

Abstract
Alzheimer's disease (AD) is characterized by the brain accumulation of Aβ peptides and by the presence of neurofibrillary tangles. Aβ is believed to play an important role in AD and it has been shown that certain flavonoids can affect Aβ production. Recently, it was suggested that the Aβ lowering properties of flavonoids are mediated by a direct inhibition the β-secretase (BACE-1) activity, the rate limiting enzyme responsible for the production of Aβ peptides. Westernblots and ELISAs were employed to monitor the impact of flavonoids on amyloid precursor protein processing and Aβ production. A cell free chemoluminescent assay using human recombinant BACE-1 was used to assess the effect of flavonoids on BACE-1 activity. The effect of flavonoids on NFκB activation was determined by using a stable NFκB luciferase reporter cell line. Molecular docking simulations were performed to predict the binding of flavonoids to the BACE-1 catalytic site. Real time quantitative PCR was used to determine the effect of flavonoids on BACE-1 transcription. We show in a cell free assay that flavonoids are only weak inhibitors of BACE-1 activity. Docking simulation studies with different BACE-1 structures also suggest that flavonoids are poor BACE-1 inhibitors as they appear to adopt various docking poses in the active site pocket and have weak docking scores that differ as a function of the BACE-1 structures studied. Moreover, a weak correlation was observed between the effect of flavonoids on Aβ production in vitro and their ability to lower BACE-1 activity suggesting that the Aβ lowering properties of flavonoids in whole cells are not mediated via direct inhibition of BACE-1 activity. We found however a strong correlation between the inhibition of NFκB activation by flavonoids and their Aβ lowering properties suggesting that flavonoids inhibit Aβ production in whole cells via NFκB related mechanisms. As NFκB has been shown to regulate BACE-1 expression, we show that NFκB lowering flavonoids inhibit BACE-1 transcription in human neuronal SH-SY5Y cells. Altogether, our data suggest that flavonoids inhibit Aβ and sAPPβ production by regulating BACE-1 expression and not by directly inhibiting BACE-1 activity.
Background
Alzheimer's disease (AD) is a major health concern among the aging population and is the most prevalent form of dementia. While the cause of the disease remains uncertain, the extracellular senile plaques and the intracellular neurofibrillary tangles constitute the two major neuropathological hallmarks present in the brains of AD patients. Neurofibrillary tangles contain hyperphosphorylated microtubule-associated protein tau, while senile plaques contain a core of β-amyloid (Aβ) peptides. Although the central role of Aβ remains to be proven in clinical trials, data accumulated during the past two decades place Aβ peptides and in particular soluble forms of the peptide as being the main molecule triggering the pathological cascade that eventually leads to AD and initiates tau pathology [1]. Aβ peptides are derived from the cleavage of the β-amyloid precursor protein (APP) by β- and γ-secretases. The major β-secretase is an aspartyl protease termed BACE-1 (β-site APP cleaving enzyme) [2–4]. BACE-1 cleaves APP within the extracellular domain of APP, resulting in the secretion of the large ectodomain (APPsβ) and generating a membrane-tethered C-terminal fragment CTFβ or C99 which serves as a substrate for γ-secretase [5]. The multimeric γ-secretase complex cleaves at multiple sites within the transmembranous CTFβ generating C-terminally heterogeneous Aβ peptides ranging between 38 to 43 amino-acid residues in length that are secreted [6]. In addition to BACE-1 and γ-secretase, APP can be cleaved by α-secretase within the Aβ domain between Lys16 and Leu17, releasing APPsα and generating CTFα or C83 which is further cleaved by γ- secretase to generate an N-terminally truncated Aβ termed p3. Genetic ablation of BACE-1 completely abolishes Aβ production, establishing BACE-1 as the major neuronal enzyme responsible for initiating the amyloidogenic processing of APP [7].
Current treatments for AD include cholinesterase inhibitors and glutamate antagonists. Although useful, these symptomatic treatments do not stop the disease process or prevent neuronal degeneration. There is an on-going need for the development of new treatments for AD. It has been suggested that a diet rich in polyphenols including flavonoids may have beneficial effects in AD [8]. Flavonoids are plant metabolites that are dietary antioxidant, and it has been hypothesized that this activity may account for their beneficial effects against dementia [9]. The Ginkgo biloba extract EGb761 which contains essentially flavonoids (quercetin, kaempferol and isorhamnetin) and terpene lactones (ginkgolides A,B,C and bilobalide) has also been suggested to have positive effects against dementia and AD [10, 11]. Recently, several flavonoids have been shown to regulate Aβ production and it has been suggested that these compounds act by directly inhibiting BACE-1 activity [12]. As BACE-1 is the rate limiting enzyme responsible for Aβ production and is considered to be a prime target for AD, we further investigated whether flavonoids can lower Aβ production in whole cells by directly inhibiting BACE-1 activity. We tested the effects of different flavonoids on Aβ production and APP processing using a cell line overexpressing human APP and attempted to correlate the Aβ lowering activity of the flavonoids with their BACE-1 inhibitory activity. Moreover, we investigated the binding affinity of flavonoids for the BACE-1 catalytic site using thorough docking simulations to determine whether flavonoids hold promise as BACE-1 inhibitors.
Methodology
Flavonoids
Daidzein (4',7-Dihydroxyisoflavone, 7-Hydroxy-3-(4-hydroxyphenyl)-4H-1- benzopyran-4-one, 7-Hydroxy-3-(4-hydroxyphenyl)chromone), genistein (4',5,7-Trihydroxyisoflavone, 5,7-Dihydroxy-3-(4-hydroxyphenyl)-4H-1- benzopyran-4-one), luteolin (3',4',5,7-Tetrahydroxyflavone), kaempferol (3,4',5,7-Tetrahydroxyflavone, 3,5,7-Trihydroxy-2-(4-hydroxyphenyl)-4H-1- benzopyran-4-one), apigenin (4',5,7-Trihydroxyflavone), quercetin (2-(3,4- Dihydroxyphenyl)-3,5,7-trihydroxy-4H-1-benzopyran-4-one, 3,3',4',5,6- Pentahydroxyflavone), α-naphthoflavone (7,8-Benzoflavone), β- naphthoflavone (5,6-Benzoflavone), acacetin (5,7-Dihydroxy-4'- methoxyflavone), taxifolin (3,3',4',5,7-Pentahydroxyflavanone hydrate), PD98059 (2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) were obtained from Sigma Chemicals (MO, USA). Aminogenistein (4'-Amino-6- hydroxyflavone) and baicalein (5,6,7-Trihydroxyflavone) were obtained from Calbiochem (EMD Chemicals, CA, USA).
Aβ enzyme-linked immunosorbent assay (ELISA)
7W CHO cells stably transfected with human APP751 [13] were maintained in DMEM (ATCC, VA, USA) medium containing 10% fetal bovine serum (Invitrogen, CA, USA), 1X mixture of penicillin/streptomycin/fungizone mixture (Cambrex, ME, USA) and 0.3% geneticin (Invitrogen, CA, USA) as a selecting agent. Cells were cultured in 96-wells culture plates and treated for 24 hours with different doses of flavonoids as indicated in the figure legend. All flavonoids were diluted in DMSO before being exposed to confluent 7W CHO cells so that the final concentration of DMSO in the culture medium was 0.1%. The control wells were treated with 0.1% DMSO. Human Aβ1-40 and Aβ1-42 were analyzed in the culture medium by using commercially available sandwich ELISAs (Invitrogen, CA) according to the manufacturer's instructions. All experiments were repeated 3-4 times.
Evaluation of APP processing by Western-blots
The impact of flavonoids on APP processing was evaluated using 7W CHO cells as we previously published [14]. Briefly, confluent 7W CHO cells were treated for 24 hours with 10 and 20 µM of flavonoids in 24-well plates. Cellular proteins were extracted with 80 µL of ice-cold M-PER Reagent (Pierce Biotechnology, Rockford, IL, USA) containing 1mM phenylmethanesulfonyl fluoride, 1X of protease cocktail inhibitor (Roche, Inc., USA) and 1mM sodium orthovanadate. Samples were sonicated, denatured by boiling in Laemmli buffer (Bio-Rad, Hercules, CA, USA) and resolved onto 4− 20% gradient polyacrylamide gels (Bio-Rad, Hercules, CA, USA). After electrotransfering onto polyvinylidene difluoride membranes, western-blots were immunoprobed with a 1:1000 dilution of an anti-APP C-terminal (751− 770) antibody (EMD Biosciences Inc., San Diego, CA, USA), with an antiactin antibody (Chemicon, Temecula, CA, USA) used as a reference antibody to ensure that equal amount of proteins were electrotransferred. Additionally, sAPPα was detected by Western-blot in the culture medium surrounding 7W CHO cells using the antibody 6E10 (Signet Laboratories Inc., MA, USA) which recognizes amino acids 1−17 of Aβ and sAPPβ was detected in the culture medium using an anti-human sAPPβ antibody (Immuno-Biological Laboratories Co. Ltd., Gunma, Japan).
β-secretase activity measurements
β-secretase activity was measured using human recombinant BACE-1 (Calbiochem, CA, USA) with a commercially available chemoluminescent assay (Discoverix, CA, USA) following the recommendations of the manufacturer. The β-secretase inhibitor IV (BACE IV) inhibitor was used as a positive control in the assay and was purchased from EMD Chemicals (CA, USA). Briefly, BACE-1 enzymatic reactions were carried out for 2 hours at room temperature with 10 ng/µl of BACE-1 recombinant enzyme in 96-well plates in a final volume of 100 µl. Chemoluminescent signals were quantified on a HTS Synergy multiplate reader from Biotek (VT, USA).
NFκB luciferase activity
NFκB activation was quantified using a stable NFκB luciferase reporter cell line of HEK293 cells with chromosomal integration of a luciferase reporter construct regulated by 6 copies of the NFκB response element (Panomics, CA, USA). Cells were grown in DMEM containing 10% serum, 1% penicillin/streptomycin/fungizone and 100 µg/ml of hygromycin B. Confluent cells were treated with 20 ng/ml of TNFα (Sigma, MO, USA) to induce NFκB activation and with a dose range of the different flavonoids for 3 hours. Luciferase activity was detected with the Luc-Screen Extended-Glow from Applied Biosystems (CA, USA) and a Synergy HT Biotek chemoluminescent reader (VT, USA) as we previously described [14].
Evaluation of BACE-1 transcription
Confluent SH-SY5Y cells (ATCC, VA, USA) grown in DMEM/F12 medium supplemented with 10% fetal bovine serum and 1X mixture of penicillin/streptomycin/fungizone mixture were treated with 20 ng/ml of TNFα, or with 20 µM of apigenin, luteolin, quercetin and daidzein either alone or in combination with 20 ng/ml of TNFα for 30 minutes at 37°C, 5% CO2 (control wells received the same volume of vehicle used to dissolved the flavonoids). After 30 minutes of incubation, RNA was extracted as we previously described [15]. The quality and purity of the RNA obtained were tested on agarose gels and spectrophotometrically at 260 nm and 280 nm. All RNA samples had an A260/280 absorbance ratio between 1.9 and 2.1. Real time quantitative PCR (RT-qPCR) were performed as we previously described [15]. Briefly, to quantify the transcript levels of the BACE-1 gene by RTqPCR, a protocol using FastStart TaqMan Prob Master Mix (Roche) reaction was performed in duplicates. All reactions contained 2 µL of cDNA (20 ng), 10 µL of the 2× Master Mix and 0.5 µL of 20 µM of each BACE-1 primer (ttcatcaacggctccaact and ctccagggagtcgtcagg), 250 nM of BACE-1 gene specific probe (#04688058001), 500 nM reference (HPRT) gene primer mix (cgtgattagtgatgatgaaccag and cgagcaagacgttcagtcct), 250nM reference HPRT gene probe (# 05046157001) and DEPC-treated water to a final volume of 20µl. The reaction protocol started with a 2-min activation step at 50°°C, a 10 min template denaturation step at 95°C, followed by 50 cycles of 95°C for 15 sec and 60°C for 20 sec, BACE-1 mRNA fold change (relative to the hypoxanthine-phosphoribosyl-transferase (HPRT) mRNA was calculated as previously described [15].
Molecular docking study of flavonoids
Information regarding selected Flavonoids 2D structure was obtained as SMILES notation from Pubchem database (http://pubchem.ncbi.nlm.nih.gov/) for: Genistein, Quercetin, Taxifolin, Kaemferol, PD98059, Luteolin, Apigenin, Daizein, Aminogeneistein, alpha- and beta-napthofalvone. Each of the 2D ligand string was converted into 3D conformers using LigPrep tool and each conformer was further energy minimized by applying OPLS_2005 force field. Low energy conformer for each ligand was retained and subsequently used for docking experiments. We used GLIDE XP (eXtra Precision release 2010, Schordinger Inc, USA) to perform docking against two different crystal structures of BACE-1 from the PDB files 2B8L and 2QMF. The ligand bound active site pocket in the two crystal structures were identified and a grid map was generated using the centroid of the bound ligand. We used a Vander-Waals radius scaling factor of 1.0 and a partial charge cutoff of 0.25 to soften the potential of non-polar atoms in the BACE pocket and no other explicit constraints were specified. Each docking run samples a variety of poses for the ligand in the BACE active site pocket. An energetic and empirical scoring criterion is used to rank several thousand docked conformations. The scoring algorithm rewards (favorable interactions) or penalizes (unfavorable interactions) the Glide XP score (scoring function) which can be used to further rank the docked pose. A Van der Waals scaling factor of 0.8 for the ligand was used in order to soften the potential for non-polar ligand atoms. We also performed a post-dock minimization run to obtain favorable conformations and to reduce any strain. We applied a strain correction factor (to account for excessive strain due to conformational restriction upon binding to the pocket) to the Glide XP score whenever the observed strain energy of bound ligand exceeds 4 kcal/mol. A docked pose with a low Glide XP score was considered to be the most favorable state and ranked as the top conformer.
Statistical analysis
Data were expressed as means ± S.E.M of n experiments. The statistical significance of the differences between treatment groups was determined by one-way ANOVA and post-hoc comparisons where appropriate using SPSS 12.0.1 for Windows. The half maximal inhibitory concentration (IC50) for the different assays was determined using GraphPad Prism 5 (GraphPad Software, Inc., CA, USA).
Results
Effect of flavonoids on Aβ, sAPPβ and sAPP± production
The impact of flavonoids on Aβ production was evaluated using 7W CHO cells overexpressing wild-type human APP. Following 24 hours of treatment with a dose range of flavonoids, the amount of human Aβ1-40 and Aβ1-42 in the culture media surrounding the 7W CHO cells was measured by ELISAs. A dose dependent inhibition of Aβ1-40 and Aβ1-42 production was observed for β-naphtoflavone, aminogenistein, luteolin, apigenin, quercetin, acacetin and PD98059 whereas genistein, ±-naphtholavone, kaempferol baicalein, daidzein and taxifolin were inefficient for the dose range tested (Figure 1). The calculated IC50 for Aβ1-40 and Aβ1-42 were as follows for the most potent flavonoids identified; β-naphtoflavone (0.9 and 1.1 µM)<aminogenistein (5.7 and 5.8 µM)<luteolin (6.5 and 8.8 µM)<apigenin (7.1 and 10.8 µM)<quercetin (10.2 and 10.2 µM). Interestingly, a statistically significant correlation was observed between Aβ1-40 and Aβ1-42 values showing that active flavonoids are inhibiting Aβ1-40 and Aβ1-42 with the same potency (Figure 1C).
Figure 1.
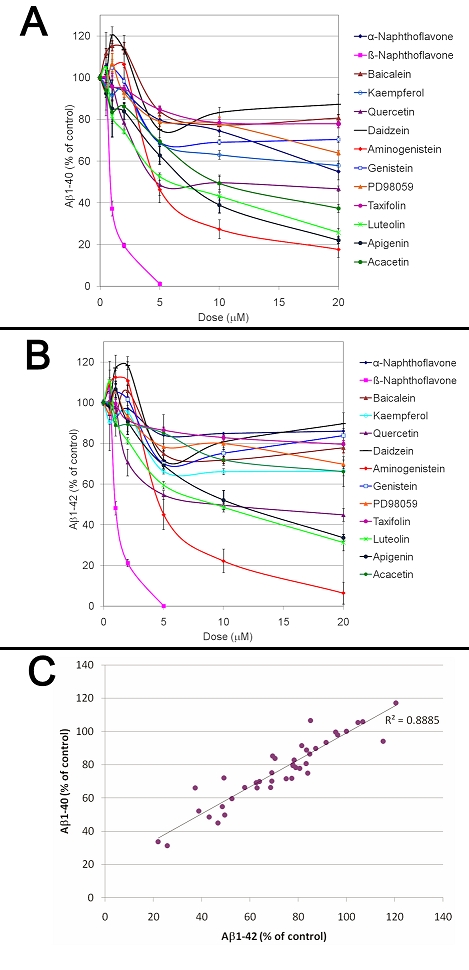
Dose dependent effects of flavonoids on Aβ production. The effect of a dose range of flavonoids on Aβ1-40 (A) and Aβ1-42 (B) production was investigated using 7W CHO cells overexpressing human APP after 24 hours of treatment. ANOVA reveals a significant main effect of the treatments (P<0.001) and doses (P<0.001) on both Aβ1-40 and Aβ1-42 production. Posthoc comparisons show significant differences for Aβ1-40 production between the control conditions and the β-naphthoflavone (P<0.001), kaempferol (P<0.04), quercetin (P<0.001), aminogenistein (P<0.001), luteolin (P<0.001), apigenin (P<0.001) and acacetin (P<0.001) treatments but no significant differences between the control conditions and baicalein, α-naphthoflavone, daidzein, genistein, PD98059 and taxifolin (P>0.05) treatments. Post-hoc comparisons show significant differences for Aβ1-42 production between the control conditions and the β-napthoflavone (P<0.001), quercetin (P<0.001), aminogenistein (P<0.001), luteolin (P<0.001) and apigenin (P<0.008) but no significant differences between the control conditions and baicalein, α- naphthoflavone, kaempferol, daidzein, genistein, PD98059, taxifolin and acacetin (C). Graph showing a correlation between Aβ1-40 and Aβ1-42 values obtained following treatments with different doses of the Aβ lowering flavonoids. A strong correlation between Aβ1-40 and Aβ1-42 values was observed (Pearson correlation coefficient=0.911; P<0.001) showing that flavonoids inhibit Aβ1-40 and Aβ1-42 with similar potency.
We next investigated the effects of flavonoids on sAPPβ and sAPP± secretion by 7W CHO cells. None of the flavonoids tested appear to stimulate sAPP± production suggesting that these compounds do not stimulate the ±-secretase cleavage of APP (Figure 2). Among the flavonoids tested, quercetin, kaempferol, luteolin, apigenin and β-naphtoflavone appear to reduce sAPPβ production suggesting an inhibition of the β-cleavage of APP (Figure 2). We did not observe a significant impact of PD98059 and aminogenistein (at 10 and 25 µM) on sAPPβ production, however a reduction in sAPPβ secretion was observed with a 50 µM dose of PD98059 but not for aminogenistein, daidzein, genistein and aminogenistein (Figure 2) suggesting that among the flavonoids tested only luteolin, apigenin, quercetin, β-naphthoflavone, taxifolin, kaempferol and PD98059 impact the β-cleavage of APP. We analyzed the effect of some of the flavonoids on APP C-terminal fragments and observed that daidzein, aminogenistein and genistein which were unable to inhibit sAPPβ production do not inhibit APP-CTFβ generation, and to the contrary induce an accumulation of APP-CTF± and CTFβ suggesting inhibition of the γ-secretase cut (Figure 2). A decreased APP-CTFβ production as well as an inhibition of sAPPβ secretion was observed for luteolin, quercetin, apigenin and kaempferol (Figure 2) suggesting that these flavonoids lower Aβ production by impacting the β-cleavage of APP.
Figure 2.

Effects of different flavonoids on APP processing in 7W CHO cells overexpressing human APP. (A) Effect of a 24 hour treatment with different flavonoids (10 and 25 µM) on sAPPβ and sAPPα production by 7W CHO cells. Luteolin, apigenin, β-naphthoflavone, quercetin and kaempferol appear to reduce sAPPβ production. (B) Effect of a 24 hour treatment with different flavonoids at a concentration of 50 µM on sAPPβ and sAPPα production by 7W CHO cells. An inhibition of sAPPβ production was observed with quercetin, taxifolin, kaempferol, PD98059, luteolin, apigenin and β- naphtoflavone, however genistein, aminogenistein, baicalein and daidzein did not impact sAPPβ production. (C) Impact of a 24 hour treatment on APP Cterminal fragments with different flavonoids at a concentration of 20 µM. A reduction in APP-CTFβ production was observed for luteolin, quercetin, apigenin whereas an accumulation of APP-CTFα and APP-CTFβ (suggesting an impact on γ-secretase activity) was observed for daidzein, aminogenistein and genistein. Altogether these data suggest that quercetin, taxifolin, kaempferol, PD98059, luteolin, apigenin and β-naphtoflavone inhibit the β- cleavage of APP.
Effects of flavonoids on BACE-1 activity
As many of the flavonoids tested appear to inhibit the β-cleavage of APP, we determined whether these flavonoids were direct inhibitors of the β-secretase (BACE-1) using a chemoluminescent cell free assay. We selected a chemoluminescent assay over a fluorescent based assay for measuring BACE-1 activity since flavonoids are fluorescent (data not shown) and may interfere with the detection of BACE-1 activity by fluorescence. Among the flavonoids tested, baicalein appears the most potent for inhibiting BACE-1 activity with an IC50 around 10 µM (Figure 3), however this compound is unable to significantly lower Aβ or sAPPβ production in whole cells (Figure 2). Luteolin, quercetin, kaempferol and apigenin dose dependently inhibited BACE-1 activity (Figure 3). Overall, the flavonoids tested appear weak inhibitors of BACE-1 activity in the cell free assay and the calculated IC50 values were as follows; baicalein (10.2 µM)<kaempferol (27.7 µM)<quercetin (30.2 µM)<apigenin (34.2 µM)<luteolin (56.5 µM). To further evaluate the possible interaction of the flavonoids with BACE-1, we performed in silico docking simulations. The results of our docking experiments along with the Glide XP score is summarized in Table 1. We observed that the independent docking experiments based on two X-ray structures gave different rank orders of the flavonoids tested. As such, we observed a correlation coefficient of 0.33 (p =0.12) showing no significant relationship between the Glide XP score of the docked flavonoids from the two different docking experiments (Figure 4). We further aligned the docked structure of flavonoids from the different docking experiments and found that the docked poses do not align. We observed that the highest docking score difference was for apigenin which had a Glide XP score of -9.01 while docking to 2B8L and -3.18 while docking to 2QMF. The best score among flavonoids observed while docking to 2B8L was for taxifolin (XP score -10.49) and worst for daizein (XP score -2.91). While docking flavonoids to 2QMF, we found that the best score was observed for baicalein (-8.81) and the worst for apigenin (-3.18). Based on the observed BACE-1 IC50 in a cell free assay, we found that the docking experiment with 2QMF had a better score for active flavonoids that inhibited BACE-1 at submicromolar range. Overall the flavonoids tested appear to be only weak BACE-1 inhibitors. In addition, only a weak correlation between the inhibition of BACE-1 activity and the inhibition of Aβ production by flavonoids in whole cells (Figure 5) was observed and their IC50 for inhibiting BACE-1 activity in a cell free assay is higher than their IC50 for inhibiting Aβ production in whole cells suggesting these compounds mitigate Aβ production via additional mechanisms independently of direct BACE-1 inhibition.
Figure 3.

Effects of flavonoids on BACE-1 activity in a cell free assay. ANOVA reveals a significant main effect of the treatment groups across the doses tested (P<0.001). Post-hoc comparisons show statistical significant BACE-1 activity inhibition across the doses tested for baicalein (P<0.001), apigenin (P<0.02), luteolin (P<0.02), quercetin (P<0.001) and kaempferol (P<0.001) but no significant effect of α-naphthoflavone, β-naphthoflavone, genistein, daidzein, taxifolin, PD98059 and aminogenitein on BACE-1 activity (P>0.05).
Figure 4.

(A) Graph showing the docking scores (Observed Glide XP score) correlation for the flavonoids between two crystal structures for BACE-1 (2B8L and 2QMF). The Different flavonoids used in this study are indicated on the graph by the first two letters as bolded here: Genistein, Quercetin, Taxifolin, Kaemferol, PD98059, Luteolin, Apigenin, Daizein, Aminogeneistein, alpha- and beta-Napthofalvone. No significant correlation (P>0.1) was observed between the two scores. In addition no correlation between the different docking scores and the amount of BACE-1 inhibition induced by flavonoids was observed showing that docking simulations do not predict accurately the affinity of flavonoids for the BACE-1 catalytic site. (B) Overlay of several different BACE-1 PDB structures revealing flexible loops (Loop1 and Loop2) in the active site pocket. Different ligand binding sites (using the following PDB structures: 2B8L, 2QMF, 3CIB, 3L5F, 3KMX) in the BACE-1 catalytic site are indicated by space fill dot representation. The active site aspartyl residues (Asp 32 and Asp 228) are represented in red space fill in the left. (C) Docking of kaemferol to BACE-1 structures. Overlay of crystal structure of BACE-1 from PDB file 2B8L (brown ribbon) and 2QMF (green ribbon) along with the top ranked docked pose of kaemferol. Left Side is a zoom of the active site showing differences in the docking orientation of kaempferol in 2B8L (ligand represented by pink sticks) and 2QMF (white sticks). (D) Docking of quercetin to BACE-1 structures. Overlay of crystal structure of BACE-1 from PDB file 2B8L (brown ribbon) and 2QMF (green ribbon) along with top ranked docked pose of quercetin. Left Side is a zoom of the active site showing differences in the docking orientation of quercetin in 2B8L (ligand represented by pink sticks) and 2QMF (white sticks).
Figure 5.

Graphs representing the correlation between the inhibition of BACE- 1 activity observed in a cell free assay and the inhibition of Aβ1-40 (A) and Aβ1-42 (B) production in whole cells for the different flavonoids tested. Only a weak correlation was observed between Aβ production and BACE-1 inhibition (Pearson correlation coefficient=0.471; P<0.05).
Effect of flavonoids on NFκB activity and BACE-1 transcription
We next investigated the effect of flavonoids on NFκB activity since flavonoids are known to display anti-inflammatory properties [16] and since we have shown previously that compounds of unrelated structure that inhibit NFκB activation can lower Aβ production by targeting the β-cleavage of APP [17, 18]. We observed that all of the flavonoids unable to significantly lower Aβ level in whole cells for the dose range tested (PD98059, taxifolin, genistein, daidzein and baicalein) were also unable to reduce NFκB activation induced by TNF±, whereas Aβ and APPsβ lowering flavonoids (kaempferol, quercetin, acacetin, apigenin and luteolin) dose dependently inhibited NFκB activation (Figure 6). Interestingly, we found a strong correlation between the amount of NFκB inhibition and the level of Aβ1-40 and Aβ1-42 inhibition for the different doses of the flavonoids tested, suggesting the Aβ lowering activity of the flavonoids is mediated via NFκB (Figure 6). We next investigated the possible impact of daidzein, apigenin, luteolin and quercetin on BACE-1 transcription using human neuronal SHSY cells and confirmed that NFκB lowering flavonoids (apigenin, luteolin and quercetin) inhibit BACE-1 transcription stimulated by TNF± whereas daidzein, which does not significantly inhibit NFκB activity, does not affect BACE-1 transcription (Figure 7).
Figure 6.

(A) Dose dependent inhibition of NFκB activity by Aβ lowering flavonoids. HEK293 cells stably expressing an NFκB luciferase reporter construct were co-treated with 20 ng/ml of TNFα and different doses of the flavonoids for 3 hours before measuring NFκB luciferase activity. ANOVA reveals a significant main effect of the flavonoid treatments (P<0.001) and of the doses used (P<0.001) on NFκB activity. Post-hoc comparisons show statistically significant differences between the control conditions and the treatments with kaempferol, quercetin, luteolin, apigenin and acacetin (P<0.001) on NFκB activity. Graphs representing the correlation between the inhibition of NFκB activity and Aβ1-40 (B) and Aβ1-42 (C) production observed for the different flavonoids tested. A strong correlation between Aβ values and the amount of NFκB inhibition induced by flavonoids was observed (Pearson correlation coefficient=0.872; P<0.001).
Figure 7.

Effect of daidzein, apigenin, luteolin and quercetin on BACE-1 transcription in human neuronal SH-SY5Y cells. BACE-1 transcription was quantified by RT-PCR in SHSY cells following treatment with flavonoids for 30 minutes in presence and abscence of 20 ng/ml of TNFα. A strong induction of BACE-1 mRNA was observed in SH-SY5Y cells challenged with the NFκB inducer TNFα and an approximate 10 fold reduction in BACE-1 transcription was observed following treatments with apigenin, luteolin and quercetin showing that these flavonoids can mitigate BACE-1 expression induced by NFκB stimulation. No inhibition of TNFα induced BACE-1 transcription was observed with daidzein. ANOVA reveals a statistically significant main effect of TNFα (P<0.001), apigenin (P<0.001), luteolin (P<0.001) and quercetin (P<0.001) on BACE-1 mRNA level but no significant main effect of daidzein (P=0.323). Post-hoc comparisons show significant differences between TNFα and TNFα+apigenin (P<0.001), TNFα+luteolin (P<0.001) or TNFα+quercetin treatments (P<0.001) but no statistically significant difference between TNFα and TNFα+daidzein (P=0.805) treatments on BACE-1 mRNA level.
Discussion
Several studies have revealed that natural flavonoids can reduce Aβ neurotoxicity [19–21] possibly via an antioxidative mechanism and inhibition of Aβ oligomerization [22]. In addition, several flavonoids have been shown to lower brain Aβ accumulation in transgenic mouse models of AD [23, 24]. Flavonoids have been proposed to act as BACE-1 inhibitors, the rate limiting enzyme responsible for the production of Aβ peptides [12, 25]. In our study, some of the Aβ lowering flavonoids that we tested decreased sAPPβ production suggesting an inhibition of the β-cleavage of APP. However, these flavonoids only weakly inhibited BACE-1 activity in a cell free assay and we found only a weak correlation between their effect on BACE-1 activity and their ability to lower Aβ in whole cells suggesting their Aβ lowering properties are mediated principally via other mechanisms. We also investigated the binding of flavonoids to the BACE-1 catalytic site employing docking simulations. Careful mining of the Protein Data Bank (PDB) for BACE-1 revealed that there are 144 entries including apo and complexed structures with several different inhibitors. When we analyzed these structures using a molecular visualization tool, we observed that the active site of BACE-1 possesses a high conformational flexibility upon ligand binding. The active site consists of aspartyl residues (32 and 228) encompassed by two highly flexible loop regions (Loop1: 9-13 KSGQG and Loop2: 71-74 YTQG) that undergo both side chain and backbone rearrangements. For example, in the structure 2B8L the SER10 and its corresponding residue SER71 in the structure 2QMFA, the distance between the backbone atom C-± is 6.83 Ǻ whereas the distance between the side chain atom O-γ is 7.68 Ǻ. The distance between 2B8L:72THR and 3KMXA:133THR for CG2 atom is 5.74 Ǻ and between the backbone C-± atoms is 4.38 Ǻ. The active site pocket is plastic and different structures can be aligned to understand conformational changes due to induced fitting of the ligand. BACE-1 possesses a large active site pocket typical of aspartyl proteases and further adopts conformational changes upon ligand binding that may include both side-chain rearrangement and backbone movement of residues in the active site [26–28]. Due to this fact, docking studies do not adequately reproduce ligand binding and may possibly over or under rank a ligand upon docking resulting in false positives. In our study, we used two different structures for BACE-1 and performed multiple docking studies. We observed that as a function of the different structures selected for BACE-1, the docking score/rank for the flavanoids was different. As a matter of fact, we found no correlation between the observed IC50 for BACE-1 inhibition in the cell-free assay and the Glide XP Score in the docking experiments. Energetic calculations from our BACE-1 docking studies also reveal that the flavonoids only weakly bind the BACE-1 catalytic site; in agreement with the weak inhibition of BACE-1 activity we observed with flavonoids in a BACE-1 cell free assay. Docking studies of BACE-1 do not adequately reproduce the native structure when ligands are cross-docked and hence further studies that combine docking with Monte Carlo/Molecular Dynamics methods may be required [29]. Our data suggest the Aβ lowering flavonoids we tested are only poor BACE-1 inhibitor and must therefore regulate Aβ production in whole cells by other mechanisms. This conclusion is different from past studies [12] in which docking results on a single BACE-1 structure were presented as evidence to support the hypothesis that flavonoids directly inhibit BACE-1 activity explaining their Aβ lowering properties in whole cells. Using docking experiments with two different crystal structures for BACE-1, we demonstrate that the docking score of flavonoids is different for the two BACE-1 structures tested and do not correlate with the inhibition of BACE-1 activity observed with flavonoids in a cell free assay . It is therefore inaccurate to classify active from inactive flavonoids as BACE-1 inhibitors by simply using docking simulations alone. The Aβ lowering activity of flavonoids cannot be explained solely by their effect on BACE-1 activity. We found a lack of correlation between the amount of BACE-1 inhibition observed and the ability of the compounds to lower Aβ production in whole cells. In addition, their IC50 for inhibiting BACE-1 activity in a cell free assay was higher than their IC50 for inhibiting Aβ in whole cells suggesting that additional mechanisms besides direct BACE-1 inhibition also contribute to their Aβ lowering properties. We assessed the impact of flavonoids on NFκB activation since we have previously identified that compounds inhibiting NFκB activation can prevent the β-cleavage of APP and Aβ production [18]. We observed a strong correlation between the NFκB inhibition potency of the flavonoids and their ability to inhibit Aβ production suggesting that the preponderant mechanism responsible for the Aβ lowering properties of the flavonoids is related to their effect on NFκB activation. Interestingly, many flavonoids are known to inhibit tyrosine kinases and hence may regulate NFκB activity via their inhibitory action on multiple tyrosine kinases upstream of NFκB. NFκB has been shown previously to regulate the production of Aβ by regulating the β-cleavage of APP [18]. Others studies have shown that NFκB inhibition can directly regulate BACE-1 expression level [14, 30–32]. We effectively observed that apigenin, luteolin and quercetin inhibit BACE-1 transcription in human neuronal SHSY cells.
Conclusion
Altogether our data suggest a multimodal mechanism of action of flavonoids towards Aβ production as the direct inhibition of BACE-1 activity by flavonoids appears to only marginally account for their Aβ lowering properties. We suggest that the Aβ lowering properties of flavonoids are mainly mediated via their effect on NFκB signaling which in turn affects the regulation of BACE-1 expression.
Authors' contributions
DP conceived of the study, developed the methodology for studying APP processing and NFκB activity, performed BACE-1 activity measurements, the statistical analyzes and drafted the manuscript. VM performed the molecular docking simulations. GA realized the RTQ-PCR experiments. DB and NP performed the western-blots and ELISAs. CB contributed to the NFκB luciferase activity measurements and helped in writing the manuscript. MM critically evaluated the manuscript. All authors read and approved the final manuscript.
Competing interest
The authors declare that they have no competing interests.
Supplementary material
Acknowledgments
We would like to thank Diane and Robert Roskamp for their generosity in helping to make this work possible. We thank Dr. Michael Wolfe (Harvard Medical School, Boston, Massachusetts, USA) for providing the 7W CHO APP overexpressing cells.
Footnotes
Citation:Paris et al, Bioinformation 6(6): 229-236 (2011)
References
- 1.DJ Selkoe. Behav Brain Res. 2008;192:106. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.R Vassar, et al. Science. 1999;286:735. [Google Scholar]
- 3.S Sinha, et al. Nature. 1999;402:537. [Google Scholar]
- 4.R Yan, et al. Nature. 1999;402:533. [Google Scholar]
- 5.SL Cole, R Vassar. J Biol Chem. 2008;283:29621. doi: 10.1074/jbc.R800015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.T Iwatsubo. Rinsho Shinkeigaku. 2004;44:768. [PubMed] [Google Scholar]
- 7.H Cai, et al. Nat Neurosci. 2001;4:233. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 8.Q Dai, et al. Am J Med. 2006;119:751. doi: 10.1016/j.amjmed.2006.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D Commenges, et al. Eur J Epidemiol. 2000;16:357. doi: 10.1023/a:1007614613771. [DOI] [PubMed] [Google Scholar]
- 10.O Napryeyenko, et al. Arzneimittelforschung. 2007;57:4. [Google Scholar]
- 11.M Mazza, et al. Eur J Neurol. 2006;13:981. [Google Scholar]
- 12.Y Shimmyo, et al. Biochim Biophys Acta. 2008;1780:819. doi: 10.1016/j.bbagen.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 13.EH Koo, SL Squazzo. J Biol Chem. 1994;269:17386. [PubMed] [Google Scholar]
- 14.D Paris, et al. J Neuroinflammation. 2010;7:17. doi: 10.1186/1742-2094-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.G Ait-Ghezala, et al. Brain Res Mol Brain Res. 2005;140:73. doi: 10.1016/j.molbrainres.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 16.A Garcia-Lafuente, et al. Inflamm Res. 2009;58:537. doi: 10.1007/s00011-009-0037-3. [DOI] [PubMed] [Google Scholar]
- 17.G Ait-Ghezala, et al. Eur J Neurosci. 2007;25:1685. doi: 10.1111/j.1460-9568.2007.05424.x. [DOI] [PubMed] [Google Scholar]
- 18.D Paris, et al. Neurosci Lett. 2007;415:11. [Google Scholar]
- 19.S Bastianetto, et al. Eur J Neurosci. 2006;23:55. doi: 10.1111/j.1460-9568.2005.04532.x. [DOI] [PubMed] [Google Scholar]
- 20.T Iuvone, et al. J Pharmacol Exp Ther. 2006;317:1143. doi: 10.1124/jpet.105.099317. [DOI] [PubMed] [Google Scholar]
- 21.JK Kim, et al. Biosci Biotechnol Biochem. 2010;74:397. doi: 10.1271/bbb.90585. [DOI] [PubMed] [Google Scholar]
- 22.K Ono, et al. J Neurochem. 2003;87:172. doi: 10.1046/j.1471-4159.2003.01976.x. [DOI] [PubMed] [Google Scholar]
- 23.H Onozuka, et al. J Pharmacol Exp Ther. 2008;326:739. doi: 10.1124/jpet.108.140293. [DOI] [PubMed] [Google Scholar]
- 24.K Rezai-Zadeh, et al. J Cell Mol Med. 2009;13:574. doi: 10.1111/j.1582-4934.2008.00344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.YH Choi, et al. Planta Med. 2008;74:1405. doi: 10.1055/s-2008-1081301. [DOI] [PubMed] [Google Scholar]
- 26.M Citron, et al. J Neurosci Res. 2002;70:373. doi: 10.1002/jnr.10393. [DOI] [PubMed] [Google Scholar]
- 27.S Patel, et al. J Mol Biol. 2004;343:407. doi: 10.1016/j.jmb.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 28.N Moitessier, et al. J Med Chem. 2006;49:5885. doi: 10.1021/jm050138y. [DOI] [PubMed] [Google Scholar]
- 29.MA Eriksson, et al. J Med Chem. 1999;42:868. doi: 10.1021/jm980277y. [DOI] [PubMed] [Google Scholar]
- 30.S Wang, J Jia. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:159. doi: 10.1002/ajmg.b.30968. [DOI] [PubMed] [Google Scholar]
- 31.K Sambamurti, et al. FASEB J. 2004;18:1034. [Google Scholar]
- 32.V Buggia-Prevot, et al. J Biol Chem. 2008;283:10037. doi: 10.1074/jbc.M706579200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.