

Abstract
Microbial-induced inflammation is important for eliciting humoral immunity. Genetic defects of NADPH oxidase 2–based proteins interrupt phagocyte superoxide generation and are the basis for the human immunodeficiency chronic granulomatous disease (CGD). Hyperinflammation is also a significant clinical manifestation of CGD. Herein, we evaluated humoral immunity in the phagocyte oxidase p47phox-deficient model of CGD and found that UV-inactivated Streptococcus pneumoniae and Listeria monocytogenes (Lm) elicited higher specific antibody (Ab) titers in p47phox-/- mice than wild-type (WT) mice. Both organisms elicited robust and distinct antigen-presenting cell maturation phenotypes, including IL-12 hypersecretion, and higher major histocompatibility complex II and costimulatory protein expression in Lm-stimulated p47phox-/- dendritic cells (DCs) relative to WT DCs. Furthermore, p47phox-/- DCs pulsed with Lm and adoptively transferred into naïve WT mice elicited Ab titers, whereas Lm-pulsed WT DCs did not elicit these titers. The observed robust p47phox-/- mouse humoral response was recapitulated with live Lm and sustained in vivo in p47phox-/- mice. Notably, anti–serum samples from p47phox-/- mice that survived secondary Lm infection were protective in WT and p47phox-/- mice that were rechallenged with secondary lethal Lm infection. These findings demonstrate a novel benefit of NADPH oxidase 2 deficiency (ie, dependent inflammation in antigen-presenting cell–mediated humoral immunity) and that anti-Lm Ab can be protective in an immunodeficient CGD host.
Antigen-presenting cells (APCs), such as dendritic cells (DCs) and macrophages, are important cellular mediators of inflammation. APCs also bridge innate and humoral immunity to combat microbial infection. Bacteria induce APC maturation. In turn, mature APCs instruct adaptive immunity by presenting bacteria-derived peptides, along with costimulatory signals, to T cells and secreting inflammatory cytokines that drive T-cell activation and consequent T-cell–mediated and/or humoral immunity.
Both Listeria monocytogenes (Lm), a facultative intracellular bacterium, and Streptococcus pneumoniae (Pn), an anaerobic extracellular bacterium, have been used to study the fundamental components of innate and adaptive immunity.1–3 Lm and Pn each elicit strong APC-mediated inflammatory and cellular responses that are important for initiating protective immune responses.
Pn elicits antigen-specific antibody (Ab) production and anti-Pn humoral immunity.3 In contrast, early Lm research4 indicated that Lm-induced Ab production was not specific for Lm resistance, and experimental evidence1,2 showed that T-cell–mediated immunity is most critical for eliminating Lm. However, subsequent studies5,6 have shown that humoral immunity can play a significant role in the elimination of Listeria infection.
The local oxidative environment, reactive oxygen species (ROS), and free radical responses are widely postulated to promote inflammation as part of the adaptive response to restoring tissue homeostasis after acute infection and tissue injury. However, recent observations that phagocytes, and nonphagocytic cells, generate ROS as they orchestrate adaptive immune responses raise questions about the source and relative role of ROS in modulating inflammatory responses that are important for eliciting humoral immunity.7,8 Patients with chronic granulomatous disease (CGD) have heterogeneous genetic defects of phagocytic oxidase NADPH oxidase 2 (Nox2)–based proteins and an absent or reduced phagocyte respiratory burst.9–12 CGD is a multifaceted clinical disease that manifests clinically as life-threatening bacterial and fungal infections.9,13 Interestingly, noninfectious hyperinflammation is also a common occurrence in patients with CGD.14 Because one of the clinical manifestations of CGD is increased inflammation, we investigated the ability of p47phox (Ncf1)–deficient (p47phox-/-) APCs to secrete cytokines and up-regulate receptors important for initiating humoral immunity against Lm and Pn. We demonstrate that Pn and Lm stimulation leads to dissimilar p47phox-/- DC maturation. We also show that, although Pn predictably induces humoral immunity, including memory Ab production in p47phox-/- mice, anti-Pn humoral immunity is enhanced in p47phox-/- mice compared with wild-type (WT) control mice. Interestingly, we found that Lm similarly elicits enhanced and protective humoral immunity in p47phox-/- mice.
Materials and Methods
Mice
Nox p47phox-deficient (p47phox-/-) mice have been described.15,16 Congenic p47phox-/- mice on a C57BL/6NTac background were generated by backcrossing over 10 generations with WT C57BL/6NTac. gp91phox-/-/Nox2-/- B6.129S6-Cybbtm1Din/J mice17 were obtained from The Jackson Laboratory (Bar Harbor, ME). Animal care was provided in accordance with Institutional Animal Care and Use Committee procedures, approved by the National Institute of Allergy and Infectious Diseases, NIH (Bethesda, MD). All mice used were between the ages of 3 and 6 weeks.
Preparation of Pn Type 2 (R36A) and Lm
Frozen stocks of nonencapsulated variant (strain R36A) of virulent Pn capsular type 2 (strain D39) were thawed and subcultured on BBL agar plates (VWR International, West Chester, PA). Similarly, recombinant Lm strain 10403S expressing ovalbumin; a gift from Dr. Hao Chen18 (University of Pennsylvania School of Medicine, Philadelphia, PA) was subcultured on Difco Brain Heart Infusion Agar (BD, Franklin Lakes, NJ). Isolated colonies were collected and UV inactivated (UVi) (UV Stratalinker 1880; Artisan Scientific, Champagne, IL) at 1000 mJ for 1 hour. Sterility was confirmed by subculture on blood agar plates for Pn and Brain Heart Infusion Agar for Lm. After extensive washings, the bacterial suspensions were adjusted with PBS to provide an OD of 0.6, which corresponded to 109 colony-forming units (CFUs)/mL of Pn; and 0.1, which corresponded to 108 CFUs/mL of Lm. Bacteria were then divided at 1010 CFUs/mL and frozen at -80°C until their use.
Preparation of Bone Marrow–Derived Dendritic Cells
Bone marrow–derived DCs (BMDCs) were prepared as previously described.19 Briefly, bone marrow was flushed with PBS, resuspended in 1 mL ACK Lysing Buffer (BioWhittaker, Walkersville, MD), and incubated at room temperature for 5 minutes to eliminate red cells. The single-cell suspension was filtered through a 40-μm cell strainer (BD Falcon/BD Biosciences, San Jose, CA), resuspended at a density of 1.25 × 106 cells/mL (24-well plates) in RPMI 1640 plus 5% fetal bovine serum, 10,000 IU penicillin, 10 mg/mL streptomycin, 1 mmol/L sodium pyruvate, 2 mmol/L L-glutamine, 0.1 mmol/L nonessential amino acids, and 25 mmol/L HEPES (culture medium), supplemented with 10 ng/mL murine recombinant granulocyte macrophage colony-stimulating factor (M-CSF) (Sigma-Aldrich, St. Louis, MO). After 7 days of culture, nonadherent cells were harvested.
Preparation of Bone Marrow–Derived Macrophages
Bone marrow–derived macrophages (BMMs) were obtained using a similar approach as used for BMDCs, with slight modifications. Bone marrow cells were cultured at 1 × 106 cells/mL in cell culture medium supplemented with 10 ng/mL murine M-CSF (Sigma-Aldrich). Cells were plated in six-well plates in a volume of 4 to 5 mL per well. On days 3 and 5, three-fourths culture medium was removed; and fresh culture medium was added. On day 7, BMMs were harvested by washing plates with sterile PBS to remove nonadherent cells. Cells were detached from the plate as previously described.19 Briefly, 2 mL of detachment buffer (4 mg/mL lidocaine, 5 mmol/L EDTA, and PBS) was added for 3 to 5 minutes. After incubation, detachment buffer was pipetted until the adherent cells detached.
Reagents
Recombinant pneumococcal surface protein A (PspA; family 1, sero clade 2) was provided by Clifford M. Snapper (Uniformed Services University of the Health Sciences, Bethesda). Purified listeriolysin-O (LLO) was purchased from Abcam (Cambridge, MA).
Histopathological and Immunohistochemical Features
A necropsy of all mice was performed at the ages noted. All tissues were examined grossly, and most were fixed in 10% neutral-buffered formalin, embedded in paraffin, and sectioned. Slides containing formalin-fixed, paraffin-embedded tissue sections (3 to 4 μm) were deparaffinized in xylene and rehydrated by processing them through alcohols. Pretreatment of tissues before incubation with the Listeria primary Ab consisted of bleaching with Peroxidazed 1 (Biocare Medical, Concord, CA) for 5 minutes, digesting with proteinase K (Dako, Carpentaria, CA) for 5 minutes, and pageing with Background Sniper (Biocare Medical) for 10 minutes. Sections were incubated with a goat polyclonal Ab against Listeria (KPL, Gaithersburg, MD) for 60 minutes at a dilution of 1:1500. The bound Ab was detected using a goat polymer detection system (Biocare Medical) and Vulcan Fast Red chromogen (Biocare Medical). Sections were counterstained with CAT hematoxylin (Biocare Medical), air dried, and mounted using Permount mounting medium (Fisher Scientific, Pittsburgh, PA). Negative controls included replacing the primary Ab with normal goat serum at a comparable protein concentration and testing noninfected tissues with the primary Ab. Slides were imaged using Aperio ScanScope software (Aperio Technologies Inc., Vista, CA).
Flow Cytometric Analysis
All steps were performed on ice. Fc receptors were pageed with 10 μg/mL purified rat anti-mouse CD16/CD32 mouse Fc page (clone 2.4G2). Cells were stained for 30 minutes with fluorescein isothiocyanate–mouse IgG2a and κ anti-mouse major histocompatibility complex (MHC) class IIb (clone AF6-120.1), phosphatidylethanolamine-mouse IgG2a and κ anti-mouse CD40 (clone 3/23), phosphatidylethanolamine-mouse IgG2a and κ anti-mouse CD86 (clone GL1), and Armenian hamster IgG2 and κ anti-mouse CD80 (clone 16-10A1). All monoclonal antibodies were purchased from BD Pharmingen (Franklin Lakes, NJ). Irrelevant isotype- and species-matched monoclonal antibodies (Abs) were used as staining controls. Cells were analyzed on a BD FacsCanto flow cytometer.
In Vitro Incubation of BMDCs and BMMs with Bacteria
BMDCs, cultured in granulocyte M-CSF, and BMMs, cultured in M-CSF, were pulsed in vitro with UVi Pn (108 CFUs) or UVi Lm (108 CFUs) overnight. IL-6, IL-12, and tumor necrosis factor (TNF)-α were measured from supernatant by sandwich enzyme-linked immunosorbent assay (ELISA).
In Vivo Bacterial Challenge
WT, p47phox-/-, and/or Nox2-/- mice were immunized by i.p. injection with 2 × 108 CFUs of UVi Pn or Lm on day 0; on day 14, a secondary challenge of UVi bacteria was given to assess the potential generation of memory. Serum was harvested 7, 14 (before rechallenge), and 21 days after bacterial challenge. For live Lm infection, WT and p47phox-/- mice were infected i.v. with 5 × 104 CFUs (0.1 LD50) of Lm.
Measurement of Serum Ig Titers
ELISA plates (Immunolon 4) were coated with 5 μg/mL (50 μL/well) PspA, 3 μg/mL (50 μL/well) LLO, and 3 μg/mL (50 μL/well) UVi Pn (107 CFUs/well) or UVi Lm (107 CFUs/well) in PBS overnight at 4°C. Plates were pageed with PBS plus 1% bovine serum albumin (BSA) for 30 minutes at 37°C and washed three times with PBS plus 0.1% Tween 20. Threefold dilutions of serum samples, starting at a 1:50 serum dilution, in PBS plus 0.05% Tween 20 were then added and left overnight at 4°C. Plates were washed three times with PBS plus 0.1% Tween 20. Alkaline phosphatase–conjugated polyclonal goat anti-mouse IgM or IgG Abs (200 ng/mL final concentration in PBS plus 0.05% Tween 20) were then added, and plates were incubated for 37°C for 1 hour. Plates were washed five times with PBS plus 0.1% Tween 20. Substrate (p-nitrophenyl phosphate, disodium) at 1 mg/mL in TM buffer [1 mol/L Tris plus 0.3 mmol/L MgCl2 (pH 9.8)] was then added for 30 minutes at room temperature for color development. Color was read at an absorbance of 405 nm.
Measurement of Cytokine Concentrations in Culture Supernatant by ELISA
Concentrations of specific cytokines released into the medium of cell cultures were measured using optimized standard sandwich ELISA. ELISA (Immunolon 4) plates were coated with IL-6 (2 μg/mL), IL-12 (6 μg/mL), and TNF-α (10 μg/mL) capture Ab in PBS overnight at 4°C. Plates were pageed with PBS plus 1% BSA for 30 minutes at 37°C and washed three times with PBS plus 0.1% Tween 20. Twofold dilutions of supernatant samples and standards, including recombinant (r) IL-6 (4 ng/mL), rIL-12 (8 ng/mL), or rTNF-α (8 ng/mL) in PBS plus 0.05% Tween 20, were then added and left overnight at 4°C. Plates were washed three times with PBS plus 0.1% Tween 20. Secondary IL-6 (1 μg/mL), IL-12 (1 μg/mL), and TNF-α (1 μg/mL) antibodies with PBS plus 1% BSA were added for 1 hour at 37°C and washed three times with PBS plus 0.1% Tween 20. Streptavidin-alkaline phosphatase with PBS plus 1% BSA was added at a 1:1000 concentration for 1 hour at 37°C. Substrate (p-nitrophenyl phosphate, disodium; at 1 mg/mL in TM buffer [1 mol/L Tris plus 0.3 mmol/L MgCl2 (pH 9.8)] was then added for 30 minutes at room temperature for color development. Color was read at an absorbance of 405 nm.
Immune Serum Transfer
Twenty WT and p47phox-/- mice were i.v. infected with 5 × 104 Lm, then were rechallenged with 5 × 106 Lm 28 days later. Serum was harvested from the surviving WT (n = 20) and p47phox-/- (n = 16) mice 6 days after reinfection and was adoptively transferred i.v. into a second set of WT and p47phox-/- recipients that were infected with 5 × 104 Lm 28 days previously. One day after immune serum transfer, WT and p47phox-/- mice were reinfected with 5 × 106 Lm and monitored for survival.
Statistics
Differences between the group means were analyzed by the Student's t-test (Prism 5; GraphPad Software, Inc., San Diego, CA). P ≤ 0.05 was considered statistically significant.
Results
Lm and Pn Elicit Distinct Maturation Phenotypes in p47phox-/- and WT APCs
For initial investigations, we used UVi Lm or Pn to compare proinflammatory cytokine induction in WT and p47phox-/- BMMs and BMDCs propagated in vitro. In contrast to heat-killed inactivation, which can cause bacterial rupture, UV-irradiated bacteria are replication incompetent20 and remain structurally intact. As illustrated in Table 1, Pn-pulsed p47phox-/- APCs hypersecrete IL-6 and TNF-α; in contrast, Lm-stimulated p47phox-/- APCs hypersecrete IL-12 relative to WT APCs. Interestingly, although Lm-pulsed p47phox-/- DCs secrete less TNF-α than WT DCs, p47phox-/- macrophages propagated in M-CSF secreted at least twofold more IL-6, IL-12, and TNF-α in response to both Pn and Lm relative to WT macrophages (Table 1).
Table 1.
APC Cytokine Secretion from WT and p47phox-/- Mice Cultured with 1 × 108 CFUs/mL of UVi Pn or Lm for 24 Hours in Vitro
| Variable | IL-6 |
IL-12 |
TNF-α |
|||
|---|---|---|---|---|---|---|
| WT | p47phox-/- | WT | p47phox-/- | WT | p47phox-/- | |
| BMDCs | ||||||
| Pn | 5.68 ± 0.63 | 10.27 ± 0.88⁎ | 1.25 ± 0.04 | 1.37 ± 0.06 | ND | 0.89 ± 0.03 |
| Lm | 24.39 ± 0.63 | 21.24 ± 0.88 | 3.12 ± 0.14 | 4.82 ± 0.03† | 5.25 ± 0.30 | 3.62 ± 0.50 |
| Medium | 0.625 ± 0.03 | 0.646 ± 0.002 | 0.263 ± 0.001 | 0.237 ± 0.57 | ND | 0.393 ± 0.17 |
| BMMs (M-CSF) | ||||||
| Pn | 0.75 ± 0.09 | 2.05 ± 0.08† | 1.40 ± 0.09 | 4.70 ± 0.10† | ND | 1.84 ± 0.09 |
| Lm | 2.81 ± 0.39 | 5.16 ± 0.33⁎ | 1.89 ± 0.07 | 6.43 ± 0.12† | 1.50 ± 0.20 | 3.87 ± 0.14† |
| Medium | ND | ND | ND | ND | ND | 1.42 ± 0.38 |
Data are given as mean ± SEM (in ng/mL). n = 4. Concentrations of IL-6, IL-12, and TNF-α in the culture supernatant were determined by ELISA. The limit of detection for IL-6 was <31 pg/mL; IL-12, <62 pg/mL; and TNF-α, <250 pg/mL.
ND, not detectable.
P = 0.04.
P = 0.0004.
Data are from Vasilevsky et al.19
Next, we compared MHCs with costimulatory molecule expression in Lm- and Pn-stimulated APCs (Table 2). After overnight culture with UVi microorganisms, p47phox-/- DCs expressed higher surface levels of MHC class II and CD80 in media alone and in response to Lm stimulation compared with WT DCs (Figure 1A). In addition, surface CD40 and CD86 is significantly up-regulated in Lm-stimulated p47phox-/- DCs relative to similarly treated WT DCs. In contrast, WT DCs expressed higher surface levels of MHC class II, CD86, and CD40 in response to Pn stimulation compared with p47phox-/- DCs (Figure 1B). WT and p47phox-/- macrophages propagated in M-CSF and pulsed with Lm or Pn similarly up-regulated MHC class II, CD80, CD86, and CD40 (Figure 1, C and D). Thus, in addition to finding that p47phox differentially regulates proinflammatory cytokine secretion in Pn- and Lm-stimulated APCs, these observations show that Pn and Lm induce distinct maturation immunophenotypes in p47phox-/- and WT APCs. Notably, only Lm-pulsed p47phox-/- DCs expressed higher levels of MHC class II, CD80, CD86, and CD40 compared with WT, suggesting a role for p47phox in DC antigen presentation.
Table 2.
APC MFI Values
| Variable | BMDCs |
BMMs |
||||||
|---|---|---|---|---|---|---|---|---|
| WT |
p47phox-/- |
WT |
p47phox-/- |
|||||
| gMFI | No. of cells | gMFI | No. of cells | gMFI | No. of cells | gMFI | No. of cells | |
| MHC Class II | ||||||||
| Medium | 72 | 5481 | 123 | 5749 | 230 | 3195 | 342 | 3970 |
| Pn | 123 | 4630 | 100 | 6976 | 1241 | 6377 | 1877 | 6590 |
| Lm | 141 | 4568 | 210 | 6466 | 958 | 7242 | 895 | 7418 |
| Control | 56 | 6521 | 60 | 7119 | 366 | 4140 | 343 | 7518 |
| CD80 | ||||||||
| Medium | 503 | 4550 | 1033 | 6477 | 462 | 4955 | 384 | 5017 |
| Pn | 621 | 5900 | 1147 | 6383 | 558 | 5824 | 721 | 7163 |
| Lm | 511 | 6059 | 1656 | 5743 | 451 | 6550 | 582 | 7501 |
| Control | 130 | 6461 | 127 | 7843 | 324 | 8207 | 290 | 7375 |
| CD86 | ||||||||
| Medium | 336 | 5590 | 266 | 6848 | 278 | 44,879 | 351 | 4280 |
| Pn | 751 | 6129 | 309 | 7344 | 439 | 4969 | 529 | 7198 |
| Lm | 427 | 4438 | 1035 | 5859 | 677 | 7490 | 725 | 7516 |
| Control | 114 | 5807 | 86 | 8522 | 245 | 4140 | 232 | 7375 |
| CD40 | ||||||||
| Medium | 208 | 5764 | 130 | 6673 | 821 | 3586 | 446 | 4513 |
| Pn | 379 | 6122 | 255 | 7494 | 2142 | 6699 | 2028 | 7040 |
| Lm | 355 | 4435 | 755 | 6069 | 2592 | 7353 | 1374 | 6979 |
| Control | 114 | 5807 | 127 | 7843 | 73 | 7375 | 75 | 7375 |
APCs from WT and p47phox-/- mice were cultured with 1 108 CFUs/mL of UVi Pn or Lm for 24 hours in vitro.
gMFI, geometric mean fluorescence intensity.
Figure 1.
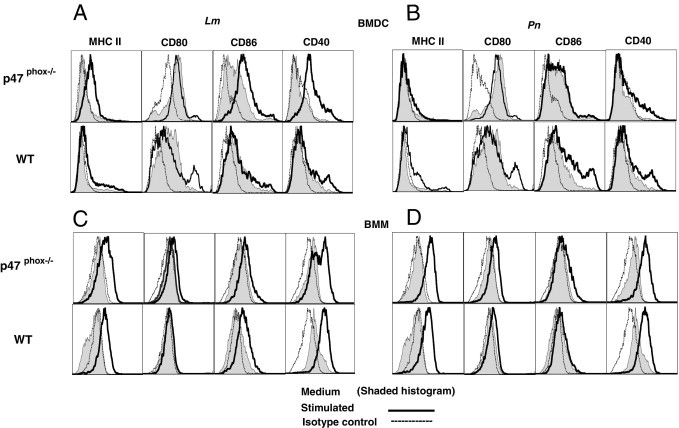
Pn- and Lm-induced maturation of p47phox-/- and WT APCs in vitro. WT and p47phox-/- BMDCs (A and B) and BMMs (C and D) (1 × 106 cells/mL) were stained with fluorochrome-conjugated monoclonal Abs specific for the indicated cell surface proteins after 24 hours in culture in medium alone or in the presence of UVi Pn or Lm (1 × 109 CFUs/mL) and analyzed by flow cytometry. Results are representative of two independent experiments with pooled cells from three to four of each genotype per experiment.
p47phox-/- DC–Mediated Regulation of Anti-Lm Humoral Immunity
Although Pn-elicited humoral immunity in mice is well characterized,3,21 few investigations5,6 have shown a benefit for Lm-induced humoral immunity and Ab production. Next, to clarify whether p47phox deficiency affected anti-Lm Ab production, we used ELISA to compare specific Ab titers for intact bacteria and Pn and Lm virulence factors, PspA and LLO, respectively, after UVi Pn or Lm challenge. Although LLO is a heat-liable protein,22 we exploited our UVi challenge model to examine this parameter. For these investigations, we immunized p47phox-/- and WT mice with UVi bacteria on day 0; on day 14, a secondary challenge of UVi bacteria was given to assess the generation of memory Ab production. As illustrated in Figure 2A, Pn predictably induced potent Ab production in both WT and p47phox-/- mice. However, the p47phox-/- anti-Pn IgM titers were twofold higher than WT 7 and 14 days after UVi Pn challenge. In addition, p47phox-/- anti-Pn IgG titers were threefold higher on day 7 before rechallenge and twofold higher than WT after UVi Pn rechallenge (Figure 2A, day 21). Similarly, p47phox-/- mice had higher IgM and IgG isotype PspA-specific titers than WT mice after UVi Pn challenge (Figure 2C). Notably, serum from UVi-Lm–challenged p47phox-/- mice also exhibited increased anti-Lm IgM (twofold to threefold higher on days 7 and 14) and IgG (33-fold higher on day 14 and 15-fold higher on day 21) titers relative to WT mice (Figure 2B). Moreover, p47phox-/- anti-LLO IgM titers were elevated 11-fold on day 21 and anti-LLO IgG titers were elevated twofold higher than WT on day 14; serum anti-LLO IgG titers in WT and p47phox-/- mice were similar on day 21 after a secondary UVi Lm boost on day 14 (Figure 2C). Collectively, these results demonstrate that humoral immunity is enhanced in inflammation-prone p47phox-/- mice and thereby reveal a complex and unforeseen benefit of phagocyte oxidase deficiency for enhancing anti-Pn and Lm humoral immunity.
Figure 2.
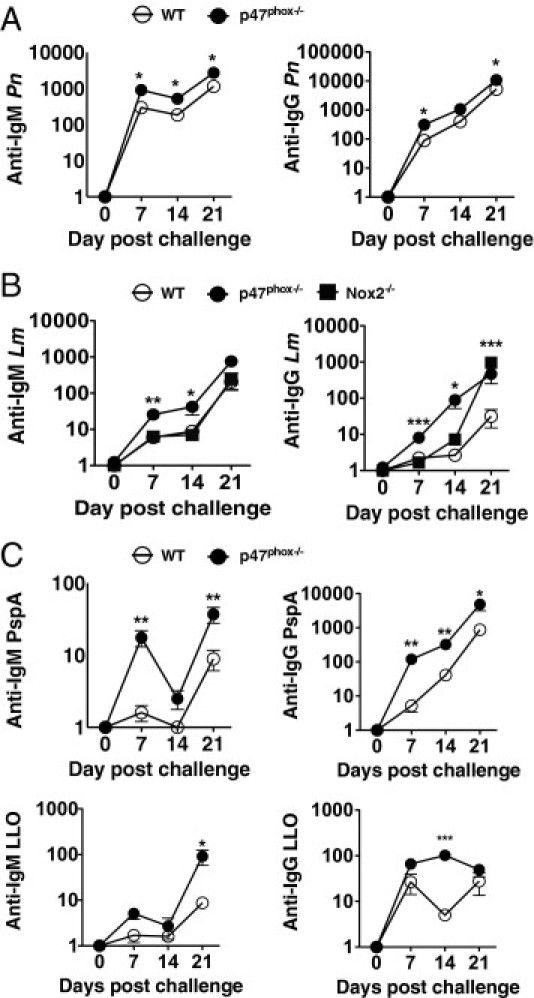
Ab production in response to UVi Pn and Lm challenge. WT, p47phox-/-, and Nox2-/- mice were immunized by i.p. injection with 2 × 108 CFUs of UVi Pn or Lm on days 0 and 14 before serum collection. Serum was harvested on the indicated days. Serum titers for IgG and IgM specific for whole Pn (A), Lm (B), and PspA and LLO (C) were determined by ELISA. Absorbance values (Titer-1) represent the mean ± SEM. *P ≤ 0.05, **P ≤ 0.005, and ***P ≤ 0.0005. Results are representative of two individual experiments with five of each genotype per experiment.
Previous investigations23,24 showed that, although gp91phox-deficient (Nox2-/-) mice have increased susceptibility to Lm infection and that Nox2-/- macrophages cannot kill virulent Lm, primary Lm infection is not fatal in p47phox-/- mice.25 Thus, to discern the role of p47phox independently or as part of the multicomponent phagocytic Nox in Lm-elicited humoral immunity, we examined Lm-induced Ab production in Nox2-/- catalytic subunit mice. As shown in Figure 2B, compared with p47phox-/- mice, anti-Lm IgM-specific titers were not elevated from Lm-challenged Nox2-/- mice. However, anti–Lm-specific IgG titers were elevated 2.5-fold on day 14 in Nox2-/- mice compared with Lm-challenged WT mice and twofold and 31-fold compared with p47phox-/- and WT mice, respectively, on day 21 after a secondary Lm boost on day 14 (Figure 2B). These data show that, although the kinetics of anti–Lm-specific IgG induction are different in Nox2-/- and p47phox-/- mice, Lm challenge elicits an equally robust recall IgG Ab response in Nox2-/- mice as in p47phox-/- mice, therefore indicating that Nox2 enzymatic activity is important for controlling Lm-elicited Ab production.
These observations lead us to question the relevance of the observed enhanced Lm-induced p47phox-/- DC maturation phenotype to the robust UVi Lm-elicited humoral response in p47phox-/- mice and whether the anti-Lm humoral immune response may be protective in p47phox-/- mice. Therefore, to further investigate these parameters, we adoptively transferred 1 × 106 Lm-pulsed p47phox-/- and WT BMDCs into naïve WT recipient mice. As controls, unpulsed p47phox-/- and WT DCs were also transferred into a separate group of WT recipients. There was no difference in anti-Lm IgM or IgG titers in WT recipients of Lm-pulsed or unpulsed control DCs 7 and 14 days after DC transfer (data not shown). In contrast, there was a threefold difference in anti-Lm IgM titers in WT mice that received Lm-pulsed p47phox-/- DCs compared with recipients that received unpulsed p47phox-/- DCs 7 and 14 days after DC transfer (Figure 3). However, there was no difference in anti-Lm IgG titers in these mice. To assess whether the adoptively transferred p47phox-/- DCs could prime for a recall response, we challenged the DC recipients with UVi Lm after serum harvest on day 14. As shown in Figure 3, there was a 14-fold increase on day 21 in anti-Lm IgG titers of recipients of Lm-pulsed p47phox-/- DCs compared with day 14 titers. Thus, Lm-pulsed p47phox-/- DCs elicit similar IgM titers to those seen in WT and p47phox-/- mice challenged with UVi (Figure 2) and are primed for anti-Lm IgG recall humoral immunity.
Figure 3.
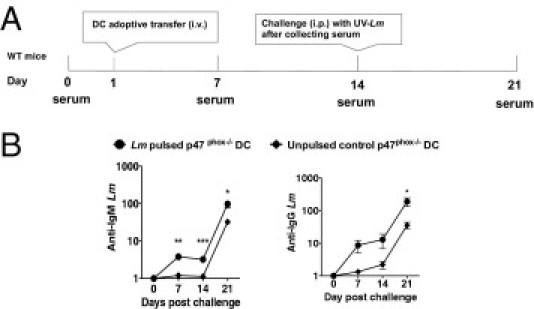
p47phox in DCs is critical for IgM and IgG Abs in response to Lm. WT or p47phox-/- BMDCs (1 × 106), untreated or pulsed with 1 × 108 CFUs of UVi Lm for 5 hours, were i.v. injected into WT recipient mice. The recipients also received a booster injection, 1 × 108 CFUs of UVi Lm i.p., on day 14 after serum collection. A: Schematic timeline of adoptive transfer and serum collection. B: Serum titers for IgG and IgM specific for whole Lm were determined by ELISA. Absorbance values (Titer-1) represent the mean ± SEM. *P ≤ 0.05, **P ≤ 0.005, and ***P ≤ 0.0005. Results are representative of two individual experiments, with five and nine of each genotype in serial experiments.
Immune Serum from Lm-Infected p47phox-/- Mice Is Protective against Secondary Lm Challenge
Next, we immunized p47phox-/- and WT mice with a sublethal dose of live Lm and collected serum after 7, 14, and 120 days to determine whether live Lm infection would also elicit higher Ab production in p47phox-/- mice. Histological examination results of spleens from p47phox-/- and WT mice 3 and 14 days after infection revealed diffuse inflammation with histiocytosis. In p47phox-/- mice, splenic lesion necrosis was more severe and consisted of diffuse severe inflammation with histiocytosis (Figure 4). Thus, p47phox-/- mice have enhanced inflammatory cytokine secretion and extensive tissue inflammation during primary Lm infection.
Figure 4.
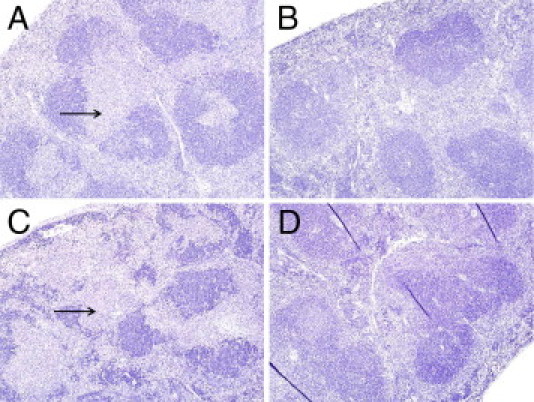
Histological characteristics of Lm-infected spleens. A to D: H&E and immunohistochemistry labeling for Lm in infected spleens. A: WT spleen, immunostaining against Lm 3 days after infection. Original magnification, ×8. B: WT spleen, mild increase in lymphocytes, 14 days after infection. Original magnification, ×8 (H&E). C: p47phox-/- spleen, immunostaining against Lm 14 days after infection. Original magnification, ×8 (severe inflammation with lymphoid necrosis). D: p47phox-/- spleen, diffuse inflammation with histiocytosis, 3 days after infection. Original magnification, ×8.
As illustrated in Figure 5A, live Lm stimulated significantly enhanced anti–Lm-specific IgM (25-fold) and IgG (45-fold) titers in p47phox-/- mice on day 120 compared with similarly challenged WT mice. Interestingly, the kinetics of IgM production showed that, although both WT and p47phox-/- titers peaked on day 7, the anti–Lm-specific IgM was sustained in p47phox-/- mice but decreased in WT mice over time. Strikingly, the p47phox-/- anti-Lm IgG titer increased 225-fold from day 14 to 120 compared with fivefold in WT mice. Furthermore, unlike UVi Lm, live Lm infection induced 24-fold higher anti–LLO-specific IgG titers in p47phox-/- mice than WT mice over time (Figure 5B). Thus, live Lm triggers a robust anti-Lm and anti-LLO IgG response in p47phox-/- mice.
Figure 5.
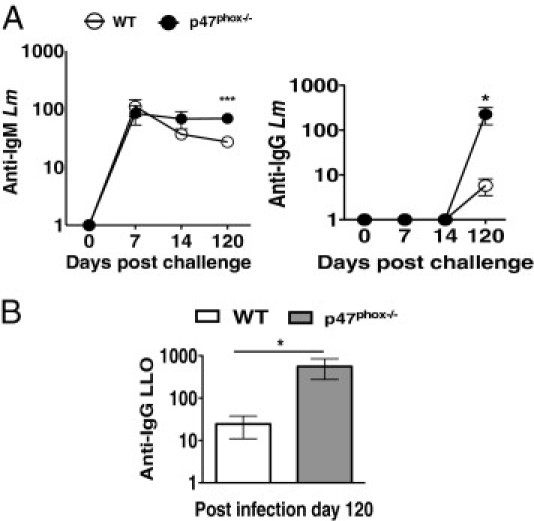
Ab production in response to live Lm challenge. Ten WT and p47phox-/- mice were infected i.v. with 5 × 104 CFUs (0.1 LD50) of Lm. A: Anti-Lm IgM and IgG. B: Anti-LLO IgG-specific serum titers were determined by ELISA on the indicated days. Absorbance values (Titer-1) represent the mean ± SEM. *P ≤ 0.05 and ***P ≤ 0.0005.
Parallel investigations in our laboratory have found that nearly 75% of p47phox-/- mice became moribund during the first week after Lm rechallenge compared with 25% of Nox2-/- and WT mice (Q. Liu, L. Yi, S. Sadiq-Ali, S.M. Walker, N. Zhu, and S.H. Jackson, unpublished data). Given these results and finding that both UVi and live Lm trigger enhanced anti-Lm and anti-LLO Ab production in p47phox-/- mice, we speculated that the observed humoral response may be protective against Lm in p47phox-/- mice. Therefore, to elucidate whether Lm elicited protective anti-Lm Ab production, we harvested immune serum from Lm-infected WT and p47phox-/- mice 6 days after Lm reinfection (see Materials and Methods) for passive Ab transfer into WT and p47phox-/- recipients 1 day before rechallenge with a lethal dose of Lm. As shown in Figure 6, all WT immune serum recipient mice survived lethal secondary Lm reinfection. Likewise, all of the p47phox-/- mice that received immune serum from secondarily reinfected p47phox-/- donors also survived lethal secondary Lm reinfection. In contrast, only 60% of the p47phox-/- mice that received WT immune serum samples survived lethal Lm secondary reinfection. These results demonstrate that, although both WT and p47phox-/- immune serum samples conveyed protection against secondary Lm infection, only the p47phox-/- immune serum samples rescued both WT and p47phox-/- mice and are, therefore, most effective.
Figure 6.
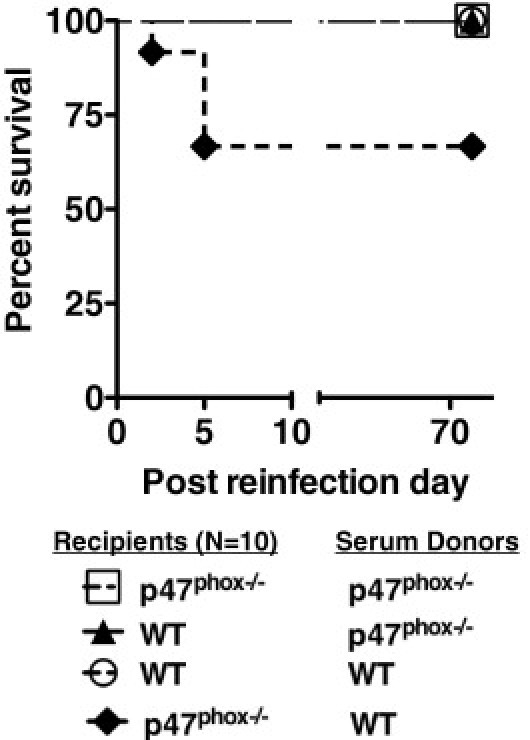
Serum from Lm-infected p47phox-/- mice is protective against secondary Lm challenge. Ten WT and p47phox-/- mice that survived primary i.v. Lm infection received 200 mL of pooled serum from the WT and p47phox-/- mice that survived Lm secondary reinfection, before reinfection with 10× LD50 of Lm. This figure represents survival of WT and p47phox-/- mice treated with pooled serum before secondary Lm reinfection. P = 0.005.
Discussion
We studied Ab production in p47phox-deficient (p47phox-/-) mice,15 a murine model of CGD,9 to examine the relative role of Nox2-dependent ROS deficiency in mediating antimicrobial humoral immunity. Similar to patients with CGD, genetically engineered murine models of the most common genetic variants of CGD each recapitulate that the phagocyte oxidase respiratory burst is critical for combating microbial infection15,17 and that hyperinflammation is a consequence of ROS deficiency. In addition, these models allow for complex analyses of microbial-induced Nox2-dependent innate and adaptive immune responses that are not possible in humans with CGD.
Chronic disease states, such as persistent and recurrent infection in an immunodeficient host, autoimmunity, and cancer, each pose distinct tissue insults that drive chronic, and often aberrant, inflammation that is not restorative. Furthermore, the mechanisms that drive systemic inflammation during these chronic disease states are not well defined. DC- and macrophage-derived inflammatory cytokines, such as IL-6, IL-12, and TNF-α, released early after infection play a key role in innate and adaptive host defenses against both Lm and Pn.26,27 TNF-α, IL-6, and IL-12 have also been important for protein-specific anti-Pn IgG production26 and critical for T-cell–mediated Lm clearance.28 Lm-activated APCs also stimulate CD4+ and CD8+ T cells to secrete interferon-γ, which further enhances macrophage bactericidal activity by preventing Lm escape from the phagosome29 and by triggering macrophages to produce antimicrobial-reactive Nox2-dependent oxygen and inducible nitric oxide synthase–dependent nitrogen intermediates.2,30 Interferon-γ also induces CD8+ T cells to lyse Lm-infected host cells.31,32 Consistent with previous reports33,34 using other microorganisms and Toll-like receptor ligands, we found that p47phox-/- APCs are hyperresponsive to stimulation with UVi Pn and Lm in vitro and that p47phox-/- APC proinflammatory cytokine secretion is exaggerated compared with WT APCs. We found that the expression of selective cell surface stimulatory molecules that regulate innate and adaptive immune cell function is exaggerated in Lm-stimulated p47phox-/- APCs. In addition, our findings indicate the novel observation that in vitro Lm-pulsed DCs from phagocyte oxidase p47phox-/- mice elicit a more robust humoral immune response against Lm in naïve recipient mice while similarly treated WT DCs did not. Collectively, these findings indicate a role for ROS modulation of proinflammatory cellular pathways in APCs. Furthermore, the data indicate that ROS deficiency modulates an inflammatory response that to leads enhanced humoral immunity.
Although ROS are critical inflammatory mediators, interestingly, hyperinflammation is a prominent disease manifestation in phagocyte oxidase–deficient patients with CGD. In addition, hypergammaglobulinemia has been reported to be a common occurrence in patients with CGD since the first clinical description by Charles A. Janeway, Jr., M.D., and colleagues in 1954.35,36 Notably, previous investigations of humoral immunity in murine models with reduced or change ascent to elevated Nox2 catalytic activity showed contrasting results. Richards and Clark37 reported that phagocyte oxidase–deficient gp91phox/Nox2-/- mice develop enhanced nonspecific IgG titers and dinitrophenyl hapten–specific anti–IgM and IgG titers. In contrast, anti–collagen-specific IgG titers were reduced in gp91phox/Nox2-/- mice with enhanced collagen-induced arthritis.34 Anti–collagen-specific IgG titers and collagen-induced arthritis were enhanced in Ncf1-mutant mice that have reduced ROS production.38–40 Our findings demonstrate that phagocyte oxidase–deficient p47phox-/- mice mount a more robust humoral immune response against UVi bacteria, Pn and Lm. Furthermore, our findings demonstrate that primary and recall Ab production against Pn and Lm is enhanced in p47phox-/- mice.
Consistent with our findings, Dreskin et al41 reported that patients with CGD and recently documented Staphylococcus aureus infection, defined as acute infection and infection within 2 years of analysis, had higher anti–S. aureus titers than normal controls and patients with CGD without a history of recent S. aureus infection. Thus, our findings implicate a role for Nox2-dependent ROS as inflammatory mediators that regulate Ab production and humoral immunity. The data also suggest that Nox2-dependent ROS influence critical processes in APC differentiation and function that attenuate proinflammatory cytokine production in combination with surface-stimulatory molecule expression, which activate Ab production.
During infection, Lm is absorbed by phagocytic cells and most bacteria are eliminated within phagosomes. However, occasionally, Lm escapes the phagosome by secreting LLO, a virulence factor that destroys the phagosomal membrane.42 The released Lm then enters the cytosol, where it is able to replicate. The consequent Lm cytosolic invasion triggers an innate inflammatory response and induces adaptive immunity.2,30 Although early Lm research4,43 indicated that Lm-induced Ab production was not specific for Lm resistance, Edelson et al5,6 demonstrated that antibodies against LLO provide resistance to Lm by neutralizing bacterial growth and pageing Lm escape into the cytosol. We found that IgG titers against Lm and LLO were significantly higher in p47phox-/- mice that were previously infected with Lm than similarly treated WT mice. In addition, we found that anti–serum samples from secondarily Lm-infected WT and p47phox-/- mice conveyed protection against Lm reinfection. Moreover, our findings indicate the novel observation that both WT and p47phox-/- recipients of serum from reinfected p47phox-/- mice were protected. In contrast, only 60% of p47phox-/- recipients of WT anti–serum samples were protected from secondary Lm infection. Thus, our findings indicate that, although ROS-deficient–dependent inflammation is not restorative, it can modulate an inflammatory response that is ultimately protective.
Acknowledgments
We thank Harry L. Malech and Clifford M. Snapper for careful review of the manuscript.
Footnotes
Supported by the Division of Intramural Research of the National Institutes of Health/National Institute of Allergy and Infectious Diseases and partially supported by the National Institutes of Health/National Center on Minority Health and Health Disparities.
References
- 1.Zenewicz L.A., Shen H. Innate and adaptive immune responses to Listeria monocytogenes: a short overview. Microbes Infect. 2007;9:1208–1215. doi: 10.10110/2/076/j.micinf.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pamer E.G. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 3.AlonsoDeVelasco E., Verheul A.F., Verhoef J., Snippe H. Streptoccocus pneumoniae: virulance factors, pathogenesis, and vaccines. Microbiol Rev. 1995;59:591–603. doi: 10.1128/mr.59.4.591-603.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackaness G.B. Cellular resistance to infection. J Exp Med. 1962;116:381–406. doi: 10.1084/jem.116.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edelson B.T., Cossart P., Unanue E.R. Paradigm revisited: antibody provides resistance to Listeria infection. J Immun. 1999;163:4087–4090. [PubMed] [Google Scholar]
- 6.Edelson B.T., Unanue E.R. Intracellular antibody neutralizes Listeria growth. Immunity. 2001;14:503–512. doi: 10.1016/s1074-7613(01)00139-x. [DOI] [PubMed] [Google Scholar]
- 7.Bokoch G.M., Knaus U.G. NADPH oxidases: not just for leukocytes anymore! Trends Biochem Sci. 2003;28:502–508. doi: 10.1016/S0968-0004(03)00194-4. [DOI] [PubMed] [Google Scholar]
- 8.Geiszt M., Leto T.L. The Nox family of NAD(P)H oxidases: host defense and beyond. J Biol Chem. 2004;279:51715–51718. doi: 10.1074/jbc.R400024200. [DOI] [PubMed] [Google Scholar]
- 9.Heyworth P.G., Cross A.R., Curnutte J.T. Chronic granulomatous disease. Curr Opin Immunol. 2003;15:578–584. doi: 10.1016/s0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 10.Williams D.A., Tao W., Yang F., Kim C., Gu Y., Mansfield P., Levine J.E., Petryniak B., Derrow C.W., Harris C., Jia B., Zheng Y., Ambruso D.R., Lowe J.B., Atkinson S.J., Dinauer M.C., Boxer L. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood. 2000;96:1646–1654. [PubMed] [Google Scholar]
- 11.Ambruso D.R., Knall C., Abell A.N., Panepinto J., Kurkchubasche A., Thurman G., Gonzalez-Aller C., Hiester A., deBoer M., Harbeck R.J., Oyer R., Johnson G.L., Roos D. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci U S A. 2000;97:4654–4659. doi: 10.1073/pnas.080074897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matute J.D., Arias A.A., Wright N.A., Wrobel I., Waterhouse C.C., Li X.J., Marchal C.C., Stull N.D., Lewis D.B., Steele M., Kellner J.D., Yu W., Meroueh S.O., Nauseef W.M., Dinauer M.C. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114:3309–3315. doi: 10.1182/blood-2009-07-231498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Assari T. Chronic granulomatous disease: fundamental stages in our understanding of CGD. Med Immunol. 2006;5:4. doi: 10.1186/1476-9433-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenzweig S.D. Inflammatory manifestations in chronic granulomatous disease (CGD) J Clin Immunol. 2008;28(Suppl 1):S67–S72. doi: 10.1007/s10875-007-9160-5. [DOI] [PubMed] [Google Scholar]
- 15.Jackson S.H., Gallin J.I., Holland S.M. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Q., Cheng L.I., Yi L., Zhu N., Wood A., Changpriroa C.M., Ward J.M., Jackson S.H. p47phox deficiency induces macrophage dysfunction resulting in progressive crystalline macrophage pneumonia. Am J Pathol. 2009;174:153–163. doi: 10.2353/ajpath.2009.080555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollock J.D., Williams D.A., Gifford M.A., Li L.L., Du X., Fisherman J., Orkin S.H., Doerschuk C.M., Dinauer M.C. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 18.Foulds K.E., Rotte M.J., Seder R.A. IL-10 is required for optimal CD8 T cell memory following Listeria monocytogenes infection. J Immunol. 2006;177:2565–2574. doi: 10.4049/jimmunol.177.4.2565. [DOI] [PubMed] [Google Scholar]
- 19.Vasilevsky S., Colino J., Puliaev R., Canaday D.H., Snapper C. Macrophages pulsed with Streptococcus pneumoniae elicit a T cell-dependant antibody response upon transfer into naive mice. J Immunol. 2008;181:1787–1797. doi: 10.4049/jimmunol.181.3.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zavilgesky G.B., Gurzadyan G.G., Nikogosyan D.N. Pyrimidine dimers, single-strand breaks and crosslinks induced DNA by powerful laser UV irradiation. Photochem Photobiol. 1984;8:175–187. [Google Scholar]
- 21.Colino J., Shen Y., Snapper C.M. Dendritic cells pulsed with intact Streptococcus pneumoniae elicit both protein- and polysaccharide-specific immunoglobulin isotype responses in vivo through distinct mechanisms. J Exp Med. 2002;195:1–13. doi: 10.1084/jem.20011432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhunia A.K. Antibodies to Listeria monocytogenes. Crit Rev Microbiol. 1997;23:77–107. doi: 10.3109/10408419709115131. [DOI] [PubMed] [Google Scholar]
- 23.Dinauer M.C., Deck M.B., Unanue E.R. Mice lacking reduced nicotinamide adenine dinucleotide phosphate oxidase activity show increased susceptibility to early infection with Listeria monocytogenes. J Immunol. 1997;158:5581–5583. [PubMed] [Google Scholar]
- 24.Shiloh M.U., MacMicking J.D., Nicholson S., Brause J.E., Potter S., Marino M., Fang F., Dinauer M., Nathan C. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 1999;10:29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- 25.Endres R., Luz A., Schulze H., Neubauer H., Futterer A., Holland S.M., Wagner H., Pfeffer K. Listeriosis in p47(phox-/-) and TRp55-/- mice: protection despite absence of ROI and susceptibility despite presence of RNI. Immunity. 1997;7:419–432. doi: 10.1016/s1074-7613(00)80363-5. [DOI] [PubMed] [Google Scholar]
- 26.Khan A.Q., Shen Y., Wu Z.-Q., Wynn T.A., Snapper C.M. Endogenous pro- and anti-inflammatory cytokines differentially regulate an in vivo humoral response to Streptococcus pneumoniae. Infect Immun. 2002;70:749–761. doi: 10.1128/iai.70.2.749-761.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tam M.A., Wick M.J. Dendritic cells and immunity to Listeria: tipDCs are a new recruit. Trends Immunol. 2004;25:335–339. doi: 10.1016/j.it.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Nakane A., Numata A., Minagawa T. Endogenous tumor necrosis factor, interleukin-6, and gamma interferon levels during Listeria monocytogenes infection in mice. Infect Immun. 1992;60:523–528. doi: 10.1128/iai.60.2.523-528.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Portnoy D.A., Schreiber R.D., Connely P., Tilney L.G. Gamma interferon limits access of Listeria monocytogenes to the macrophage cytoplasm. J Exp Med. 1989;170:2141–2146. doi: 10.1084/jem.170.6.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaughnessy L.M., Swanson J.A. The role of the activated macrophage in clearing Listeria monocytogenes infection. Front Biosci. 2007;12:2683–2692. doi: 10.2741/2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gregory S.H., Liu C.C. CD8+ T-cell-mediated response to Listeria monocytogenes taken up in the liver and replicating within hepatocytes. Immunol Rev. 2000;174:112–122. doi: 10.1034/j.1600-0528.2002.017405.x. [DOI] [PubMed] [Google Scholar]
- 32.Jiang X., Gregory S.H., Wing E.J. Immune CD8+ T lymphocytes lyse Listeria monocytogenes-infected hepatocytes by a classical MHC class I-restricted mechanism. J Immunol. 1997;158:287–293. [PubMed] [Google Scholar]
- 33.Brown K.L., Bylund J., MacDonald K.L., Song-Zhao G.X., Elliott M.R., Falsafi R., Hancock R.E., Speert D.P. ROS-deficient monocytes have aberrant gene expression that correlates with inflammatory disorders of chronic granulomatous disease. Clin Immunol. 2008;129:90–102. doi: 10.1016/j.clim.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 34.George-Chandy A., Nordstrom I., Nygren E., Jonsson I.M., Postigo J., Collins L.V., Eriksson K. Th17 development and autoimmune arthritis in the absence of reactive oxygen species. Eur J Immunol. 2008;38:1118–1126. doi: 10.1002/eji.200737348. [DOI] [PubMed] [Google Scholar]
- 35.Carnide E.G., Jacob C.A., Castro A.M., Pastorino A.C. Clinical and laboratory aspects of chronic granulomatous disease in description of eighteen patients. Pediatr Allergy Immunol. 2005;16:5–9. doi: 10.1111/j.1399-3038.2005.00225.x. [DOI] [PubMed] [Google Scholar]
- 36.Bridges R.A., Berendes H., Good R.A. A fatal granulomatous disease of childhood: the clinical, pathological, and laboratory features of a new syndrome. AMA J Dis Child. 1959;97:387–408. [PubMed] [Google Scholar]
- 37.Richards S.M., Clark E.A. BCR-induced superoxide negatively regulates B-cell proliferation and T-cell-independent type 2 Ab responses. Eur J Immunol. 2009;39:3395–3403. doi: 10.1002/eji.200939587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hultqvist M., Olofsson P., Holmberg J., Backstrom B.T., Tordsson J., Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci U S A. 2004;101:12646–12651. doi: 10.1073/pnas.0403831101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hultqvist M., Backlund J., Bauer K., Gelderman K.A., Holmdahl R. Lack of reactive oxygen species breaks T cell tolerance to collagen type II and allows development of arthritis in mice. J Immunol. 2007;179:1431–1437. doi: 10.4049/jimmunol.179.3.1431. [DOI] [PubMed] [Google Scholar]
- 40.Hagenow K., Gelderman K.A., Hultqvist M., Merky P., Backlund J., Frey O., Kamradt T., Holmdahl R. Ncf1-associated reduced oxidative burst promotes IL-33R+ T cell-mediated adjuvant-free arthritis in mice. J Immunol. 2009;183:874–881. doi: 10.4049/jimmunol.0900966. [DOI] [PubMed] [Google Scholar]
- 41.Dreskin S.C., Goldsmith P.K., Gallin J.I. Immunoglobulins in the hyperimmunoglobulin E and recurrent infection (Job's) syndrome: deficiency of anti-Staphylococcus aureus immunoglobulin A. J Clin Invest. 1985;75:26–34. doi: 10.1172/JCI111683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bielecki J., Youngman P., Connelly P., Portnoy D.A. Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature. 1990;345:175–176. doi: 10.1038/345175a0. [DOI] [PubMed] [Google Scholar]
- 43.Osebold J.W., Sawyer M.T. Immunization studies on listeriosis in mice. J Immunol. 1957;78:262–268. [PubMed] [Google Scholar]