun et al., 2008; Zhong et al., 2009). We found that these events are not affected by thapsigargin, either under control or starvation conditions (data not shown).
Previous studies suggest fusion of autophagosomes with lysosomes requires the conventional cellular fusion machinery consisting of Rab GTPases, tethers and SNARE proteins (Cai et al., 2007; Pfeffer, 2007). Rab7 is responsible for coordinating fusion between late endosomes and lysosomes by recruiting tethers and SNARE proteins, and it has also been implicated in the fusion of autophagosomes with the endocytic system (Gutierrez et al., 2004; Jager et al., 2004; Pankiv et al., 2010). We thus looked at autophagosome and Rab7 localization under thapsigargin treatment. Upon starvation, there was significant co-localization of Rab7 with LC3 positive puncta (Figure 7A and 7B). In contrast, this colocalization was almost completely abolished in thapsigargin-treated cells, even though extensive LC3 puncta were observed (Figure 7A and 7B). This result was supported by subsequent subcellular fractionation experiments using discontinuous iodixonal density gradient centrifugation. On these gradients we found that starvation and thapsigargin treatment had little effect on the density of the membrane-bound LC3-II, although both led to higher levels of LC3-II as expected (Figure 7C). Under normal growth conditions, the majority of Rab7 was in fraction 5. Upon starvation, it shifted to the denser fraction 6 (Figure 7C and 7D). Importantly, treatment with thapsigargin alone did not change Rab7 behavior but it blocked the starvation-induced shift of Rab7 to fraction 6. Given that the Rab7 fractions also contain LC3-II, it is likely that the starvation-induced increase in co-fractionation of Rab7 with LC3-II (as in fraction 6) represents, at least in part, the membrane compartments showing co-localization in Figure 7A. These results suggest that upon autophagy, Rab7 is recruited to autophagosomes (or an intermediate compartment between autophagosomes and lysosomes) to aid in their fusion with the late endocytic pathway, an event blocked by thapsigargin.
Figure 7. Rab7, but not Vps16, is required for autophagosome fusion with lysosomes.

(A) MEF cells were starved for 1 h in amino acid-free media or treated with 3 μM thapsigargin as indicated. Cells stained with DAPI (blue), antibodies against Rab7 (green) and LC3 (red). Shown is a combined confocal image stack. The boxed region is reproduced below the main image displaying a single section highlighting the localization of Rab7 and LC3. Bar, 10 μm. (B) Quantitation of data represented in (A) showing the mean percentage ± SD, from 20 cells in two independent experiments, of LC3 structures co-localizing with Rab7. Rab7 positive structures per cell remained relatively constant with mean values of 38 ± 4 under starvation conditions, compared to 45 ± 9 under thapsigargin treatment. (C) MEF cells were incubated in complete media (control), amino acid-free media for 1 h (starve), complete media with 3 μM thapsigargin for 5 h (TG), or complete media with 3 μM thapsigargin for 5 h amino with acid starvation for the final hour (TG + starve). Cell homogenates were subject to Iodixonal density gradient centrifugation (10%-50% gradient) and fractions immunoblotted. The boxed regions highlight the thapsigargin block of Rab7 increase in fraction 6. (D) Quantitation of the data presented in (C). Values represent mean Rab7 levels in the indicated fractions ± SEM from three independent experiments. (E) EGFR degradation in MEF cells stably expressing control shRNA, Rab 7 shRNA or Vps16 shRNA (the left panel (ii) shows the RNAi efficiency). (F) Quantitation of EGFR degradation. Values represent EGFR levels relative to the start of chase from 4 independent experiments ± SEM. (G) MEF cells stably expressing GFP-LC3 and control shRNA, Rab 7 shRNA or Vps16 shRNA were incubated in complete media or amino acid-free media for 1 h in the presence or absence of 10 nM Bafilomycin A1. (H) Quantitation of the GFP-LC3 immunoblot in (G) showing autophagic flux upon starvation (change in LC3-II levels upon bafilomycin treatment). Values represent means normalized to tubulin from 3 independent experiments ±SEM. (I) MEF cells stably expressing GFP-LC3 and control shRNA, Rab 7 shRNA or Vps16 shRNA were incubated in complete media, amino acid-free media for 1 h (starve) or amino acid-free media for 1 h followed by addition of 50 nM wortmannin for 1h (starve → wort). Bar, 10-μm. (J) Quantitation of data in (I) showing mean number of GFP-LC3 puncta per cell ± SEM (n=20 cells per condition). See also Figure S5.
To investigate further the mechanism of the thapsigargin effect, we used RNAi to deplete Rab7 and Vps16. Vps16 is a component of the HOPS complex, a multisubunit tether involved in Rab7-mediated fusion within the endocytic system. Loss of Rab7 or Vps16 from MEF cells resulted in impairment of EGFR degradation (Figure 7E and F), in agreement with their critical roles in the fusion of endosomes with lysosomes. Surprisingly, we found that while Rab7 is required for a complete autophagy flux, Vps16 is not (Figure 7G and H). Loss of Rab7 led to accumulation of LC3-II even under basal conditions, which was further increased upon starvation; however, under either condition LC3-II levels were not further increased by addition of bafilomycin (Figure 7G and H). This indicates that Rab7 depletion can block the lysosomal degradation of autophagosomes, under both basal and stimulated autophagy conditions, consistent with the published role for Rab7 in the process of autophagosomal-lysosomal fusion (Gutierrez et al., 2004; Jager et al., 2004; Pankiv et al., 2010). In contrast, Vps16 depletion had little effect on LC3 levels or its autophagic flux (Figure 7G and H). Consistently, Vps16 does not colocalize with LC3-II in cells upon starvation or thapsigargin treatment (Figure S5). We next analyzed GFP-LC3 puncta formation (Figure 7I and J); the phenotype in control-depleted and Vps16-deplted cells was essentially the same, with a low level of GFP-LC3 puncta, which increased upon starvation-induced autophagy. In both cases, addition of wortmannin following starvation resulted in loss of LC3 puncta due to lysosomal fusion of the preexisting autophagosomes and simultaneous block in formation of new autophagosomes. In contrast and in line with Figure 7G and H, cells depleted of Rab7 had more GFP-LC3 puncta under basal conditions that was further augmented upon starvation; addition of wortmannin did not eliminate the formed LC3 puncta, as fusion of autophagosomes with lysosomes is already blocked in Rab7-depleted cells. These Rab7 data closely resemble the effect of thapsigargin on autophagy (compare Figure 7G-J with Figure 1B, D and Figure 2A-C), strengthening our conclusion that thapsigargin functions to block autophagosome–lysosome fusion, at least partially by interfering with Rab7 function (Figure 7A-D). These experiments demonstrate that the molecular basis of autophagosomal-lysosomal fusion is fundamentally distinct from that of endosomal-lysosomal fusion in that (1) different molecular components are involved in these two processes; and (2) common factors, such as Rab7, can be differentially regulated.
DISCUSSION
The role of thapsigargin in autophagy was somewhat controversial with previous reports claiming both inducing (Grotemeier et al., 2010; Hoyer-Hansen et al., 2007; Ogata et al., 2006; Sakaki et al., 2008) or inhibitory (Gordon et al., 1993; Williams et al., 2008) effects. Recently, based on use of LC3 as the molecular marker of autophagy, it is generally believed that thapsigargin induces autophagy. The evidence presented here unambiguously demonstrates that thapsigargin inhibits autophagy by blocking autophagosomal fusion with lysosomes. Therefore, accumulation of LC3-II and autophagosomes upon thapsigargin treatment, phenomena previously interpreted as thapsigargin-induced autophagy, is in fact a consequence of blockage of autophagic flux. To reach this conclusion, we used several approaches including LC3 flux measurement, live tandem-tagged LC3 microscopy, EM coupled with immuno-gold labeling, and long-lived protein degradation (Figures 1–5). Further, inhibition of autophagic flux via a block in the autophagosome–lysosome fusion is most likely a general function of thapsigargin instead of a cell type-specific phenomenon, as it caused the same inhibitory effect in all four cell lines we tested (Figures 1, 4, and S3).
Thapsigargin caused clustering of autophagosomes (Figure 5C and Movie S6); this may be a result of failure of autophagic flux, leading to accumulation and subsequent clustering. Alternatively, this clustering may be responsible for the blockage of fusion, perhaps by preventing recruitment of the fusion machinery or simply by a steric-type hindrance.
The effect of thapsigargin on autophagy block is independent of a functional ER stress response pathway (Figure S1) but could be related to the activity of thapsigargin to inhibit SERCA and thus to influence cellular calcium (Ca) signaling. Ca is an essential factor for autophagy as we and others have shown that its removal by chelation blocks autophagy at an early stage prior to LC3 conversion and autophagosome formation (Figure S1 and Fader et al., 2008; Hoyer-Hansen et al., 2007; Sakaki et al., 2008). However, we could not find any Ca modulating compounds or ionophores that mimic the thapsigargin block (Figure S1). Possibly Ca is needed in the autophagosomal lumen for fusion, which it receives from the ER when forming. We do note a close association of the ER with the forming autophagosome (Figure 5A and B and Movies S4 and S5) and Ca has previously been detected in LC3 positive structures (Fader et al., 2008). Ca is known to be important for membrane fusion (Hay, 2007) and it may be that a localized release of Ca from the ER, in the vicinity of autophagosomes and lysosomes is needed to drive their fusion. Indeed, inositol triphosphate receptors, sites of Ca release from the ER, have been implicated in autophagy (Giusti et al., 2009; Khan and Joseph, 2010; Lam and Golstein, 2008; Lam et al., 2008; Sarkar et al., 2005; Vicencio et al., 2009; Williams et al., 2008). Further experimental work is needed to determine which, if any, of these possibilities is the case.
What is perhaps most profound and significant about the thapsigargin-induced block of autophagosomal fusion with endosomes and lysosomes is that it is specific. The endocytic system itself, as revealed by endocytosis-mediated EGFR degradation and dextran uptake and lysosomal acidification (Figure 7A-D), is unaltered by thapsigargin. We also note that we could not detect any changes in the actin or tubulin cytoskeleton upon thapsigargin treatment (Figure S4). Autophagy has a close relationship with endocytosis and both share the common end-point of lysosomal degradation. A functional endocytic pathway is required for autophagy (Eskelinen, 2005; Filimonenko et al., 2007; Lee et al., 2007; Razi et al., 2009) and recent work has suggested that the protein Rubicon, a negative modulator of autophagy, also affects endocytosis, implying linked regulation (Matsunaga et al., 2009; Zhong et al., 2009). Some of the components required for the fusion of autophagosomes with the endocytic system are known, ranging from Rab7 (Gutierrez et al., 2004; Jager et al., 2004; Pankiv et al., 2010), the class C/HOPS tethering complex (Wurmser et al., 2000) and SNAREs (Fader et al., 2009; Furuta et al., 2010; Ishihara et al., 2001). However, these components are required for normal endocytic fusion, a process unimpaired during thapsigargin treatment. Therefore, there must be a fundamental difference between endosome–lysosome fusion and that of autophagosomes with lysosomes. In support of this, we saw that Rab7 is required for both endosome and autophagosome trafficking to lysosomes; yet Vps16, a subunit of the Rab7-binding HOPS complex, was only required for the endosomal pathway (Figure 7). Does autophagosomal-lysosomal fusion utilize other tethering machinery (Cai et al., 2007) instead of the HOPS complex? Although the HOPS complex is implicated in autophagy mainly through studies in yeast (Darsow et al., 1997; Wurmser et al., 2000), these studies relied on morphological features and did not distinguish autophagy with the yeast Cvt pathway at the molecular level. Alternatively, does autophagosomal-lysosomal fusion engage a more specialized HOPS complex with distinct subunit arrangement? These are the future mechanistic questions critical for the trafficking and autophagy fields. Further, autophagosomes have been proposed to enter the endocytic system at multiple points (early endosomes, late endosomes and lysosomes themselves) (Orsi et al., 2010). Fusion between different compartments requires a specific subset of molecular components (if the same machinery were used then compartment identity would be lost), which suggests that autophagosomes would require different components to fuse with early endosomes than to fuse with lysosomes. The fact that thapsigargin blocks autophagosomal fusion with the endocytic system at multiple steps implies that there is most likely a common factor, unique to autophagosomes, required for autophagosomes to become fusion competent. Our data support a model in which Rab7 is recruited to the mature autophagosome directly or indirectly by this thapsigargin-sensitive factor, leading to the further recruitment of autophagy-specific fusion machinery. How this recruitment occurs and the identity of this fusion machinery is under further investigation.
EXPERIMENTAL PROCEEDURES
For full Experimental Procedures see Supplemental Information
Autophagy assays
Cells were washed and incubated in amino acid-free DMEM for 1 h (or complete media as a control) unless indicated. For thapsigargin treatment, complete or amino acid-free media supplemented with 3 μM thapsigargin for indicated time. For thapsigargin treatment in combination with starvation or bafilomycin A1, cells were pretreated with 3 μM thapsigargin for 2 h, unless stated.
Long-lived protein degradation assay
MEF cells were incubated in complete media containing 0.2 μCi/ml [14C] valine for 21 h. Cells were washed and incubated in complete media supplemented with 10 mM unlabeled valine for 2 h to allow degradation of fast turnover proteins. Cells were then chased in complete media, amino acid-free media or complete media containing 3 μM thapsigargin or 10 nM bafilomycin A1 for 5 h 30 min. Next, cells were washed and collected in PBS with 1mM DTT and protease inhibitors. Proteins were precipitated by addition of 20% TCA and incubated on ice overnight. Samples were spun at 4°C, 14000 rpm for 30 min and the pellets resuspended in 0.2 N NaOH and counted in a scintillation counter (Beckman Coulter). Autophagic degradation was calculated as a percentage of the bafilomycin-treated samples.
EGF receptor degradation assay
MEF cells were incubated in serum-free DMEM for 3 h then 3 μM thapsigargin (or DMSO control) was added to indicated plates for an additional 2 h. Media was then replaced with serum-free DMEM containing 40 ng/ml EGF and 20 μg/ml cycloheximide, with/without 3 μM thapsigargin or 10 nM bafilomycin A1 for the indicated times. Cells were lysed and samples normalized for protein concentration and subject to SDS-PAGE and immunoblot.
Density gradient centrifugation
MEF cells were treated with/without complete media containing 3 μM thapsigargin for 5 h. For the final hour, the indicated cells were washed and incubated in amino acid-free media. Cells were placed on ice, washed in PBS and collected in HB buffer [25 mM Tris, pH 7.5, 250 mM sucrose, 50 mM NaCl, 1 mM EDTA] containing protease inhibitors. Samples were homogenized by 10 passes through 25 gauge needle and post nuclear supernatant (PNS) obtained by centrifugation at 1000xg for 5 min. PNS was loaded on the top of a discontinuous Iodixonal (Optiprep) gradient ranging from 10% iodixonal to 50% iodixonal (in 25 mM Tris, pH 7.5 and 1 mM EDTA). Samples were centrifuged for 16 h at 35,000 rpm, 4°C in a MLS-50 rotor (Beckman Coulter). Following centrifugation fractions were collected from the top of the gradient and subject to SDS-PAGE and immunoblotting.
Supplementary Material
HIGHLIGHTS.
Thapsigargin specifically blocks autophagosome fusion with the endocytic system
Thapsigargin blocks autophagosomal recruitment of the small GTPase Rab7
Vps16, is required for endocytic fusion, but not for autophagosomal fusion
Autophagosomal-lysosomal fusion is distinct from endosomal-lysosomal fusion
Acknowledgments
We thank N. Lampen and Dr. M. Overholtzer for assistance on electron microscopy and confocal time-lapse microscopy respectively and members of the X. J. laboratory for critical reading of the manuscript and discussion. We also thank Drs. Ron, Mizushima, Debnath, and Overholtzer for sharing precious reagents. This work was partially supported by NIH R01 CA113890 (to XJ), and a Catherine and Frederick Adler Chair fund (to XJ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- Cai H, Reinisch K, Ferro-Novick S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell. 2007;12:671–682. doi: 10.1016/j.devcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. 2009;2:pe51. doi: 10.1126/scisignal.284pe51. [DOI] [PubMed] [Google Scholar]
- Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell. 2009;20:2004–2014. doi: 10.1091/mbc.E08-12-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darsow T, Rieder SE, Emr SD. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–4710. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- Fader CM, Sanchez D, Furlan M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9:230–250. doi: 10.1111/j.1600-0854.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- Fader CM, Sanchez DG, Mestre MB, Colombo MI. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta. 2009;1793:1901–1916. doi: 10.1016/j.bbamcr.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179:485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer von Mollard G, Stevens TH. The Saccharomyces cerevisiae v-SNARE Vti1p is required for multiple membrane transport pathways to the vacuole. Mol Biol Cell. 1999;10:1719–1732. doi: 10.1091/mbc.10.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta N, Fujita N, Noda T, Yoshimori T, Amano A. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol Biol Cell. 2010;21:1001–1010. doi: 10.1091/mbc.E09-08-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Carroll K, Bittova L, Pfeffer S. Rab9 GTPase regulates late endosome size and requires effector interaction for its stability. Mol Biol Cell. 2004;15:5420–5430. doi: 10.1091/mbc.E04-08-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giusti C, Tresse E, Luciani MF, Golstein P. Autophagic cell death: analysis in Dictyostelium. Biochim Biophys Acta. 2009;1793:1422–1431. doi: 10.1016/j.bbamcr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Gordon PB, Holen I, Fosse M, Rotnes JS, Seglen PO. Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem. 1993;268:26107–26112. [PubMed] [Google Scholar]
- Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, Viollet B, Wesselborg S, Proikas-Cezanne T, Stork B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal. 2010;22:914–925. doi: 10.1016/j.cellsig.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay JC. Calcium: a fundamental regulator of intracellular membrane fusion? EMBO Rep. 2007;8:236–240. doi: 10.1038/sj.embor.7400921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Hamasaki M, Yokota S, Suzuki K, Kamada Y, Kihara A, Yoshimori T, Noda T, Ohsumi Y. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell. 2001;12:3690–3702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9:574–587. doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T, Inagi R, Takano H, Sato S, Ingelfinger JR, Fujita T, Nangaku M. Endoplasmic reticulum stress induces autophagy in renal proximal tubular cells. Nephrol Dial Transplant. 2009;24:2665–2672. doi: 10.1093/ndt/gfp215. [DOI] [PubMed] [Google Scholar]
- Khan MT, Joseph SK. The role of inositol trisphosphate receptors in autophagy in DT40 cells. J Biol Chem. 2010 doi: 10.1074/jbc.M110.114207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Moretti L, Mitchell LR, Jung DK, Lu B. Endoplasmic reticulum stress mediates radiation-induced autophagy by perk-eIF2alpha in caspase-3/7-deficient cells. Oncogene. 2010 doi: 10.1038/onc.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147:435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Kochl R, Hu XW, Chan EY, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–145. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- Lam D, Golstein P. A specific pathway inducing autophagic cell death is marked by an IP3R mutation. Autophagy. 2008;4:349–350. doi: 10.4161/auto.5521. [DOI] [PubMed] [Google Scholar]
- Lam D, Kosta A, Luciani MF, Golstein P. The inositol 1,4,5-trisphosphate receptor is required to signal autophagic cell death. Mol Biol Cell. 2008;19:691–700. doi: 10.1091/mbc.E07-08-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol. 2007;17:1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10:776–787. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008;455:396–400. doi: 10.1038/nature07208. [DOI] [PubMed] [Google Scholar]
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi A, Polson HE, Tooze SA. Membrane trafficking events that partake in autophagy. Curr Opin Cell Biol. 2010;22:150–156. doi: 10.1016/j.ceb.2009.11.013. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjorkoy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253–269. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer SR. Unsolved mysteries in membrane traffic. Annu Rev Biochem. 2007;76:629–645. doi: 10.1146/annurev.biochem.76.061705.130002. [DOI] [PubMed] [Google Scholar]
- Razi M, Chan EY, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol. 2009;185:305–321. doi: 10.1083/jcb.200810098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaki K, Wu J, Kaufman RJ. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J Biol Chem. 2008;283:15370–15380. doi: 10.1074/jbc.M710209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–1111. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TK, Rehling P, Peterson MR, Emr SD. Class C Vps protein complex regulates vacuolar SNARE pairing and is required for vesicle docking/fusion. Mol Cell. 2000;6:661–671. doi: 10.1016/s1097-2765(00)00064-2. [DOI] [PubMed] [Google Scholar]
- Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2008;105:19211–19216. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321:117–120. doi: 10.1126/science.1154822. [DOI] [PubMed] [Google Scholar]
- Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, Joza N, Vitale I, Morselli E, Tailler M, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009;16:1006–1017. doi: 10.1038/cdd.2009.34. [DOI] [PubMed] [Google Scholar]
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman RL, Zhang Y, Lee KP, Harding HP, Haynes CM, Price J, Sicheri F, Ron D. Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Mol Cell. 2010;38:291–304. doi: 10.1016/j.molcel.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurmser AE, Sato TK, Emr SD. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J Cell Biol. 2000;151:551–562. doi: 10.1083/jcb.151.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2009 doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.